
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning through-space interactions via the secondary coordination sphere of an artificial metalloenzyme leads to enhanced Rh(III)-catalysis†
Isra S.
Hassan‡
a,
Jack T.
Fuller‡
b,
Vanessa N.
Dippon
a,
Angeline N.
Ta
c,
Michael W.
Danneman
a,
Brian R.
McNaughton§
*c,
Anastassia N.
Alexandrova
 *b and
Tomislav
Rovis
*a
*b and
Tomislav
Rovis
*a
aDepartment of Chemistry, Columbia University, New York, NY 10027, USA. E-mail: tr2504@columbia.edu
bDepartment of Chemistry & Biochemistry, University of California Los Angeles, Los Angeles, CA 90095, USA. E-mail: ana@chem.ucla.edu
cDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523, USA. E-mail: bmcnaughton@desu.edu
First published on 29th July 2022
Abstract
We report computationally-guided protein engineering of monomeric streptavidin Rh(III) artificial metalloenzyme to enhance catalysis of the enantioselective coupling of acrylamide hydroxamate esters and styrenes. Increased TON correlates with calculated distances between the Rh(III) metal and surrounding residues, underscoring an artificial metalloenzyme's propensity for additional control in metal-catalyzed transformations by through-space interactions.
Artificial metalloenzymes (ArMs) can be made by anchoring a non-natural (metal) cofactor into a protein scaffold, with the goal of imbuing new-to-nature reactivity.1 One of the most common ArM platforms is the biotin-tetrameric(strept)avidin (biotin-tSav) system pioneered by Whitesides and Ward.2,3 These ArMs utilize high-affinity (up to KD ∼10−14 M) interactions between tSav and biotin–metal conjugates. tSav-based ArMs have appeared in an increasing number of transition-metal catalyzed transformations.4–6 In collaboration with the Ward group, we have previously described a tetrameric streptavidin (tSav) system containing a biotinylated Rh(III) cofactor for the asymmetric synthesis of dihydroisoquinolones using benzhydroxamate esters and acrylate partners.7 Monomeric streptavidin (mSav), a streptavidin/rhizavidin hybrid designed to resist tetramerization, retains its high affinity for biotin (KD ∼10−9 M).8,9 We recently described the use of mSav as a new ArM,10 whose simpler topology encourages protein engineering via a site-directed mutagenesis approach.
Traditional manipulation of a metal's reactivity has been accomplished by modification of the electronic and steric properties of the bound ligands (Fig. 1a).11,12 For example, we have documented and parsed the impact of Cp electronics and sterics on a number of Rh(III) catalyzed transformations, by structural changes to the ligand in the primary coordination sphere of Rh.13 On the other hand, ArMs have traditionally been used as modifiers of a metal's steric environment largely focusing on inducing asymmetry in the bond-forming events. Less broadly appreciated is the fact that any mutations in residues proximal to the active site may also impact the metal's electronic properties via changes to the secondary coordination sphere (Fig. 1b), with the prospect of delivering more active catalysts for a given transformation.
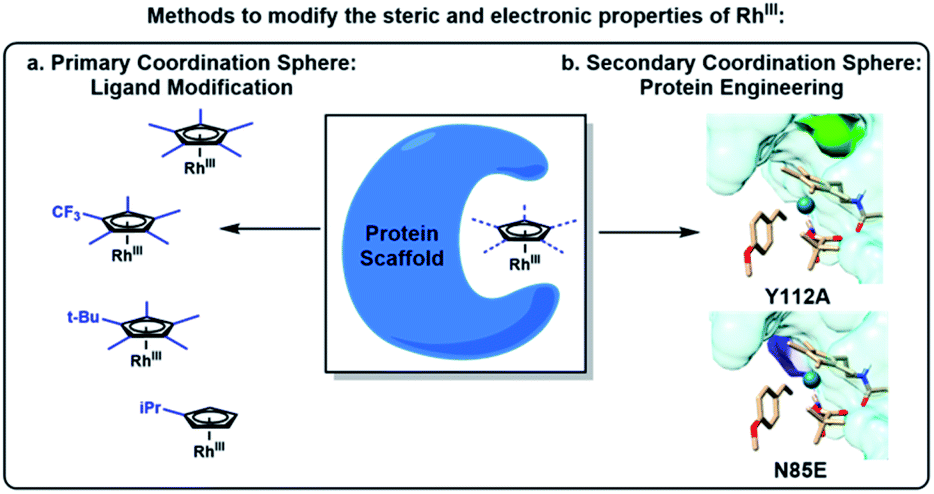 | ||
| Fig. 1 Methods to modify the (a) primary and (b) secondary coordination sphere of a Rh(III) catalyst. | ||
Previously, we described a mSav·Rh(III) catalyst and demonstrated its use in the direct enantioselective coupling of acrylamide hydroxamate esters and styrenes.10 The reaction allows rapid access to piperidines – the most common N-heterocycle found in FDA-approved pharmaceuticals.14 One of the most interesting aspects of this reaction was our observation of a 7-fold increase in turnover number (TON) by embedding the cofactor into mSav's active site.15 It has been a long-standing goal of ArMs to not only enable new-to-nature reactivity, but also for them to achieve the stellar kinetics of a native metalloenzyme. As these systems lack the evolutionary privilege of a natural metalloenzyme, extensive mutation of the protein scaffold may be required to find the optimal environment of the metal cofactor.
Predicting the effects of specific mutations can prove very challenging, as any alterations to the protein conformation and charge distribution can impact reactivity regardless of the mutation's distance from the active site.16–19 In order to design a better mutant, we embarked on a collaborative experimental and computational study to define the role of the protein scaffold and how single point mutations affect reactivity. We identified two key residues that play a pivotal role in mSav·Rh(III) ArM's secondary coordination sphere, and have used this insight to design a more active mutant.
For the purposes of this study, we focused on the mSav·Rh(III) ArM-catalyzed coupling of methacrylamide with 4-methoxystyrene as our model reaction (Fig. 2a). Using a small model of the catalyst, the lowest energy pathway of this reaction's proposed mechanism was generated (Fig. S9†). The calculations were performed in Turbomole20–32 with the M06 density functional.33 Geometries were optimized with the def2-SVP basis set, and final electronic energies were calculated with the def2-TZVP basis set.34 The conductor-like screening model (COSMO)35 was used as implicit solvent with a dielectric of 80 to simulate water. These calculations predicted similar barriers for the N–H activation, the C–H activation, and the migratory insertion (differences less than 3 kcal mol−1). Isotope-exchange experiments revealed that the C–H activation step is reversible, implicating the migratory insertion step as turnover-limiting.10
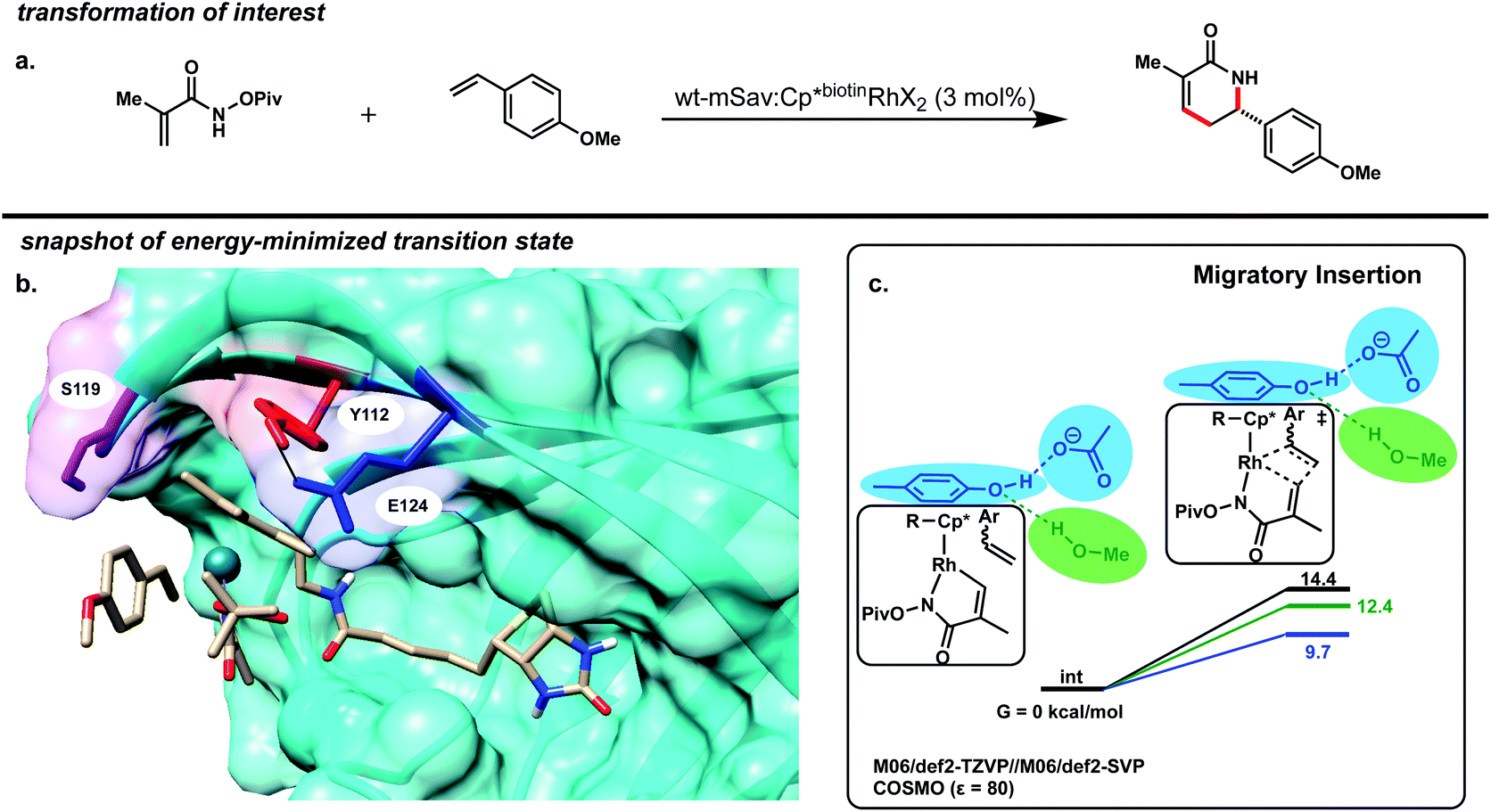 | ||
| Fig. 2 (a) Model transformation. (b) Snapshot of the transition state for alkene insertion illustrating key nearby residues Y112 (red), E124 (blue), and S119 (purple). (c) Computed barrier to alkene insertion in the presence and absence of phenol and acetate (shown in blue). | ||
The Cp* moiety of the Cp*biotinRhX2 cofactor is non-covalently localized in the active site likely due to a π–π stacking interaction with Y112 (Fig. 2b). This assignment is supported by the observation that mutant Y112A leads to lower yield and enantioselectivity.10 We hypothesized that we could further manipulate both the sterics and electronics of the Cp* moiety by either directly mutating Y112 or indirectly by mutating other residues that affect the Y112-Cp* interaction.
To generate a model of mSav's protein scaffold and active-site we used QM/DMD36 – a hybrid quantum mechanics/molecular mechanics method that simulates proteins piecewise. Discrete molecular dynamics (DMD) equilibrates the entire system except for the metal and part of the substrate.36 After a trajectory of ∼0.5 ns, quantum mechanics (QM) is used to optimize the metal region plus sidechains and residues immediately surrounding it. This process is repeated, providing efficient sampling of the entire protein scaffold while treating the metal environment quantum-mechanically. For this study, the migratory insertion transition state was modeled in WT by freezing the coordinates of the rhodium atom and the two carbon atoms forming a bond. For each system, five replicate simulations were run for ∼20 ns each.
Residues E124 and S119 both hydrogen bond to Y112 and are in close proximity to the RhCp* catalytic site (Fig. 2b).37 To estimate the electronic effects of these three residues on the reaction, an acetate ion, methanol molecule, and 4-methylphenol (p-cresol) molecule were added to a small catalyst model without constraints but initially positioned to mimic the sidechains of these residues (Fig. 2c). The migratory insertion energy barrier decreases by 2 kcal mol−1 with incorporation of the three residues. However, this energy barrier decreases by an additional 3 kcal mol−1 upon the deletion of the methanol molecule representing S119. Not only does this imply that these amino acid sidechains may be the primary reason for the increased activity of the protein-installed catalyst, but also suggest that a longer Y112–S119 distance is favorable, so long as no water can insert in this region and replace S119 in its H-bond with Y112. We hypothesize that the carboxylate group of E124 acts as a hydrogen bond acceptor, donating electron density to the Y112 phenol ring, which in turn donates electron density to the catalyst via π–π charge transfer. This could enhance the electron donation of the metal and decrease the energy barrier to the migratory insertion step. On the other hand, S119 acts as a hydrogen bond donor which would remove electron density from Y112 and subsequently the Rh(III) moiety.
Unfortunately, mutation of Y112 (Y112F and Y112W) results in negligible protein yields. We thus identified three flanking residues (T111, E113, H87) that may be expected to have a significant impact on Y112's position, and one distal (T32) residue, chosen as distal mutations sometimes have significant impact (Fig. 3). Through this subset of mutants, we attempted to increase TON and establish a correlation between the Y112–Rh distance and Y112–S119 distance of the mutants and their reactivities.
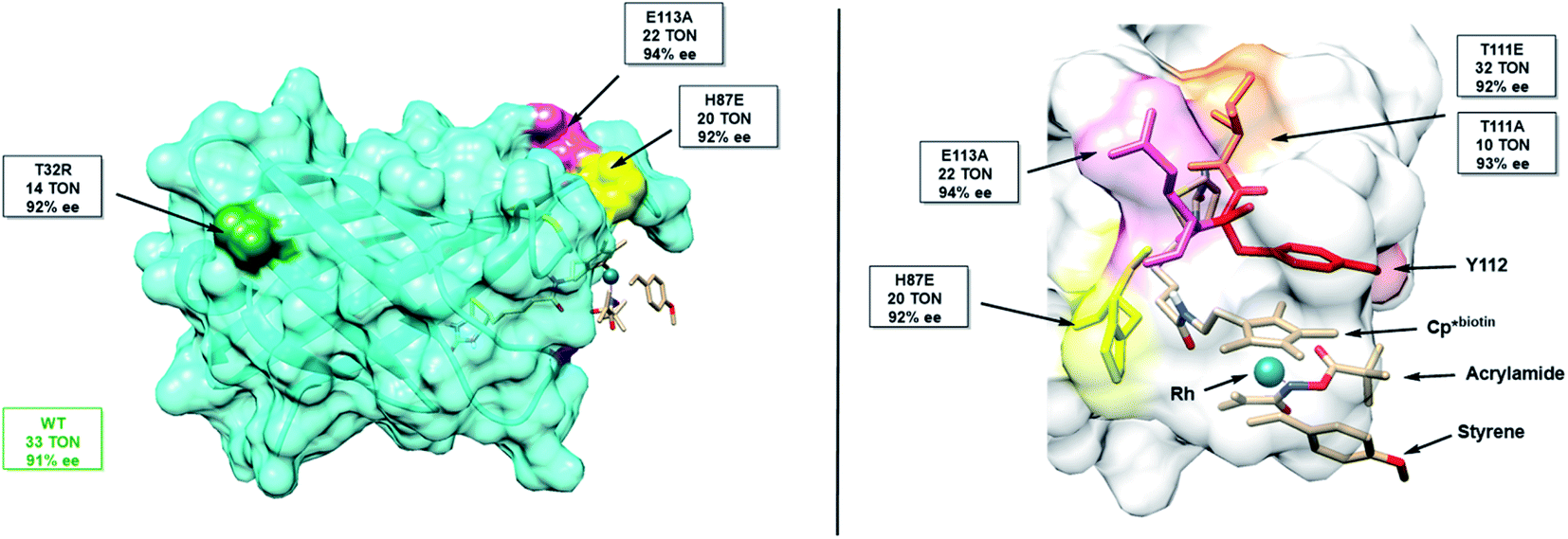 | ||
| Fig. 3 Structure of mSav from two different views highlighting some of the mutated residues including their TON and enantioselectivity. | ||
We used QM/DMD to simulate a representative set of these mutants spanning a wide range of TONs measured in the experiment. The Y112–Rh and Y112–S119 distances were measured every ∼0.5 ps for every simulation. The results can be represented by a 3-dimensional plot with Y112–Rh distance on the X axis, Y112–S119 distance on the Y axis, and probability density on the Z axis (Fig. 4). We find the best correlation between TON and probability density in the conformational region where the Y112–Rh distance is the shortest and the Y112–S119 interaction is not energetically relevant.38
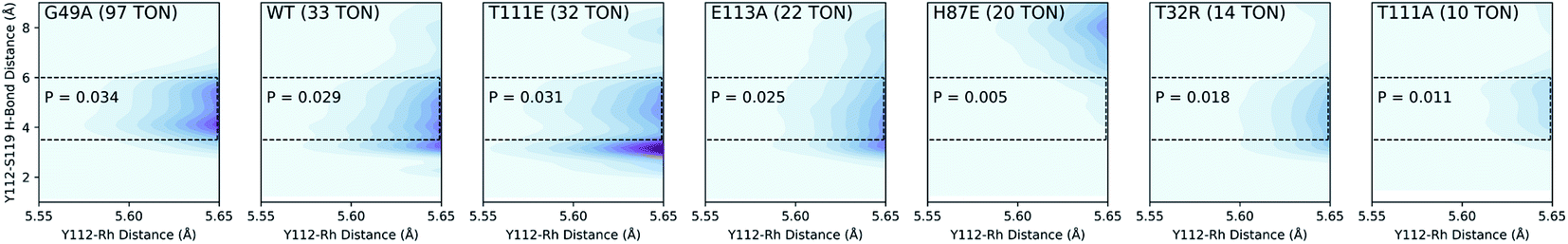 | ||
| Fig. 4 Three-dimensional probability distributions from select mutants by simultaneous sampling of Rh–Y112 and S119–Y112 distances. Probabilities for the outlined regions are also shown. | ||
To clarify this correlation, we calculated the probability of having a Y112–S119 distance between 3.5–6 Å and a Y112–Rh distance less than 5.65 Å. This Y112–S119 distance corresponds to negligible hydrogen bonding.39 Additionally, we constrained the small model catalyst shown in Fig. 5b (ref. 40) and calculated the corresponding energy barriers at different Y112–Rh distances (Fig. 5a). Since rate increases exponentially as the barrier decreases,35 differences in probabilities in the region where the Y112–Rh distance is between 5.4–5.65 Å have the greatest impact on the relative TONs of our model methacrylamide styrene coupling. We conclude that mutants with increasing probability in this region provide increasing TON.
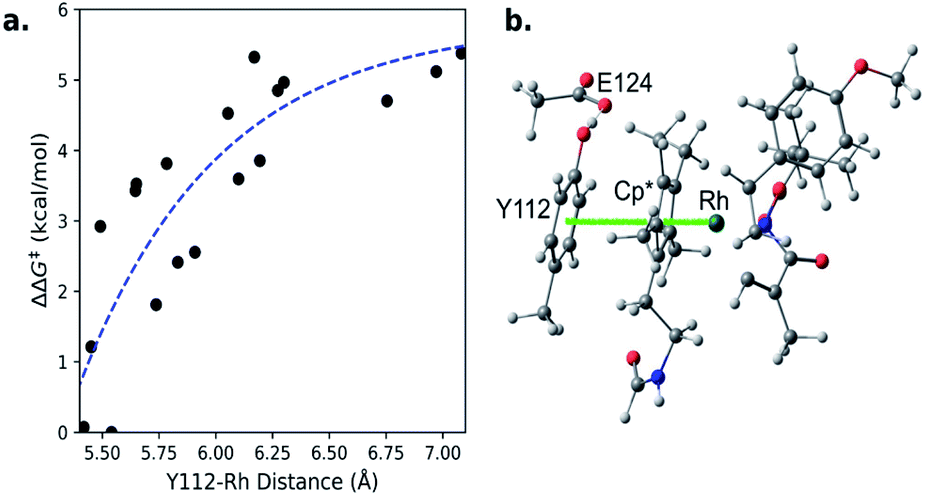 | ||
| Fig. 5 a) Theoretical dependence of migratory insertion barrier on Rh-phenol distance. (b) Small-model catalyst. | ||
Theoretically, a shorter Y112–Rh distance relative to WT would result in increased reactivity. Residue G49 is located under the Rh(III) moiety (Fig. 6). We hypothesize that by mutating the glycine into an alanine, steric congestion would force the biotinylated Rh(III) cofactor to shift upwards closer to the electron donating phenol side chain of residue Y112. Analyzing the critical portions of the Y112–Rh and Y112–S119 distances in tandem reveals that G49A has the highest probability density in this region (Fig. 4). Indeed, experimentally, this mutant gives 97 TON and 91% ee (Fig. 6). The combination of a short Y112–Rh distance and long Y112–S119 distance leads to an increase in reactivity. This is an approximate 3-fold improvement in the TON relative to WT. The G49A mutant serves as an experimental proof of concept that a computational analysis of an ArMs secondary coordination sphere can lead to the design of a more efficient ArM.
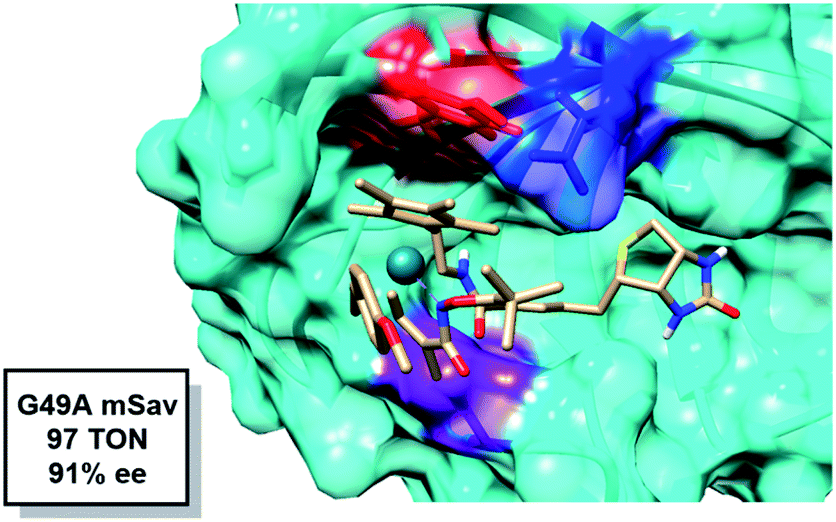 | ||
| Fig. 6 Snapshot of the transition state for alkene insertion highlighting the position of G49 (purple) relative to Rh. Y112 is shown in red and E124 is shown in blue. | ||
In summary, we have identified three key residues that contribute to accelerating the rate of a Rh(III)-catalyzed reaction by electronic communication to the metal via the secondary coordination sphere. E124 hydrogen bonds to Y112 transferring electron density via π–π charge transfer, an effect that is attenuated by hydrogen bonding from S119. Optimal interaction of these residues can be described computationally by finding mutants that have multiple conformations bearing short Y112–Rh distances coupled with negligible bonding between Y112 and S119. This hypothesis was experimentally verified by a mutant that enforces a closer Y112–Rh distance leading to improved TON. This result demonstrates the use of a hypothesis-based site-directed mutagenesis of the secondary sphere residues, to optimize the metal's electronic environment within the protein scaffold and enhance an ArM's activity.
Author contributions
B. R. M., A. N. A. and T. R. conceived the study. I. S. H., V. N. D., A. N. T. and M. W. D. designed and executed the experiments. J. T. F. and A. N. A. designed and executed the computational studies. All authors contributed to writing the manuscript, and given it final approval.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
NIGMS grants GM80442 to T. R., GM107520, B. R. M., and 1R01GM134047 to A. N. A., NSF predoctoral fellowship to I. S. H., Societe de Chemie summer fellowship to V. N. D., and UCLA-IDRE cluster Hoffman2 and XSEDE are acknowledged.Notes and references
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Leburn, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142 CrossRef CAS.
- M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306 CrossRef CAS.
- M. Skander, N. Humbert, J. Collot, J. Gradinaru, G. Klein, A. Loosli, J. Sauser, A. Zocchi, F. Gilardoni and T. R. Ward, J. Am. Chem. Soc., 2004, 126, 14411 CrossRef CAS PubMed.
- C. Letondor, N. Humbert and T. R. Ward, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4683 CrossRef CAS PubMed.
- J. Zhao, D. C. Bachmann, M. Lenz, D. G. Gillingham and T. R. Ward, Catal. Sci. Technol., 2018, 8, 2294 RSC.
- M. Hestericova, T. Heinisch, L. Alonso-Cotchico, J.-D. Marechal, P. Vidossich and T. R. Ward, Angew. Chem., Int. Ed., 2018, 57, 1863 CrossRef CAS PubMed.
- T. K. Hyster, L. Knorr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS PubMed.
- D. Demonte, C. M. Dundas and S. Park, Appl. Microbiol. Biotechnol., 2014, 98, 6285 CrossRef CAS.
- M. H. Qureshi and S. L. Wong, Protein Expression Purif., 2002, 25, 409 CrossRef CAS PubMed.
- I. S. Hassan, A. N. Ta, M. W. Danneman, N. Semakul, M. Burns, C. H. Basch, V. N. Dippon, B. R. McNaughton and T. Rovis, J. Am. Chem. Soc., 2019, 141, 4815 CrossRef CAS PubMed.
- R. L. Lucas, M. K. Zart, J. Murkerjee, T. N. Sorrell, D. R. Powell and A. S. Borovik, J. Am. Chem. Soc., 2006, 128, 15476 CrossRef CAS PubMed.
- H. Jung, M. Schrader, K. Dongwook, M. Baik, Y. Park and S. Chang, J. Am. Chem. Soc., 2019, 141, 15356 CrossRef CAS PubMed.
- T. Piou, F. Romanov-Michailidis, M. Romonova-Michaelides, K. E. Jackson, N. Semakul, T. D. Taggart, B. S. Newell, C. D. Rithner, R. S. Paton and T. Rovis, J. Am. Chem. Soc., 2017, 139, 1296 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- A 15% yield was observed using 3 mol% of the biotinylated Rh(III) cofactor (5 TON) while a 99% yield was obtained using 3 mol% of the corresponding ArM (33 TON).
- A. Shimotohno, S. Oue, T. Yano, S. Kuramitsu and R. Kagamiyama, J. Biochem., 2001, 129, 943 CrossRef CAS PubMed.
- B. Spiller, A. Gershenson, F. H. Arnold and R. Stevens, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12305 CrossRef CAS PubMed.
- M. W. Fraaije, J. Wu, D. P. Heuts, E. W. van Hellemond, J. H. Spelberg and D. B. Janssen, Biotechnol, 2005, 66, 393 CAS.
- P. A. Romero and F. H. Arnold, Nat. Rev. Mol. Cell Biol., 2009, 10, 866 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- M. Häser and R. Ahlrichs, J. Comput. Chem., 1989, 10, 104 CrossRef.
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119 Search PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136 CrossRef CAS.
- P. Deglmann, K. May, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2004, 384, 103 CrossRef CAS.
- P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511 CrossRef CAS.
- P. Deglmann and F. Furche, J. Chem. Phys., 2002, 117, 9535 CrossRef CAS.
- M. v. Arnim and R. Ahlrichs, J. Comput. Chem., 1998, 19, 1746 CrossRef.
- M. v. Arnim and R. Ahlrichs, J. Chem. Phys., 1999, 111, 9183 CrossRef.
- R. Ahlrichs, Phys. Chem. Chem. Phys., 2004, 6, 5119 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 5, 799 RSC.
- M. Sparta, D. Shirvanyants, F. Ding, N. V. Dokholyan and A. N. Alexandrova, Biophys. J., 2012, 103, 767 CrossRef CAS.
- F. Ding, D. Tsao, H. Nie and N. V. Dokholyan, Structure, 2008, 16, 1010 CrossRef CAS PubMed.
- Deletion of E124 (E124A) results in a significant decrease in reactivity (TON 7, 90% ee). We considered similar tests of S119 (S119A, e.g.) but conformational analysis of these changes revealed detrimental structural alterations arising from modifications of the 1,2-loop bearing the S119 residue, and these were not further pursued.
- We note an additional factor that may affect reactivity: the active site is partially exposed to solvent, potentially allowing water to hydrogen bond with Y112 instead of S119. Longer Y112–S119 distances may mean the active site is too exposed to water. We hypothesize this is why correlation degrades if the probability density is integrated beyond 6 Å for the Y112–S119 distance, although this improves correlation in the case of the H87E mutant.
- G. A. Jeffrey An introduction to hydrogen bonding. Oxford University Press, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03674f |
| ‡ These authors contributed equally. |
| § Present address: Delaware Institute for Science & Technology, Delaware State University, Dover, DE 19901. |
| This journal is © The Royal Society of Chemistry 2022 |