
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning reactivity in trimetallic dual-atom alloys: molecular-like electronic states and ensemble effects†
Shengjie
Zhang
a,
E. Charles H.
Sykes
 b and
Matthew M.
Montemore
*a
b and
Matthew M.
Montemore
*a
aDepartment of Chemical and Biomolecular Engineering, Tulane University, New Orleans, LA 70118, USA. E-mail: mmontemore@tulane.edu
bDepartment of Chemistry, Tufts University, Medford, MA 02155, USA
First published on 18th November 2022
Abstract
Single-atom alloys (SAAs) have drawn significant attention in recent years due to their excellent catalytic properties. Controlling the geometry and electronic structure of this type of localized catalytic active site is of fundamental and technological importance. Dual-atom alloys (DAAs) consisting of a heterometallic dimer embedded in the surface layer of a metal host would bring increased tunability and a larger active site, as compared to SAAs. Here, we use computational studies to show that DAAs allow tuning of the active site electronic structure and reactivity. Interestingly, combining two SAAs into a dual-atom site can result in molecular-like hybridization by virtue of the free-atom-like electronic d states exhibited by many SAAs. DAAs can inherit the weak d–d interaction between dopants and hosts from the constituent SAAs, but exhibit new electronic and reactive properties due to dopant–dopant interactions in the DAA. We identify many heterometallic DAAs that we predict to be more stable than either the constituent SAAs or homometallic dual-atom sites of each dopant. We also show how both electronic and ensemble effects can modify the strength of CO adsorption. Because of the molecular-like interactions that can occur, DAAs require a different approach for tuning chemical properties compared to what is used for previous classes of alloys. This work provides insights into the unique catalytic properties of DAAs, and opens up new possibilities for tailoring localized and well-defined catalytic active sites for optimal reaction pathways.
1. Introduction
Transition metals and alloys are common active sites in heterogeneous catalysts; however, due to the complexity of supported heterogeneous catalysts, atomic-scale design of stable alloy active sites with high catalytic performance for a particular reaction remains difficult. Many catalytic surfaces exhibit a roughly mean-field behavior that restricts their ability to overcome linear scaling relations that can limit catalyst performance.1–4 As an important class of single-atom catalysts,5,6 single-atom alloys (SAAs) feature isolated metal atom dopants in the surface of a different host metal, which have been shown to deviate from scaling relations through differences in binding site preference of the transition states and intermediates7,8 or through spillover.9 In recent years, notable progress in catalysis has been made with this strategy.10,11 Furthermore, localized electron density at the dopant as characterized by free-atom-like d states are considered to play an important role in some SAAs' chemical reactivity.12 These free-atom-like states result from the minimal interaction between the d states of the dopant atom and the host metal and manifest on the dopant atom as a narrow d state compared to the pure bulk material.Although SAAs have shown excellent catalytic performance for many reactions, some larger reactants or intermediates and multi-step reactions may require an active site larger than one atom. Supported dual-atom catalysts, which provide larger active sites than single-atom catalysts, have been shown to enable enhanced catalytic performance for many reactions.13–15 Currently, most studies focus on homo-dual-atom catalysts, including Fe,16 Pt,17 Co18 and Cu19 dimers in carbon-based materials and on other supports. The success of supported dual-atom catalysts suggests that dual-atom alloys (DAAs) may also have interesting chemical properties. Indeed, previous work has suggested that dimers or small localized clusters of the same element can have electronic structures more akin to localized molecular orbitals, which has important implications for reactivity.20–22 However, these monometallic dimers offer a limited structure space and would be difficult to create experimentally in metal surfaces because most SAAs are thermodynamically stable with respect to aggregation, i.e. dimerization of the dopants.
Hetero-dual-atom catalysts contain atomic pairs of two different elements in the support. This brings improved tunability and possibly easier synthesis of the dual atom sites because thermodynamic mixing enthalpies can drive the formation of the heteroatom pairs. Both computational and experimental studies have demonstrated that this type of surface structure can bring unique catalytic properties.23 So far, hetero-dual atom catalysts have been reported in carbon-based materials,24–26 metal oxides27 and metal organic frameworks (MOFs),28,29 but studies of heterodimers in a metal host to form a trimetallic alloy are lacking.21,30 These DAAs would likely have desirable catalytic properties based on analogies to both SAAs and dual-atom catalysts. Herein, we elucidate unique and fundamental properties of dual-atom sites in metal hosts, particularly in their electronic structure. Specifically, we raise the fundamental question: if two dopants exhibit free-atom-like d states as SAAs, will the combination of these electronic states in a dual-atom site lead to hybridization and formation of molecular-like states on the dopant atom pair, analogous to the formation of molecular orbitals when two atoms are within bonding distance (Fig. 1(a) and (b))?
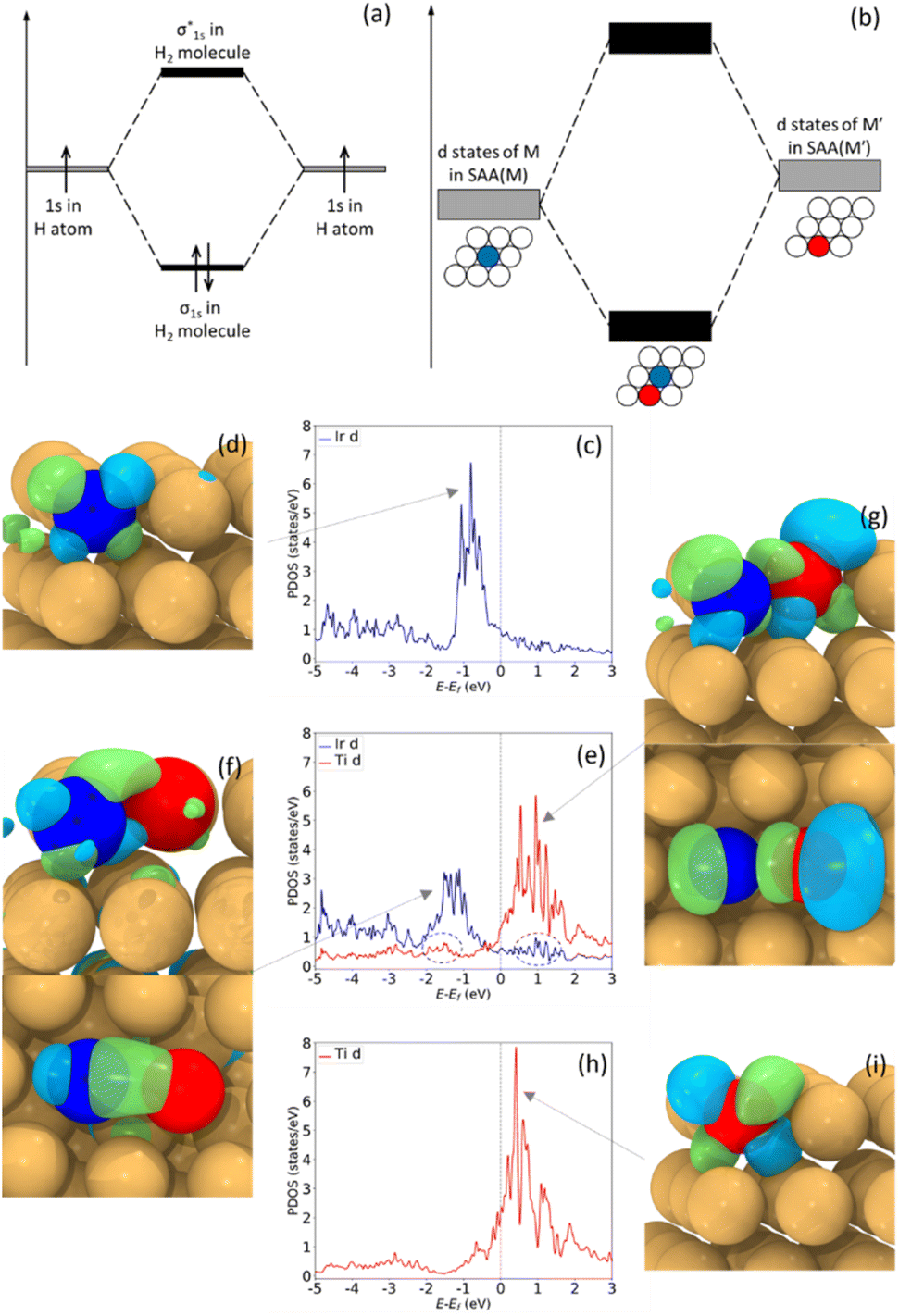 | ||
| Fig. 1 (a) H2 molecular orbitals result from combining two H 1s orbitals. (b) Molecular-like states may result from combining free-atom-like d states of two different dopants in a metal host. (c–i) pDOS of d states and wavefunctions of Ir1Cu (c and d), Ir1Ti1Cu (e–g), and Ti1Cu (h and i). (c) and (h) show narrowed d states of Ir and Ti atoms in their SAA form in a Cu host. (e) shows shifted and broadened d states and two new small peaks, all due to Ir–Ti hybridization in the Ir1Ti1Cu DAA. (d) and (i) are dxz orbitals of Ir at −0.8 eV and of Ti at 0.4 eV. The combination of (d) and (i) result in a d–d π orbital (f) at −1.7 eV and a d–d π* orbital (g) at 1.1 eV. | ||
More generally, controlling the geometric and electronic structure of catalytic active sites is of fundamental and technological importance. Often, reactivity of alloy surfaces is interpreted in terms of ensemble and electronic effects. Ensemble effects describe the changes in catalytic properties due to the local structure and composition of the active site.31 Electronic effects (or ligand effects) refer to the changes in the electronic structure of the active site caused by metal–metal interactions, usually associated with hybridization and/or charge transfer.32 In most cases, both effects exist in an alloy surface and can be difficult to separate because modifying the surface structure or composition typically also affects the electronic structure. Computational33,34 and experimental35 studies on these two effects have been reported. These studies focused on bimetallic systems with different dopant concentrations. It has been shown that ensemble effects dominate when a bimetallic alloy consists of a strongly adsorbing metal and a weakly adsorbing metal, such as Rh, Pd and Pt alloyed with Au, Ag and Cu, with electronic effects becoming more important when both metals bind adsorbates strongly.34
Herein, we demonstrate that combining the dopant atoms of two different SAAs into a dual-atom site can result in molecular-like hybridization. Many of these dual-atom sites are predicted to be stable by consideration of the alloying energetics. We demonstrate that the molecular-like hybridization has a significant influence on the reactivity of the dual site, as demonstrated by changes in the binding strength of CO. Even in dual-atom sites without significant rehybridization, ensemble effects appear to affect the binding of CO to the active site. In this way we demonstrate cases in which ensemble or electronic effects can affect the reactivity of heterometallic dual-atom sites. This provides a strategy to controllably tune the catalytic properties and opens up new possibilities in the design of catalytic active sites with tunable electronic structure in a way that is qualitatively different from alloys that exhibit a more mean-field behavior.
2. Method
All periodic, plane-wave density functional theory (DFT)36,37 calculations were performed with the Vienna Ab initio Simulation Package (VASP)38,39 using the Perdew–Burke–Ernzerhof generalized gradient (GGA-PBE)40 exchange-correlation functional with the Tkatchenko–Scheffler method41 for van der Waals interactions. The cutoff energy in this work was set to 400 eV. A 7 × 7 × 1 Monkhorst–Pack k-point mesh42 was used for relaxation calculations with a 19 × 19 × 1 mesh for non-self-consistent projected density of states (pDOS) calculations. Different initial spin states for each dopant-host system were tested to determine the most stable one. The post-calculation analysis (pDOS and real-space wavefunctions generated from the planewave coefficients of a Kohn–Sham orbital) was done by VASPKIT43 and the wavefunctions were visualized by VMD.44In this work, we used 3 × 3 × 4 unit cells to model the (111) surfaces of face-centered cubic (FCC) structures, with the top two layers and adsorbates relaxed and the bottom two layers fixed. The lattice constant calculations and convergence tests were done in our previous work.21 A ∼16 Å vacuum gap was set along the z direction. The formation energy of a dimer in a trimetallic alloy, Eform, was calculated as:
| Eform = Etri + Ehost − ESAA(M) − ESAA(M′) | (1) |
For the CO adsorption studies, CO was initially placed either on top of a dopant or at the bridge site (μ2-CO) between two dopants. The CO adsorption energy, Eads, was calculated as:
| Eads = Etot − Etri(or ESAA) − ECO(g) | (2) |
3. Results and discussion
3.1 Molecular states in a dual-atom site
To gain insight into molecular-like electronic states in DAAs, we first study Ir1Cu, Ti1Cu, and the dual site Ir1Ti1Cu, as our calculations suggest that Ir1Cu and Ti1Cu form stable SAAs with free-atom-like d states on the Ir and Ti atoms respectively. For example, in the Ir1Cu SAA, the Ir d states have a sharp peak centered at ∼−0.8 eV (Fig. 1(c)), while the neighboring Cu has a broad d band from −1.5 to −4.0 eV (Fig. S1(a)†). Although the Ir d states and Cu d band have some minor overlap between −2 to −5 eV, the narrow d states indicate Ir has minimal interaction with Cu. The free-atom-like electronic structure of Ir is also supported by the symmetry-resolved pDOS, where the t2g and eg states of Ir show negligible splitting, as in Fig. S1(c).† The calculated real-space wavefunction of one state at −0.8 eV clearly shows it is primarily composed of the dxz orbital of Ir (Fig. 1(d)). Although there is some contribution from Cu, the interaction is small and the clover shape of the d orbital is clear. Some other representative wavefunctions are shown in Fig. S2.†Although the Ir–Cu d–d interaction is weak, the Ir–Cu interaction is not zero; otherwise Ir would not be bonded to Cu and would not be stable in the alloy surface. In particular, there is significant sp hybridization between Ir and Cu, as s and p orbitals tend to be more delocalized than d states (Fig. S1(g)†). This sp interaction likely stabilizes Ir in the surface. However, d states generally have a larger effect on variations in adsorption and reaction energies,45 and as such their behavior is crucial in determining surface chemistry.
Similar to Ir1Cu, Ti atoms exhibit free-atom-like d-states in Ti1Cu (Fig. 1(h)). The d states of Ti are somewhat broader than those of Ir, although there is almost no overlap between the Ti d states and the Cu d band from −1 to 3 eV (Fig. S1(b)†). One possible reason for the somewhat broader states is that the Ti d states have some interaction with Cu s or p states with similar energies. The Ti–Cu d–d interaction is also visible around −2 to −5 eV, but is much weaker compared to Ir. The symmetry-resolved pDOS also indicates degenerate Ti t2g and eg states (Fig S1(d)†). Unlike Ir, we find that the Ti d states lie almost completely above the Fermi level. The wavefunction of one state at 0.4 eV shows the Ti dxz orbital retains its shape, similar to the Ir case (Fig. 1(i)).
Therefore, in the Ir1Cu and Ti1Cu SAAs, the isolated Ir and Ti atoms each have free-atom-like electronic structure for their d states. This raises the question of whether we can tune the chemical properties by creating a Ir1Ti1Cu dual-atom site without interfering with the minimal interaction of the Ir and Ti d states with Cu. Additionally, if we combine Ir and Ti into a DAA site, will Ir and Ti retain their free-atom-like electronic structures, or will there be a strong Ir–Ti interaction leading to a different electronic structure and chemical reactivity? In a molecular-orbital picture, hybridization between two states results in a new state higher in energy than the original states, and a new state lower in energy than the original states (Fig. 1(a) and (b)). This same behavior is observed for the Ir1Ti1Cu DAA shown in Fig. 1(e): the narrow Ir d-state peak shifts down while the narrow Ti d-state peak shifts slightly up. In both cases, the states broaden somewhat, as would be expected from tight-binding.46,47 The Ti d states are also observed in the Ir pDOS as a small peak at ∼1.2 eV, the same position as Ti's most intense DOS peak. Similarly, there is a small peak at ∼ −1.7 eV in the Ti pDOS, the same position as Ir's largest DOS peak. These electronic interactions and subsequent changes in the shape and d-state location as compared to the two isolated atoms provide clear evidence for a significant interaction between Ir and Ti, demonstrating that it is possible to create a DAA site with new electronic, and hence catalytic, properties based on two SAAs. This interaction also leads to significant changes in the CO adsorption energies on the top site of each dopant, which will be discussed in Section 3.3.
The localized interaction between Ir and Ti in Ir1Ti1Cu is also confirmed by the spatial extent of the wavefunctions. Fig. 1(f) shows the wavefunction of one state at −1.7 eV, which shows the Ir dxz orbital extending to Ti to form a d–d π bond. Since the d states of Ir have shifted to lower energy, in Fig. 1(f) we can also find more significant contributions from Cu. However, though the energies of Ir and Cu d states are closer than in the SAA, the wavefunctions have little spatial overlap because Cu's contributions are mainly from lower-energy states. This suggests that the interaction between Ir and Cu in the Ir1Ti1Cu DAA is still quite small. At 1.2 eV, the wavefunction shows a well-defined d–d π* bond made of Ir and Ti dxz orbitals (Fig. 1(g)), which has the same shape as in Fig. 1(f), but out of phase. This result indicates that the overlaps at −1.7 eV and 1.2 eV are molecular-like bonding and antibonding states resulting from Ir–Ti hybridization and state splitting. There are very few contributions from Cu to the antibonding state, again indicating that the Cu host metal does not participate significantly in the hybridization.
In contrast to the nearly complete degeneracy of the t2g and eg states in the SAAs in Fig. S1(c) and (d),† there are deviations between SAA and dual-atom states for both Ir and Ti after combination in the dual-atom site (Fig. S1(e) and (f)†). Ir's t2g states near −1.8 eV shift to lower energies, while Ti's t2g states near 2.0 eV shift to higher energies. These results are consistent with the wavefunction showing hybridization between dxz orbitals; further, the states' splitting shifts the bonding states to lower energies and the antibonding states to higher energies. Bonding and antibonding wavefunctions contributed by other orbitals are shown in Fig. S2(d)–(f) and (g)–(i),† including d–d π and δ (anti)bonding orbitals. Therefore, the case of Ir1Ti1Cu demonstrates that dual-atom sites in trimetallic alloys can have unique electronic properties that differ from either of the bulk alloys or SAAs and resemble the molecular orbitals formed by two interacting atoms. This raises the questions: how many other trimetallic combinations are likely to form dual-atom sites, and what is the chemical reactivity of these sites?
3.2 Stability screening: predictions of synthesizable dual-atom sites
If heterometallic DAA sites are not thermodynamically stable with respect to other configurations, they will be difficult to experimentally synthesize or maintain under reaction conditions. Previous work on single-atom alloys has suggested that aggregation is a crucial factor in determining stability.51 To predict whether specific dual-atom sites can be synthesized, we performed DFT calculations to study the formation energies of a range of dimers in various hosts. A DAA site can either be a homodimer or heterodimer. We are particularly interested in heterodimers because for surface alloys, it is experimentally difficult to control the selective formation of homodimers, and one is typically left with a mix of single atoms and a variety of larger ensembles. That is, for alloy surfaces, it is difficult to selectively create dimers, as either single atoms or larger ensembles will often be favored, depending on the thermodynamics of the specific system.11,52 We propose two criteria to determine whether a particular heterodimer–host combination is likely to be synthesizable experimentally. First, the formation energy Eform for a heterodimer (M ≠ M′) in eqn (1) should be negative. This means that the heterodimer is energetically favored over the two separate SAAs. Second, the heterodimer formation energy should be more negative than the homodimer formation energy. This ensures a dopant will tend to form a heterodimer with the other dopant metal rather than forming homodimers with itself.Based on previous work,48,49 we chose Pd1Ag, Pt1Ag, Pd1Au and Ti1Cu as the starting bimetallic systems (i.e., first dopant M′ = Pd, Pt or Ti). Many metals were tested as the second dopant (i.e., M = Co, Cr, Fe, Hf, etc.) to be added to the four bimetallic systems. The formation energies of each trimetallic are plotted in Fig. 2. The light pink regions signify trimetallics for which either the heterodimer formation energy is positive, or it is not as negative as homodimer formation energy. The dopants in these areas are unlikely to form a significant number of heterodimers. In the white areas, though heterodimers are favored, there still could be small amounts of homodimers. The other area (shaded in light blue) signifies stable heteroatom dual-atom sites but unstable homodual-atom sites and SAAs. It is clear from this survey that there are many DAAs that are likely to be thermodynamically stable and hence synthesizeable. For example, Sc, Mn, Sn, Mg, Cr, Cd and Zn are promising to form M-Pd heterodimers when added to a Pd1Ag SAA (Fig. 2(a)). Sc shows the strongest tendency for selective heterodimer formation, while the others are closer to the origin. Hf has the most negative heterodimer formation energy, though its homodimer formation energy is also slightly negative.
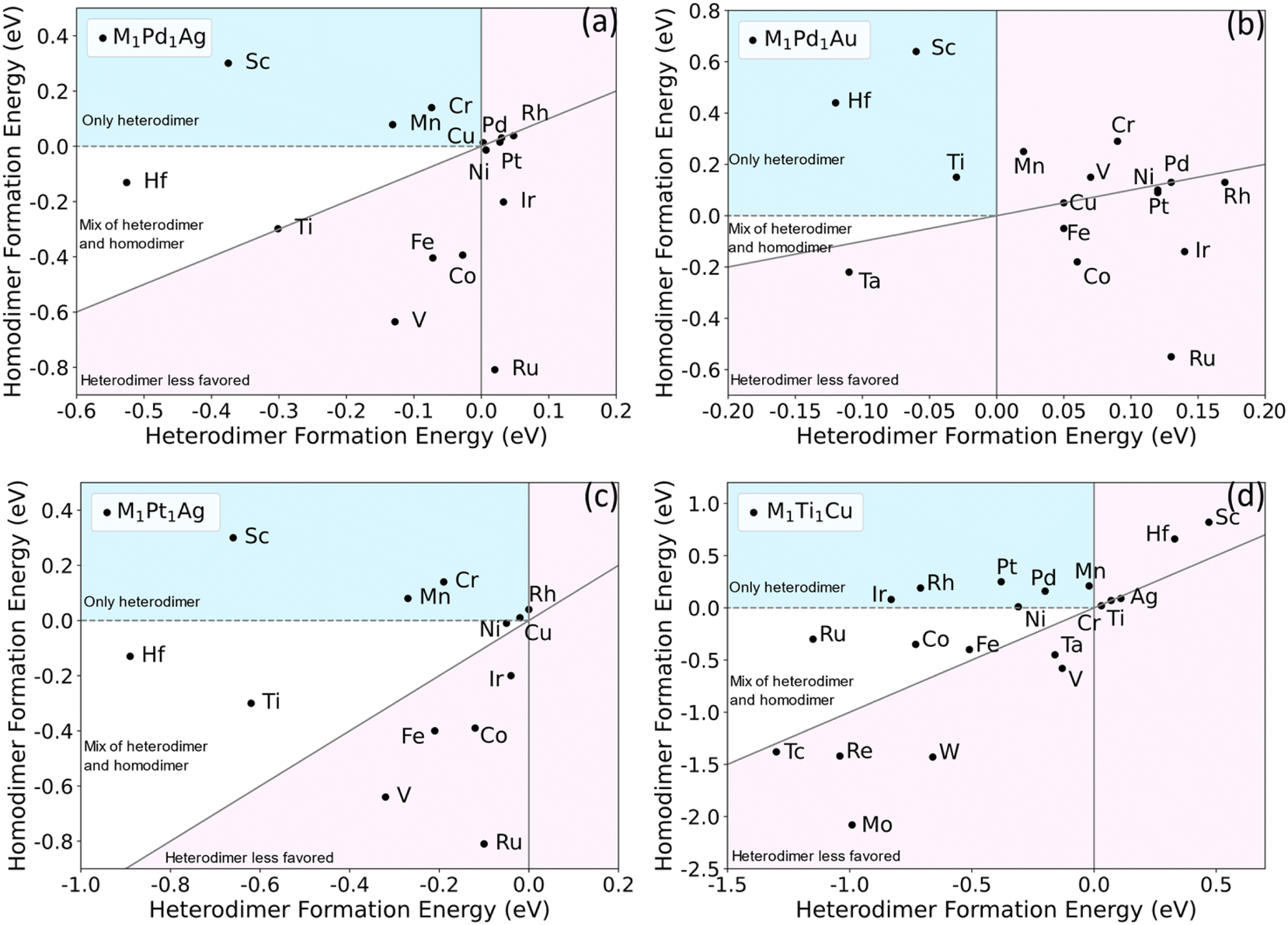 | ||
| Fig. 2 Screening of homodimer and heterodimer formation energies for M1Pd1Ag (a), M1Pd1Au (b), M1Pt1Ag (c) and M1Ti1Cu (d). The dopants in light blue areas are predicted to only form heterodimers; those in the white area are predicted to give predominantly heterodimers with some homodimers; those in the light pink area are predicted to form few or no heterodimers. Some dopants are omitted due to their high homodimer formation energies, resulting in values far below the diagonals. Complete results are listed in Table S1.† | ||
Qualitatively, M1Pt1Ag exhibits the same stabilities as M1Pd1Ag, in that the second dopants in M1Pt1Ag have similar energetics to M1Pd1Ag. Considering the similarity between Pd and Pt, this suggests that similar dopant metals, e.g., those in the same group, are likely to give qualitatively similar dimer formation energetics in a given host. For M1Pd1Au, the metals reside within a small area near the origin, and most dopants have qualitatively similar positions as M1Pd1Ag. For example, in Fig. 2(a) and (b), Sc and Hf are on the top left, while Pd, Rh, Pt, Ir and Ru are on the right side from top to bottom. On the other hand, some metals change position significantly, e.g., Ti and V. For M1Ti1Cu, both host and the first dopant M′ have been changed significantly, and the plots seem to be quite different from the other three. In this case, Ir, Rh, Pt, Pd and Mn are the most promising to form heterodimers. For both homodimers and heterodimers, the formation energies span a wider range and seem more correlated to each other than the other three systems, i.e., the homodimer and heterodimer energies roughly scale with each other for M1Ti1Cu.
From inspection of the data, we hypothesize that the strength of correlation between the homodimer and heterodimer formation energies is related to the host metal reactivity. In inert hosts, such as Ag and Au, the dopants are more likely to exhibit free-atom like electronic structures;50 therefore, the position of each element on the screening map is more dependent on the details of the dopant–dopant hybridization. In more reactive hosts, such as Cu, the dopants can more easily electronically hybridize with the host. This leads to more metallic behavior in most cases; therefore, the difference in dimerization energy between homodimers and heterodimers is reduced. Hence, in more reactive hosts that tend to electronically mix with dopants, the formation energies of homodimers and heterodimers are likely to be more correlated and may not be as likely to show a strong preference for heterodimers over homodimers.
Our two criteria—that the heterometallic dual-atom site be more stable than the corresponding SAAs and homometallic dual-atom sites—provide a convenient method to predict experimentally synthesizable hetero-dual-atom sites. Similar criteria for SAAs have proven quite successful in guiding experiments.51 However, the presence of these dual-atom sites in a given experiment depends on other factors as well, such as entropy and the effect of adsorbates on stability. For instance, some dual sites can be stabilized by bridge-bound CO molecules.52 For those dopants that have some tendency towards aggregation, keeping the SAAs as dilute as possible is key, as increasing the dopant concentration will lead to the formation of trimers and tetramers. Furthermore, in certain cases some energetically unfavorable structures can be observed in experiments through kinetic trapping.53 Therefore, while our results are a useful guide to possible dual-atom sites, changing dopant concentrations, temperature, and the reactive atmosphere will also affect the stability of these sites and this is best examined on a case-by-case basis experimentally.
For the dopant atoms to significantly affect catalytic performance, they must reside in the surface layer under reaction conditions. Previous work suggests that for Cu-, Ag-, and Au-based SAAs, most dopant atoms prefer the surface layer over the subsurface when adsorbed species are present, as would be expected under reaction conditions.57 As an additional test, we compared the stability of an Ir1Ti1 dimer in the subsurface layer and the surface layer of Cu, with and without an adsorbed CO molecule. Without CO, the dimer is 0.07 eV more stable in the subsurface, but in the presence of adsorbed CO the dimer is more stable in the surface by 1.19 eV. Therefore, while surface segregation will depend strongly on the conditions, it is likely that many of the dimers will prefer the surface over the subsurface given the appropriate reaction conditions.
3.3 Chemical properties of dual-atom sites: CO adsorption
To investigate the chemical reactivity of the dual-atom sites that are likely to be stable, we calculated the CO adsorption energy Eads for dopants that meet the two criteria when added to Pd1Ag, Pd1Au and Ti1Cu. We use CO adsorption as a simple probe for chemical reactivity because (1) CO adsorption on various surfaces has been extensively studied;54,55 (2) CO is involved in many SAA-catalyzed reactions, and the adsorption energies are often related to reaction barriers and hence catalytic performance;3 (3) strong CO adsorption often leads to catalyst poisoning, which greatly affects their performance.56,57 In addition to the adsorption energy, the CO stretching frequency is often used to study the surface environment and active site.58 Therefore, CO is a widely used probe both in experiment and theoretically, and the study of CO adsorption is a simple but effective approach to shed light on the potential catalytic properties of alloy surfaces.To understand ensemble and electronic effects, we compared CO adsorption on these dual-atom sites to CO adsorption on the corresponding SAAs. For the SAAs, the CO was placed on the top site of the dopant (Eads(SAA)), while for dual-atom sites, we calculated the CO adsorption energy on the top site of each dopant (Eads(DAA,M)) and Eads(DAA,M′)), and in the bridge site between the two dopants (Eads(DAA,Bri)), as shown in Fig. 3(a). By comparing top-site adsorption energies between the SAA and the dual-atom site, we can probe electronic effects, and by examining the bridge-site adsorption energies we can probe ensemble effects (Fig. 3(b)).
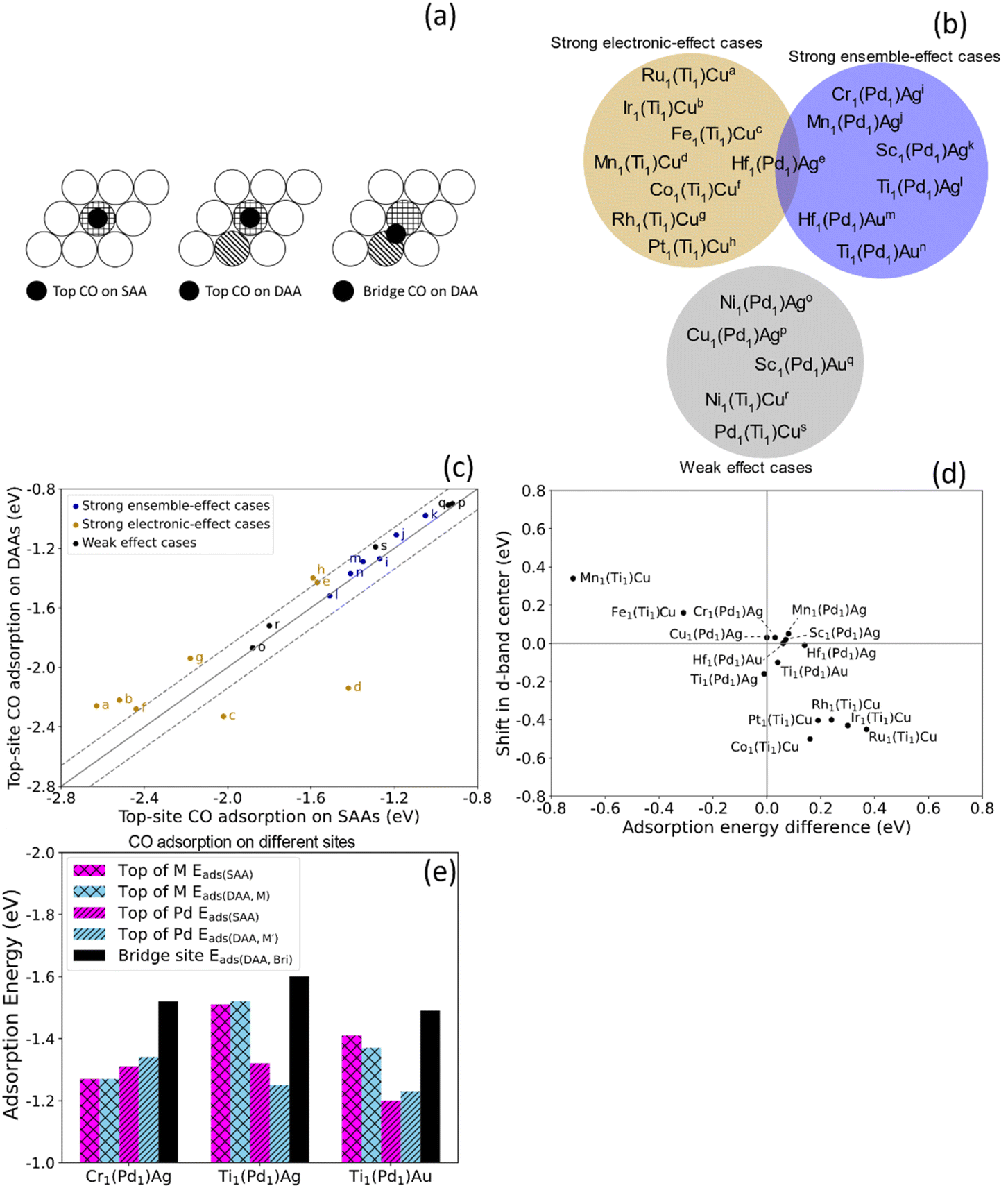 | ||
| Fig. 3 (a) Top views of CO on a top adsorption site of M in SAA and DAAs, and on a bridge site between M and M'. (b) M1Pd1Ag, M1Pd1Au, and M1Ti1Cu trimetallic alloys from the white and light blue areas in Fig. 2 were divided into three groups based on the CO adsorption energies in Table S2.† The alloys in the dark yellow area have strong electronic effects while the alloys in the blue area have strong ensemble effects. The alloys in the gray area show weaker electronic and ensemble effects. (c) CO adsorption energy on top of the dopant in SAAs (Eads(SAA)) vs. in DAAs (Eads(DAA,M)). The labels correspond to the alloys in (b). The ensemble-effect cases are close to the diagonal, indicating little difference in the top-site adsorption energies. The strong electronic-effect cases (|(Eads(DAA,M)) − Eads(SAA)| > 0.14 eV; outside dashed lines) exhibit a large deviation from the diagonal, indicating large changes in reactivity. (d) Trend for CO top-site adsorption energy difference and shift in d-band center. All strong effect cases listed in (b) are shown. Some weak-effect cases are omitted. The inverse correlation agrees with the d-band model. (e) CO adsorption energies on different sites. For these three ensemble-effect cases, while there is a only small difference between Eads(SAA) and Eads(DAA,M), the Eads(DAA,Bri) is stronger. | ||
While ensemble and electronic effects coexist, we focus on the cases where one of them dominates the change in CO adsorption energy between the SAA and the corresponding dual-atom site. We first look at the cases dominated by electronic effects, i.e., cases where the CO adsorption energy atop the dopant atom changes significantly due to the dopant–dopant interaction described in Section 3.1. Above we discussed how Ir1Ti1Cu exhibits significant hybridization between Ir and Ti. This results in an increase in the top-site CO adsorption energy on Ir of 0.30 eV, as compared to the Ir1Cu SAA. In addition to this case, which we denote Ir1(Ti1)Cu to indicate CO adsorption atop the Ir atom in this dual atom alloy, there are significant changes in CO adsorption energy for Ru1(Ti1)Cu, Fe1(Ti1)Cu, and Mn1(Ti1)Cu, etc. Specifically, |(Eads(DAA,M)) − Eads(SAA)| > 0.14 eV for these cases, meaning that CO binds much stronger or weaker to the top-site of the dual site than the isolated atom. In Fig. 3(c), we show these cases, as well as other cases that have similar top-site adsorption energies between the SAA and the DAA, which indicates minor electronic effects. Ru1(Ti1)Cu and Ir1(Ti1)Cu are above the diagonal, indicating stronger top-site CO adsorption on the SAAs, while Fe1(Ti1)Cu and Mn1(Ti1)Cu are below the diagonal, indicating stronger adsorption on the DAAs.
To understand why dopant–dopant hybridization affects top-site adsorption energies significantly in some cases but not in others, we examined the electronic structure changes between the dual-atom sites and the corresponding SAAs. Cases where the top-site CO adsorption energy changes significantly also show large changes in their electronic structure. That is, changes in top-site CO adsorption energies between SAA and DAA sites can be traced back to differences in the electronic structure of the sites. The pDOS of M d states of Ru1(Ti1)Cu, Fe1(Ti1)Cu and Mn1(Ti1)Cu are shown in Fig. S3† (Ir1(Ti1)Cu was shown in Fig. 1). Ru1(Ti1)Cu is similar to Ir1(Ti1)Cu, also showing broadened d states in the dual-atom site, indicating increased hybridization. The shifts in d-band center from SAAs to dual-atom sites are negative (to values further below Ef), at −0.45 and −0.43 eV for Ru in Ru1(Ti1)Cu and Ir in Ir1(Ti1)Cu, respectively. For Fe1(Ti1)Cu and Mn1(Ti1)Cu, which have nonzero spin, the spin-up states are narrower in the dual-atom sites, while the spin-down states are broader. The overall shifts in d-band center for these two are positive (toward Ef), 0.16 and 0.34 eV, suggesting that the d states in these particular dual-atom sites become more free-atom-like.
To more quantitatively examine the link between adsorption energy changes and electronic structure changes, we studied the correlation between the shift in d-band center and the top-site adsorption energy difference (Eads(DAA,M) − Eads(SAA)). Fig. 3(d) shows an inverse correlation between these quantities; i.e., a shift of the d-band towards Ef results in stronger adsorption.46,47 Cases with little change in top-site adsorption energy are very close to the origin, indicating small changes in electronic structure and small changes in top-site adsorption energies, consistent with our description above. Two electronic-effect cases are in the upper left quadrant and the other two are in the lower right quadrant in Fig. 3(d). These results suggest that electronic effects can either lead to more free-atom-like d-states, shifts in the d-band center towards Ef, and stronger adsorption (e.g., Mn and Fe); or to less free-atom-like states, shifts away from Ef, and weaker adsorption (e.g., Ru and Ir). Qualitatively, these results are broadly consistent with the d-band model,46,47 and generally the M1(Ti1)Cu alloys have larger differences in adsorption energy and d-band center than the M1(Pd1)Ag and M1(Pd1)Au alloys.
Thus, the interaction between dopant atoms makes it possible to change the electronic structure and hence tune the reactivity of each atom in the dual site. At the same time, ensemble effects can also affect the reactivity in cases where the electronic effects are small. When ensemble effects dominate, CO adsorption on the top sites of the dual-atom site is similar in strength to CO adsorption on the corresponding SAA top sites (Eads(SAA) ≈ Eads(DAA,M)), because the small electronic interaction between the dopant atoms leads to almost the same chemical reactivity of the individual atoms. At the same time, ensemble effects can lead to CO adsorption on the bridge site in M1M′1H being stronger than either top site (i.e., Eads(DAA,Bri) is lower than Eads(DAA,M) and Eads(DAA,M′)). Cr1(Pd1)Ag, Ti1(Pd1)Ag and Ti1(Pd1)Au all show strong ensemble effects but weak electronic effects. Compared with the electronic-effect cases in Fig. 3(b), these ensemble-effect cases have negligible deviations from the diagonal. Fig. 3(e) shows the CO adsorption energy on these three surfaces, among which Cr1(Pd1)Ag has the smallest difference between Eads(SAA) and Eads(DAA,M), as well as the largest decrease from Eads(DAA,M) and Eads(DAA,M′) to Eads(DAA,Bri), indicating ensemble effects have the most influence on Cr1(Pd1)Ag. The pDOS of M d states of the three ensemble-effect cases generally show small differences between the SAAs and the DAA (Fig. S4†). For Cr1(Pd1)Ag and Ti1(Pd1)Au in particular, although there are differences in the pDOS due to inevitable small electronic effects, the changes are very small. For Ti1(Pd1)Ag, on the other hand, the d states are more prominent for the SAA than the dual-atom site, and small DOS peaks appear around −2 to −3 eV, indicating somewhat increased hybridization. However, the overall band shape and position for Ti1(Pd1)Ag are not drastically altered, which is consistent with the small change in top-site adsorption energy. Therefore, small changes in electronic structure result in small changes to reactivity.
Whether the top site or the bridge site is favored for CO adsorption on a DAA depends on the nature of the dopants, the host, and their interactions. Our calculations show that one cannot predict the site preference only based on CO adsorption on the corresponding SAA and homodimer. For example, CO prefers the bridge site in some homodimers (Cr2Ag, Mn2Ag, Ti2Au, Rh2Cu, Mn2Cu, etc.) but the preference in the corresponding DAAs varies: the bridge site is favored for Cr1Pd1Ag, Mn1Pd1Ag, Ti1Pd1Au, while the top site is favored for Rh1Ti1Cu and Mn1Ti1Cu. The calculations indicate the top site is generally preferred when the two top-site adsorption energies in a DAA are very different, while either the bridge site or the top site can be preferred if the adsorption energies are close. However, a detailed understanding of the site preference of CO in all cases is beyond the scope of this work.
Compared to what is known for bimetallics,33,34 our calculations demonstrate that there are additional considerations that must be taken into account to understand the reactivity of trimetallic DAAs. Although our host metals (Cu, Ag, and Au) are weakly adsorbing and most heterometallic dimers embedded in host metals exhibit a mixture of ensemble and electronic effects, some cases show obvious ensemble effects, like Cr1Pd1Ag, while electronic effects can dominate in other cases, like Ru1(Ti1)Cu. Electronic hybridization can result in electronic structures that resemble the molecular orbitals of gas phase molecules. This demonstrates that even in a trimetallic alloy, which conventional wisdom would suggest should have delocalized electronic states, it is possible to form what could be termed a “metal molecule”, e.g., the Ir1Ti1 pair site in Cu which exhibits bonding and antibonding localized molecular states as seen in Fig. 1(f) and (g). We note that strong electronic effects do not always coincide with a strong bond between the two dopants, as measured by our stability calculations, but instead only indicate a large change in the CO top-site adsorption energy, which tends to correlate with the change in the d states. Our example in Section 3.1 shows free-atom-like dopants in SAAs and strong electronic interaction between the two dopants in DAAs, resulting in molecular-like electronic states. However, the details of hybridization between different atoms, orbitals, and spin channels can be complex in some cases. This can lead to the counterintuitive result of more free-atom-like d orbitals in DAAs as compared to SAAs (e.g., Mn1Ti1Cu vs. Mn1Cu). Another aspect that we did not study in detail here is the contribution of s and p bands to either dopant–dopant bonding or to hybridization. While they usually show less influence on molecular adsorption than d bands, in some cases s and p bands can play an important role in the electronic structure of alloys and adsorption energies.21,59
4. Conclusions
As a first example, we computationally studied the electronic structure of the Ir1Ti1Cu trimetallic surface, showing that molecular-like electronic states can be obtained in a DAA created from the thermodynamically favorable combination of two SAAs. The pDOS and wavefunctions illustrate that Ir1Cu and Ti1Cu SAAs have free-atom-like states, while in the Ir1Ti1 DAA site there is hybridization between some of these localized Ir and Ti d states resulting in hybridization into a molecular-like “orbital” that is highly localized at the dual-atom site. We then identified many other thermodynamically preferred dual-atom sites that are more stable than either isolated dopants or homodimers, and are therefore likely to be synthesizable. Our results show that when both SAAs contain dopants with free-atom-like electronic states, the newly formed dual-atom sites often inherit the minimal interaction between the dopant and the host metal, while molecular-like bonding and antibonding states arise from hybridization and state splitting within the heterodimer itself. In most close-packed alloy surfaces, changing one of the nine nearest neighbors of a surface atom would not be expected to have a large electronic effect; however, combining “electronically insoluble” atoms, that is dopant atoms with d states that do not hybridize with their host, can result in molecular-orbital-like interactions between dopants that can significantly affect the electronic structure of the dual-atom active site. This modifies the CO binding strength of each top site of the dual-atom pair. In other cases, the creation of a new bridge site between the dopant atom pair can bind CO strongly. Importantly, our results reveal that, depending on the dopant, moving from the SAA case to the DAA site can either weaken or strengthen CO adsorption, highlighting the tunable chemistry offered by this approach.Overall, the DAA sites described in this work have unique structural and electronic properties that are expected to offer new and useful catalytic properties that differ from either traditional bimetallic alloys or SAAs. These metal dual-atom active sites have electronic structures that are somewhat analogous to molecules, in which atomic orbital overlap leads to localized and quantized bonding and antibonding states, which in turn define the chemical properties of the molecule. The catalytic properties of DAA sites can be tuned by controlling their ensemble and electronic effects, providing a new strategy for catalytic active site design. Furthermore, study of these new dilute trimetallic alloy materials should provide new fundamental insights into electronic and ensemble effects. Given the large combinatorial structure space of trimetallics, there are many other possible DAA sites to consider. By combining atoms with different kinds of chemical reactivity (e.g., oxophilic vs. carbophilic60,61) and further tuning their reactivity via electronic hybridization, DAA sites can be rationally designed for particular reactions of interest. Therefore, these DAA sites enable tuning of the electronic structure, and hence chemical reactivity, beyond what is possible in SAAs. Importantly, these DAA active sites preserve some of the important benefits of SAAs, such as a localized, well-defined active site with molecular binding energies that are different from the individual elements, while bringing much greater tunability than SAAs due to the much larger trimetallic compositional space.
Data availability
The data associated with this article is available in the ESI.†Author contributions
S. Z.: investigation, writing – original draft, writing – review & editing. E. C. H. S.: conceptualization, writing – review & editing. M. M. M.: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported as part of the Integrated Mesoscale Architectures for Sustainable Catalysis (IMASC), an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Basic Energy Sciences under Award #DE-SC0012573. This research was supported in part using high performance computing (HPC) resources and services provided by Technology Services at Tulane University, New Orleans, LA. Portions of this research were conducted with high performance computational resources provided by the Louisiana Optical Network Infrastructure (https://www.loni.org).References
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skúlason, T. Bligaard and J. K. Nørskov, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS PubMed.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS PubMed.
- M. M. Montemore and J. W. Medlin, Catal. Sci. Technol., 2014, 4, 3748–3761 RSC.
- A. Vojvodic and J. K. Nørskov, Natl. Sci. Rev., 2015, 2, 140–143 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- M. M. Montemore and J. W. Medlin, J. Phys. Chem. C, 2013, 117, 20078–20088 CrossRef CAS.
- C. F. Nwaokorie and M. M. Montemore, J. Phys. Chem. C, 2022, 126, 3993–3999 CrossRef CAS.
- F. R. Lucci, M. T. Darby, M. F. G. Mattera, C. J. Ivimey, A. J. Therrien, A. Michaelides, M. Stamatakis and E. C. H. Sykes, J. Phys. Chem. Lett., 2016, 7, 480–485 CrossRef CAS PubMed.
- G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Acc. Chem. Res., 2019, 52, 237–247 CrossRef CAS PubMed.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed.
- M. T. Greiner, T. E. Jones, S. Beeg, L. Zwiener, M. Scherzer, F. Girgsdies, S. Piccinin, M. Armbruster, A. Knop-Gericke and R. Schlogl, Nat. Chem., 2018, 10, 1008–1015 CrossRef CAS PubMed.
- Y. Zhou, E. Song, W. Chen, C. U. Segre, J. Zhou, Y.-C. Lin, C. Zhu, R. Ma, P. Liu and S. Chu, et al. , Adv. Mater., 2020, 32, 2003484 CrossRef CAS PubMed.
- Y. Ying, X. Luo, J. Qiao and H. Huang, Adv. Funct. Mater., 2021, 31, 2007423 CrossRef CAS.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- Z. He, K. He, A. W. Robertson, A. I. Kirkland, D. Kim, J. Ihm, E. Yoon, G.-D. Lee and J. H. Warner, Nano Lett., 2014, 14, 3766–3772 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu and W. Zhang, et al. , Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- M. Xiao, H. Zhang, Y. Chen, J. Zhu, L. Gao, Z. Jin, J. Ge, Z. Jiang, S. Chen and C. Liu, et al. , Nano Energy, 2018, 46, 396–403 CrossRef CAS.
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu and S.-F. Hung, et al. , Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed.
- R. A. Hoyt, M. M. Montemore, I. Fampiou, W. Chen, G. Tritsaris and E. Kaxiras, J. Chem. Inf. Model., 2019, 59, 1357–1365 CrossRef CAS PubMed.
- A. P. Monasterial, C. A. Hinderks, S. Viriyavaree and M. M. Montemore, J. Chem. Phys., 2020, 153, 111102 CrossRef CAS PubMed.
- T. D. Spivey and A. Holewinski, J. Am. Chem. Soc., 2021, 143, 11897–11902 CrossRef CAS PubMed.
- T. He, A. R. P. Santiago, Y. Kong, M. A. Ahsan, R. Luque, A. Du and H. Pan, Small, 2022, 18, 2106091 CrossRef CAS PubMed.
- Y. Li, Q. Zhang, C. Li, H.-N. Fan, W.-B. Luo, H.-K. Liu and S.-X. Dou, J. Mater. Chem. A, 2019, 7, 22242–22247 RSC.
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song and J. Li, et al. , Nat. Commun., 2019, 10, 4936 CrossRef PubMed.
- X. Peng, H.-X. Liu, Y. Zhang, Z.-Q. Huang, L. Yang, Y. Jiang, X. Wang, L. Zheng, C. Chang, C. Au, L. Jiang and J. Li, Chem. Sci., 2021, 12, 7125–7137 RSC.
- J. Fu, J. Dong, R. Si, K. Sun, J. Zhang, M. Li, N. Yu, B. Zhang, M. G. Humphrey and Q. Fu, et al. , ACS Catal., 2021, 11, 1952–1961 CrossRef CAS.
- W. Cheng, X. F. Lu, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2020, 59, 18234–18239 CrossRef CAS PubMed.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao and S. Wei, et al. , J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- J. W. M. Crawley, I. E. Gow, N. Lawes, I. Kowalec, L. Kabalan, C. R. A. Catlow, A. J. Logsdail, S. H. Taylor, N. F. Dummer and G. J. Hutchings, Chem. Rev., 2022, 122, 6795–6849 CrossRef CAS PubMed.
- J. W. A. Sachtler and G. A. Somorjai, J. Catal., 1983, 81, 77–94 CrossRef CAS.
- U. Schneider, H. Busse, R. Linke, G. R. Castro and K. Wandelt, J. Vac. Sci. Technol. A, 1994, 12, 2069–2073 CrossRef CAS.
- P. Liu and J. K. Nørskov, Phys. Chem. Chem. Phys., 2001, 3, 3814–3818 RSC.
- H. Li, K. Shin and G. Henkelman, J. Chem. Phys., 2018, 149, 174705 CrossRef PubMed.
- D. A. Slanac, W. G. Hardin, K. P. Johnston and K. J. Stevenson, J. Am. Chem. Soc., 2012, 134, 9812–9819 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kresse, J. Non-Cryst. Solids, 1995, 192–193, 222–229 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- G. O. Kayode, S. Zhang, and M. M. Montemore, Linking electronic structure to adsorption energies: Metal surfaces and single-atom catalysts, in Catalysis, ed. J. Spivey, Y.-F. Han and D. Shekhawat, The Royal Society of Chemistry, 2022, vol. 34 Search PubMed.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- B. Hammer, Y. Morikawa and J. K. Nørskov, Phys. Rev. Lett., 1996, 76, 2141–2144 CrossRef CAS PubMed.
- J. D. Lee, J. B. Miller, A. V. Shneidman, L. Sun, J. F. Weaver, J. Aizenberg, J. Biener, J. A. Boscoboinik, A. C. Foucher and A. I. Frenkel, et al. , Chem. Rev., 2022, 122, 8758–8808 CrossRef CAS PubMed.
- J. Shi, C. J. Owen, H. T. Ngan, S. Qin, V. Mehar, P. Sautet and J. F. Weaver, J. Chem. Phys., 2021, 154, 234703 CrossRef CAS PubMed.
- H. Thirumalai and J. R. Kitchin, Top. Catal., 2018, 61, 462–474 CrossRef CAS.
- R. T. Hannagan, G. Giannakakis, R. Réocreux, J. Schumann, J. Finzel, Y. Wang, A. Michaelides, P. Deshlahra, P. Christopher and M. Flytzani-Stephanopoulos, et al. , Science, 2021, 372, 1444–1447 CrossRef CAS.
- M. Ouyang, K. G. Papanikolaou, A. Boubnov, A. S. Hoffman, G. Giannakakis, S. R. Bare, M. Stamatakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Nat. Commun., 2021, 12, 1549 CrossRef CAS PubMed.
- B. Zugic, M. A. van Spronsen, C. Heine, M. M. Montemore, Y. Li, D. N. Zakharov, S. Karakalos, B. A. J. Lechner, E. Crumlin and M. M. Biener, et al. , J. Catal., 2019, 380, 366–374 CrossRef CAS.
- J. Hulva, M. Meier, R. Bliem, J. Z. akub, F. Kraushofer, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Science, 2021, 371, 375–379 CrossRef CAS PubMed.
- S. E. Mason, I. Grinberg and A. M. Rappe, J. Phys. Chem. C, 2008, 112, 1963–1966 CrossRef CAS.
- J. Liu, F. R. Lucci, M. Yang, S. Lee, M. D. Marcinkowski, A. J. Therrien, C. T. Williams, E. C. H. Sykes and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2016, 138, 6396–6399 CrossRef CAS PubMed.
- M. T. Darby, E. C. H. Sykes, A. Michaelides and M. Stamatakis, Top. Catal., 2018, 61, 428–438 CrossRef CAS PubMed.
- K. I. Hadjiivanov and G. N. Vayssilov, Advances in Catalysis, Academic Press, 2002, vol. 47, pp. 307–511 Search PubMed.
- M. M. Montemore and J. W. Medlin, J. Phys. Chem. C, 2014, 118, 2666–2672 CrossRef CAS.
- G. O. Kayode and M. M. Montemore, J. Mater. Chem. A, 2021, 9, 22325–22333 RSC.
- Y. Ouyang, L. Shi, X. Bai, Q. Li and J. Wang, Chem. Sci., 2020, 11, 1807–1813 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc03650a |
| This journal is © The Royal Society of Chemistry 2022 |