
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the selectivity descriptors of heterogeneous metal phthalocyanine electrocatalysts for hydrogen peroxide production†
Yubo
Yuan
,
Huan
Li
,
Zhan
Jiang
,
Zhichao
Lin
,
Yirong
Tang
,
Hongxuan
Wang
and
Yongye
Liang
 *
*
Department of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: liangyy@sustech.edu.cn
First published on 9th September 2022
Abstract
The electrocatalytic 2e− oxygen reduction reaction (2e− ORR) provides an appealing pathway to produce hydrogen peroxide (H2O2) in a decentralized and clean manner, which drives the demand for developing high selectivity electrocatalysts. However, current understanding on selectivity descriptors of 2e− ORR electrocatalysts is still insufficient, limiting the optimization of catalyst design. Here we study the catalytic performances of a series of metal phthalocyanines (MPcs, M = Co, Ni, Zn, Cu, Mn) for 2e− ORR by combining density functional theory calculations with electrochemical measurements. Two descriptors (ΔG*O − ΔG*OOH and ΔG*H2O2) are uncovered for manipulating the selectivity of H2O2 production. ΔG*O − ΔG*OOH reflects the preference of O–O bond breaking of *OOH, affecting the intrinsic selectivities. Due to the high value of ΔG*O − ΔG*OOH, the molecularly dispersed electrocatalyst (MDE) of ZnPc on carbon nanotubes exhibits high selectivity, even superior to the previously reported NiPc MDE. ΔG*H2O2 determines the possibility of further H2O2 reduction to affect the measured selectivities. Enhancing the hydrophobicity of the catalytic layer can increase ΔG*H2O2, leading to selectivity improvement, especially under high H2O2 production rates. In the gas diffusion electrode measurements, both ZnPc and CoPc MDEs with polytetrafluoroethylene (PTFE) exhibit low overpotentials, high selectivities, and good stability. This study provides guidelines for rational design of 2e− ORR electrocatalysts.
Introduction
Hydrogen peroxide (H2O2) has widespread applications in industry, health care, and environmental protection.1 The electrochemical two-electron oxygen reduction reaction (2e− ORR) is a promising approach to produce this important chemical in a green way.2 Its application relies on the development of electrocatalysts with high peroxide selectivity to lower electric energy consumption. Various electrocatalysts for this process have been developed recently, including noble metals, carbon-based non-metal materials, transition metal single-atom catalysts (SACs), metal complexes, and metal oxides.3 Noble metal catalysts with isolated active sites have achieved excellent 2e− ORR performances. For example, the PtHg4 electrocatalyst exhibited selectivities up to 96% in acid.4 Nevertheless, low-cost alternatives but also possessing high selectivities remain to be developed for satisfying requirements of practical applications. Recently, transition metal SACs doped with heteroatoms (i.e., N, O, or P) on carbon matrix (M–X–C) have been found promising for 2e− ORR.5–12 Co–N–C structures exhibited better 2e− ORR performances than other metal active sites.5,7 In contrast, Hyeon et al. found that the Co–N4 catalyst exhibited lower selectivities below 50%, while the Co–N4 structure with surrounding oxygen species could show enhanced selectivities up to 82%.6 A recent study showed that ORR selectivity could be tuned beyond the adsorption site by a molecular confinement strategy.13To further optimize the peroxide selectivities, it is important to understand the structure–property relationship of active sites and reveal selectivity descriptors. However, it is still challenging to define them often due to complicated catalyst structures. For example, Chen et al. used the electronic energy difference between *O2 and *H2O2 intermediates (E*O2 − E*H2O2) as a selectivity descriptor in a porphyrin-containing Co SAC and found that the 2e− pathway was dominant.9 In contrast, Xia et al. indicated that the 4e− pathway was dominant for a similar structure by using a crystal field stabilization energy descriptor.14 Thus, model systems with well-defined active centers are highly required for identifying universal selectivity descriptors.
Metal macrocyclic complexes with well-defined structures have been studied as 2e− ORR electrocatalysts in homogeneous systems and showed high selectivities. A series of N4-ligated and N2O2-ligated cobalt complexes exhibited over 90% selectivities in homogeneous organic electrolytes.15,16 However, heterogeneous macrocyclic complex electrocatalysts, which can show higher reduction current densities and easy catalyst recovery for practical applications, are still rarely reported with high selectivities.17 Metal phthalocyanines (MPcs) have been mainly considered as electrocatalysts for 4e− ORR before.18–23 Recently, we found that the molecularly dispersed electrocatalyst (MDE) of NiPc anchored on carbon nanotubes (CNTs) showed over 80% selectivities on rotating ring-disk electrodes (RRDEs), which was superior to the aggregated NiPc sample.24
In this work, we study a series of MPc MDEs with various metal centers (M = Co, Ni, Zn, Cu, Mn) toward 2e− ORR and examine the performance descriptors. The difference in the binding energy of *O and *OOH intermediates (ΔG*O − ΔG*OOH) and the *H2O2 binding energy (ΔG*H2O2) are found as the selectivity descriptors. ZnPc and CuPc MDEs with more positive ΔG*O − ΔG*OOH showed even higher selectivities than NiPc MDE. A hydrophobic environment can further enhance the measured selectivities by increasing ΔG*H2O2. In the gas diffusion electrode (GDE) measurements, CoPc and ZnPc MDEs can produce HO−2 stably and rapidly with high faradaic efficiency of H2O2 (FE(H2O2)).
Results and discussion, experimental
Theoretical ORR performances
A series of MPcs with different metal centers (M = Co, Ni, Zn, Cu, Mn) were first studied using the computational hydrogen electrode model.25 As depicted in Fig. 1a, oxygen molecules are absorbed on the metal centers of MPcs as active sites and are further reduced through the 2e− or 4e− ORR pathway.26 The calculated free energy profiles in these MPcs are plotted in Fig. 1b at 0.7 V (theoretical equilibrium potential for 2e− ORR) to analyze the ORR selectivities and activities of MPcs.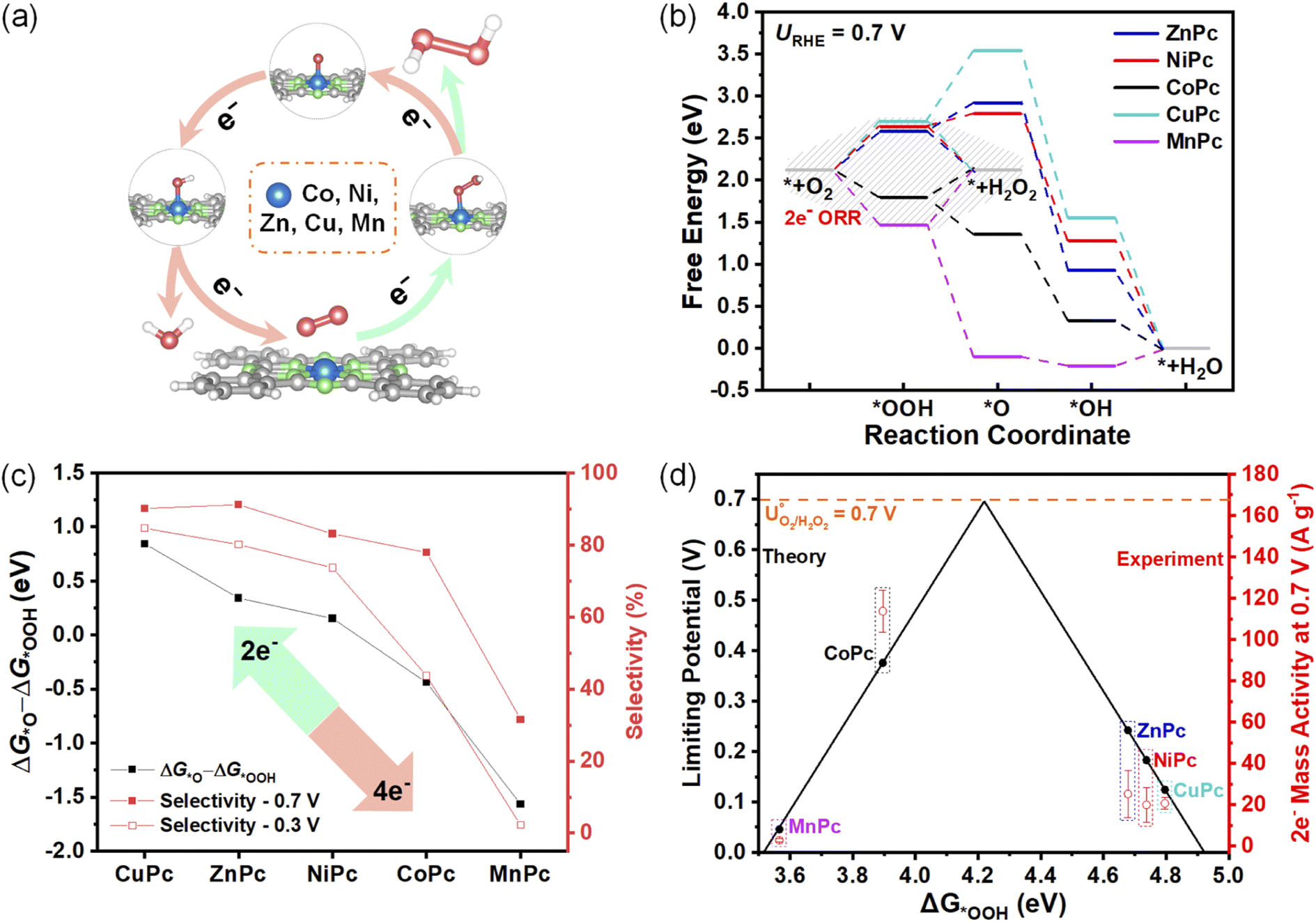 | ||
| Fig. 1 (a) Schematic illustration of the 2e− and 4e− pathway on MPcs (M = Co, Ni, Zn, Cu, Mn). Blue, green, grey, and white spheres represent metal, nitrogen, carbon, and hydrogen, respectively. (b) DFT-calculated free energy profiles of the 2e− ORR pathway to H2O2 (highlight in a dashed region) and the 4e− ORR pathway to H2O on MPcs at 0.7 V. (c) Correlation between ΔG*O − ΔG*OOH and the measured selectivities at 0.7 V and 0.3 V. (d) Correlation between ΔG*OOH and the measured 2e− mass activity at 0.7 V. | ||
Further reactions of the *OOH intermediate determine which pathway proceeds. The *–O bond breaks to directly generate H2O2 for the 2e− pathway, while the O–O bond breaks to produce H2O for the 4e− pathway (details are given in ESI Methods†). Therefore, the next proton-coupled electron transfer processes of *OOH determine the selectivities. The catalysts with high selectivities should be more inclined thermodynamically to form H2O2 relative to H2O. Several possible selectivity descriptors are extracted according to the trade-off relationship between the 4e− and the 2e− pathway. Higher ΔG*O − ΔG*OOH (Fig. 1c) and ΔG*O − ΔG*+H2O2 (Fig. S2a†) could inhibit the 4e− pathway to achieve higher peroxide selectivities. The values of ZnPc and CuPc are higher than the previously reported NiPc, suggesting their higher preference for the 2e− pathway. On the other hand, a higher value of ΔG*OOH − ΔG*+H2O2 (Fig. S2b†), which lowers the free energy required for H2O2 formation, could also facilitate the 2e− pathway.
As *OOH is the key intermediate in the 2e− ORR pathway, the binding energy of *OOH (ΔG*OOH) should be neither too strong nor too weak.7 Hence, ΔG*OOH is used as an activity descriptor to calculate limiting potentials (details are given in ESI Methods†) for the 2e− ORR pathway.27 We constructed an activity-volcano plot based on the limiting potential as a function of ΔG*OOH in Fig. 1d. It predicts that CoPc has the lowest overpotential for the 2e− ORR pathway, indicating its highest 2e− activity. It can also be seen that the overpotential of MnPc is the largest among these five MPcs.
Electrochemical ORR performances
We prepared a series of MPc MDEs (M = Co, Ni, Zn, Cu, Mn) by anchoring MPc molecules on CNTs according to our previous approach.28–31 The metal contents of all MPc MDEs were adjusted to around 0.6 wt% (Table S2†). MPc MDEs show nanotubular structures of CNTs and no noticeable aggregations of MPc molecules are observed in transmission electron microscope (TEM) (Fig. S3a–e†). Discrete bright spots can be seen on the side walls of CNTs in HAADF-STEM (Fig. S3f†), suggesting the dispersion of MPc molecules in MDEs.Then, we evaluated their electrochemical ORR performances by using RRDEs in 0.1 M KOH. The ORR polarization curves of MPc MDEs vary with the metal centers and are significantly different from that of CNT (Fig. 2a), suggesting MPc molecules are catalytically active in MDEs. The disk currents mainly compose two parts of currents originating from 2e− and 4e− ORR pathway, while the ring currents are produced by the oxidation of HO−2 on the ring electrodes of RRDEs. The peroxide selectivities can be calculated from the disk currents and the ring currents (details are given in ESI Methods†), which are plotted in Fig. 2b. The selectivities of ZnPc and CuPc MDEs reach over 90% in a wide potential range from 0.5 to 0.75 V, which are superior to the previously reported NiPc MDE. CoPc MDE shows around 78% selectivities above 0.5 V, but it drops sharply below 0.5 V and reaches 35% at 0.2 V. In contrast, MnPc MDE exhibits large limiting disk reduction current density and small ring currents, indicating that it mainly facilitates the 4e− pathway. The highest selectivity of MnPc MDE is only 36% at 0.75 V, and a sharp decline is observed with the increase of overpotentials.
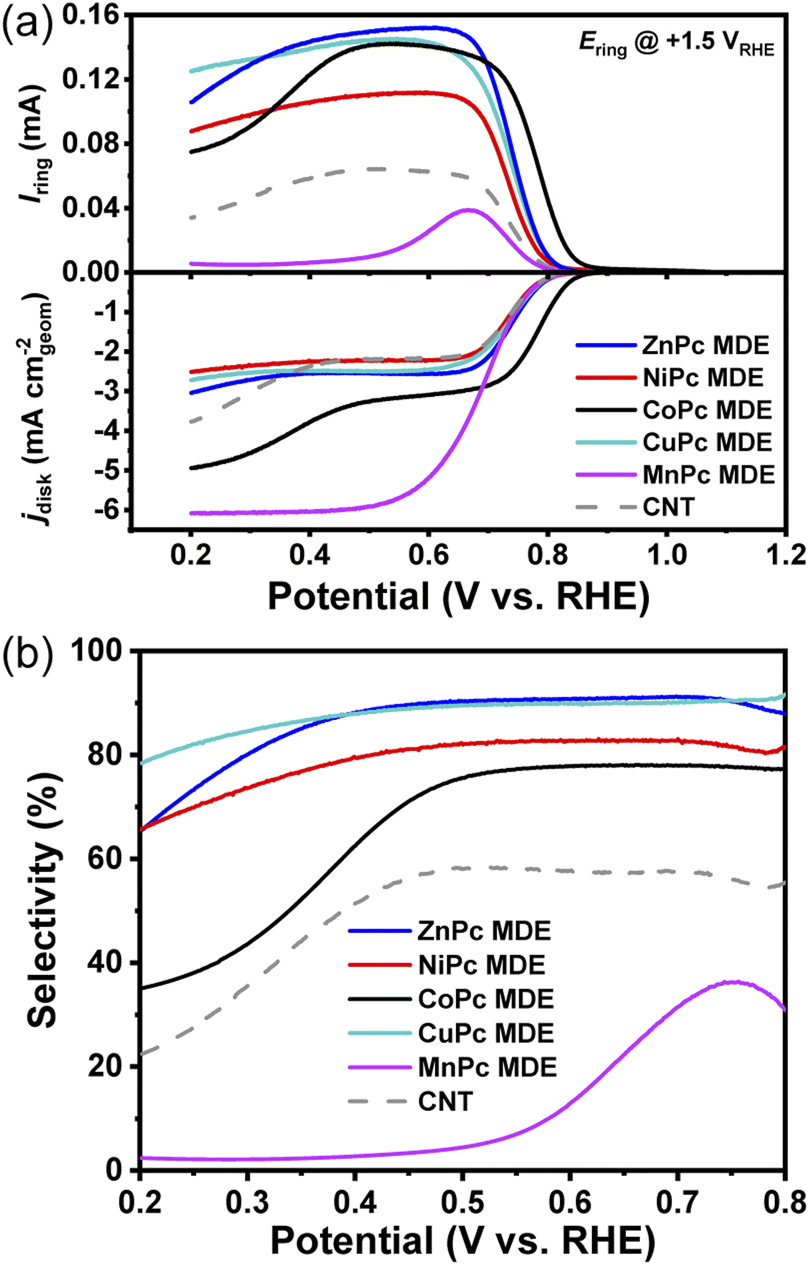 | ||
| Fig. 2 (a) ORR polarization plots for MPc MDEs (M = Co, Ni, Zn, Cu, Mn) with the loading of 0.2 mg cm−2 on RRDEs at 1600 rpm in 0.1 M KOH saturated with O2. (b) Corresponding selectivities of MPc MDEs calculated from RRDE measurments. | ||
To reveal the selectivity descriptor, we plotted the selectivities of MPc MDEs at a high potential of 0.7 V and a low potential of 0.3 V in Fig. 1c, which show similar tendencies as the theoretical predictions of ΔG*O − ΔG*OOH. The sharply reduced selectivities of MnPc MDE compared to others are attributed to the very negative ΔG*O − ΔG*OOH of MnPc. However, the other descriptors showed certain contradiction with the experiment data. For example, there is a huge difference in ΔG*O − ΔG*+H2O2 between NiPc and CoPc, suggesting the CoPc is supposed to have substantially lower selectivities than NiPc (Fig. S2a†). And NiPc is supposed to exhibit higher selectivities than ZnPc deduced from the descriptors of ΔG*OOH − ΔG*+H2O2 (Fig. S2b†). CoPc would show much lower selectivities based on the previously reported descriptor of E*O2 − E*H2O2 (Fig. S2c†).9 Therefore, the ΔG*O − ΔG*OOH descriptor can better describe the experimental selectivities of these MPc MDEs.
Onset potential is often used to describe ORR activities. CoPc MDE exhibits the most positive onset potential (defined as the potential at the reduction current density of 0.1 mA cm−2) of 0.85 V (Fig. 2a) compared to other MPc MDEs. However, to verify the 2e− activity descriptor, it should be noted that the experimental activities cannot be simply extracted from ORR onset potentials due to the interference of the 4e− pathway. Therefore, 2e− mass activity calculated from the product of mass activity and FE(H2O2) is proposed to evaluate the activity of 2e− ORR. As shown in Fig. 1d, the trend of experimental 2e− mass activity at 0.7 V is consistent with the theoretical activity volcano plot. Moreover, after a two-hour chronoamperometric measurement under 0.6 V, the UV-vis adsorption profiles show that the molecular structure in CoPc MDE remains unchanged, suggesting its good structural stability (Fig. S5†).
We also measured the electrochemical performances of aggregated molecule samples. We prepared the mixture samples of MPc and CNTs by drop-drying from the ethanol dispersion (denoted as MPc + CNT). Significant aggregates of MPc molecules can be observed in MPc + CNT from the scanning electron microscope (SEM) (Fig. S6†). In Fig. S7a,† the onset potentials of CoPc + CNT, NiPc + CNT, and MnPc + CNT are positively shifted compared to their MDE counterparts, whereas those of CuPc + CNT and ZnPc + CNT are negatively shifted. Moreover, the selectivities of NiPc + CNT and CuPc + CNT (Fig. S7b†) show opposite trends when compared to the theoretical predictions. We also prepared neat MPc samples without CNTs. All of them are less active than their MDE counterparts, showing similar onset potentials at ∼0.7 V (Fig. S7c†) and lower reduction current densities. In brief, the performances of MPc electrocatalysts in aggregated states (MPc + CNT and neat MPcs) are inconsistent with the theoretical calculations, revealing the interference of molecular aggregation on studies of intrinsic electrocatalytic performances.
H2O2 reduction performances of MDEs
The produced H2O2 from 2e− ORR could be trapped inside the catalyst layer and be further reduced.6 It is found that the selectivities of all MPc MDEs show sharp drops below certain potentials (Fig. 2b). For example, the selectivity of CoPc MDE decreases dramatically below 0.5 V. When we reduced the catalyst loading amount of CoPc MDE from 0.2 to 0.05 mg cm−2, the selectivity also became higher below around 0.5 V (Fig. S8†). A similar phenomenon was also observed in the reported Co SAC.6 The diffusion pathways of the produced H2O2 would be shortened as the catalyst loading amount becomes lower, decreasing its chance for further reduction. The H2O2 reduction reaction (H2O2RR) performances of MPc MDEs on rotating disk electrodes (RDEs) were evaluated in 0.1 M KOH containing 1 mM H2O2 (Fig. 3a). The H2O2RR onset potential of MnPc MDE is the most positive, followed by CoPc MDE. The H2O2RR activities of the other three samples are much lower. The onset potentials of H2O2RR highly coincide with the potentials at which selectivities begin to sharply decrease (Fig. 3b), and the currents of H2O2RR are almost positively correlated with the extent of selectivity reduction. These results indicate that the further reduction of H2O2 could lower the measured selectivities.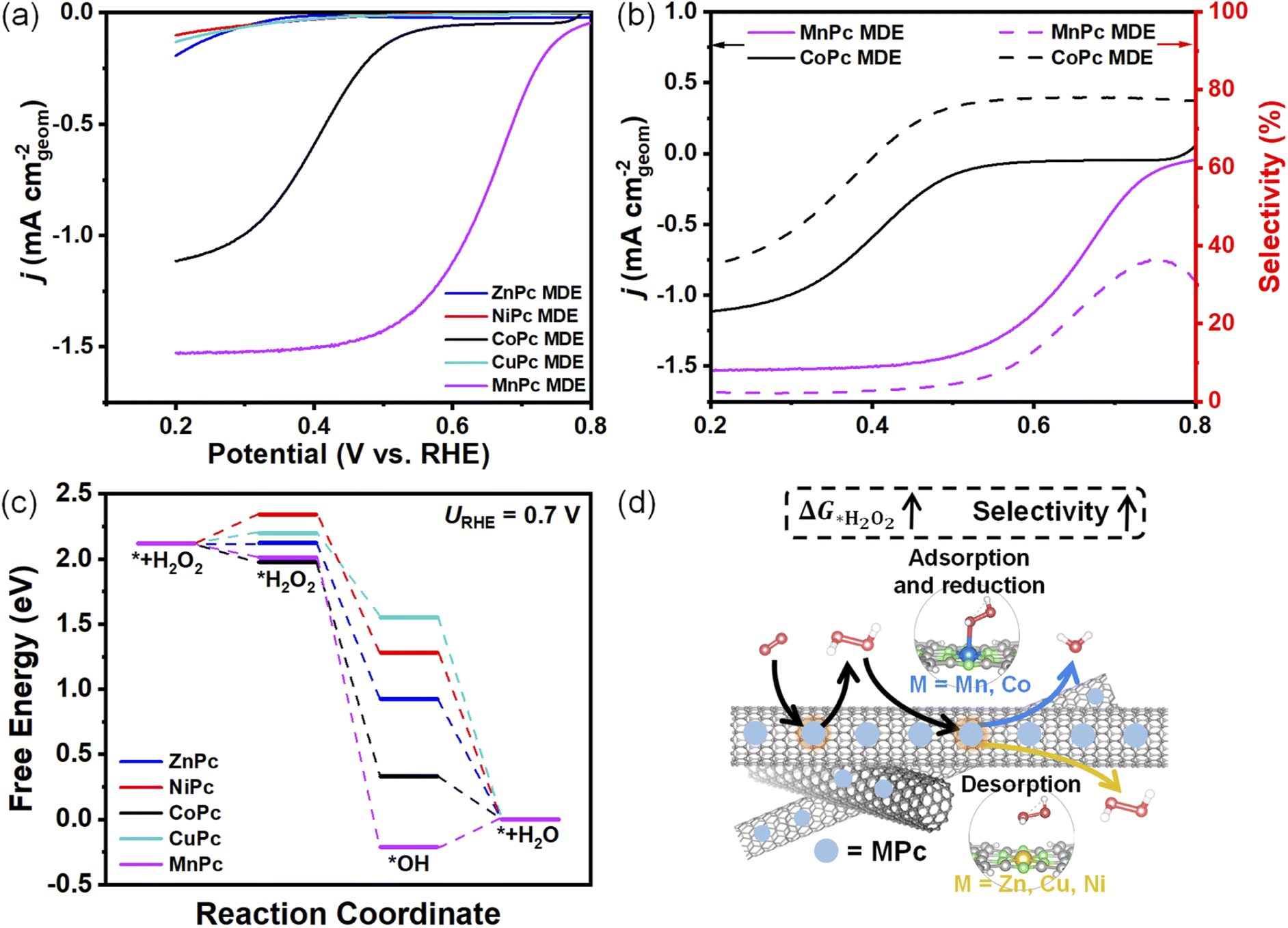 | ||
| Fig. 3 (a) H2O2RR performances of MPc MDEs loaded on RDEs at 1600 rpm in Ar saturated 0.1 M KOH (containing 1 mM H2O2). (b) Comparison of H2O2RR performances (solid line) and peroxide selectivities (dash line) of CoPc and MnPc MDEs. (c) DFT-calculated free energy profiles of H2O2RR on MPcs at 0.7 V. (d) Schematic illustrations of H2O2RR on two different types of MPc MDEs. | ||
To investigate the reason for high H2O2 reduction abilities of CoPc and MnPc MDEs, we carried out theoretical calculations for H2O2RR (Fig. 3c). The adsorption pathway of H2O2 molecules on NiPc, CuPc, and ZnPc is endothermic, while that for CoPc and MnPc is exothermic. The differences in the H2O2 reduction ability of these two types of MPc MDEs are depicted in Fig. 3d. It indicates that catalysts in real production conditions should have higher ΔG*H2O2 to hinder H2O2RR from diffusive H2O2 adsorption for higher measured selectivities. Therefore, ΔG*H2O2 is another selectivity descriptor.
H2O2 production in gas diffusion electrodes
To further explore practical application of MPc MDEs toward 2e− ORR, we measured MPc MDEs in GDE devices (Fig. S10†), which could afford higher reduction current densities by increasing oxygen concentration on catalyst layers.32 The catalyst inks of MPc MDEs with polytetrafluoroethylene (PTFE) were loaded on gas diffusion layers and tested in flowed 1 M KOH. CoPc MDE can still exhibit the highest activity among all MPc MDEs (Fig. 4a). The chronoamperometry measurements at 10 to 225 mA cm−2 were conducted (Fig. S11†), and the determined FE(H2O2)s at varied applied voltages are shown in Fig. 4b. NiPc, CuPc, and ZnPc MDEs can exhibit over 95% FE(H2O2)s in the whole current density range, which are even higher than the RRDE measurements (Fig. S12†). Interestingly, the FE(H2O2)s of CoPc MDE are around 90%, which are substantially higher than those of CoPc MDE without PTFE. The H2O2RR performances of CoPc MDE with and without PTFE are compared in Fig. S13.† CoPc MDE with PTFE shows much smaller H2O2 reduction current densities than the one without PTFE. These results suggest that H2O2RR is inhibited in CoPc MDE with PTFE, leading to higher FE(H2O2)s.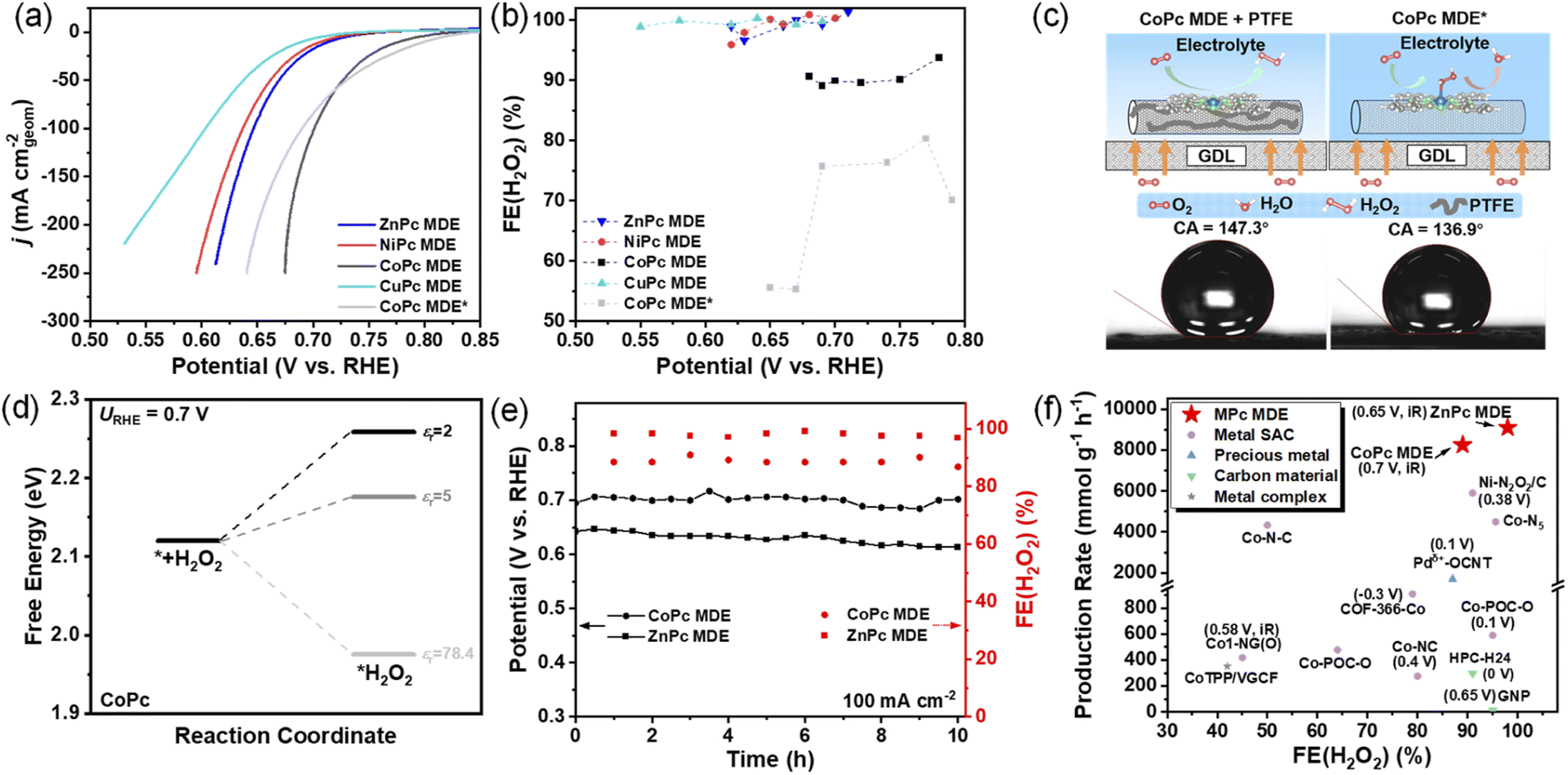 | ||
| Fig. 4 (a) Linear sweep voltammetry of MPc MDEs (M = Co, Ni, Zn, Cu) with PTFE and CoPc MDE* without PTFE in 1 M KOH tested by GDEs. (b) The measured FE(H2O2)s at different applied potentials. (c) Schematic illustrations of the hydrophobic effect of PTFE on inhibiting further reduction of H2O2 and contact angles of CoPc MDE with and without PTFE loaded on carbon paper. (d) DFT-calculated free energy profiles of the H2O2 adsorption process on CoPc with different relative permittivity. (e) Long-term stability tests of CoPc and ZnPc MDEs at the reduction current density of 100 mA cm−2. (f) Comparison of the FE(H2O2)s and production rates of CoPc and ZnPc MDEs with other reported electrocatalysts. | ||
As shown in Fig. 4c, PTFE increases the hydrophobicity of catalyst layers. For simulating the influence of the hydrophobic environment with PTFE in the implicit solvation model, we assumed that the access of water around active sites is limited and the relative permittivity (εr) in the DFT calculations becomes lower than that of bulk water (78.4).33,34 When εr decreased from 78.4 to 2–5, the calculated ΔG*H2O2 at 0.7 V increases from 1.97 eV to 2.26–2.18 eV (Fig. 4d), respectively, suggesting the inhibition of H2O2 adsorption. However, the value of another selectivity descriptor ΔG*O − ΔG*OOH remains basically unchanged (less than 0.05 eV) with decreasing relative permittivity (Table S4†). These results indicate that the hydrophobic effect (low εr) increases FE(H2O2)s by decreasing H2O2RR.
In the stability test for 10 hours (Fig. 4e), CoPc MDE can produce HO−2 stably with a high reduction current density of 100 mA cm−2 at ∼0.7 V. The FE(H2O2) maintained 89% and the production rate is calculated to be 8200 mmol gcat−1 h−1 on the average (calculation details are provided in ESI†). ZnPc MDE can also deliver ∼98% FE(H2O2) and a production rate of 9100 mmol gcat−1 h−1 at around 0.65 V. CoPc and ZnPc MDEs outperform the reported catalysts with relatively high FE(H2O2)s under higher production rates (Fig. 4f and Table S5†). The cathodic energy efficiency (calculation details are provided in the ESI†) was also analyzed. Although ZnPc MDE has lower activity than CoPc MDE, it exhibits similar cathodic energy efficiency of around 80% to CoPc MDE owning to its higher FE(H2O2)s.
Conclusions
With the advantages of well-defined active sites and no influence from molecular aggregation, MPc MDEs are beneficial for studying intrinsic performances. Two selectivity descriptors toward 2e− ORR are uncovered. The first descriptor ΔG*O − ΔG*OOH emphasizes the importance of O–O bond breaking for intrinsic selectivity. The second descriptor ΔG*H2O2 determines the extent of further H2O2 reduction that affects measured selectivity. The hydrophobic environment in the catalyst layer can increase ΔG*H2O2 to inhibit H2O2 reduction. In GDE devices, ZnPc and CoPc MDEs can continuously produce HO−2 at over 8200 mmol gcat−1 h−1 with 98% FE(H2O2) and 89% FE(H2O2), respectively. This work can benefit the development of high-performance electrocatalysts toward 2e− ORR, and facilitate the rational design of noble-metal-free electrocatalysts.Data availability
All supporting data can be found in the ESI.†Author contributions
Y. L. conceived the project and designed the experiments. Y. Y. carried out the DFT calculations and electrochemical measurements. H. L. and Z. J. performed the material characterizations. Y. L. and Y. Y. analyzed the data and prepared the manuscript with input from all the authors. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by National Natural Science Foundation of China (22075125).Notes and references
- Y. Y. Jiang, P. J. Ni, C. X. Chen, Y. Z. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Adv. Energy Mater., 2018, 8, 25 Search PubMed.
- K. Jiang, J. J. Zhao and H. T. Wang, Adv. Funct. Mater., 2020, 30, 11 Search PubMed.
- N. Wang, S. Ma, P. Zuo, J. Duan and B. Hou, Adv. Sci., 2021, 8, 2100076 CrossRef CAS PubMed.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS.
- Y. Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. K. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. L. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS PubMed.
- E. Jung, H. Shin, B.-H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. Hooch Antink, S. Park, K.-S. Lee, S.-P. Cho, J. S. Yoo, Y.-E. Sung and T. Hyeon, Nat. Mater., 2020, 19, 436–442 CrossRef CAS PubMed.
- J. J. Gao, H. B. Yang, X. Huang, S. F. Hung, W. Z. Cai, C. M. Jia, S. Miao, H. M. Chen, X. F. Yang, Y. Q. Huang, T. Zhang and B. Liu, Chem, 2020, 6, 658–674 CAS.
- Q. Zhang, X. Tan, N. M. Bedford, Z. Han, L. Thomsen, S. Smith, R. Amal and X. Lu, Nat. Commun., 2020, 11, 4181 CrossRef.
- C. Liu, H. Li, F. Liu, J. Chen, Z. Yu, Z. Yuan, C. Wang, H. Zheng, G. Henkelman, L. Wei and Y. Chen, J. Am. Chem. Soc., 2020, 142, 21861–21871 CrossRef CAS PubMed.
- Q. L. Zhao, Y. Wang, W. H. Lai, F. Xiao, Y. X. Lyu, C. Z. Liao and M. H. Shao, Energy Environ. Sci., 2021, 14, 5444–5456 RSC.
- C. Tang, Y. Jiao, B. Shi, J.-N. Liu, Z. Xie, X. Chen, Q. Zhang and S.-Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 9171–9176 CrossRef CAS PubMed.
- B.-Q. Li, C.-X. Zhao, J.-N. Liu and Q. Zhang, Adv. Mater., 2019, 31, 1808173 CrossRef PubMed.
- X. Li, S. Tang, S. Dou, H. J. Fan, T. S. Choksi and X. Wang, Adv. Mater., 2022, 34, 2104891 CrossRef CAS PubMed.
- C.-Y. Lin, L. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2017, 29, 1606635 CrossRef PubMed.
- A. Rana, Y.-M. Lee, X. Li, R. Cao, S. Fukuzumi and W. Nam, ACS Catal., 2021, 11, 3073–3083 CrossRef CAS.
- Y.-H. Wang, M. L. Pegis, J. M. Mayer and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 16458–16461 CrossRef CAS PubMed.
- Y. Wang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Energy Mater., 2021, 11, 2003323 CrossRef CAS.
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS.
- J. Guo, H. Li, H. He, D. Chu and R. Chen, J. Phys. Chem. C, 2011, 115, 8494–8502 CrossRef CAS.
- R. M. Reis, R. B. Valim, R. S. Rocha, A. S. Lima, P. S. Castro, M. Bertotti and M. R. V. Lanza, Electrochim. Acta, 2014, 139, 1–6 CrossRef CAS.
- H. He, Y. Lei, C. Xiao, D. Chu, R. Chen and G. Wang, J. Phys. Chem. C, 2012, 116, 16038–16046 CrossRef CAS.
- R. R. Chen, H. X. Li, D. Chu and G. F. Wang, J. Phys. Chem. C, 2009, 113, 20689–20697 CrossRef CAS.
- Z. Zhang, S. Yang, M. Dou, H. Liu, L. Gu and F. Wang, RSC Adv., 2016, 6, 67049–67056 RSC.
- Y. Wang, Z. Zhang, X. Zhang, Y. Yuan, Z. Jiang, H. Zheng, Y.-G. Wang, H. Zhou and Y. Liang, CCS Chem., 2022, 4, 228–236 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- G. F. Wang, N. Ramesh, A. Hsu, D. Chu and R. R. Chen, Mol. Simul., 2008, 34, 1051–1056 CrossRef CAS.
- G.-F. Han, F. Li, W. Zou, M. Karamad, J.-P. Jeon, S.-W. Kim, S.-J. Kim, Y. Bu, Z. Fu, Y. Lu, S. Siahrostami and J.-B. Baek, Nat. Commun., 2020, 11, 2209 CrossRef CAS PubMed.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- Z. Jiang, Y. Wang, X. Zhang, H. Zheng, X. Wang and Y. Liang, Nano Res., 2019, 12, 2330–2334 CrossRef CAS.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- Z. Lin, Z. Jiang, Y. Yuan, H. Li, H. Wang, Y. Tang, C. Liu and Y. Liang, Chin. J. Catal., 2022, 43, 104–109 CrossRef CAS.
- Y. L. Wang, R. Shi, L. Shang, G. I. N. Waterhouse, J. Q. Zhao, Q. H. Zhang, L. Gu and T. R. Zhang, Angew. Chem., Int. Ed., 2020, 59, 13057–13062 CrossRef CAS PubMed.
- B. W. Noffke, Q. Li, K. Raghavachari and L.-s. Li, J. Am. Chem. Soc., 2016, 138, 13923–13929 CrossRef CAS.
- K. Takeyasu, M. Furukawa, Y. Shimoyama, S. K. Singh and J. Nakamura, Angew. Chem., Int. Ed., 2021, 60, 5121–5124 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03714a |
| This journal is © The Royal Society of Chemistry 2022 |