
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand-enabled oxidation of gold(I) complexes with o-quinones†
György
Szalóki
a,
Julien
Babinot
a,
Vlad
Martin-Diaconescu
b,
Sonia
Mallet-Ladeira
c,
Yago
García-Rodeja
 d,
Karinne
Miqueu
d and
Didier
Bourissou
*a
d,
Karinne
Miqueu
d and
Didier
Bourissou
*a
aLaboratoire Hétérochimie Fondamentale et Appliquée (LHFA, UMR 5069), CNRS, Université Toulouse III – Paul Sabatier, 118 Route de Narbonne, Toulouse 31062, Cedex 09, France. E-mail: didier.bourissou@univ-tlse3.fr
bALBA Synchrotron – CELLS, Carrer de la Llum 2-26, Cerdanyola del Vallès 08290, Barcelona, Spain
cInstitut de Chimie de Toulouse (UAR 2599), 118 Route de Narbonne, Toulouse 31062, Cedex 09, France
dInstitut des Sciences Analytiques et Physico-Chimie pour l’Environnement et les Matériaux (IPREM, UMR 5254), CNRS, Université de Pau et des Pays de l’Adour E2S UPPA, Hélioparc, 2 Avenue du Président Angot, Pau 64053, Cedex 09, France
First published on 5th August 2022
Abstract
Chelating P^P and hemilabile P^N ligands were found to trigger the oxidation of Au(I) complexes by o-benzoquinones. The ensuing Au(III) catecholate complexes have been characterized by NMR spectroscopy, single crystal X-ray diffraction and X-ray absorption spectroscopy. They adopt tetracoordinate square-planar structures. Reactivity studies substantiate the reversibility of the transformation. In particular, the addition of competing ligands such as chloride and alkenes gives back Au(I) complexes with concomitant release of the o-quinone. DFT calculations provide insight about the structure and relative stability of the Au(I) o-quinone and Au(III) catecholate forms, and shed light on the 2-electron transfer from gold to the o-quinone.
Introduction
Long considered too inert and useless in chemistry, gold complexes have proven in fact very powerful and they are attracting much interest. Besides unique carbophilic properties which make gold complexes extremely efficient in π-acid catalysis, 2-electron redox transformations at gold have emerged over the past 10–15 years.1 In line with the very high redox potential of the Au(I)/Au(III) couple, Au(I) complexes are inherently reluctant to 2-electron oxidation into well-defined Au(III) complexes. Strong oxidants are thus required. Known examples include aqua regia, X2 and surrogates, F+ equivalents, diazonium and iodonium salts, I(III) derivatives…2 Oxidative addition of S–S, Si–Si, Sn–Sn3 and strained C–C4 bonds to gold(I) have also been reported, as well as C(sp2) and C(sp)–I/Br bonds, most recently.5 Depending on the gold complex and the oxidant, the Au(I) → Au(III) transformation occurs spontaneously or it is promoted by a photoredox catalyst, by UV-vis irradiation or by a bidentate ligand. The ensuing Au(III) complexes are interesting on their own,6 with applications ranging from catalysis, materials science to medicinal chemistry. Au(I)/Au(III) redox cycles also open new avenues in gold catalysis, and major achievements have been reported recently in cross-coupling and alkene difunctionalization reactions in particular.1,7To develop this chemistry further, it is highly desirable to identify and study new routes to cycle between Au(I) and Au(III). In this context, we questioned here the possibility to use o-benzoquinones to oxidize Au(I) complexes and obtain catecholate Au(III) complexes. Precedents for Au(III) catecholate complexes are rare. They mainly derive from C,N-cyclometallated ligands8 and have all been accessed by reacting Au(III) dihalo complexes with catecholates (ligand exchange). Another well-established route to prepare catecholate complexes consists in the 2-electron oxidation of transition metals by o-benzoquinones.9 This alternative strategy is known for many metals, from group 6 to group 12 (Mo, Fe, Co, Rh, Ir, Ni, Pd, Pt, Cu, Zn…), but to the best of our knowledge, it is unprecedented with gold.10,11
Here we report that strongly oxidizing o-benzoquinones, i.e. o-chloranil and its bromo/fluoro congeners, do react with Au(I) complexes to give the corresponding Au(III) catecholate complexes (Chart 1). P^P chelating or P^N hemilabile ligands enable the reaction to proceed. The structure of the obtained Au(III) catecholate complexes has been thoroughly analyzed by NMR, X-ray diffraction and absorption (XRD and XAS) means. Reactivity studies substantiate the reversibility of the transformation. DFT calculations shed light on the 2-electron transfer from gold to the o-quinone.
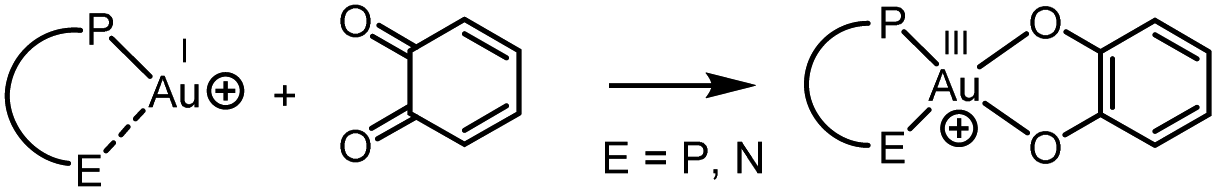 | ||
| Chart 1 Ligand-enabled 2-electron oxidation of gold(I) complexes by o-quinones, as studied in this work. | ||
Results and discussion
First, the oxidation of LAuCl complexes featuring simple phosphines and NHC ligands (PPh3 and IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) ligands by o-benzoquinones was studied. All our attempts remained unsuccessful, whatever the o-benzoquinone (Cl4, F4 or 3,5-tBu2-substituted) and the conditions (with/without a silver salt present). No reaction was observed within hours at room temperature and forcing the conditions resulted only in degradation, the corresponding L2Au+ complex being systematically detected within the obtained reaction mixtures. To promote the oxidation of Au(I) and stabilize the ensuing Au(III) species, we then turned to o-carboranyl diphosphines. We wondered if the bending strategy we initially developed to trigger oxidative addition to gold4b,5a could also work here. Gratifyingly, following halogen abstraction with AgNTf2 or AgOTf, the tetrachloro o-benzoquinone (o-chloranil) was found to rapidly and cleanly react with the o-carboranyl diphosphine complex 1 to give 2a (Scheme 1).12 The related tetrabromo and tetrafluoro o-benzoquinones behave similarly, but no reaction occurred with the less oxidizing parent and 3,5-tBu2-substituted o-benzoquinones. The (P^P) Au (catecholate) complexes 2a–c were characterized by multi-nuclear NMR spectroscopy. They all display a31P NMR signal at about δ 88–89 ppm. A good reporter for the oxidation of gold from Au(I) to Au(III) is the methyl 1H NMR signal for the iPr group at N. As for the oxidative addition products of aryl iodides,5a it is found at δ ∼ 1.45 ppm for complexes 2a–cvs. 1.23 ppm for 1.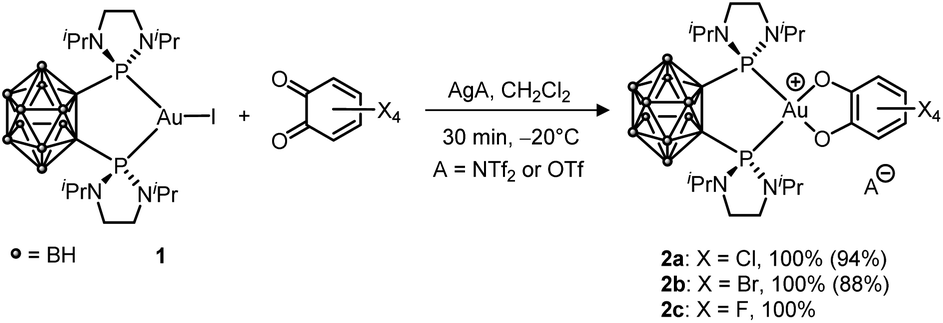 | ||
| Scheme 1 Oxidation of the (P^P)-chelated Au(I) complex 1 with tetrahalo o-benzoquinones (isolated yields in parentheses). | ||
Attempts to grow crystals suitable for X-ray diffraction analysis invariably led to some decomposition and only the corresponding neutral B-decapped complex 3b featuring a nido-carborane cage could be structurally authenticated (Fig. 1 and Table 1). The gold center is tetracoordinate, with chelating P^P and O^O ligands. It sits in a quasi-ideal square-planar environment (τ4 = 0.07).13 The C–O and C–C bond lengths (1.33–1.34 and 1.406–1.143 Å, respectively) fall in the typical range for catecholate complexes14 and unambiguously report for the 2-electron transfer from gold to the o-quinone (the metrical oxidation state (MOS), as introduced by Brown,14b is actually – 1.892). A few crystals of 3b were redissolved in d2-DCM and analyzed by NMR. In line with B-decapping, a broad 1H NMR signal was observed at δ −2.7 ppm for the BHB bridge, while the 11B NMR spectrum displayed five signals between δ −8.6 and −32.8 ppm.
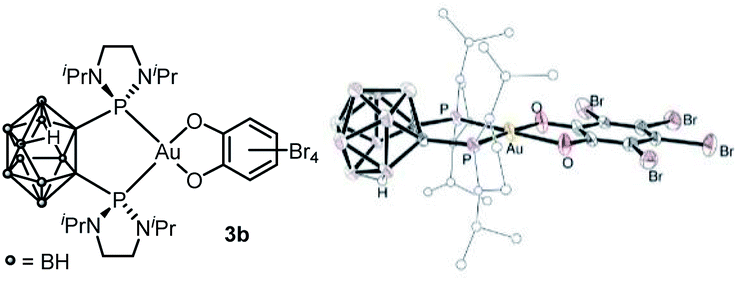 | ||
| Fig. 1 Molecular structure of the nido-P^P Au(III) catecholate complex 3b (For sake of clarity, the substituents at phosphorus are simplified and the hydrogen atoms are omitted, except the bridging one within the nido-carborane cage). | ||
| 3b | 5a | 5b | 5c | |
|---|---|---|---|---|
| τ 4 13 | 0.07 | 0.03 | 0.04 | 0.04 |
| AuP | 2.270 (2) | 2.279 (1) | 2.278 (1) | 2.274 (1) |
| AuP/AuN | 2.272 (2) | 2.065 (2) | 2.066 (3) | 2.053 (4) |
| PAuP/PAuN | 90.03 (5) | 87.71 (5) | 87.60 (7) | 87.77 (12) |
| CO | 1.334 (7) | 1.363 (3) | 1.341 (4) | 1.367 (6) |
| 1.337 (7) | 1.350 (3) | 1.351 (4) | 1.350 (6) | |
| CC | 1.405 (9) | 1.394 (3) | 1.398 (5) | 1.408 (6) |
| MOS14b | −1.89 (6) | −2.03 (7) | −1.89 (10) | −1.99 (10) |
The o-carborane framework survives the oxidation of gold by the o-quinones to give complexes 3a–c, but it slowly degrades afterwards from a closo to a nido form.15 With the aim to increase the stability of the Au(III) catecholate complex and generalize the approach of ligand-enabled oxidation of Au(I) with o-quinones, we then moved to the hemilabile P^N ligand MeDalphos we showed to be very efficient to promote the oxidative addition of aryl/alkynyl/vinyl-iodides as well as aryl-bromides to gold.5b–d
Treatment of (MeDalphos)AuCl with o-chloranil in the presence of a chloride abstractor (AgOTf, AgPF6, AgSbF6, NaBArF24 with ArF = 3,5-(CF3)2C6H3) was found to immediately and quantitatively give the corresponding Au(III) catecholate complex 5a (Scheme 2).12 The reaction proceeds similarly with the related tetrabromo and tetrafluoro o-benzoquinones to give the corresponding Au(III) catecholate complexes 5b,c. Again, no reaction was observed with the less oxidizing parent and 3,5-tBu2-substituted o-benzoquinones. Diagnostic of the Au(I) to Au(III) oxidation and P^N chelation in 5a–c are the low field shifts of the 31P (δ ∼ 88 ppm), 1H (NMe2, δ ∼ 4.0 ppm) and 13C(NMe2, δ ∼ 60 ppm) NMR resonances, as well as the 15N chemical shift (δ 68.5 ppm for 5a, as determined by an HSQC 15N–1H experiment). Very similar data were observed for the related [(P^N)AuCl2]SbF6 complex (δ31P: 109.8 ppm; 1H: 3.92 ppm; 13C: 59.1 ppm; 15N: 82.3 ppm).16
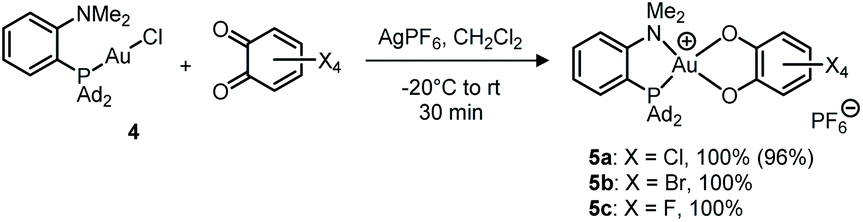 | ||
| Scheme 2 Oxidation of the (P^N)-ligated Au(I) complex 4 with tetrahalo o-benzoquinones (isolated yields in parentheses). | ||
The higher stability of the P^N-ligated complexes 5a–c enabled to grow crystals and carry out X-ray diffraction analyses (Fig. 2 and Table 1). The molecular structures of 5a–c are very similar. The three complexes adopt discrete ion-pair structures with tetracoordinate square-planar Au centers (τ4 = 0.03).13 The short N–Au distances (2.05–2.07 (2) Å) and small PAuN bite angles (87.6–87.8°) testify to the strong coordination of N to gold and thus to the chelating behavior of the P^N ligand. Oxidation from Au(I) to Au(III) and concomitant o-quinone to catecholate reduction is again clearly apparent from the C–O/C–C bond lengths (1.34–1.37/1.39–1.41 Å, respectively) and MOS values (from −1.887 to −2.029).14
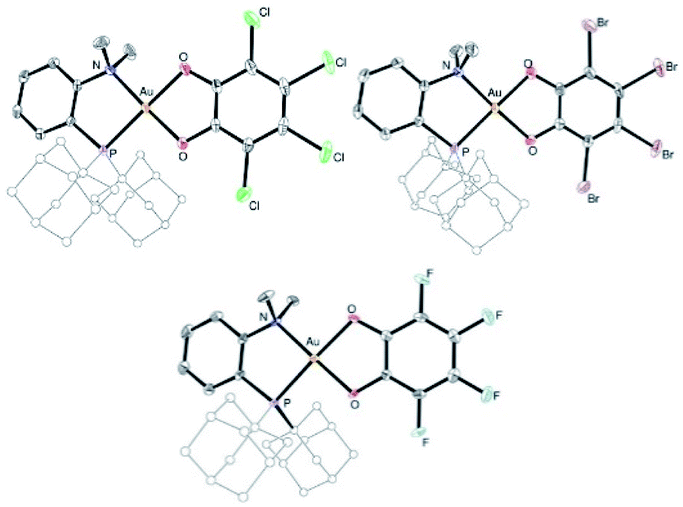 | ||
| Fig. 2 Molecular structures of the P^N Au(III) catecholate complexes 5a–c (for sake of clarity, the substituents at phosphorus are simplified and the hydrogen atoms are omitted). | ||
The assignment of 5a as a Au(III) catecholate complex was further supported by X-ray absorption spectroscopy (Fig. 3). The XANES (X-ray Absorption Near-Edge Structure) profile at the Au L3-edge is dominated by 2p → 5d dipole allowed transitions, and is particularly sensitive to metal oxidation state. Gold in Au foil (Au(0)) and (MeDalphos)AuCl (Au(I), d10 complex) do not exhibit intense whiteline features due to the lack of vacant d acceptor orbitals. Nevertheless, the rising edge for the Au(I) species at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 920.7 eV is higher in energy relative to Au0 at 11.919 eV reflecting the change in oxidation state. In contrast, 5a shows an intense whiteline peak with a maximum centered at 11921.5 eV consistent with a Au(III) d8 gold center.17 Furthermore, EXAFS (extended X-ray absorption fine structure) analysis of 5a clearly describes a tetracoordinate Au center with 1 P/Cl scattering atom at 2.27 Å,3 N/O atoms distributed at 1.96 Å and 2.05 Å. For comparison, (MeDalphos)AuCl has a much lower intensity in the Fourier transformed spectra and only exhibits two Au–P/Cl scattering interactions at ∼2.30 Å. Both EXAFS models are consistent with crystallographic data and theoretical models.
920.7 eV is higher in energy relative to Au0 at 11.919 eV reflecting the change in oxidation state. In contrast, 5a shows an intense whiteline peak with a maximum centered at 11921.5 eV consistent with a Au(III) d8 gold center.17 Furthermore, EXAFS (extended X-ray absorption fine structure) analysis of 5a clearly describes a tetracoordinate Au center with 1 P/Cl scattering atom at 2.27 Å,3 N/O atoms distributed at 1.96 Å and 2.05 Å. For comparison, (MeDalphos)AuCl has a much lower intensity in the Fourier transformed spectra and only exhibits two Au–P/Cl scattering interactions at ∼2.30 Å. Both EXAFS models are consistent with crystallographic data and theoretical models.
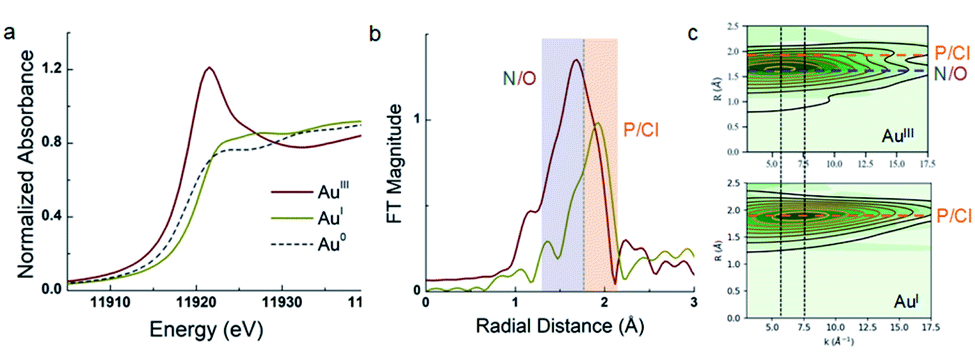 | ||
| Fig. 3 (a) Comparison of the Au L3-edge profiles for 5a (red), (MeDalphos)AuCl (yellow) and Au foil (grey), having +3, +1, and 0 oxidation states, respectively. (b) Fourier Transformed (FT) spectra of 5a (red) and (MeDalphos)AuCl (yellow) with k-range 3–17.5 Å−1, Hannings window dk = 1. (c) Cauchy wavelet transforms of data (green density) and fit (contour line) showing signal dependence on both r-space and k-space. | ||
The reactivity of the (P^N)Au (tetrachloro-catecholate) complex 5a was then interrogated (Scheme 3). Upon treatment with 1 equivalent of tetra-n-butylammonium chloride, 5a was immediately and quantitatively converted back into the (MeDalphos)AuCl complex 4. With ethylene, a large excess was needed to drive the reaction to completion. Under vacuum, the resulting ethylene complex 6a18 gave back 5a, demonstrating the possibility to easily cycle between Au(I) π-alkene and Au(III) catecholate complexes. The addition of 5 equivalents of styrene to 5a resulted in an equilibrium with partial formation of the corresponding Au(I) π-complex 6b (∼25:75 ratio based on 31P NMR spectroscopy).18 With norbornene, the reaction is shifted forward and the Au(III) catecholate complex was quantitatively transformed into the new Au(I) π-complex 6c with only one equivalent of alkene. As unambiguously established by X-ray diffraction analysis (Fig. 4), the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond is side-on coordinated to gold via its exo face and the P^N ligand is strongly chelating, inducing significant Au to alkene back-donation.18 Of note, the reduction of gold from 5a to complexes 4 and 6c was accompanied by the oxidation of the catecholate moiety and o-chloranil was released, as apparent from 13C NMR spectroscopy.12 The oxidation of gold by o-quinones is thus reversible and it can be shifted backward by adding a competing ligand for gold(I), such as chloride or alkenes.
C double bond is side-on coordinated to gold via its exo face and the P^N ligand is strongly chelating, inducing significant Au to alkene back-donation.18 Of note, the reduction of gold from 5a to complexes 4 and 6c was accompanied by the oxidation of the catecholate moiety and o-chloranil was released, as apparent from 13C NMR spectroscopy.12 The oxidation of gold by o-quinones is thus reversible and it can be shifted backward by adding a competing ligand for gold(I), such as chloride or alkenes.
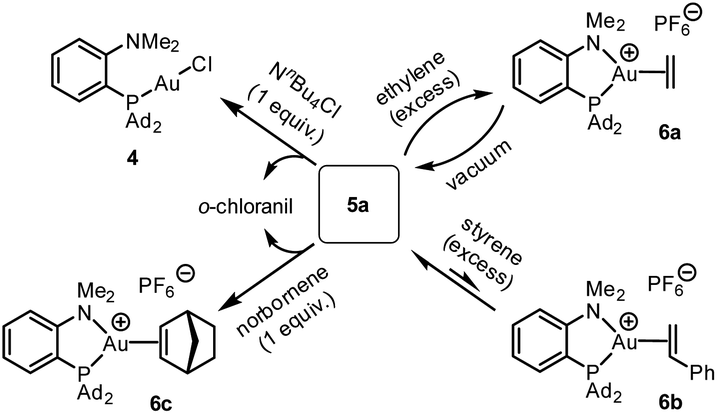 | ||
| Scheme 3 Reactivity of the (P^N)Au(III) catecholate complex 5a towards ammonium chloride and alkenes. | ||
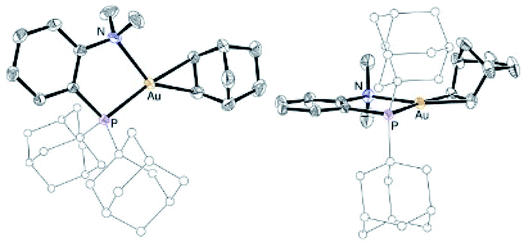 | ||
| Fig. 4 Molecular structure of the P^N Au(I) norbornene complex 6c, top view (left) and side view (right) (for sake of clarity, the substituents at phosphorus are simplified and the hydrogen atoms are omitted). | ||
To gain more insight into the structure and stability of the Au(III) catecholate complexes, DFT calculations were performed at the B3PW91-D3(BJ)/SDD + f (Au), 6-31G** (H, C, N, O, P, Cl) level of theory. The counter-anion was not included, but solvent (PCM–DCM) and dispersion effects (Grimme's D3BJ corrections) were taken into account. First, complexes featuring the actual P^N ligand MeDalphos were investigated, to compare with the experimental results. Both o-chloranil and the parent o-benzoquinone were considered. In both cases, two energy minima were located on the potential energy surface (Fig. 5 for o-chloranil, Fig. S44† for o-benzoquinone). The ground state structure corresponds to the square-planar Au(III) form, as obtained experimentally upon gold oxidation with o-chloranil. The optimized geometry reproduces nicely the solid-state structure determined crystallographically. The other form (5′a) is a Au(I) complex with the o-benzoquinone unsymmetrically coordinated (Au–O distances of 2.116 and 2.614 Å) and seesaw-type geometry (bond angles: PAuO 159.73 and 114.56°, PAuN 82.67°).19 The 31P, 1H and 13C NMR chemical shifts computed for the Au(III) form 5a match nicely those observed experimentally, but differ noticeably from those of the corresponding Au(I) form 5′a (Table S2†), corroborating our structural assignment.
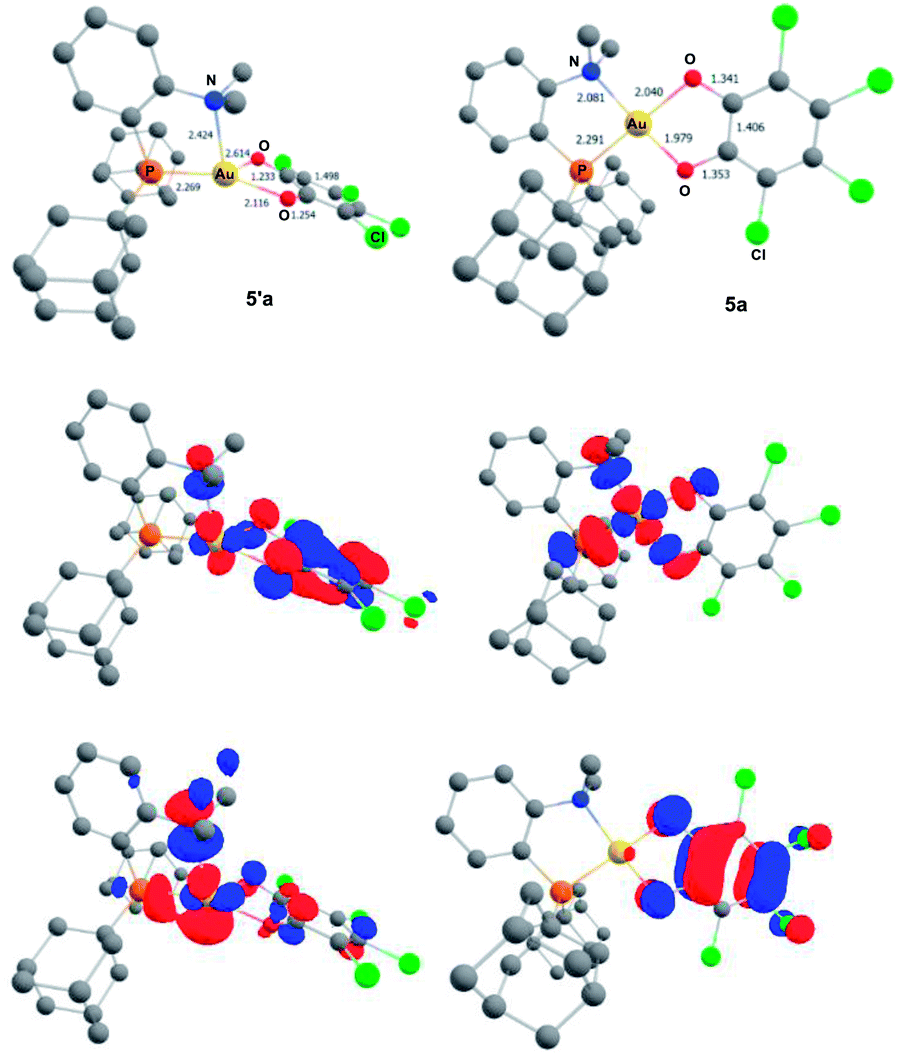 | ||
| Fig. 5 Optimized geometries of the Au(I) o-benzoquinone (5′a, left) and Au(III) catecholate (5a, right) valence isomers of the (P^N)Au(O^O)Cl4+ complex, computed at the PCM(DCM)-B3PW91-D3(BJ)/SDD + f (Au), 6-31G** (C, H, N, O, P, Cl) level of theory. Distances in Å. Plot of the frontier orbitals with cutoff: 0.05. Hydrogen atoms have been omitted for clarity. | ||
With o-chloranil, the Au(III) catecholate complex 5a is 10.8 kcal mol−1 lower in energy than the corresponding Au(I) valence isomer 5′a. This energy gap is reduced to 1.1 kcal mol−1 for the parent o-benzoquinone, the Au(I) form being only slightly above the Au(III) form in this case. To compare the propensity of the two o-quinones to react with the (P^N)Au+ fragment, the thermodynamic balance of the isodesmic reaction (1) was computed (Chart 2). It was found uphill in energy by 7.2 kcal mol−1. This is consistent with experimental observations. With less oxidizing o-quinones, no reaction occurs.
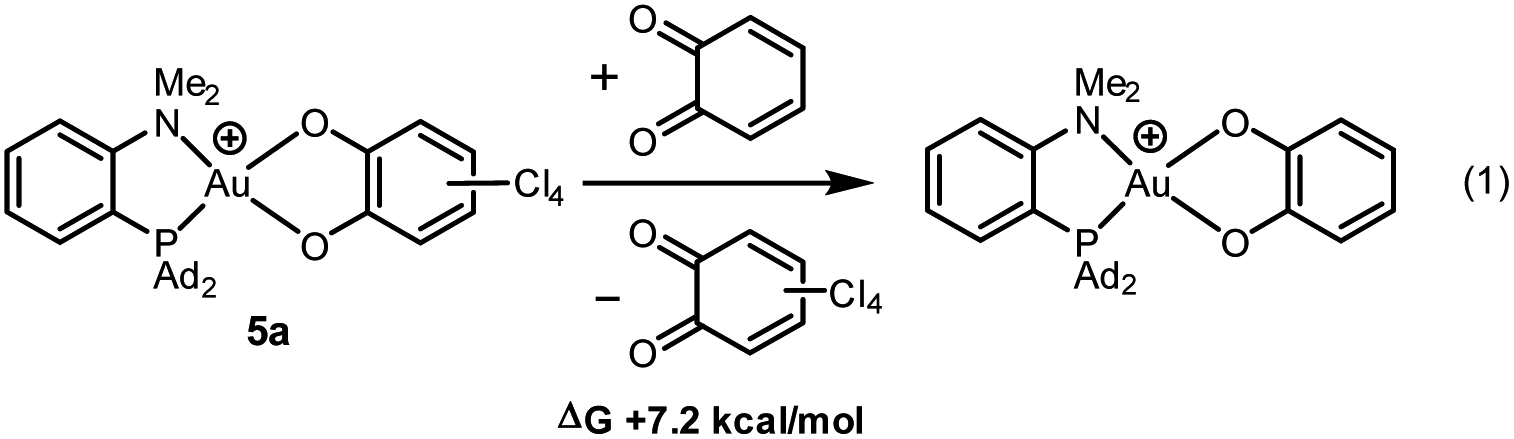 | ||
| Chart 2 Catechol/o-quinone exchange (isodesmic reaction) considered to compare the reactivity of the parent and tetrachloro o-quinones towards Au(I) oxidation. | ||
Besides the geometric features, the changes in the redox states of the Au center and O^O ligand between 5′a and 5a is clearly apparent from the respective frontier orbitals (Fig. 5). For the Au(I) valence isomer, the HOMO is centered on Au (with some parentage of the N lone pair) and the LUMO corresponds to the π* of the o-quinone moiety, while for the Au(III) form, the HOMO is centered on the catecholate ligand and the LUMO is centered on gold (in-plane 5d orbital in anti-bonding interaction with O, P and N).
The formation of the Au(III) catecholate complex 5a from the corresponding Au(I) o-quinone 5′a was then investigated (Fig. 6). 2-Electron transfer from gold to the o-quinone was found to be about barrierless (1.6 kcal mol−1) in line with the facile and instantaneous formation of 5a. The corresponding transition state (TS1) connecting 5′a to 5a is actually very early. It resembles the Au(I) o-quinone form and retains d10 configuration. The Au to o-quinone electron transfer occurs later, when falling down to the Au(III) catecholate form, as apparent from IBO analysis.12 From TS1 to 5a, close to the transition state, one of the in-plane 5d (Au) orbital evolves into a π(catecholate) orbital (Fig. 7), while the two πCO(o-quinone) orbitals turn into nOπ (catecholate) orbitals (Fig. S46†). In the mean time, the electron configuration of gold changes from d10 to d8 according to the occupancy of the 5d (Au) orbitals (NBO calculations).
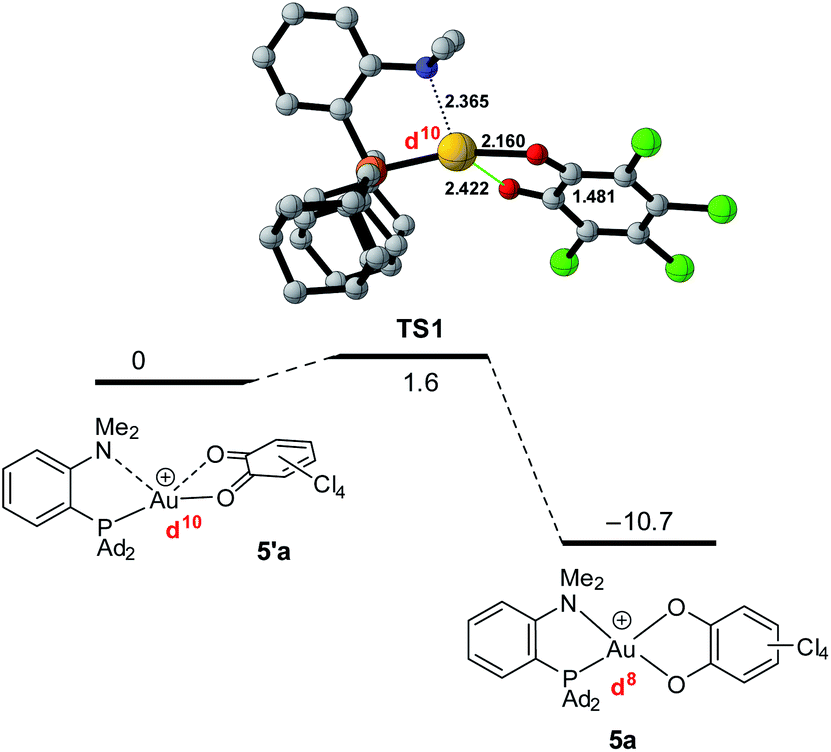 | ||
| Fig. 6 Conversion of the Au(I) o-quinone valence isomer 5′a into the Au(III) catecholate complex 5a, computed at the PCM(DCM)-B3PW91-D3(BJ)/SDD + f (Au),6-31G**(C, H, N, O, P, Cl) level of theory. | ||
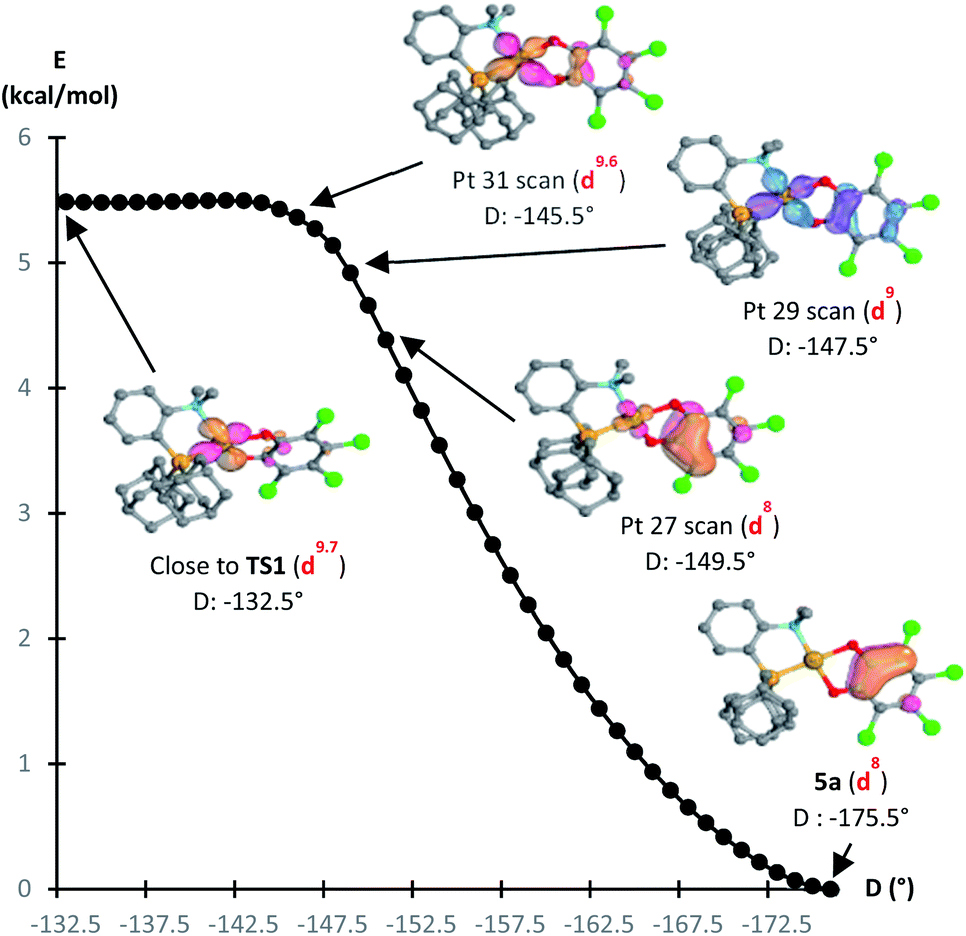 | ||
| Fig. 7 Reaction path from TS1 to 5a by integrating the intrinsic reaction coordinate (D = bond dihedral angle of Ocis-to-PAuPCPh in °), computed at the B3PW91-D3(BJ)/SDD + f (Au), 6-31G** (C, H, N, O, P, Cl) level of theory. IBO orbitals of the Au to o-quinone electron transfer along the reaction coordinate from TS1 to 5a, showing the change from an in-plane 5d (Au) to a π (catecholate) orbital. Electron configuration at Au from NBO calculations. | ||
The related gold complexes featuring the chelating o-carboranyl diphosphine were also computed. At the same level of theory, taking into account dispersion and solvent effects, only the Au(III) catecholate forms could be located as energy minima on the potential energy surface (Fig. 8). Au(I) o-quinone structures could be found as a transition state on PES for the parent o-benzoquinone and only by imposing geometric constraints (torsion of the o-quinone moiety with respect to the (P^P)Au coordination plane) for the chlorinated derivative. They sit 6.4 and 14.2 kcal mol−1 above the Au(III) catecholate forms for the parent o-benzoquinone and o-chloranil, respectively.20 The optimized geometry and frontier orbitals of the experimentally prepared Au(III) catecholate complex 2a are displayed in Fig. 8. The P^P ligand is chelating gold which sits in a quasi-perfect square-planar environment (PAuP bite angle = 93.3°, τ4 = 0.07).13 The C–O and C–C bond lengths (1.343 and 1.410 Å, respectively) are diagnostic for the reduction of o-chloranil into the corresponding catecholate. Moreover, the HOMO/LUMO very much resemble those of the related P^N-ligated complex 5a.
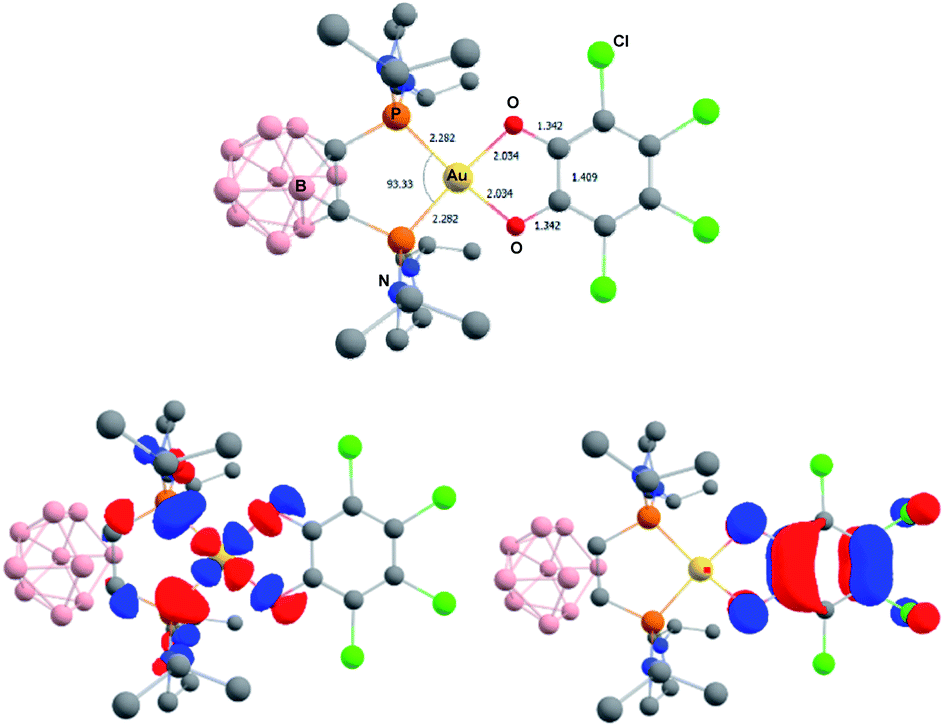 | ||
| Fig. 8 Optimized geometry of the Au(III) catecholate complex 2a, computed at the PCM(DCM)-B3PW91-D3(BJ)/SDD + f (Au), 6-31G** (H, B, C, N, O, Cl, P) level of theory. Distances in Å, angles in °. Plot of the frontier orbitals with cutoff: 0.05. Hydrogen atoms have been omitted for clarity. | ||
Conclusions
Chelating P^P and hemilabile P^N ligands were found to promote the reaction of Au(I) complexes with o-benzoquinones to give Au(III) catecholate complexes. While single crystals suitable for X-ray diffraction analysis could not be obtained with the o-carboranyl diphosphine ligand due to slow B-decapping, the P^N-ligand MeDalphos provided highly stable and crystalline complexes. NMR spectroscopy, X-ray diffraction and X-ray absorption spectroscopy unambiguously establish the 2-electron process, i.e. oxidation of gold and reduction of the o-quinone. The reaction requires strongly oxidizing o-quinones such as o-chloranil and the related tetrabromo/tetrafluoro o-benzoquinones. It is reversible, as substantiated by reactivity studies. The addition of competing ligands such as chloride and alkenes gives back Au(I) complexes with concomitant release of the o-quinone. The structure and stability of the Au(I) o-quinone and Au(III) catecholate forms were thoroughly compared thanks to DFT calculations. 2-Electron transfer from gold to the o-quinone was found to be indeed favored by the strongly oxidizing o-chloranil and to occur at a late stage, after the transition state connecting the Au(I) o-quinone and Au(III) catecholate forms.Complexes 2a–c and 5a–c represent a new type of Au(III) catecholate complexes, deriving from P-containing ligands, and the ligand-enabled oxidation of Au(I) precursors with o-quinones oxidation stands as a new synthetic route. Future work will aim to extend the approach to other chelating/hemilabile ligands and increase further the chemical diversity of Au(III) catecholate complexes. Such complexes are expected to exhibit versatile chemical, redox, photophysical properties that will be thoroughly studied and tuned by ligand design. On mid term, Au(III) catecholate complexes may open new avenues in materials science (luminescence, optoelectronic applications), medicinal chemistry (cytotoxicity) and catalysis. Au(III) complexes,7 including complexes deriving from O,O-ligands (catechols, β-diketones, bis-naphthols, bis-phenols),8a,21 have been recently shown to be quite promising in these areas and they attract increasing interest.
Data availability
The ESI† contains the experimental procedures, the analytical data for the obtained products, the NMR spectra, the XRD and XAS data, the computational details and Z-matrices for the optimized structures.Conflicts of interest
There are no conflicts to declare.Author contributions
G. S. and D. B. designed the experiments. G. S. and J. B. conducted the experiments. G. S. processed and interpreted the analytical data. V. M.-D. and S. M.-L. performed and interpreted the XAS and XRD analyses, respectively. Y. G.-R. and K. M. performed and analyzed the DFT calculations. The manuscript was written and reviewed by all authors. D. B. designed and directed the project.Acknowledgements
This work has been supported financially by the Centre National de la Recherche Scientifique (CNRS) and the Université Toulouse III – Paul Sabatier (UPS). The NMR service of ICT (M. Vedrenne) is acknowledged for assistance with the HSQC 15N-1H NMR experiment. The X-ray absorption experiments were performed in part at CLAESS beamline at ALBA Synchrotron with the collaboration of ALBA staff and in part at BM23 beamline at ESRF Synchtotron with the assistance of Cesare Atzori (local contact). The “Direction du Numérique” of the Université de Pau et des Pays de l’Adour, CINES under allocation A011080045 made by Grand Equipment National de Calcul Intensif (GENCI) and Mésocentre de Calcul Intensif Aquitain (MCIA) are acknowledged for computational facilities. D. Vesseur (LHFA) is warmly acknowledged for independent synthesis and characterization of the (P^N)Au(nbe),PF6 complex 6c.Notes and references
- (a) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261 CrossRef CAS PubMed; (b) M. O. Akram, S. Banerjee, S. S. Saswade, V. Bedi and N. T. Patil, Chem. Commun., 2018, 54, 11069 RSC; (c) B. Huang, M. Hu and F. D. Toste, Trends Chem., 2020, 2, 707 CrossRef CAS PubMed; (d) P. Font and X. Ribas, Eur. J. Inorg. Chem., 2021, 2556 CrossRef CAS.
- For representative examples, see: (a) S. Vanicek, J. Beerhues, T. Bens, V. Levchenko, K. Wurst, B. Bildstein, M. Tilset and B. Sarkar, Organometallics, 2019, 38, 4383 CrossRef CAS PubMed; (b) T. S. Teets and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 7411 CrossRef CAS PubMed; (c) S. Gaillard, A. M. Z. Zlawin, A. T. Bonura, E. D. Stevens and S. P. Nolan, Organometallics, 2010, 29, 394 CrossRef CAS; (d) A. C. Reiersølmoen, S. Battaglia, A. Orthaber, R. Lindh, M. Erdélyi and A. Fiksdahl, Inorg. Chem., 2021, 60, 2847 CrossRef PubMed; (e) G. Kleinhans, A. K.-W. Chan, M.-Y. Leung, D. C. Liles, M. A. Fernandes, V. W.-W. Yam, I. Fernández and D. I. Bezuidenhout, Chem.–Eur. J., 2020, 26, 6993 CrossRef CAS PubMed; (f) A. Tlahuext-Aca, M. N. Hopkinson, C. G. Daniliuc and F. Glorius, Chem.–Eur. J., 2016, 22, 11587 CrossRef CAS PubMed; (g) L. Huang, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2016, 52, 6435 RSC; (h) E. O. Asomoza-Solís, J. Rojas-Ocampo, R. A. Toscano and S. Porcel, Chem. Commun., 2016, 52, 7295 RSC; (i) A. Tabey, M. Berlande, P. Hermange and E. Fouquet, Chem. Commun., 2018, 54, 12867 RSC; (j) M. Hofer, T. de Haro, E. Gómez-Bengoa, A. Genoux and C. Nevado, Chem. Sci., 2019, 10, 8411 RSC; (k) Y. Yang, L. Eberle, F. F. Mulks, J. F. Wunsch, M. Zimmer, F. Rominger, M. Rudolph and A. S. K. Hashmi, J. Am. Chem. Soc., 2019, 141, 17414 CrossRef CAS PubMed.
- (a) R. E. Bachman, S. A. Bodolosky-Bettis, C. J. Pyle and M. A. Gray, J. Am. Chem. Soc., 2008, 130, 14303 CrossRef CAS PubMed; (b) P. Gualco, S. Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2011, 50, 8320 CrossRef CAS PubMed; (c) N. Lassauque, P. Gualco, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2013, 135, 13827 CrossRef CAS PubMed; (d) M. Joost, P. Gualco, Y. Coppel, K. Miqueu, C. E. Kefalidis, L. Maron, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2014, 53, 747 CrossRef CAS PubMed.
- (a) C.-Y. Wu, T. Horibe, C. Borch Jacobsen and F. D. Toste, Nature, 2015, 517, 449 CrossRef CAS PubMed; (b) M. Joost, L. Estévez, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5236 CrossRef CAS PubMed; (c) J. Chu, D. Munz, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 7884 CrossRef CAS PubMed.
- (a) M. Joost, A. Zeineddine, L. Estévez, S. Mallet−Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654 CrossRef CAS PubMed; (b) A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565 CrossRef PubMed; (c) J. Rodriguez, A. Zeineddine, E. D. Sosa Carrizo, K. Miqueu, N. Saffon-Merceron, A. Amgoune and D. Bourissou, Chem. Sci., 2019, 10, 7183 RSC; (d) J. Rodriguez, A. Tabey, S. Mallet-Ladeira and D. Bourissou, Chem. Sci., 2021, 12, 7706 RSC; (e) M. J. Harper, C. J. Arthur, J. Crosby, E. J. Emmett, R. L. Falconer, A. J. Fensham-Smith, P. J. Gates, T. Leman, J. E. McGrady, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2018, 140, 4440 CrossRef CAS PubMed; (f) J. A. Cadge, H. A. Sparkes, J. F. Bower and C. A. Russell, Angew. Chem., Int. Ed., 2020, 59, 6617 CrossRef CAS PubMed; (g) J. A. Cadge, J. F. Bower and C. A. Russell, Angew. Chem., Int. Ed., 2021, 60, 24976 CrossRef CAS PubMed.
- (a) R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1994 CrossRef CAS PubMed; (b) B. Bertrand, B. M. Bochmann, J. Fernandez-Cestau and L. Rocchigiani, in Pincer Compounds − Chemistry and Applications, ed. D. Morales-Morales, Elsevier, Amsterdam, 2018, pp. 673–699 Search PubMed; (c) L. Rocchigiani and M. Bochmann, Chem. Rev., 2021, 121, 8364 CrossRef CAS PubMed; (d) C. M. Che and R. W.-Y. Sun, Chem. Commun., 2011, 47, 9554 RSC; (e) B. Bertrand, B. M. R. M. Williams and M. Bochmann, Chem.–Eur. J., 2018, 24, 11840 CrossRef CAS PubMed; (f) J. Rodriguez and D. Bourissou, Angew. Chem., Int. Ed., 2018, 57, 386 CrossRef CAS PubMed; (g) V. W.-W. Yam and A. S.-Y. Law, Coord. Chem. Rev., 2020, 414, 213298 CrossRef CAS.
- V. W. Bhoyare, A. G. Tathe, A. Das, C. C. Chintawar and N. T. Patil, Chem. Soc. Rev., 2021, 50, 10422 RSC.
- (a) C. H. A. Goss, W. Henderson, A. L. Wilkins and C. Evans, J. Organomet. Chem., 2003, 679, 194 CrossRef CAS; (b) P. Byabartta and M. Laguna, Inorg. Chem. Commun., 2007, 10, 666 CrossRef CAS; (c) K. J. Kilpin, W. Henderson and B. K. Nicholson, Inorg. Chim. Acta, 2009, 362, 3669 CrossRef CAS; (d) K. J. Kilpin, B. P. Jarman, W. Henderson and B. K. Nicholson, Appl. Organomet. Chem., 2011, 25, 810 CrossRef CAS; (e) T. S. Smith, J. R. Lane, M. R. Mucalo and W. Henderson, Transition Met. Chem., 2016, 41, 581 CrossRef CAS.
- For selected references, see: (a) J. S. Valentine and D. Valentine Jr, J. Am. Chem. Soc., 1970, 92, 5795 CrossRef CAS; (b) Y. S. Sohn and A. L. Balch, J. Am. Chem. Soc., 1971, 93, 1290 CrossRef; (c) Y. S. Sohn and A. L. Balch, J. Am. Chem. Soc., 1972, 94, 1144 CrossRef CAS; (d) C. Floriani, R. Henzi and F. Calderazzo, J. Chem. Soc., Dalton Trans., 1972, 2640 RSC; (e) A. L. Balch, J. Am. Chem. Soc., 1973, 95, 2723 CrossRef CAS; (f) C. G. Pierpont, H. H. Downs and T. G. Rukavina, J. Am. Chem. Soc., 1974, 96, 5573 CrossRef CAS; (g) C. G. Pierpont and H. H. Downs, J. Am. Chem. Soc., 1975, 97, 2123 CrossRef CAS; (h) C. G. Pierpont and R. M. Buchanan, J. Am. Chem. Soc., 1975, 97, 4912 CrossRef CAS; (i) C. G. Pierpont and R. M. Buchanan, J. Am. Chem. Soc., 1975, 97, 6450 CrossRef CAS; (j) C. G. Pierpont and R. M. Buchanan, Coord. Chem. Rev., 1981, 38, 45 CrossRef CAS; (k) C. G. Pierpont and C. W. Lange, Prog. Inorg. Chem., 1994, 41, 331 CAS; (l) C. G. Pierpont, Coord. Chem. Rev., 2001, 219–221, 415 CrossRef CAS; (m) M. Glavinović, F. Qi, A. D. Katsenis, T. Friščić and J.-P. Lumb, Chem. Sci., 2016, 7, 707 RSC.
- Tetrachloro-1,2-benzoquinone was reported not to react with Ph3PAuCl, see ref. 9b..
- Elemental Cu was reported to react with o-benzoquinones, affording Cu(I)/semiquinone or Cu(II)/catecholate complexes, depending on the ancillary ligand, see: (a) O. Kahn, R. Prins, J. Reedijk and J. S. Thompson, Inorg. Chem., 1987, 26, 3557 CrossRef CAS; (b) G. Speier, S. Tisza, A. Rockenbauer, S. R. Boone and C. G. Pierpont, Inorg. Chem., 1992, 31, 1017 CrossRef CAS; (c) J. Rall, M. Wanner, M. Albrecht, F. M. Hornung and W. Kaim, Chem.–Eur. J., 1999, 5, 2802 CrossRef CAS; (d) B. Tapodi, G. Speier, M. Giorgi, M. Réglier, T. Funabiki, L. Korecz and A. Rockenbauer, Inorganica Chim. Acta., 2006, 9, 367 CAS.
- See ESI† for details.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- (a) P. Zanello and M. Corsini, Coord. Chem. Rev., 2006, 250, 2000 CrossRef CAS; (b) S. N. Brown, Inorg. Chem., 2012, 51, 1251 CrossRef CAS PubMed.
- To the best of our knowledge, two Au(III) complexes deriving from nido-o-carboranyl diphosphines have been reported previously. They were prepared by reacting the corresponding closo-o-carboranyl diphosphines with AuCl3, see: (a) F. Teixidor, C. Viñas, M. M. Abad, R. Kivekiis and R. Sillanpää, J. Organomet. Chem., 1996, 509, 139 CrossRef CAS; (b) P. G. Jones, M. D. Villacampa, O. Crespo, M. C. Gimeno and A. Laguna, Acta Crystallogr., Sect. C:, 1997, 53, 570 CrossRef.
- M. Navarro, A. Tabey, G. Szalóki, S. Mallet-Ladeira and D. Bourissou, Organometallics, 2021, 40, 1571 CrossRef CAS.
- H. Duggal, P. Rajput, I. Alperovich, T. Asanova, D. Mehta, S. N. Jha and S. Gautam, Vacuum, 2020, 176, 109294 CrossRef CAS.
- (a) M. Navarro, A. Toledo, M. Joost, A. Amgoune, S. Mallet-Ladeira and D. Bourissou, Chem. Commun., 2019, 55, 7974 RSC; (b) M. Navarro, A. Toledo, S. Mallet-Ladeira, E. Daiann Sosa Carrizo, K. Miqueu and D. Bourissou, Chem. Sci., 2020, 11, 2750 RSC; (c) M. Navarro and D. Bourissou, Adv. Organomet. Chem., 2021, 76, 101 CrossRef.
- For a diphosphine-ligated Cu (I) o-quinone complex, see: S. Roy, B. Sarkar, D. Bubrin, M. Niemeyer, S. Záliš, G. Kumar Lahiri and W. Kaim, J. Am. Chem. Soc., 2008, 130, 15230 CrossRef CAS PubMed.
- True energy minima could be located for both the Au(III) catecholate and Au(I) o-quinone forms at the PCM(DCM)-B3PW91/SDD+f(Au),6-31G**(H, B, C, N, O, P, Cl) level of theory, not taking into account dispersion effects. With o-chloranil, the Au (III) form is more stable than the Au(I) form by 6.8 kcal mol−1 (vs 6.0 kcal mol−1 for the MeDalphos ligand). The difference in energy drops to 0.4 kcal mol−1 for the parent o-benzoquinone (with MeDalphos, the Au(I) form is more stable than the Au(III) form by 2.4 kcal mol−1), in line with its less oxidizing character.12.
- (a) K. T. Chan, G. S. M. Tong, Q. Wan, G. Cheng, C. Yang and C.-M. Che, Chem.–Asian J., 2017, 12, 2104 CrossRef CAS PubMed; (b) J.-F. Cui, H.-M. Ko, K.-P. Shing, J.-R. Deng, N. C.-H. Lai and M.-K. Wong, Angew. Chem., Int. Ed., 2017, 56, 3074 CrossRef CAS PubMed; (c) J.-J. Jiang, J.-F. Cui, B. Yang, Y. Ning, N. Chun-Him Lai and M.-K. Wong, Org. Lett., 2019, 21, 6289 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, NMR spectra, XRD and XRD data, computational details and Z-matrices. CCDC 2180464–2180468. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03724f |
| This journal is © The Royal Society of Chemistry 2022 |