
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystallographic interface control of the plasmonic photocatalyst consisting of gold nanoparticles and titanium(IV) oxide†
Shin-ichi
Naya
a,
Atsunobu
Akita
b,
Yoko
Morita
b,
Musashi
Fujishima
bc and
Hiroaki
Tada
 *bc
*bc
aEnvironmental Research Laboratory, Kindai University, 3-4-1, Kowakae, Higashi-Osaka 577-8502, Osaka, Japan
bGraduate School of Science and Engineering, Kindai University, 3-4-1, Kowakae, Higashi-Osaka 577-8502, Osaka, Japan. E-mail: h-tada@apch.kindai.ac.jp
cDepartment of Applied Chemistry, Faculty of Science and Engineering, Kindai University, 3-4-1, Kowakae, Higashi-Osaka 577-8502, Osaka, Japan
First published on 14th October 2022
Abstract
A big question in the field of plasmonic photocatalysis is why a typical photocatalyst consisting of gold nanoparticles and rutile titanium(IV) oxide (Au/R-TiO2) usually exhibits activity much higher than that of Au/anatase TiO2 (Au/A-TiO2) under visible-light irradiation. Shedding light on the origin should present important guidelines for the material design of plasmonic photocatalysts. Au nanoparticles (NPs) were loaded on ordinary irregular-shaped TiO2 particles by the conventional deposition precipitation method. Transmission electron microscopy analyses for the Au/TiO2 particles ascertain that faceting of Au NPs is induced on R-TiO2 by using a domain-matching epitaxial junction with the orientation of (111)Au//(110)R-TiO2, whereas non-faceted hemispherical Au NPs are exclusively formed on A-TiO2. The faceting probability of Au NPs (Pf) on R-TiO2 increases with decreasing Au particle size (dAu) to reach 14% at dAu = 3.6 nm. A clear positive correlation between the photocatalytic activity and Pf in several test reactions indicates that the heteroepitaxial junction-induced faceting of Au NPs is the principal factor for governing the plasmonic photocatalytic activity of Au/TiO2. In light of this finding, R-TiO2 nanorods with a high percentage (95%) of {110} facets were hydrothermally synthesized and used for the support of Au NPs. Consequently, the Pf value increases to as much as 94% to enhance the photocatalytic activity with respect to that of Au/R-TiO2 with Pf = 14% by factors of 2.2–4.4 depending on the type of reaction.
Introduction
Photocatalysts are the key materials for the solar-to-chemical transformations, and their development is pivotal for dealing with increasing global energy demands and environmental issues. While research on semiconductor photocatalysts has been ongoing for many years, gold nanoparticle (NP) based-plasmonic photocatalysts represented by Au/TiO2 have recently attracted much attention as a new type of visible-light responsive photocatalysts.1–6 According to the action mechanism, plasmonic photocatalysts consisting of Au NPs and semiconductors can be classified into the hot-electron transfer (HET) type and local electric field enhancement (LEFE) or plasmon-resonance energy transfer type.7 The former possesses an outstanding feature that photons with the energy below the band gap of the semiconductor can be utilized as the energy source for chemical reactions,8,9 whereas the available photons are basically limited to the ones with the energy above the band gap in the latter.10 HET-type plasmonic photocatalysis is initiated by the excitation of the localized surface plasmon resonance (LSPR) of Au NPs and/or the interband transition (light harvesting or hot-carrier generation). Part of the hot electrons generated in the Au NP are injected into the conduction band (CB) of the semiconductor (hot electron injection).11 The CB-electrons are kept away from the holes in the Au NPs by using the Schottky junction between Au NPs and the semiconductor (charge separation).12 Consequently, the electrons escaping from recombination are used for reduction on the semiconductor surface, while the holes in the Au NPs cause oxidation on the Au surface or the interface with the semiconductor (surface chemical reactions). So far, much effort has been devoted to increase the activity of HET-type plasmonic photocatalysts by separately enhancing each physical and chemical process through the control of the size and shape of Au NPs,13 and the assembled structure.14 In contrast, the effect of the semiconductor on the activity is not fully understood, and research on the Au NP-semiconductor interface is still in its infancy.15 In this study, we focus on an unresolved issue that Au/rutile TiO2 (Au/R-TiO2) exhibits plasmonic photocatalytic activity much higher than that of Au/anatase TiO2 (Au/A-TiO2) for various reactions (Table S1†) in contrast to the UV-light activity of R-TiO2, which is lower than that of A-TiO2.16 Among the reactions, the oxygen evolution reaction (OER) has a large activation energy and is usually the bottle neck in the artificial photosynthetic reactions.17 Recently, Au/R-TiO2 has also been reported to show much higher plasmonic photocatalytic activity than Au/A-TiO2.18 Density functional theory calculations for a Au8/TiO2 model suggested that the rate-determining step of the plasmonic OER is the formation of *OOH, and the activation energy for the process in the Au/R-TiO2 system is lower than that in the Au/A-TiO2 system, where the symbol * expresses the adatom. However, the general superiority of Au/R-TiO2 to Au/A-TiO2 in the HET-induced plasmonic reactions suggests that the TiO2 crystal form mainly affects the physical processes involving light harvesting, hot electron injection, and charge separation rather than the chemical reaction process. The understanding of this origin would provide important and versatile guidelines for material design because the physical processes are common to the plasmonic photocatalytic reactions.Experimental
Materials
Hydrogen tetrachloroaurate(III) tetrahydrate (HAuCl4·4H2O > 99%), urea (CH4N2O > 99.0%), benzylamine (C7H9N >98.0%), and 2-naphthol (C10H8O > 99.0%) were purchased from Kanto Chemical Co. Cinnamyl alcohol (C9H10O > 97.0%) was purchased from Tokyo Chemical Industry Co. Titanium bis(ammonium lactate) dihydroxide ([CH3CH(O−)CO2NH4]2Ti(OH)2, 50 wt% in H2O) solution was purchased from Sigma-Aldrich Co. Irregular-shaped anatase TiO2 particles (mean particle size = 150 nm, specific surface area = 8.1 m2 g−1, A-100, Ishihara Sangyo) and rutile-TiO2 particles (mean particle size = 80 nm, specific surface area = 18 m2 g−1, MT-700B, TAYCA) were used as the TiO2 supports. All chemicals were used as received without further purification.Rutile TiO2 nanorod synthesis
Rutile TiO2 NRs with {110} side walls were synthesized by the hydrothermal method.19 Titanium bis(ammonium lactate) dihydroxide (6 mL, 50 wt% in H2O) was added to H2O (24 mL), and poured into a Teflon-lined stainless-steel autoclave (capacity 50 mL). The solution was heated at 220 °C for 72 h, and cooled in air at ambient temperature. The resulting particles were washed with distilled water and acetone, and dried in vacuo to obtain rutile TiO2 NRs.Au nanoparticle loading
Au NPs were loaded on TiO2 by the deposition–precipitation method using urea as the neutralizer.20 The TiO2 particles were pre-heated at 923 K for 4 h to avoid the influence of the change in the properties of TiO2 with heating at various temperatures during the following deposition–precipitation process. TiO2 nanoparticles (1 g) were dispersed in an aqueous solution (50 mL) of HAuCl4 (4.86 mM) and urea (1.46 g), and stirred at 353 K for 18 h. The resulting particles were washed with hot-distilled water (323 K) 10 times and acetone. After drying in vacuo, the particles were calcined by varying the heating temperature (673 K ≤ Tc ≤ 873 K) and time (1 h ≤ tc ≤ 24 h) to obtain Au/TiO2.Catalyst characterization
For quantifying the loading amount of Au, Au NPs loaded on TiO2 were dissolved by using aqua regia, and the concentration was determined by inductively coupled plasma spectroscopy. The samples for TEM and HR-TEM observation were prepared by dropping a suspension of the samples in ethanol onto a copper grid with a carbon support film (grid-pitch 150 μm, Okenshoji Co., Ltd, #10-1006). The measurements were performed by using a JEOL JEM-2100F at an applied voltage of 200 kV. The Au particle size (dAu) was determined from the image analysis of ∼200 Au particles observed in TEM images. Diffuse reflectance UV-vis-NIR absorption spectra were measured by using a UV-2600 spectrometer (Shimadzu) with an integrating sphere unit (Shimadzu, ISR-2600Plus). BaSO4 was used as a reference for the reflectance (R∞). The Kubelka–Munk function [F(R∞) = (1 − R∞)2/2R∞] was used for expressing the relative absorption coefficient. The measurement was carried out by using sample powder without dilution. X-ray photoelectron spectra (XPS) were collected by using a Kratos Axis Nova X-ray photoelectron spectrometer at 15 kV and 10 mA using Al Kα as the X-ray source. For the energy reference, the peak of C 1s (284.6 eV) was used.Cinnamyl alcohol oxidation
Au/TiO2 (5 mg) was dispersed in a solution (20 mL, H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile = 99:1 v/v) of cinnamyl alcohol (0.5 mM) in a test tube placed in a double jacket type reaction cell (80 mm in length and 15 mm in diameter). Acetonitrile was used for the complete dissolution of cinnamyl alcohol. After stirring for 0.5 h at 298 K in the dark, visible light was irradiated by means of a 300 W Xe lamp (HX-500, Wacom) with an optical filter Y-51 (λex > 490 nm, AGC TECHNO GLASS) with stirring for 600 rpm while circulating thermostated water (298 K) through an outer jacket around the cell during the reaction. The light intensity integrated from 420 to 485 nm (I420–485) was adjusted to 3.3 mW cm−2. The cinnamaldehyde yield and selectivity were determined by UV-vis spectroscopy (Shimadzu, UV-1800) and high-performance liquid chromatography (Shimadzu, LC-6 AD, SPD-6 A, C-R8A) [conditions: Shim-pack CLC-ODS (4.6 mm × 150 mm); MeOH–H2O (7:3 v/v); flow rate = 1.0 mL min−1; λ = 300 nm]. To confirm the reproducibility of the data, the photocatalytic activity was evaluated by repeating the reaction more than twice for each sample.
:acetonitrile = 99:1 v/v) of cinnamyl alcohol (0.5 mM) in a test tube placed in a double jacket type reaction cell (80 mm in length and 15 mm in diameter). Acetonitrile was used for the complete dissolution of cinnamyl alcohol. After stirring for 0.5 h at 298 K in the dark, visible light was irradiated by means of a 300 W Xe lamp (HX-500, Wacom) with an optical filter Y-51 (λex > 490 nm, AGC TECHNO GLASS) with stirring for 600 rpm while circulating thermostated water (298 K) through an outer jacket around the cell during the reaction. The light intensity integrated from 420 to 485 nm (I420–485) was adjusted to 3.3 mW cm−2. The cinnamaldehyde yield and selectivity were determined by UV-vis spectroscopy (Shimadzu, UV-1800) and high-performance liquid chromatography (Shimadzu, LC-6 AD, SPD-6 A, C-R8A) [conditions: Shim-pack CLC-ODS (4.6 mm × 150 mm); MeOH–H2O (7:3 v/v); flow rate = 1.0 mL min−1; λ = 300 nm]. To confirm the reproducibility of the data, the photocatalytic activity was evaluated by repeating the reaction more than twice for each sample.
Benzylamine oxidation
Au/TiO2 (5 mg) Au/TiO2 (5 mg) was dispersed in an acetonitrile solution (20 mL) of benzylamine (0.1 mM) in a test tube placed in a double jacket type reaction cell (80 mm in length and 15 mm in diameter). After stirring for 0.5 h at 298 K in the dark, visible light was irradiated by means of a 300 W Xe lamp (HX-500, Wacom) with an optical filter Y-45 (λex > 430 nm, AGC TECHNO GLASS) with stirring for 600 rpm while circulating thermostated water (298 K) through an outer jacket around the cell during the reaction. The light intensity (I420–485) was adjusted to 6.0 mW cm−2. The benzaldehyde yield and selectivity were determined by UV-vis spectroscopy (Shimadzu, UV-1800) and high-performance liquid chromatography (Shimadzu, LC-6 AD, SPD-6 A, C-R8A) [conditions: Shim-pack CLC-ODS (4.6 mm × 150 mm); acetonitrile; flow rate = 0.5 mL min−1; λ = 280 nm]. To confirm the reproducibility of the data, the photocatalytic activity was evaluated by repeating the reaction more than twice for each sample.Decomposition of 2-naphthol
Au/TiO2 (5 mg) was dispersed in a solution (20 mL, H2O:acetonitrile = 99:1 v/v) of 2-naphthol (10 μM) in a test tube placed in a double jacket type reaction cell (80 mm in length and 15 mm in diameter). Acetonitrile was used for the complete dissolution of 2-naphthol. After stirring for 0.25 h at 298 K in the dark, visible light was irradiated by means of a 300 W Xe lamp (HX-500, Wacom) with an optical filter Y-45 (λex > 430 nm, AGC TECHNO GLASS) with stirring for 600 rpm while circulating thermostated water (298 K) through an outer jacket around the cell during the reaction. The light intensity (I420–485) was adjusted to 6.0 mW cm−2. The concentration of 2-naphthol was quantified by high-performance liquid chromatography (Shimadzu, LC-6 AD, SPD-6 A, C-R8A) [conditions: Shim-pack CLC-ODS (4.6 mm × 150 mm); MeOH–H2O (7:3 v/v); flow rate = 1.0 mL min−1; λ = 223 nm]. To confirm the reproducibility of the data, the photocatalytic activity was evaluated by repeating the reaction more than twice for each sample.
Oxygen evolution reaction
Au/TiO2 (10 mg) and La2O3 (20 mg) was dispersed in an aqueous solution (10 mL) of AgNO3 (10 mM) in a test tube placed in a double jacket type reaction cell (80 mm in length and 15 mm in diameter). La2O3 was used for maintaining a constant pH (pH 7.45).21 After removing the dissolved O2 by Ar bubbling for 0.5 h, visible light was irradiated by using a xenon lamp with an optical filter Y-49 (λex > 470 nm, AGC TECHNO GLASS) with stirring for 600 rpm while circulating thermostated water (298 K) through an outer jacket around the cell during the reaction. The light intensity integrated from 470–780 nm was adjusted to 30 mW cm−2. The amount of O2 evolved was measured by gas chromatography (Shimadzu. GC-8APT) with a thermal conductivity detector [conditions: a molecular sieve 5A column (ϕ 3.0 mm × 1 m), carrier gas = Ar]. The injection and column temperatures were set at 60 °C. To confirm the reproducibility of the data, the photocatalytic activity was evaluated by repeating the reaction more than twice for each sample.Finite difference time domain (FDTD) simulations
Au/anatase TiO2 and Au/rutile TiO2 were modelled with an Au hemisphere (HS Au) and an Au truncated octahedron (t-Oh Au) loaded on a TiO2 slab (650 × 650 × 50 nm3), respectively. Particle sizes obtained from the TEM observations were used as the size of the modelled Au particles. The optical response of the models was reproduced on the basis of complex refractive indices for each constituent from the literature.22 Light scattering behavior of the models was simulated using a total-field scattered-field (TFSF) source. An x-polarized plane wave with a wavelength from 300 to 1200 nm (f = 250 – 1000 THz) was irradiated on the top of the Au particle from the z-axis direction perpendicular to the TiO2 slab. Local electric field images were obtained by using E-filed monitors located on the xy plane (z = 0) and the xz plane (y = 0). Considering the balance between the calculation cost and accuracy, mesh refinement and a perfectly matched layer (PML) with symmetric and/or anti-symmetric boundary conditions were imposed on the models. All of the FDTD simulations were carried out with an Ansys Lumerical FDTD simulation program package.Results and discussion
Preparation and characterization of Au/TiO2
Au NPs were loaded on irregular-shaped commercial A-TiO2 particles (mean particle size = 150 nm and specific surface area = 8.1 m2 g−1) and R-TiO2 particles (mean particle size = 80 nm and specific surface area = 18 m2 g−1) by the deposition precipitation method.20 Prior to the deposition of Au NPs, cleanness of the TiO2 surface was confirmed by X-ray photoelectron (XP) spectroscopy (Fig. S1†). The loading amount of Au (xAu) was quantified to be 4.22 ± 0.03 mass% for A-TiO2 and 4.25 ± 0.07 mass% for R-TiO2 by inductively coupled plasma spectroscopy (Table S2†). In the high resolution-transmission electron microscopy (HR-TEM) images of the samples prepared at heating temperature (Tc) = 673 K and heating time (tc) = 1 h (Fig. 1a, b and S2†), the d-spacings of the supports are in agreement with the values of A-TiO2(101) (3.53 Å) and R-TiO2(110) (3.24 Å), respectively. Also, the d-spacing of the deposits in each sample is equal to the value of Au(111) (2.35 Å). Au NPs with mean sizes (dAu) of 3.5 nm and 3.6 nm are highly dispersed on A-TiO2 and R-TiO2 particles, respectively. Intriguingly, faceted Au NPs with sharp edges and corners are observed on R-TiO2 particles, while every Au NP on A-TiO2 is hemisphere-like (HS).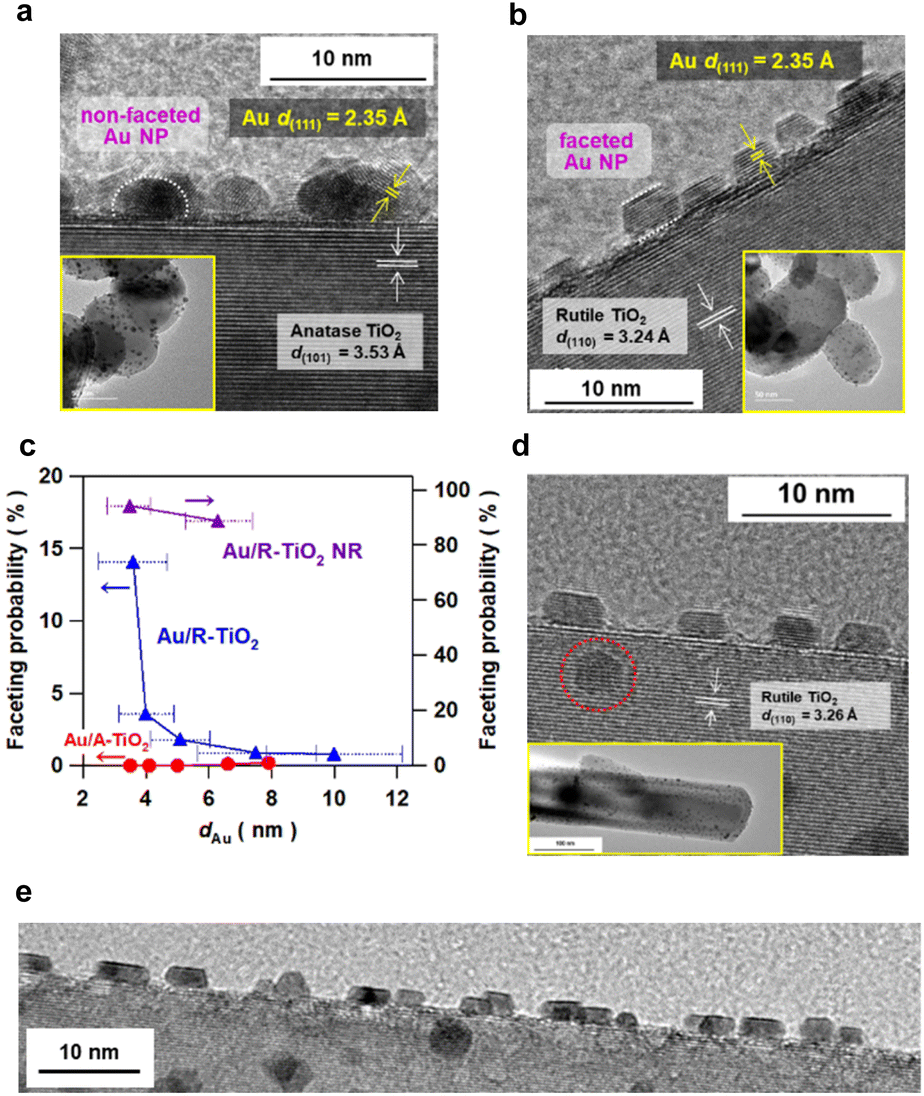 | ||
| Fig. 1 HR-TEM images of Au/anatase TiO2 (Au/A-TiO2) (a) and Au/rutile TiO2 (Au/R-TiO2) (b) prepared at heating temperature Tc = 673 K and time tc = 1 h. The insets show the TEM images of the samples. (c) Faceting probability of Au NPs on A-TiO2 (red), R-TiO2 (blue), and rutile TiO2 nanorod (R-TiO2 NR, violet) particles as a function of dAu. The length of the bars for dAu expresses the standard deviation of the Au particle size for each sample. (d) HR-TEM image of Au/R-TiO2–NR prepared at Tc = 673 K and tc = 1 h. The inset is the TEM image of the sample. (e) HR-TEM image of Au/R-TiO2–NR prepared at Tc = 673 K and tc = 1 h in a wide field of view. | ||
The size of Au NPs was controlled with the loading amount maintained constant by changing Tc and tc during the deposition precipitation process (Fig. S3–S5†).23 The dAu varies from 3.5 nm to 7.9 nm in the Au/A-TiO2 system, and from 3.6 nm to 10.0 nm in the Au/R-TiO2 system with increasing heating temperature (673 K ≤ Tc ≤ 873 K) and time (1 h ≤ tc ≤ 24 h). Faceting probability (Pf) of Au/TiO2 was defined as the ratio of the number of faceted Au NPs to the total number of Au NPs. In this study, hemispherical Au NPs (Fig. 1a, white dotted semicircle) and Au NPs with a sharp truncated top plane parallel to the interface with TiO2 (Fig. 1b, white dotted parallel lines) are assigned to the non-faceted and faceted particles, respectively. The basis for these criteria is described in the next section. To minimize the arbitrariness, the Pf was calculated by counting the numbers of faceted and non-faceted Au NPs on the edge of TiO2 particles in the TEM images over 103 particles (Fig. S6, Table S2†). While the Pf is negligibly small in the Au/A-TiO2 system (<0.2%) regardless of dAu, the Pf increases with a decrease in dAu from 0.8% at dAu = 10.0 nm to 14% at dAu = 3.6 nm in the Au/R-TiO2 system (Fig. 1c). Although NPs can appear non-faceted if imaged off-axis as TEM shows a projection on the NPs, the very small Pf for the Au/A-TiO2 system is not for this. Actually, not faceted but hemispherical Au NPs are usually observed in many TEM images of Au/A-TiO2 reported so far.
Interface analysis of Au/TiO2
The interface between Au NPs and R-TiO2 was analyzed by HR-TEM. The HR-TEM image of a typical faceted Au NP on R-TiO2 (Fig. 2a) shows that an atomically commensurate junction is formed with an orientation of (111)Au//(110)R-TiO2. The two-dimensional rectangular lattices of Au(111) and R-TiO2(110) possess the unit dimensions of 4.995 Å × 2.884 Å and 6.496 Å × 2.959 Å, respectively (Fig. 2b). The quadruple and triple of the unit cells almost match with a mismatch of +2.5% and −2.5%, which strongly suggests the formation of a domain-matching epitaxial junction. A previous study by means of electron backscattered diffraction showed the same orientation for an evaporated Au film on the (110) surface of a R-TiO2 single crystal followed by annealing at 775 K.24 The most thermodynamically stable shape of Au NPs on the R-TiO2(110) surface was constructed by using the Wulff theorem using the values for the surface energies of 0.72 J m−2 for Au(111) (ref. 25) and 0.40 J m−2 for R-TiO2(110),26 and an interfacial energy of 0.86 J m−2 for Au(111) and R-TiO2(110).27 As shown in the side view and top view (Fig. 2c and d), the Au NP takes the most stable truncated octahedral shape (t-Oh Au NP), and the side view (Fig. 2c) is very similar to that observed in the HR-TEM image (Fig. 2a). Thus, the faceting of Au NPs on R-TiO2 can be induced by the crystallographic metal-support interaction between Au(111) and R-TiO2(110).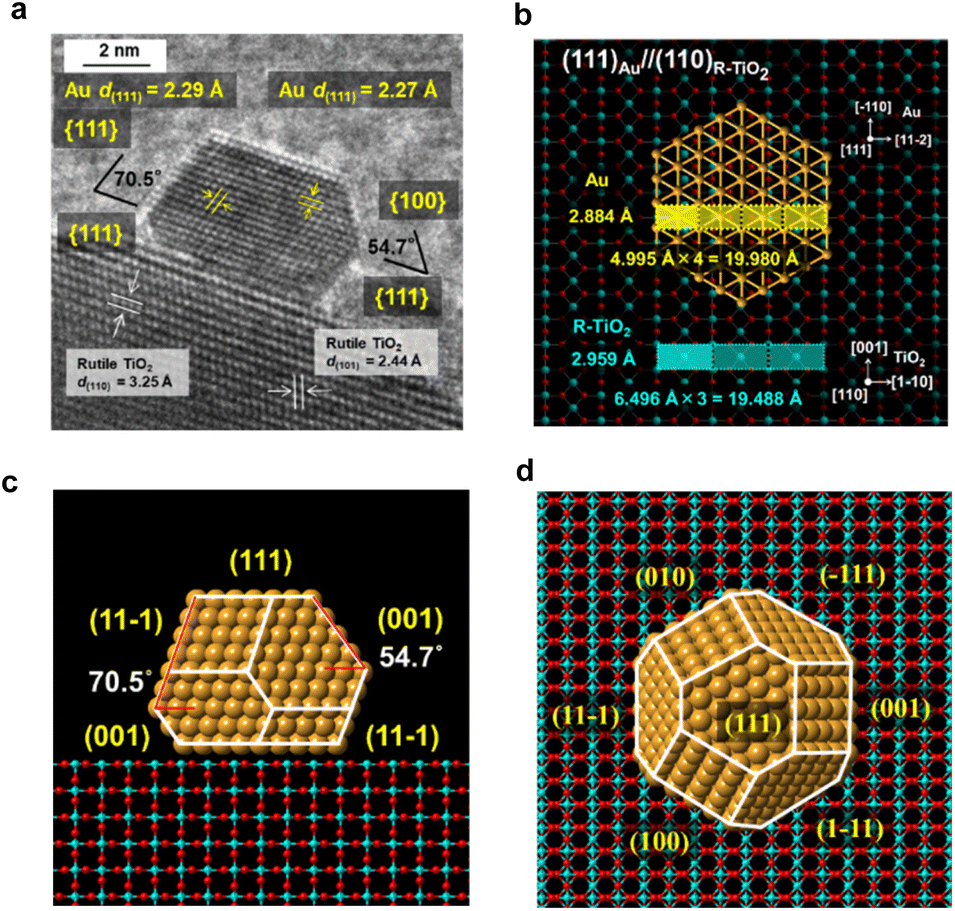 | ||
| Fig. 2 (a) HR-TEM image of Au/R-TiO2. The heteroepitaxial junction model with an orientation of (111)Au//(110)R-TiO2. (b) The domain-matching epitaxy model. The side view (c) and top view (d) of the most thermodynamically stable shape of Au NPs on rutile TiO2 constructed by using the Wulff theorem. | ||
Plasmonic photocatalytic activity of Au/TiO2
The Au NP-faceting effect of Au/A-TiO2 (red) and Au/R-TiO2 (blue) plasmonic photocatalysts on the activities was studied for the oxidation of cinnamyl alcohol to cinnamaldehyde (circles)28 and of benzylamine to benzaldehyde (triangles),29 and the oxidative degradation of 2-naphthol (squares)30 under visible-light irradiation. The action spectra previously confirmed that these reactions proceed via the LSPR excitation-driven HET mechanism.28–30 Each reaction apparently follows a zero-order rate equation (Fig. S7–S9†), and from the slope in the time course for the reaction, the reaction rate was calculated. Furthermore, the photocatalytic activity is expressed by using the relative reaction rate with respect to the value for Au/R-TiO2 with dAu = 3.6 nm and Pf = 14%. The dAu-dependence of the relative photocatalytic activity for each reaction (Fig. 3a) shows that Au/R-TiO2 (blue) possesses photocatalytic activity much higher than that of Au/A-TiO2 (red). The photocatalytic activity of Au/R-TiO2 increases with a decrease in dAu, while the dAu-dependence of the activity is weak in the Au/A-TiO2 system. Plots of the photocatalytic activity of Au/A-TiO2 (red) and Au/R-TiO2 (blue) against Pf show a clear positive correlation in every reaction system (Fig. 3b). In addition, visible-light irradiation (λex > 470 nm) of Au/TiO2 yields O2, the amount of which increases in proportion to irradiation time. The O2-generation rate for Au/R-TiO2 is 2.7 times higher as compared with the value for Au/A-TiO2 (Fig. S10†).18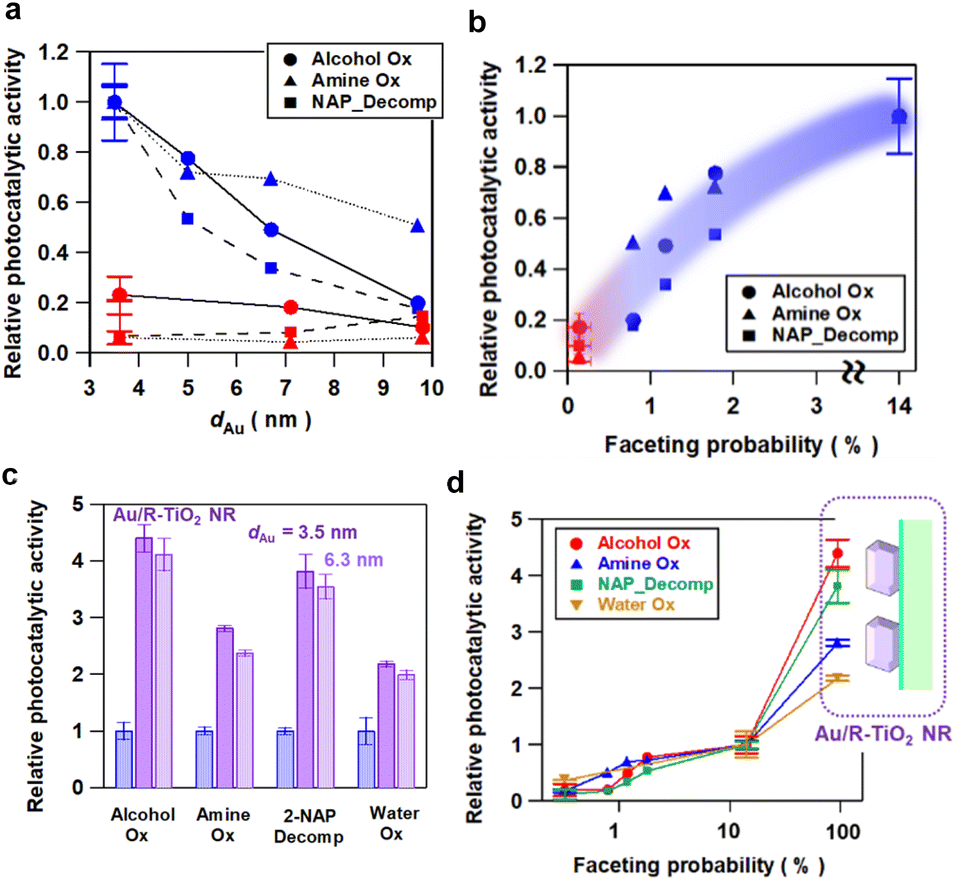 | ||
| Fig. 3 (a) The dAu dependence of the relative plasmonic photocatalytic activities of Au/A-TiO2 and Au/R-TiO2 for the oxidations of cinnamyl alcohol to cinnamaldehyde (Alcohol Ox) and benzylamine to benzaldehyde (Amine Ox), and the oxidative degradation of 2-naphthol (NAP_Decomp). The photocatalytic activity is normalized by using the value of Au/R-TiO2 with a Pf of 14.1% or dAu = 3.6 nm. (b) Relation between the photocatalytic activity ratio of Au/TiO2 and the Pf value. (c) Comparison between the photocatalytic activity of Au/R-TiO2 (dAu = 3.6 nm, xAu = 4.2 mass%, Pf = 14.1%), Au/R-TiO2 NR (dAu = 3.5 nm, xAu = 4.2 mass%, Pf = 94%) and Au/R-TiO2 NR (dAu = 6.3 nm, xAu = 4.2 mass%, Pf = 89%). (d) Relation between the photocatalytic activity of Au/TiO2 and the Pf value including the data for Au/R-TiO2 NRs with a Pf of 94%. The photocatalytic activity was evaluated by repeating the reaction more than twice for each catalyst under the same conditions. In all figures, the length in the ordinate shows the scattering of the data. | ||
As demonstrated by the interface analysis, the faceting of Au NPs on R-TiO2 can be induced by using the heteroepitaxial junction with the orientation of (111)Au//(110)R-TiO2. For the equilibrium form of TiO2 crystals, the percentage of the R-TiO2(110) facet was estimated to be 83% by using the Wulff construction.31 A domain matching epitaxial junction can also be formed between Au and A-TiO2 with an orientation of (111)Au//(001)A-TiO2.32 However, the percentage for the A-TiO2(001) facet predicted by the Wulff theorem is below 6%.33 These considerations qualitatively rationalize the large gap in the Pf between Au/A-TiO2 and Au/R-TiO2 (Fig. 1c). However, the Pf of Au/R-TiO2 is much smaller than the percentage of the R-TiO2(110) facet thermodynamically predicted. Actually, the area of the (110) domain exposed on the surface limits the Pf in the ordinary irregular shaped R-TiO2 particles, which would further be responsible for the decrease in the Pf with increasing dAu. Then, R-TiO2 nanorods (NR) with large-area (110) side walls (mean long axis = 0.84 ± 0.22 μm, mean short axis = 87 ± 32 nm, and specific surface area = 6.0 m2 g−1) were synthesized by a hydrothermal method (Fig. S11†) and used as the support of Au NPs (Au/R-TiO2 NRs). Surprisingly, the Pf at dAu = 3.5 nm on the TiO2 NRs increases to 94% close to the percentage of the {110} facet area to the total surface area for a rectangular with the R-TiO2 NR dimension (95%) (Fig. 1c and Table S2†). The top view of most Au NPs (dotted red circles in Fig. 1d and S12†) has a hexagonal shape that closely resembles that of t-Oh Au NP (Fig. 2d). Furthermore, the photocatalytic activity of Au/R-TiO2 NR for the reactions was evaluated under the same conditions. For every reaction, Au/R-TiO2 NRs exhibit photocatalytic activity higher than even that of Au/R-TiO2 with a Pf of 14% by a factor of 2.2–4.4, depending on the type of reaction (Fig. 3cand d). Also, in the Au/R-TiO2 NR system, the Pf value only decreases to 89% when the dAu increases from 3.5 nm to 6.3 nm (Fig. 1c). The photocatalytic activity of Au/R-TiO2 NRs with dAu = 6.3 nm and Pf = 89% for each reaction is slightly lower than that of Au/R-TiO2 NRs with dAu = 3.5 nm and Pf = 94% (Fig. 3c). TEM observation for Au/R-TiO2 NRs after each reaction confirmed that the structure is almost maintained (Fig. S13†).
The results of the activities of the Au/TiO2 plasmonic photocatalysts can be summarized as follows. The Au loading amount of all Au/TiO2 samples used in this study is controlled to be almost constant (∼4.2 mass%) (Table S2†). In the Au/A-TiO2 system, the Pf is below 0.2% regardless of the Au particle size (Fig. 1c), and the photocatalytic activity remains low (Fig. 3a). In the Au/R-TiO2 system, the photocatalytic activity increases with an increase in the Pf value (Fig. 3band d). Furthermore, Au/R-TiO2 NRs with dAu = 3.5 nm and Pf = 94% shows much higher photocatalytic activity than Au/R-TiO2 with dAu = 3.6 nm and Pf = 14% (Fig. 3d), while the photocatalytic activity of Au/R-TiO2 NRs with dAu = 3.5 nm and Pf = 94% is comparable with that of Au/R-TiO2 NRs with dAu = 6.3 nm and Pf = 89% (Fig. 3c). Au/R-TiO2 NRs were confirmed to be stable during each photocatalytic reaction. In the Au/TiO2 system, a positive correlation is recognized between the photocatalytic activity and the Pf value although the degree of the dependence depends on the type of reaction (Fig. 3d). Evidently, the photocatalytic activity of Au/TiO2 can be unified to the Pf value regardless of the TiO2 crystal form and Au particle size.
Physical properties of Au/TiO2
XP spectra were measured for Au/TiO2 particles, TiO2 particles and a Au film-coated glass plate for comparison (Fig. S14 and Table S3†). In the XPS spectra of the Au film/glass, two signals due to the emission from the 4f7/2 and 4f5/2 orbitals are observed at the binding energy (EB) = 84.0 eV and 87.7 eV, respectively, close to the literature values of 83.9 eV and 87.6 eV for bulk Au.34 The EB values of the Au 4f7/2 and 4f5/2 orbitals for Au/A-TiO2 (83.2 ± 0.2 eV and 86.9 ± 0.2 eV) and Au/R-TiO2 (83.2 ± 0.1 eV and 86.9 ± 0.2 eV) are significantly smaller than the values for the Au film-coated glass (Fig. S14a, b and Table S3†). On the other hand, Au/A-TiO2 and Au/R-TiO2 have the Ti 2p3/2-EB values of 458.6 ± 0.1 eV and 458.5 ± 0.1 eV, respectively, which are slightly larger than the value of 458.3 eV for unmodified R-TiO2 (Figure S14c, d and Table S3†). These results suggest the formation of a Schottky junction in the Au/A-TiO2 and Au/R-TiO2 systems with interfacial electron transfer from TiO2 to Au NPs.12Optical properties of Au/A-TiO2 and Au/R-TiO2 were studied by diffuse reflectance UV-visible-near infrared spectroscopy. In each Kubelka–Munk-transformed absorption spectrum of Au/A-TiO2 (Fig. 4a) and Au/R-TiO2 (Fig. 4b), the LSPR peak of Au NPs is observed in the 550–600 nm range with absorption due to the Au interband transition from the Au 5d band to 6sp at < ∼600 nm.35 The LSPR significantly intensifies with the peak redshifted with increasing the Au particle size from 551 nm at dAu = 3.5 nm to 572 nm at dAu = 7.9 nm in the Au/A-TiO2 system and from 583 nm at dAu = 3.6 nm to 589 nm at dAu = 10.0 nm in the Au/R-TiO2 system. Uniquely, in the Au/R-TiO2 NR system, the main LSPR peak redshifts to 620 nm with two shoulders at around 550 nm and 800 nm. Furthermore, the spatial distribution of the local electric field was calculated by the 3D-FDTD method for the models of Au(dAu = 3.5 nm)/A-TiO2 (Fig. 4c) and Au(dAu = 3.6 nm)/R-TiO2 (Fig. 4d) upon excitation by light with wavelength (λex) = 702 nm and 704 nm, respectively. In the former, the local electric field is fairly uniformly distributed along the perimeter of the Au NPs at the interface with A-TiO2 (Fig. 4e). In contrast, the LSPR excitation of the latter generates a highly concentrated electric field or “plasmonic hot spots” at the edges and corners of the faceted Au NPs near the interface with R-TiO2 (Fig. 4f). The enhancement factor (EF) defined by the square of the ratio of the local electric field intensity to the electric field intensity of incident light (|E|2/|E0|2) was calculated (Table S4†). The excited localized electromagnetic fields were monitored at the xz and xy planes of the models. Remarkably, the maximum EF in the t-Oh Au/R-TiO2 system reaches 7.4 × 104 at the xz plane and 2.1 × 105 at the xy plane, which are greater than the values of 2.5 × 103 at the xz plane and 3.6 × 103 at the xy plane in the HS-Au/A-TiO2 system by factors of 30 and 58, respectively. Clearly, the local electric field generated by the LSPR excitation is drastically enhanced by the heteroepitaxial junction-induced faceting of Au NPs on R-TiO2.
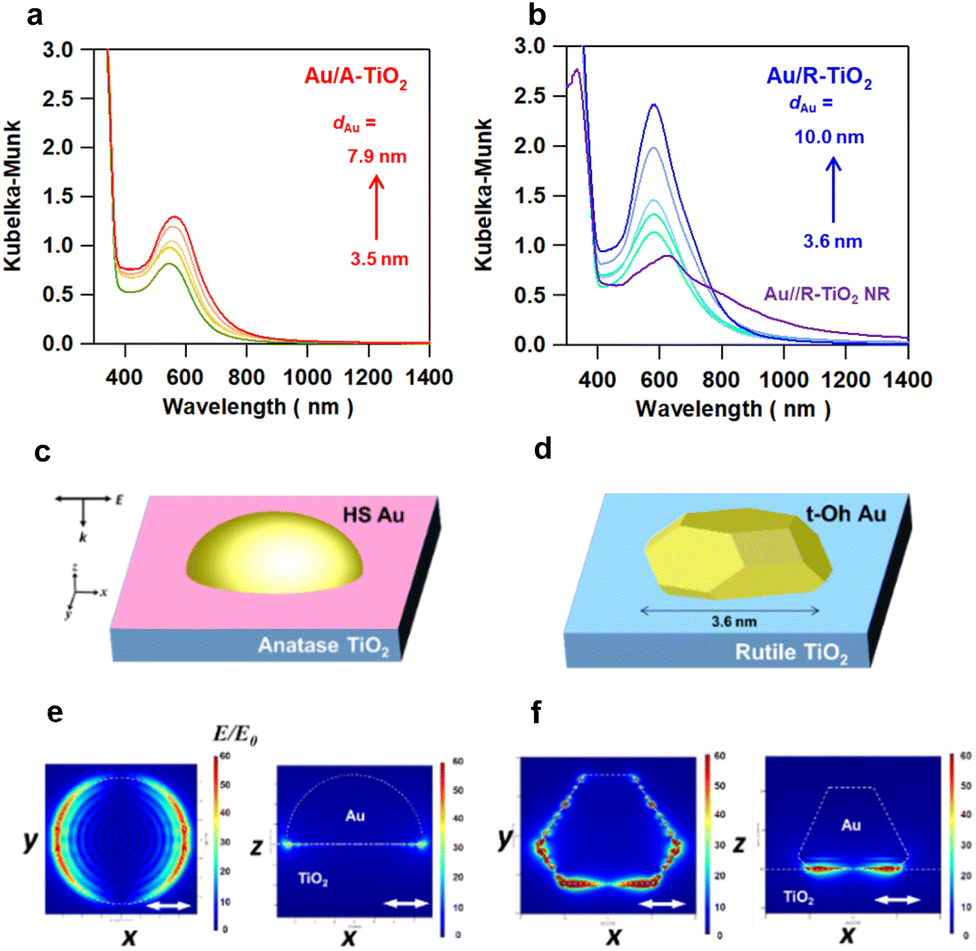 | ||
| Fig. 4 Kubelka–Munk-transformed absorption spectra of Au/A-TiO2 (a) and Au/R-TiO2 (b) with varying Au particle sizes (dAu) with Au(dAu = 6.3 nm)/R-TiO2 NRs. FDTD-simulation models for HS Au(dAu = 3.5 nm)/A-TiO2 (c) and t-Oh Au(dAu = 3.6 nm)/R-TiO2 (d). 3D-FDTD-calculated local electric field distribution for HS Au(dAu = 3.5 nm)/A-TiO2 (e) and t-Oh Au(dAu = 3.6 nm)/R-TiO2 (f) excited with x-polarized light incident from the z-axis direction (k//z, E//x) at wavelengths of 702 and 704 nm, respectively. The Au particle-TiO2 slab interface is set to the origin of the z-axis. The white double-headed arrows represent the polarization direction of the injected light. | ||
Discussion
The overall efficiency in the HET-type plasmonic photocatalytic reactions (ϕall) can be expressed by multiplication of the efficiencies of a series of physical events including light harvesting (LHE), hot-electron injection (ϕHEI), and charge separation (ϕCS), and the efficiency of the surface chemical reaction (ϕchem). Among the factors, the strength of the local electric field induced by LSPR excitation (E) is decisively important because the overall efficiency is proportional to the square of |E|(eqn (1)).36,37| ϕall = ϕphys × ϕchem ∝ (A/γrel)|E|2ϕchem | (1) |
The LSPR excitation of Au/R-TiO2 induces “plasmonic hot spots”, in which the local electric field is much more intense than that of the Au/A-TiO2 system, at the edges and corners of t-Oh Au NPs around the perimeter interface with TiO2. As a result, light harvesting or the rate of hot carrier generation in Au NPs is enhanced near the interface (LHE ↑). This has recently been experimentally evidenced by means of Kelvin probe force microscopy for Ag NPs on GaN,38 and photoconductive atomic force microscopy for the Au nanoprism on TiO2 (ref. 39) and GaN.40 In this case, it is worth noting that the absorption intensity in the absorption spectrum is not directly related to the photocatalytic activity (Fig.3a, d, 4a and b). The hot electrons are efficiently injected into the CB of TiO2 through the large-area (A ↑) and high-quality interface (ϕHEI ↑).41,42 In the Au/TiO2 system, the CB-band bending generated by the Schottky junction can assist charge separation (ϕCS ↑).12 In this manner, the finding that the activity of the Au/TiO2 plasmonic photocatalysts for various reactions is governed by the Pf of Au NPs can be explained in terms of the enhancement of the efficiencies in the photophysical processes. In a recent study on the OER by using Au/TiO2-nanotube array plasmonic electrodes, the photocurrent has been reported to increase with a decrease in the Au particle size due to the lowering in the Schottky barrier at the Au/TiO2 interface.43 In the Au/TiO2 plasmonic photocatalyst system, this effect seems to be minor because the photocatalytic activities of Au/TiO2 are completely different in spite of the comparable Au particle size, i.e., Au(dAu = 3.5 nm)/R-TiO2 NR ≫ Au(dAu = 3.6 nm)/R-TiO2 ≫ Au(dAu = 3.5 nm)/A-TiO2. On the other hand, the faceting effect of Au NPs on the plasmonic photocatalytic activity depends on the type of reaction (Fig. 3d). This fact also points to the importance of the chemical effect or ϕchem for increasing the overall reaction efficiency through optimization according to the reaction.
Conclusions
This study has provided the following important results on the typical plasmonic photocatalysts of Au/TiO2. First, the faceting of Au NPs is induced on R-TiO2 by the heteroepitaxial junction with the (111)Au//(110)R-TiO2 orientation, while hemispherical Au NPs are exclusively formed on A-TiO2. Second, the faceting probability of Au NPs, particularly on the ordinary irregular shaped R-TiO2 particle, increases with decreasing Au particle size. Third, a positive correlation is valid between the plasmonic photocatalytic activity of Au/TiO2 and the faceting probability of Au NPs on TiO2 regardless of the TiO2 crystal form and Au particle size. Most importantly, the use of R-TiO2 NR with large-area (110) side walls as the support of Au NPs increases the Pf to over 90%, drastically enhancing the photocatalytic activity. While the technique for the synthesis of various faceted semiconductor NPs is rapidly in progress,44 the importance of the intimate junction between metal NPs and semiconductors is being recognized in the field of photocatalysis.15,45 This study has shown that the combination of crystal facet engineering and atom-level-interface control can be a powerful and versatile methodology for enhancing the photocatalytic activity of metal–semiconductor nanohybrids for various solar-to-chemical transformations.Data availability
Many data have already been shown in ESI.†Author contributions
S. N., A. A. and Y. M. conducted catalyst synthesis, characterization, and photocatalytic reactions, M. F. performed FDTD simulations, and H. T. supervised the work and data analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the JSPS KAKENHI Grant-in-Aid for Scientific Research (C) no. 20K05674 and 21K05236, the Futaba Foundation, the Nippon Sheet Glass Foundation for Materials Science and Engineering, the Sumitomo Foundation, the Takahashi Industrial and Economic Research Foundation.Notes and references
- C. Wang and D. Astruc, Nanogold plasmonic photocatalysis for organic synthesis and clean energy conversion, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC
.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Plasmon-induced hot carrier science and technology, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed
- D. A. Panayotov and J. R. Morris, Review on surface chemistry of Au/TiO2 and in thermally and photolytically activated reactions, Surf. Sci. Rep., 2016, 71, 77–271 CrossRef CAS
- P. Narang, R. Sundararaman and H. A. Atwater, Plasmonic hot carrier dynamics in solid-state and chemical systems for energy conversion, Nanophotonics, 2016, 5, 96–111 CAS
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Catalytic conversion of solar to chemical energy on plasmonic metal nanostructures, Nat. Catal., 2018, 1, 656–665 CrossRef
- A. Gellé, T. Jin, L. De la Garza, G. D. Price, L. V. Besteiro and A. Moores, Applications of plasmon-enhanced nanocatalysis to organic transformations, Chem. Rev., 2020, 120, 986–1041 CrossRef PubMed
- H. Tada, Overall water splitting and hydrogen peroxide synthesis by gold nanoparticle-based plasmonic photocatalysts, Nanoscale Adv., 2019, 1, 4238–4245 RSC
- S. Mubeen, J. Lee, N. Singh, S. Kramer, G. D. Stucky and M. Moskovits, An autonomous photosynthetic device in which all charge carriers derive from surface plasmons, Nat. Nanotechnol., 2013, 8, 247–251 CrossRef CAS
- X. Shi, K. Ueno, T. Oshikiri, Q. Sun, K. Sasaki and H. Misawa, Enhanced water splitting under modal strong coupling conditions, Nat. Nanotechnol., 2018, 13, 953–958 CrossRef CAS
- E. Thimsen, F. Le Formal, M. Grätzel and S. C. Warren, Influence of plasmonic Au nanoparticles on the photoactivity of Fe2O3 electrodes for water splitting, Nano Lett., 2011, 11, 35–43 CrossRef CAS PubMed
- Y. Tian and T. Tatsuma, Mechanisms and applications of plasmon-induced charge separation at TiO2 films loaded with gold nanoparticles, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed
- M. W. Knight, H. Sobhani, P. Nordlander and N. J. Halas, Photodetection with active optical antennas, Science, 2011, 332, 702–704 CrossRef CAS PubMed
- L. Liu, X. Zhang, L. Yang, L. Ren, D. Wang and J. Ye, Metal nanoparticles induced photocatalysis, Natl. Sci. Rev., 2017, 4, 761–780 CrossRef CAS
- G. Yu, J. Qian, P. Zhang, B. Zhang, W. Zhang, W. Yan and G. Liu, Collective excitation of plasmon-coupled Au-nanochain boosts photocatalytic hydrogen evolution of semiconductor, Nat. Commun., 2019, 10, 4912 CrossRef
- H. Tada, S. Naya and M. Fujishima, Nanohybrid crystals with heteroepitaxial junctions for solar-to-chemical transformations, J. Phys. Chem. C, 2020, 124, 25657–25666 CrossRef CAS
- A. Y. Ahmed, T. A. Kandiel, T. Oekermann and D. Bahnemann, Photocatalytic activities of different well-defined single crystal TiO2 surfaces: anatase versus rutile, J. Phys. Chem. Lett., 2011, 2, 2461–2465 CrossRef CAS
- L. Yang, H. Zhou, T. Fan and D. Zhang, Semiconductor photocatalysts for water oxidation: current status and challenges, Phys. Chem. Chem. Phys., 2014, 16, 6810–6826 RSC
- S. Wang, Y. Gao, S. Miao, T. Liu, L. Mu, R. Li, F. Fan and C. Li, Positioning the water oxidation reaction sites in plasmonic photocatalysts, J. Am. Chem. Soc., 2017, 139, 11771–11778 CrossRef CAS PubMed
- T. A. Kandiel, R. Dillert, A. Feldhoff and D. W. Bahnemann, Direct synthesis of photocatalytically active rutile TiO2 nanorods partly decorated with anatase nanoparticles, J. Phys. Chem. C, 2010, 114, 4909–4915 CrossRef CAS
- R. Zanella, S. Giorgio, C. R. Henry and C. Louis, Alternative methods for the preparation of gold nanoparticles supported on TiO2, J. Phys. Chem. B, 2002, 106, 7634–7642 CrossRef CAS
- A. Kasahara, K. Nukumizu, G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Photoreactions on LaTiO2N under visible light irradiation, J. Phys. Chem. A, 2002, 106, 6750–6753 CrossRef CAS
- J. G. E. Jellison Jr, L. A. Boatner, J. D. Budai, B. S. Jeong and D. P. Nortan, Spectroscopic ellipsometry of thin film and bulk anatase (TiO2), J. Appl. Phys., 2003, 93, 9537–9541 CrossRef
-
H. Tada, T. Kiyonaga and S. Naya, Metal oxide-supported gold nanoparticles-size dependence of catalytic activities, Lambert Academic Publishing, Saabrúcken, 2012 Search PubMed
- F. Cosandey, L. Zhang and T. E. Madey, Effect of substrate temperature on the epitaxial growth of Au on TiO2(110), Surf. Sci., 2001, 474, 1–13 CrossRef CAS
- D. Holec, P. Dumitraschkewitz, D. Vollath and F. D. Fischer, Surface energy of Au nanoparticles depending on their size and shape, Nanomaterials, 2020, 10, 484 CrossRef CAS PubMed
- W. Heckel, T. Würger, S. Müller and G. Feldbauer, Van der Waals interaction really matters: energetics of benzoic acid on TiO2 rutile surfaces, J. Phys. Chem. C, 2017, 121, 17207–17214 CrossRef CAS
- F. Cosandey and T. E. Madey, Growth, morphology, interfacial effects and catalytic properties of Au on TiO2, Surf. Rev. Lett., 2011, 8, 73–93 CrossRef
- K. Kimura, S. Naya, Y. Jin-nouchi and H. Tada, TiO2 crystal form-dependence of the Au/TiO2 plasmon photocatalyst's activity, J. Phys. Chem. C, 2012, 116, 7111–7117 CrossRef CAS
- S. Naya, K. Kimura and H. Tada, One-step selective aerobic oxidation of amines to imines by gold nanoparticle-loaded rutile titanium(IV) oxide plasmon photocatalyst, ACS Catal., 2013, 3, 10–13 CrossRef CAS
- S. Naya and H. Tada, Dependence of the plasmonic activity of Au/TiO2 for the decomposition of 2-naphthol on the crystal form of TiO2 and Au particle size, J. Catal., 2018, 364, 328–333 CrossRef CAS
- A. Ali, I. Ruzybayev, E. Yassitepe, A. Karim, S. Ismat Shah and A. S. Bhatti, Phase transformations in the pulsed laser deposition grown TiO2 thin films as a consequence of O2 partial pressure and Nd doping, J. Phys. Chem. C, 2015, 119, 11578–11587 CrossRef CAS
- W. Yuan, B. Zhu, K. Fang, X.-Y. Li, T. W. Hansen, Y. Ou, H. Yang, J. B. Wagner, Y. Gao, Y. Wang and X. Zhang,
In situ manipulation of the active Au-TiO2 interface with atomic precision during CO oxidation, Science, 2021, 371, 517–521 CrossRef CAS PubMed
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Liu, Aanatase TiO2 single crystals with a large percentage of reactive facets, Nature, 2008, 453, 638–641 CrossRef CAS PubMed
-
K. Tanaka, S. Tanuma, K. Dohmae, Y. Nagoshi and A. Nisawa, X-ray photoelectron spectroscopy, ed. T. Sawada, S. Tanuma and K. Tanaka, Maruzen, Tokyo, 1998 Search PubMed
- L. Liu, P. Li, B. Adisak, S. Ouyang, N. Umezawa, J. Ye, R. Kodiyath, T. Tanabe, G. V. Ramesh, S. Ueda and H. Abe, Gold photosensitized SrTiO3 for visible-light water oxidation induced by Au interband transitions, J. Mater. Chem. A, 2014, 2, 9875–9882 RSC
- A. O. Govorov, H. Zhang, H. V. Demir and Y. K. Guńko, Photogeneration of hot plasmonic electrons with metal nanocrystals: Quantum description and potential applications, Nano Today, 2014, 9, 85–101 CrossRef CAS
- H. Tada, Rational design for gold nanoparticle-based plasmonic catalysts and electrodes for water oxidation towards artificial photosynthesis, Dalton Trans., 2022, 51, 3383–3393 RSC
- D.-B. Li, X.-J. Sun, Y.-P. Jia, M. I. Stockman, H. P. Paudel, H. Song, H. Jiang and Z.-M. Li, Direct observation of localized surface plasmon field enhancement by Kelvin probe force microscopy, Light: Sci. Appl., 2017, 6, e17038 CrossRef CAS
- H. Lee, H. Lee and J. Y. Park, Direct imaging of surface plasmon-driven hot electron flux on the Au Nanoprism/TiO2, Nano Lett., 2019, 19, 891–896 CrossRef CAS PubMed
- H. Lee, K. Song, M. Lee and J. Y. Park,
In Situ Visualization of localized surface plasmon resonance-driven hot hole flux, Adv. Sci., 2020, 7, 2001148 CrossRef CAS
- S. Naya, T. Kume, R. Akashi, M. Fujishima and H. Tada, Red-light-driven water splitting by Au(core)-CdS(shell) half-cut nanoegg with heteroepitaxial junction, J. Am. Chem. Soc., 2018, 140, 1251–1254 CrossRef CAS PubMed
- J. Liu, J. Feng, J. Gui, T. Chen, M. Xu, H. Wang, H. Dong, H. Chen, X. Li, L. Wang, Z. Chen, Z. Yang, J. Liu, W. Hao, Y. Yao, L. Gu, Y. Weng, Y. Huang, X. Duan, J. Zhang and Y. Li, Metal@semiconductor core-shell nanocrystals with atomically organized interfaces for efficient hot electron-mediated photocatalysis, Nano Energy, 2018, 48, 44–52 CrossRef CAS
- S. Y. Moon, H. C. Song, E. H. Gwag, I. I. Nedrygailov, C. Lee, J. J. Kim, W. H. Doh and J. Y. Park, Plasmonic hot carrier-driven oxygen evolution reaction on Au nanoparticles/TiO2 nanotube arrays, Nanoscale, 2018, 10, 22180–22188 RSC
- S. Wang, G. Liu and L. Wang, Crystal facet engineering of photoelectrodes for photoelectrochemical water splitting, Chem. Rev., 2019, 119, 5192–5247 CrossRef CAS
- X. Wan, J. Liu and J. Zhang, Construction of plasmonic metal@semiconductor core-shell photocatalysts: from epitaxial to nonepitaxial strategies, Small Struct., 2022, 2200045 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc03549a |
| This journal is © The Royal Society of Chemistry 2022 |