
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding new chemistry of aza-boracyclophanes with unique dipolar structures, AIE and redox-active open-shell characteristics†
Yawei
Jia
a,
Pengfei
Li
a,
Kanglei
Liu
*a,
Chenglong
Li
a,
Meiyan
Liu
a,
Jiaqi
Di
a,
Nan
Wang
 a,
Xiaodong
Yin
a,
Niu
Zhang
*b and
Pangkuan
Chen
*a
a,
Xiaodong
Yin
a,
Niu
Zhang
*b and
Pangkuan
Chen
*a
aBeijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, Key Laboratory of Cluster Science of the Ministry of Education, Key Laboratory of Medical Molecule Science and Pharmaceutics Engineering of the Ministry of Industry and Information Technology, School of Chemistry and Chemical Engineering, Beijing Institute of Technology of China, Beijing, 102488, China. E-mail: kanglei_liu@bit.edu.cn; pangkuan@bit.edu.cn
bAnalysis & Testing Centre, Beijing Institute of Technology of China, Beijing, 102488, China. E-mail: niuzhang2019@bit.edu.cn
First published on 26th September 2022
Abstract
π-Conjugated macrocycles involving electron-deficient boron species have received increasing attention due to their intriguing tunable optoelectronic properties. However, most of the reported B(sp2)-doped macrocycles are difficult to modify due to the synthetic challenge, which limits their further applications. Motivated by the research of non-strained hexameric bora- and aza-cyclophanes, we describe a new class of analogues MC-BN5 and MC-ABN5 that contain charge-reversed triarylborane (Ar3B) units and oligomeric triarylamines (Ar3N) in the cyclics. As predicted by DFT computations, the unique orientation of the donor–acceptor systems leads to an increased dipole moment compared with highly symmetric macrocycles (M1, M2 and M3), which was experimentally represented by a significant solvatochromic effect with large Stokes shifts up to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 318 cm−1. Such a ring-structured design also allows the easy peripheral modification of aza-boracyclophanes with tetraphenylethenyl (TPE) groups, giving rise to a change in the luminescence mechanism from aggregation-caused quenching (ACQ) in MC-BN5 to aggregation-induced emission (AIE) in MC-ABN5. The open-shell characteristics have been chemically enabled and were characterized by UV-Vis-NIR spectroscopy and electron paramagnetic resonance (EPR) for MC-BN5. The present study not only showed new electronic properties, but also could expand the research of B/N doped macrocycles into the future scope of supramolecular chemistry, as demonstrated in the accessible functionalization of ring systems.
318 cm−1. Such a ring-structured design also allows the easy peripheral modification of aza-boracyclophanes with tetraphenylethenyl (TPE) groups, giving rise to a change in the luminescence mechanism from aggregation-caused quenching (ACQ) in MC-BN5 to aggregation-induced emission (AIE) in MC-ABN5. The open-shell characteristics have been chemically enabled and were characterized by UV-Vis-NIR spectroscopy and electron paramagnetic resonance (EPR) for MC-BN5. The present study not only showed new electronic properties, but also could expand the research of B/N doped macrocycles into the future scope of supramolecular chemistry, as demonstrated in the accessible functionalization of ring systems.
Introduction
Macrocyclic structures have received much attention over the past two decades as they played a crucial role in host–guest chemistry,1 organic light-emitting diodes (OLEDs),2 biological science3 and supramolecular assembly.4 Macrocycles of π-electron systems exhibit particularly intriguing optical, electronic and magnetic behaviors owing to an equivalent of these cycles to well-defined open-chain polymers but with an infinite conjugation length.2b,5 The incorporation of main group heteroatoms (such as B, N, and S) has proved an efficient strategy to build π-conjugated materials with unique optoelectronic properties.6 Such a group of heteroatom-doped macrocycles are expected to show distinct electronic structures compared with all-carbon based analogues, and their frontier molecular orbitals can be tuned.Ito and Tanaka et al. first reported a cyclic hexamer of an electron-rich arylamine macrocycle M1,6g and then some other macrocyclic oligoarylamines were also investigated.6k,7 The electron-deficient triarylboron exhibits a remarkable electronic polarization and intramolecular charge transfer (ICT) in the presence of electron donors, leading to tunable emissions, enhanced charge-transport characteristics and unusual redox properties.8 The Jäkle group pioneered boracyclophane chemistry by developing a fluoreneborane cycle (MC-B6-Flu),6a followed by an ambipolar macrocycle M2 with alternating arylamines and arylboranes.6b They have recently designed a considerably electron-deficient M3 with a low-lying LUMO and strong electronic affinity regardless of the synthetic challenge associated with the enhanced electron deficiency.6d In fact, all the above-represented macrocycles exhibit a dipole moment μ = 0 in the ground state due to the highly symmetric geometry. Our group and coworkers proposed a new concept of a block-type macrocycle (M4) in which the electron-donor block (N3) and acceptor block (B3) are oriented on opposite sides (Scheme 1), leading to an increased dipole moment (μg) and a reduced HOMO–LUMO energy gap (Egap).9 Computations reveal that the specific patterns of B and N centers as well as the molecular symmetry are closely related to the μg and Egap levels (Fig. S28†), which dramatically impact the respective photophysical and electronic properties.
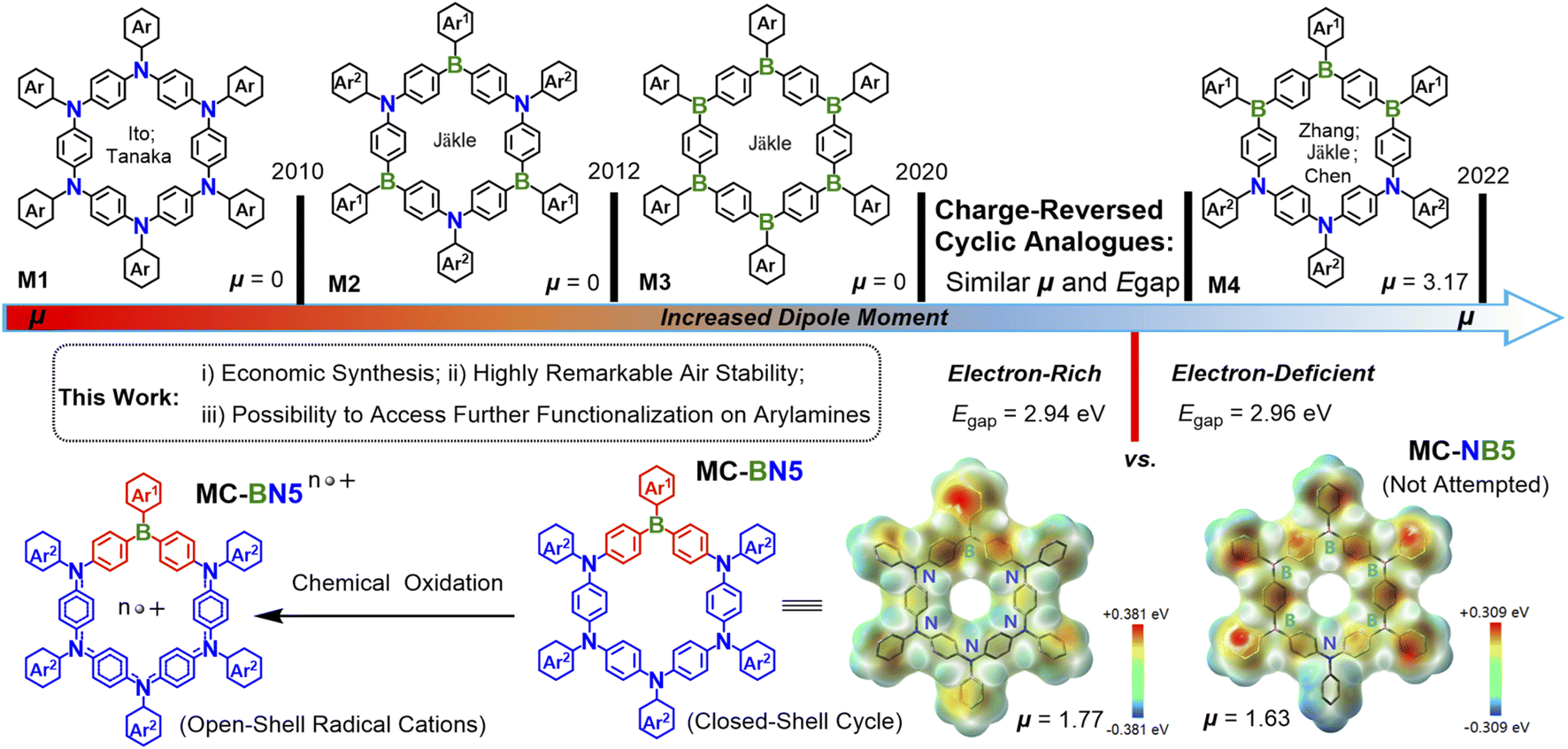 | ||
| Scheme 1 Research foundation and design principle for the dipolar aza-boracyclophane MC-BN5 in comparison to some of the π-conjugated systems of B/N-doped cyclic hexamers M1, M2, M3 and M4. | ||
To further expand the research scope of boracyclophane chemistry, we turned our attention to B/N doped macrocycles in low symmetry with accessible functionalized peripheries. In this respect, we conceive two model cycles MC-BN5 and MC-NB5 that are charge-reversed analogues and show similar dipole moments and energy gaps (MC-BN5: μg = 1.77 and Egap = 2.94 eV; MC-NB5: μg = 1.63 and Egap = 2.96 eV). However, the stepwise synthesis of pentameric triarylboranes seems to require steric shielding for the latter under strictly inert conditions given a gradually increased number of borons as Lewis acid sites. In contrast, oligotriarylamines can be readily accessible for the former cycle using some more step-economic reactions via transition-metal-catalyzed C–N cross-couplings. For future generations of aza-boracyclophanes, the donor segments of oligoarylamines selected with a high tolerance to air are of particular significance, as they allow further manipulation and processes to be reasonably achieved. Accordingly, we herein describe an efficient design strategy toward MC-BN5 and the derivative MC-ABN5. MC-BN5 strongly red-shifted the emission to orange in polar solvents from the green color for analogous boracyclophanes (M2, M3 and MC-B6-Flu), and the open-shell radical species enabled by chemical oxidation were also monitored by UV-Vis-NIR spectroscopy and electron paramagnetic resonance (EPR) spectroscopy. More importantly, peripheral substitutions of tetraphenylethyl (TPE) units at the π-ring structure of MC-ABN5 with radial p orbitals induced a transformation of aggregation-caused quenching (ACQ) to a rarely observed aggregation-induced emission (AIE) for π-conjugated macrocycles.4b,6n,10 Our strategy may provide a new possibility to develop directions in supramolecular chemistry using highly stable dipolar macrocycles.
Results and discussion
The synthetic routes and data toward MC-BN5 and MC-ABN5 are depicted in Scheme 2 and the ESI.† Precursor 1 was obtained through the gradual build-up of oligomerization according to previously reported procedures.9 Starting from the dihalogenated arylamine trimer 1, π-extended pentamers 2 and 3 were prepared by Buchwald–Hartwig coupling with 4-hexylaniline and 4-tetraphenylethenyl amine (TPE-NH2), respectively. Similar reactions in the subsequent C–N cross-couplings yielded the key intermediates 4 and 5. The lithiation of dibrominated 4 and 5 followed by the addition of TipB(OMe)2 as a protected boron source was performed in dilute ether solutions to stitch the π-extended oligoarylamines, and it resulted in aza-boracyclophanes MC-BN5 in 30% and MC-ABN5 in 15% isolated yields.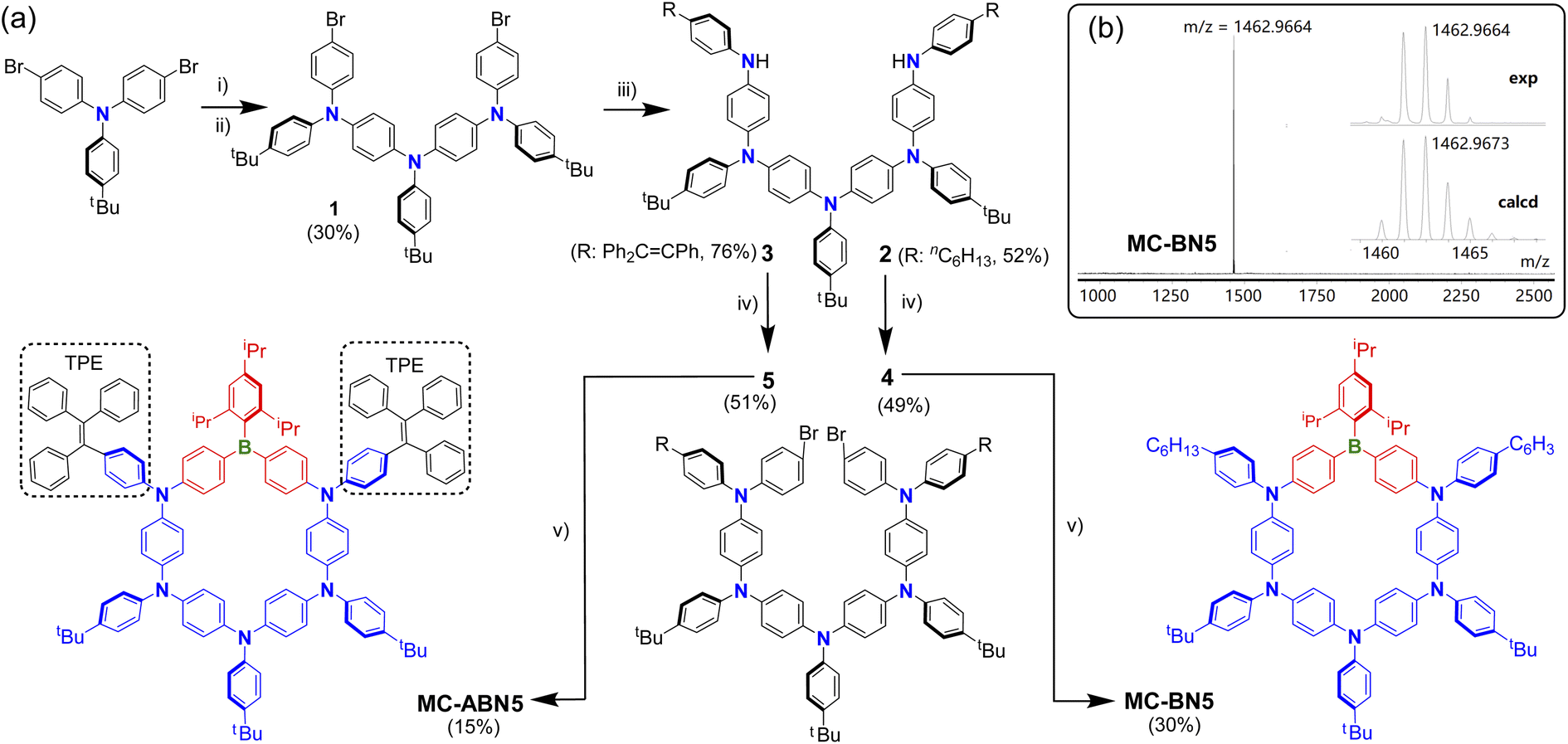 | ||
| Scheme 2 (a) Key steps in the synthesis of MC-BN5 and MC-ABN5. 1 was synthesized by similar procedures reported (see ref. 9). Reagents and conditions: (i) 4-tBu-aniline, Pd(dppf)Cl2, DPPF, tBuONa, toluene, and reflux. (ii) para-Bromo-iodobenzene, Pd(dppf)Cl2, DPPF, tBuONa, toluene, and reflux. (iii) Pd(dppf)Cl2, DPPF, tBuONa, 4-nhexyl-aniline (for 2) or 4-tetraphenylethenyl amine (TPE-NH2, for 3), toluene, and reflux. (v) nBuLi, TipB(OMe)2, ether, and −35 °C to reflux overnight. Tip = 2,4,6-triisopropylphenyl. (b) MAIDI-TOF-MS (positive-ion mode) of MC-BN5 showing experimental and simulated isotopic patterns. | ||
Both macrocycles are stable enough in air and can be easily purified by column chromatography on silica gel without particular precautions. The structures of MC-BN5 and MC-ABN5 were fully characterized by multinuclear NMR and high-resolution mass spectroscopy (ESI†). MALDI-TOF-MS spectra clearly showed a single peak (Scheme 2b, Fig. S8 and S16†), corresponding to the molecular-ion peak at m/z = 1462.9664 for MC-BN5 (calcd 1462.9673) and at 1802.9986 for MC-ABN5 (calcd 1802.9988). The broad bands centered at 31 and 56 ppm in the 11B NMR spectra for MC-BN5 MC-ABN5, respectively, confirmed the incorporation of tricoordinate arylborane species (Fig. S7 and S15†). Notably, two of the macrocycles showed no visible changes in solution after storage over a few months at ambient temperature, indicating exceptionally high structural persistence which is somewhat superior to that of M4 (Fig. S18†).
The photophysical properties of MC-BN5 and MC-ABN5 were explored by UV-Vis absorption and emission spectroscopy (Fig. 1 and ESI†). Both MC-BN5 and MC-ABN5 showed a strong absorption band at 347 and 364 nm in CH2Cl2, respectively, which were assigned to π–π* transitions (Fig. 1a). Meanwhile, they also displayed a low-energy shoulder peak at 408 (MC-BN5) and 417 nm (MC-ABN5), respectively, ascribed to an intramolecular charge transfer (ICT) from the electron-donating moieties (–N–π–N–π–N–π–N–π–N–) to the electron-accepting unit (–π–B–π–). Two molecules show similar absorption profiles but with slightly red-shifted bands for MC-ABN5, as a result of the extended π-conjugation effect by TPE substituents at the six-membered periphery of MC-BN5. The optical energy gap Egap(optical) was determined to be 2.70 eV for MC-BN5, consistent with the computed results (Egap(DFT): 3.02 eV; Egap(TDDFT): 2.73 eV, Table 1 and ESI†).
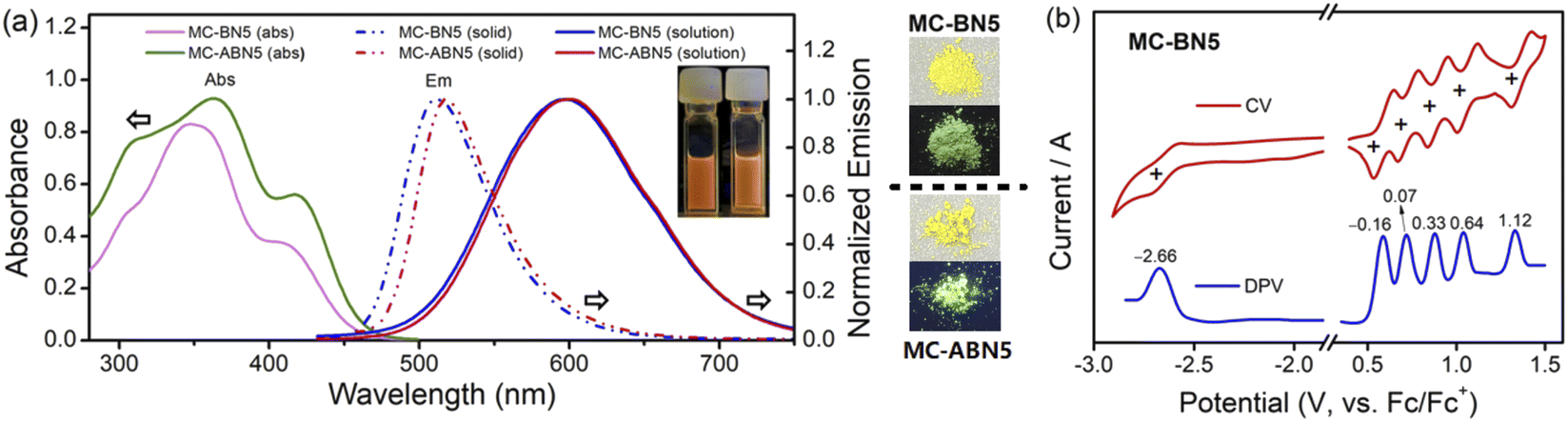 | ||
| Fig. 1 (a) UV-Vis absorption and emission spectrum of MC-BN5 (λex = 408 nm) and MC-ABN5 (λex = 417 nm) both in CH2Cl2 (c = 1 × 10−5 M) and in the solid state. Inset: photographs of solutions MC-BN5 and MC-ABN5 and respective solids under ambient light (upper) and 365 nm UV irradiation (down). (b) Cyclic and differential pulse voltammetry (CV and DPV) curves (vs. Fc+/Fc) in CH2Cl2 (oxidative potential) and THF (reductive potential), using n-Bu4NPF6 (0.1 M) as the electrolyte, and v = 100 mV s−1. | ||
MC-BN5 and MC-ABN5 emitted orange fluorescence in CH2Cl2 (MC-BN5: λem = 597 nm and ΦF = 16%; MC-ABN5: λem = 600 nm and ΦF = 10%), which is significantly red-shifted from analogous boracyclophanes involving M2,6bM3 (ref. 6d) and MC-B6-Flu.6a They showed green color emissions in the solid state (MC-BN5: 512 nm and ΦF = 13%; MC-ABN5: 517 nm and ΦF = 37%), and the emission quantum efficiency of MC-BN5 was reduced relative to that in solution, in line with a traditional ACQ mechanism. However, the blue-shifted emission of MC-ABN5 was enhanced as solids and was in accordance with AIE activity due to the TPE incorporation. As shown in the solvatochromic emissions, MC-BN5 and MC-ABN5 change their colors from blue-sky in hexane (λem: ∼490 nm) to orange in highly polar solvents (λem: ∼600 nm) with large Stokes shifts up to 12318 and 10806 cm−1, respectively (Fig. 2a and ESI†). Large slopes of the data fitting in Lippert–Mataga plots further denote an apparent ICT character that is an indication of much more polar structures in MC-BN5 and MC-ABN5 than other highly symmetric boracyclophanes such as M2, M3, MC-B6-Flu, MC-B4N26c and MC-B4N2-FMes6e (Fig. 2b and Table S4†).
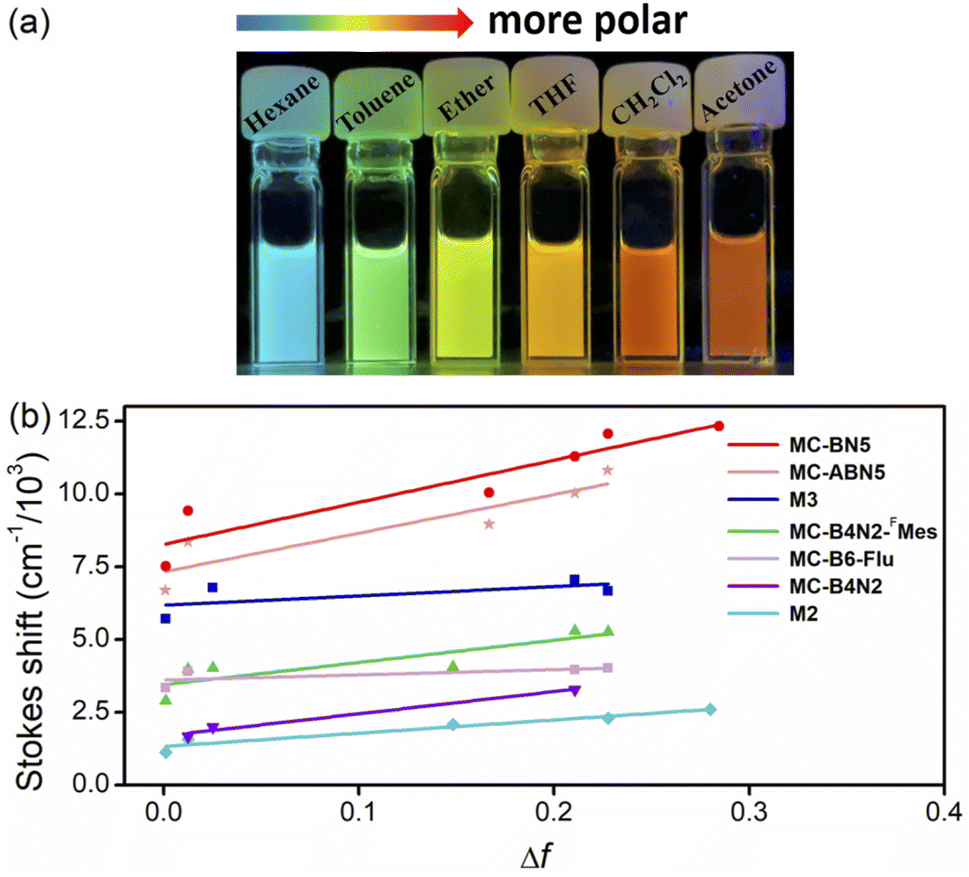 | ||
| Fig. 2 (a) Photographs of MC-BN5 in solvents of different polarity upon irritation at 365 nm (left to right: n-hexane, toluene, ether, THF, CH2Cl2 and acetone). (b) Lippert–Mataga plots for solvatochromic emissions of MC-BN5 and MC-ABN5 in comparison to some other representative B-containing macrocycles. | ||
The electrochemical properties of MC-BN5 were examined by cyclic (CV) and differential pulse voltammetry (DPV) (Fig. 1b). MC-BN5 showed five fully reversible 1e oxidation bands with the first half-wave potential at Eox1/2 = −0.16 V (vs. Fc/Fc+, CH2Cl2) and a reversible reduction band at Ered1/2 = −2.66 V (vs. Fc/Fc+, THF). These well-separated patterns are attributed to the sequential oxidations at each of the Ar3N moieties with strong electronic couplings. The six-membered aza/boracyclophane analogues show a trend of the oxidations toward higher potentials with a decreased number of arylamines: M1 (−0.28 V, N6) < MC-BN5 (−0.16 V, N5) < M4 (+0.02 V, b-N3) < M2 (+0.46 V, alt-N3), in line with the trend of reduced HOMO energy levels. A similar observation is also the case in the reductions with increasing B centers: MC-BN5 (−2.66 V, B1) < M2 (−2.53 V, alt-B3) < M4 (−2.10 V, b-B3) < M3 (−1.56 V, B6) and agrees very well with the trend of the computed LUMO energies (Table S5†). These results suggest that the number as well as the orientation of B and/or N arranged in macrocyclic architectures could effectively impact the electronic structures and molecular symmetry. MC-BN5 shows a smaller electrochemical energy gap (Egap(elec) = 2.50 eV) than the linear L-B2N5 (Egap(elec) = 2.61 eV) that involves five units of oligomeric arylamines end-capped by the two terminal arylboranes, leading to a higher oxidation potential (Eox1 = 0.01 V) in the open-chain structure.13c
MC-BN5 exhibits a more polar structure compared with the highly symmetric aza-boracyclophanes, which can be confirmed by a stronger dipole moment (μg = 1.77 D) than that of 0 D calculated for the simplified analogues M2 and M3 (i.e., alternating ambipolar MC-alt-B3N3 in D3 and electron-deficient MC-B6 in D3d, and μg = 0 D). According to computational results, MC-BN5 should be slightly less polar than the cyclic hexamer M4 (block-type MC-b-B3N3: C2, and μg = 3.17 D), but closely resembles the charge-reversed model compound MC-NB5 with μg = 1.63 D in a similar C2 symmetry (Fig. S28†). As visualized in the electrostatic potential (ESP) map, MC-BN5 shows an electronic structure with electron-donating triarylamines negatively charged and the borane acceptor observed in the positive domain (Scheme 1).
The electronic structure of simplified MC-BN5 was further elucidated by DFT (B3LYP, 6-31G*) and TD-DFT (B3LYP/CAM-B3LYP, 6-311G*) calculations (Fig. 3 and ESI†). The highest occupied molecular orbital (HOMO) is delocalized over the electron-rich pentatriarylamine block (N5), whereas the lowest unoccupied molecular orbital (LUMO) is only located B-centered with a small contribution from the adjacent N atoms. HOMO−1 and HOMO−2 are both delocalized nearly in the ring skeleton. TD-DFT reveals the lowest energy transition to S1 (f = 0.1600) due to charge transfer from the Ar3N to the Ar3B moiety, which however was usually symmetry-forbidden in highly symmetric cyclic structures including M1, M2, M3 and others.6a,c,e Such a dissymmetry-allowed HOMO–LUMO transition was also the case for the block-type macrocycle M4 with a lower symmetry.9 Vertical excitations to higher energy states up to S5 are attributed to a mixed pathway involving the π–π* transition of phenyl linkers and charge transfers from triarylamine moieties (i.e. endocyclic phenyl or exocyclic aryl) to endocyclic arylborane, and the n–π* transitions from N sites also contribute.
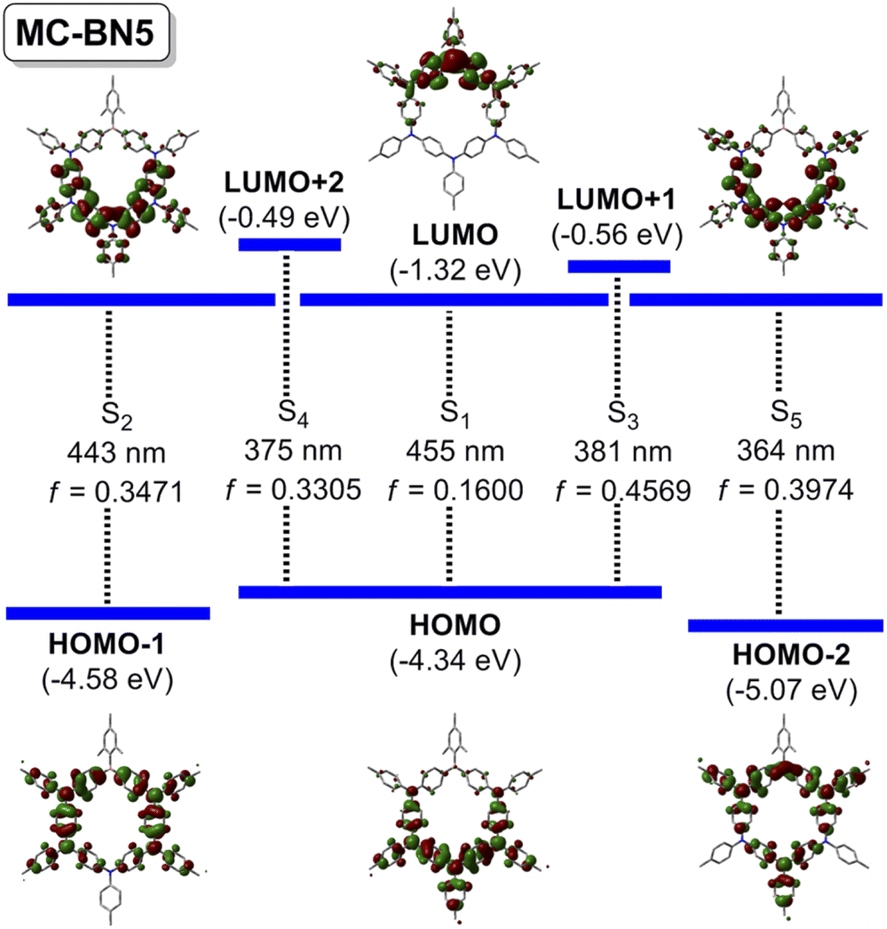 | ||
| Fig. 3 Key electronic transitions contributing to vertical excitations of simplified MC-BN5 (TD-DFT, B3LYP/6-311G*) with molecular orbital plots (iso = 0.02, B3LYP/6-31G*). The iPr, tBu and nHex groups are simplified as methyl groups. | ||
As seen in the above-mentioned electrochemical characterization, MC-BN5 exhibits fully reversible oxidation events due to sequential oxidations of all the five arylamines to radical cation species. A chemical oxidation approach for MC-BN5 using AgSbF6 was also performed to investigate its cationic states under strictly inert conditions, and in situ UV-Vis-NIR spectroscopy and electron paramagnetic resonance (EPR) were conducted.6o As shown in Fig. 4a, the addition of an approximately stoichiometric amount of AgSbF6 to MC-BN5 solution in CH2Cl2 provided a green-colored reaction mixture at room temperature. Compared with the neutral MC-BN5 (A), the oxidized species displayed two new broad absorption bands at ca. 900 and 1650 nm, and the latter band at a longer wavelength should be ascribed to the formation of the monoradical cation MC-BN5˙+ (B, Fig. 4d). This UV-Vis-NIR spectrum response to oxidation assigned to the spin-delocalized open-shell species can be confirmed by a split EPR spectrum with a 14N-hyperfine coupling constant α(14N) = 0.826 mT, α(1H) = 1.424 mT and g = 2.0037 (Fig. 4b and c).6g
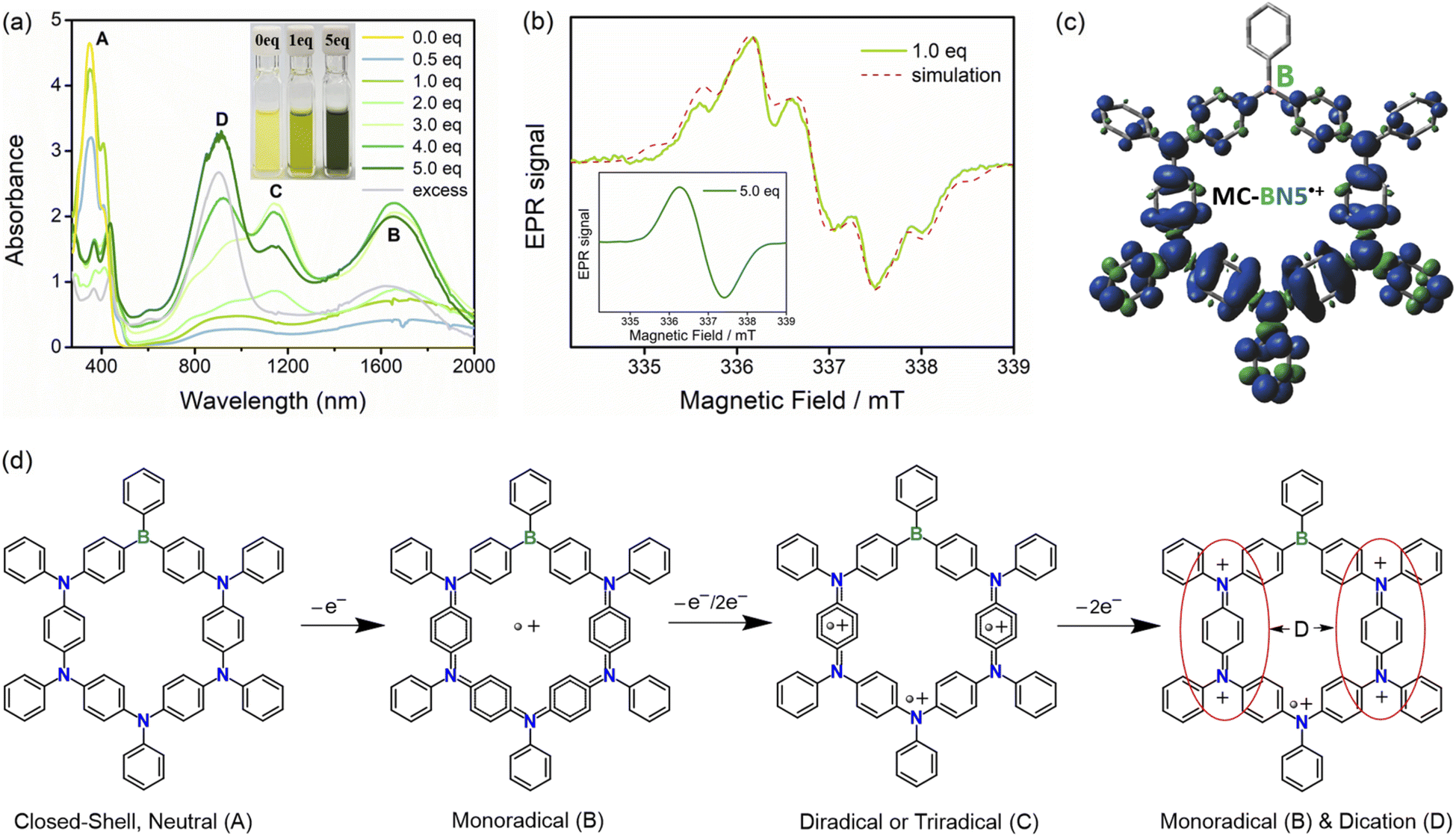 | ||
| Fig. 4 (a) Chemical oxidations of MC-BN5 (c = 5.0 × 10−5 M) monitored by UV-Vis-NIR absorption spectroscopy with different amounts of AgSbF6. Inset: photographs of MC-BN5 solutions with 0.0, 1.0 and 5.0 equiv. of AgSbF6. The EPR spectra of MC-BN5 (c = 2.0 × 10−4 M in CH2Cl2 under N2): (b) 1.0 equiv. of AgSbF6 (solid line) and simulation (dashed line). Inset: 5.0 equiv. of AgSbF6. (c) Spin density distribution of the monoradical MC-BN5˙+ (UB3LYP/6-31G(d,p); blue: positive spin and green: negative spin). (d) Schematic assignments of the oxidized species of MC-BN5 with the addition of AgSbF6. Substituents on arylboranes and arylamines are simplified as H atoms for clarity. | ||
Oxidation titration by further addition of AgSbF6 up to 5 equiv. led to a significantly increased intensity of the long wavelength band and also to a new NIR absorption peak at ca. 1150 nm, corresponding to the generation of higher oxidation states including radical dications and/or trications (C). Upon increasing the amount of AgSbF6, this new absorption band gradually disappeared, while both signals of the intense higher-energy and lower-energy bands (λabs = 900 and 1650 nm) were maintained even in the presence of an excess of AgSbF6. These observations are in agreement with the speculation that monoradicals (B) are initially formed along with a small number of diamagnetic quinoidal dications (D) and that the reaction solution can be gradually dominated by the resulting spinless dication species.7,11 The fully oxidized MC-BN5 was proved very stable in air (Fig. S26†) and showed a featureless broad signal with a g-value of 2.0037 in the EPR spectrum (Fig. 4b, inset), indicating the absence of spin–spin dipole interactions due to a higher degree of the oxidation processes.
MC-BN5 showed a reduced emission in toluene with increased concentrations, in accordance with the commonly found aggregation-caused quenching (ACQ) behavior (Fig. 5a). However, its macrocyclic derivative MC-ABN5 showed in the solid state an enhanced emission quantum efficiency (ΦS = 0.37) relative to that of ΦL = 0.10 in solution (Table 1). These differences in the emissions are significant to indicate the unique electronic properties of B/N macrocycles. As shown in Fig. 5c, MC-ABN5 maintained relatively low fluorescence profiles up to fMeOH = 70% of the solvent mixture CH2Cl2/MeOH. Further increases in MeOH fractions gave rise to a dramatically enhanced emission intensity of MC-ABN5 with a marked blue-shift of the emission bands from the orange emission (ca. 600 nm) to a bright green emission (ca. 535 nm) as well as an increased quantum yield up to ΦF = 0.43 with the emission lifetimes on the order of nanoseconds (Fig. S20a, Table S6†). This observation in agreement with the well-known aggregation-induced emission (AIE) mechanism supported by the dynamic light scattering (DLS) data (Fig. S20b†) is the consequence of structural functionalization with tetraphenylethyl (TPE) groups.10,12 To the best of our knowledge, tricoordinated organoboranes including boron-based macrocycles have rarely been reported to show AIE properties.13 Our molecular design for the optically reversed emission transition from ACQ to AIE represented by MC-BN5 and MC-ABN5 affords a new approach to aggregated macrocyclic chemistry, and it may also provide a proof of concept for supramolecular assembly using polar-structured π-conjugated cyclics.
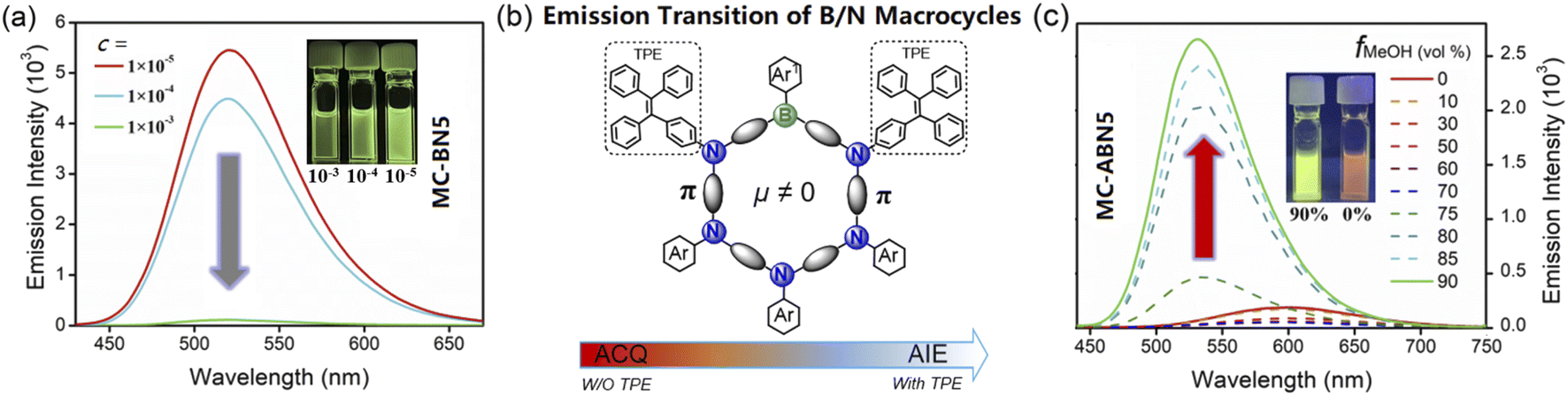 | ||
| Fig. 5 TPE-enabled emission transition of B/N macrocycles from ACQ in MC-BN5 (a) to the AIE mechanism in MC-ABN5 (b, c): fluorescence spectra recorded in CH2Cl2/MeOH with increasing MeOH fractions fMeOH (c = 1.0 × 10−5 M and λex = 408 nm). Inset: photographs under 365 nm UV light. | ||
| Compound | λ abs (nm) | λ em (nm) | λ em (nm) | Φ L (%) | Φ S (%) | τ av (ns) | E HOMO/ELUMO/Egap(DFT)f (eV)/(eV) | E TDDFT (eV) | E gap(optical) (eV) | E ox1/Ered1/Egap(elec)i (V)/(eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Recorded in CH2Cl2 (c = 1.0 × 10−5 M). b Emission in the solid state. c Emission quantum efficiency (ΦL) in CH2Cl2. d Emission quantum efficiency (ΦS) in the solid state. e Emission lifetime (τav) in CH2Cl2. f Energy gap: Egap = ELUMO − EHOMO obtained by DFT calculation (B3LYP, 6-31G*). g Vertical excitation of the lowest energy transition (S0 → S1) calculated by TD-DFT (B3LYP, 6-311G*). h E gap(optical): optical energy gap experimentally obtained from the onsets of the lowest energy absorption band in toluene. i Electrochemical energy gap determined by the first peak potential of oxidation (Eox1) and reduction (Ered1) in differential pulse voltammetry (vs. Fc+/Fc). | ||||||||||
| MC-BN5 | 347, 408 | 597 | 512 | 16 | 13 | 4 | –4.34/–1.32/3.02 | 2.73 | 2.70 | –0.16/–2.66/2.50 |
| MC-ABN5 | 364, 417 | 600 | 517 | 10 | 37 | 3 | — | — | 2.63 | — |
MC-BN5 and MC-ABN5 are also of interest as responsive materials. First, we have monitored the variable-temperature (VT) emission spectra in 2-methyltetrahydrofuran (Fig. S21†). With the temperature gradually increased, MC-BN5 was blue-shifted by ca. 60 nm in the emission from λem = 625 nm at 140 K to 567 nm at 340 K. This thermochromic response was fully reversible and displayed an excellent fatigue resistance without emission degradation when exposed to 5 cycles of alternating temperature. A similar behavior was also observed for MC-ABN5 but with a slightly stronger bathochromic shift of 70 nm. Second, given the presence of electron-deficient organoborane moieties, we performed anion binding studies of MC-BN5 with tetrabutylammonium fluoride (TBAF) in THF. MC-BN5 showed a gradual decrease of the typical charge-transfer absorption band at 408 nm, along with a gradual emission quenching in response to the titration of F− (Fig. S22†).
Conclusions
We report an efficient synthetic strategy to access new B/N doped macrocycles MC-BN5 and MC-ABN5 by structural stitching of electron-donating oligoarylamine pentamers using arylborane donor segments, leading to electronically unique low-symmetry aza-boracyclophanes (μg = 1.77 in MC-BN5). They displayed considerably stronger charge transfer properties and reduced HOMO–LUMO energy gaps in comparison to those established highly symmetric aza/boracyclophane analogues with a dipole moment μg = 0. These results suggest that the number ratio of B/N as well as their orientations can be readily tuned in macrocyclic architectures, which are essential to understand the impact of electronic structures and molecular symmetry on their optoelectronic characteristics. Beyond these findings, the environmental susceptibility of dipolar cycles was sufficiently counterbalanced to achieve exceptionally high stability that allowed chemically oxidation-induced open-shell radical species in MC-BN5 and aggregation-induced emission of its derivative MC-ABN5 under ambient conditions. We envision that the molecular derivatization of MC-BN5 is targeted not only for the emission transition between ACQ and AIE, but is truly approaching new directions in the supramolecular chemistry of polar-structured conjugated macrocycles. Pursuit for further studies on supramolecular systems related to this concept is ongoing in our laboratory.Data availability
All data supporting this study are included in the paper and provided in the ESI† accompanying this paper at the journal's website.Author contributions
Y. J., P. L. and P. C. conceived the project. Y. J. carried out all the synthetic experiments and analysed the experimental data. Y. J., P. L. and K. L. drafted the manuscript and revised the manuscript. P. L., C. L. and Y. J. performed the electrochemical measurements. M. L. obtained the solid quantum efficiency data. C. L. and J. D. acquired the MALDI-TOF-MS data. N. W. and X. Y. gave suggestions on the work. N. Z. provided help with mass spectroscopy and AIE measurements and also contributed to the write-up of the main manuscript. P. C. was responsible for the guidance of this project and finalized the manuscript as well as all calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 21772012 and 22101025). We are greatly thankful to Prof. Suning Wang at Queen's University for helpful discussions over the years. We thank the Analysis & Testing Center at the Beijing Institute of Technology for advanced facilities. The authors acknowledge the Analysis Center, Department of Chemistry at Tsinghua University for EPR experiments.Notes and references
- (a) T. Kawase and H. Kurata, Chem. Rev., 2006, 106, 5250–5273 CrossRef CAS PubMed; (b) M. Iyoda, J. Yamakawa and M. J. Rahman, Angew. Chem., Int. Ed., 2011, 50, 10522–10553 CrossRef CAS PubMed; (c) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed; (d) Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC; (e) J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC; (f) C.-F. Chen and Y. Han, Acc. Chem. Res., 2018, 51, 2093–2106 CrossRef CAS PubMed; (g) X. Ji, M. Ahmed, L. Long, N. M. Khashab, F. Huang and J. L. Sessler, Chem. Rev., 2019, 48, 2682–2697 CAS; (h) D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS PubMed; (i) S. V. Bhosale, M. A. Kobaisi, R. W. Jadhav, P. P. Morajkar, L. A. Jones and S. George, Chem. Soc. Rev., 2021, 50, 9845–9998 RSC; (j) H. Nie, Z. Wei, X.-L. Ni and Y. Liu, Chem. Rev., 2022, 122, 9032–9077 CrossRef CAS PubMed.
- (a) J. Y. Xue, T. Izumi, A. Yoshii, K. Ikemoto, T. Koretsune, R. Akashi, R. Arita, H. Taka, H. Kita, S. Sato and H. Isobe, Chem. Sci., 2016, 7, 896–904 RSC; (b) S. Izumi, H. F. Higginbotham, A. Nyga, P. Stachelek, N. Tohnai, P. Silva, P. Data, Y. Takeda and S. Minakata, J. Am. Chem. Soc., 2020, 142, 1482–1491 CrossRef CAS PubMed.
- (a) A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC; (b) R. Pinalli, A. Pedrini and E. Dalcanale, Chem. Soc. Rev., 2018, 47, 7006–7026 RSC; (c) N. Song, Z. Zhang, P. Liu, Y.-W. Yang, L. Wang, D. Wang and B. Z. Tang, Adv. Mater., 2020, 32, 2004208 CrossRef CAS PubMed; (d) Y. Qin, X. Liu, P.-P. Jia, L. Xu and H.-B. Yang, Chem. Soc. Rev., 2020, 49, 5678–5703 RSC.
- (a) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (b) S.-N. Lei, H. Xiao, Y. Zeng, C.-H. Tung, L.-Z. Wu and H. Cong, Angew. Chem., Int. Ed., 2020, 59, 10059–10065 CrossRef CAS PubMed; (c) F. Picini, S. Schneider, O. Gavat, A. V. Jentzsch, J. Tan, M. Maaloum, J.-M. Strub, S. Tokunaga, J.-M. Lehn, E. Moulin and N. Giuseppone, J. Am. Chem. Soc., 2021, 143, 6498–6504 CrossRef CAS PubMed; (d) S. Kawano, M. Nakaya, M. Saitow, A. Ishiguro, T. Yanai, J. Onoe and K. Tanaka, J. Am. Chem. Soc., 2022, 144, 6749–6758 CrossRef CAS PubMed.
- (a) Q.-H. Guo, Y. Qiu, M.-X. Wang and J. F. Stoddart, Nat. Chem., 2021, 13, 402–419 CrossRef CAS PubMed; (b) T. A. Schaub, E. A. Prantl, J. Kohn, M. Bursch, C. R. Marshall, E. J. Leonhardt, T. C. Lovell, L. N. Zakharov, C. K. Brozek, S. R. Waldvogel, S. Grimme and R. Jasti, J. Am. Chem. Soc., 2020, 142, 8763–8775 CrossRef PubMed; (c) H. Zhu, I. Badía-Domínguez, B. Shi, Q. Li, P. Wei, H. Xing, M. C. R. Delgado and F. Huang, J. Am. Chem. Soc., 2021, 143, 2164–2169 CrossRef CAS; (d) W. Fan, T. Matsuno, Y. Han, X. Wang, Q. Zhou, H. Isobe and J. Wu, J. Am. Chem. Soc., 2021, 143, 15924–15929 CrossRef CAS PubMed; (e) M. Krzeszewski, H. Ito and K. Itami, J. Am. Chem. Soc., 2022, 144, 862–871 CrossRef CAS PubMed; (f) K. Li, Z. Xu, J. Xu, T. Weng, X. Chen, S. Sato, J. Wu and Z. Sun, J. Am. Chem. Soc., 2021, 143, 20419–20430 CrossRef CAS PubMed.
- (a) P. Chen and F. Jäkle, J. Am. Chem. Soc., 2011, 133, 20142–20145 CrossRef CAS; (b) P. Chen, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2012, 51, 7994–7998 CrossRef CAS PubMed; (c) P. Chen, X. Yin, N. Baser-Kirazli and F. Jäkle, Angew. Chem., Int. Ed., 2015, 54, 10768–10772 CrossRef CAS PubMed; (d) N. Baser-Kirazli, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2020, 59, 8689–8697 CrossRef CAS PubMed; (e) N. Baser-Kirazli, R. A. Lalancette and F. Jäkle, Organometallics, 2021, 40, 520–528 CrossRef CAS; (f) D. Shimoyama, N. Baser-Kirazli, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2021, 60, 17942–17946 CrossRef CAS PubMed; (g) A. Ito, Y. Yokoyama, R. Aihara, K. Fukui, S. Eguchi, K. Shizu, T. Sato and K. Tanaka, Angew. Chem., Int. Ed., 2010, 49, 8205–8208 CrossRef CAS; (h) F. P. Gabbaï, Angew. Chem., Int. Ed., 2012, 51, 6316–6318 CrossRef; (i) X.-Y. Wang, F.-D. Zhuang, R.-B. Wang, X.-C. Wang, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2014, 136, 3764–3767 CrossRef CAS PubMed; (j) M. Iyoda and H. Shimizu, Chem. Soc. Rev., 2015, 44, 6411–6424 RSC; (k) A. Ito, J. Mater. Chem. C, 2016, 4, 4614–4625 RSC; (l) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed; (m) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed; (n) Y. Li, A. Yagi and K. Itami, J. Am. Chem. Soc., 2020, 142, 3246–3253 CrossRef CAS PubMed; (o) H. Sato, R. Suizu, T. Kato, A. Yagi, Y. Segawa, K. Awaga and K. Itami, Chem. Sci., 2022, 13, 9947–9951 RSC.
- (a) A. Ito, Y. Yamagishi, K. Fukui, S. Inoue, Y. Hirao, K. Furukawa, T. Kato and K. Tanaka, Chem. Commun., 2008, 6573–6575 RSC; (b) I. Kulszewicz-Bajer, V. Maurel, S. Gambarelli, I. Wielgus and D. Djurado, Phys. Chem. Chem. Phys., 2009, 11, 1362–1368 RSC; (c) D. Sakamaki, A. Ito, K. Furukawa, T. Kato and K. Tanaka, Chem. Commun., 2009, 4524–4526 RSC; (d) T.-F. Yang, K. Y. Chiu, H.-C. Cheng, Y. W. Lee, M. Y. Kuo and Y. O. Su, J. Org. Chem., 2012, 77, 8627–8633 CrossRef CAS PubMed; (e) D. Sakamaki, A. Ito, Y. Tsutsui and S. Seki, J. Org. Chem., 2017, 82, 13348–13358 CrossRef CAS PubMed; (f) L. Skorka, P. Kurzep, T. Chauviré, L. Dubois, J.-M. Mouesca, V. Maurel and I. Kulszewicz-Bajer, J. Phys. Chem. B, 2017, 121, 4293–4298 CrossRef CAS PubMed; (g) W. Wang, C. Chen, C. Shu, S. Rajca, X. Wang and A. Rajca, J. Am. Chem. Soc., 2018, 140, 7820–7826 CrossRef CAS PubMed.
- (a) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed; (b) A. G. Bonn and O. S. Wenger, J. Org. Chem., 2015, 80, 4097–4107 CrossRef CAS PubMed; (c) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC; (d) K. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2017, 139, 18170–18173 CrossRef CAS PubMed; (e) C. Li, Y. Liu, Z. Sun, J. Zhang, M. Liu, C. Zhang, Q. Zhang, H. Wang and X. Liu, Org. Lett., 2018, 20, 2806–2810 CrossRef CAS; (f) S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC; (g) Z.-B. Sun, J.-K. Liu, D.-F. Yuan, Z.-H. Zhao, X.-Z. Zhu, D.-H. Liu, Q. Peng and C.-H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 4840–4846 CrossRef CAS; (h) Z. Huang, S. Wang, R. D. Dewhurst, N. V. Ignat’ev, M. Finze and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 8800–8816 CrossRef CAS; (i) A. S. Scholz, J. G. Massoth, M. Bursch, J.-M. Mewes, T. Hetzke, B. Wolf, M. Bolte, H.-W. Lerner, S. Grimme and M. Wagner, J. Am. Chem. Soc., 2020, 142, 11072–11083 CrossRef CAS PubMed; (j) Y. Fu, H. Yang, Y. Gao, L. Huang, R. Berger, J. Liu, H. Lu, Z. Cheng, S. Du, H.-J. Gao and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 8873–8879 CrossRef CAS; (k) B. Adelizzi, P. Chidchob, N. Tanaka, B. A. G. Lamers, S. C. J. Meskers, S. Ogi, A. R. A. Palmans, S. Yamaguchi and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 16681–16689 CrossRef CAS PubMed; (l) X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed; (m) P.-F. Zhang, J.-C. Zeng, F.-D. Zhuang, K.-X. Zhao, Z.-H. Sun, Z.-F. Yao, Y. Lu, X.-Y. Wang, J.-Y. Wang and J. Pei, Angew. Chem., Int. Ed., 2021, 60, 23313–23319 CrossRef CAS PubMed; (n) N. Ando, T. Yamada, H. Narita, N. N. Oehlmann, M. Wagner and S. Yamaguchi, J. Am. Chem. Soc., 2021, 143, 9944–9951 CrossRef CAS; (o) L. Fritze, M. Fest, A. Helbig, T. Bischof, I. Krummenacher, H. Braunschweig, M. Finze and H. Helten, Macromolecules, 2021, 54, 7653–7665 CrossRef CAS; (p) M. Chen, K. S. Unikela, R. Ramalakshmi, B. Li, C. Darrigan, A. Chrostowska and S.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 1556–1560 CrossRef CAS PubMed; (q) J.-K. Li, X.-Y. Chen, Y.-L. Guo, X.-C. Wang, A. C.-H. Sue, X.-Y. Cao and X.-Y. Wang, J. Am. Chem. Soc., 2021, 143, 17958–17963 CrossRef CAS PubMed; (r) J. Guo, Y. Yang, C. Dou and Y. Wang, J. Am. Chem. Soc., 2021, 143, 18272–18279 CrossRef CAS; (s) Y. Zhang, D. Zhang, T. Huang, A. J. Gillett, Y. Liu, D. Hu, L. Cui, Z. Bin, G. Li, J. Wei and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 20498–20503 CrossRef CAS; (t) S. Oda, B. Kawakami, Y. Yamasaki, R. Matsumoto, M. Yoshioka, D. Fukushima, S. Nakatsuka and T. Hatakeyama, J. Am. Chem. Soc., 2022, 144, 106–112 CrossRef CAS; (u) Q. Zhu, S. Wang and P. Chen, Org. Lett., 2019, 21, 4025–4029 CrossRef CAS PubMed; (v) J.-F. Chen, X. Yin, B. Wang, K. Zhang, G. Meng, S. Zhang, Y. Shi, N. Wang, S. Wang and P. Chen, Angew. Chem., Int. Ed., 2020, 59, 11267–11272 CrossRef CAS; (w) G. Ji, N. Wang, X. Yin and P. Chen, Org. Lett., 2020, 22, 5758–5762 CrossRef CAS; (x) C. Li, Y. Shi, P. Li, N. Zhang, N. Wang, X. Yin and P. Chen, Org. Lett., 2021, 23, 7123–7128 CrossRef CAS PubMed; (y) F. Zhao, J. Zhao, Y. Wang, H.-T. Liu, Q. Shang, N. Wang, X. Yin, X. Zheng and P. Chen, Dalton Trans., 2022, 51, 6226–6234 RSC; (z) X. Tian, J. Guo, W. Sun, L. Yuan, C. Dou and Y. Wang, Chem.–Eur. J., 2022, 28, e202200045 CrossRef CAS PubMed.
- P. Li, D. Shimoyama, N. Zhang, Y. Jia, G. Hu, C. Li, X. Yin, N. Wang, F. Jäkle and P. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200612 CAS.
- (a) H.-T. Feng, Y.-X. Yuan, J.-B. Xiong, Y.-S. Zheng and B. Z. Tang, Chem. Soc. Rev., 2018, 47, 7452–7476 RSC; (b) J. Yu, C. Tang, X. Gu, X. Zheng, Z.-Q. Yu, Z. He, X.-G. Li and B. Z. Tang, Chem. Commun., 2020, 56, 3911–3914 RSC; (c) X. Zhang, H. Liu, G. Zhuang, S. Yang and P. Du, Nat. Commun., 2022, 13, 3543 CrossRef CAS.
- G. Wang, E. Dmitrieva, B. Kohn, U. Scheler, Y. Liu, V. Tkachova, L. Yang, Y. Fu, J. Ma, P. Zhang, F. Wang, J. Ge and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202116194 CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (c) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (d) R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CrossRef CAS.
- (a) W.-M. Wan, D. Tian, Y.-N. Jing, X.-Y. Zhang, W. Wu, H. Ren and H.-L. Bao, Angew. Chem., Int. Ed., 2018, 57, 15510–15516 CrossRef CAS PubMed; (b) H. Huang, L. Liu, J. Wang, Y. Zhou, H. Hu, X. Ye, G. Liu, Z. Xu, H. Xu, W. Yang, Y. Wang, Y. Peng, P. Yang, J. Sun, P. Yan, X. Cao and B. Z. Tang, Chem. Sci., 2022, 13, 3129–3139 RSC; (c) P. Li, Y. Jia, S. Zhang, J. Di, N. Zhang and P. Chen, Inorg. Chem., 2022, 61, 3951–3958 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis and characterization, DFT and TD-DFT calculations and others. See https://doi.org/10.1039/d2sc03581b |
| This journal is © The Royal Society of Chemistry 2022 |