
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of MoS2 monolayer electrocatalysts via reduction and phase control in molten sodium for selective hydrogenation of nitrogen to ammonia†
Hong
Zhang
ab,
Bin
Song
*c,
Weiwei
Zhang
 d,
Yingwen
Cheng
*e,
Qianwang
Chen
ab and
Ke
Lu
*ab
d,
Yingwen
Cheng
*e,
Qianwang
Chen
ab and
Ke
Lu
*ab
aInstitutes of Physical Science and Information Technology, School of Materials Science and Engineering, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Anhui University, Hefei, Anhui 230601, China. E-mail: luke@ahu.edu.cn
bHefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, Anhui 230026, China
cInstitute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou, Jiangsu 215123, China. E-mail: bsong@suda.edu.cn
dSchool of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, Shandong 273165, China
eDepartment of Chemistry and Biochemistry, Northern Illinois University, DeKalb, IL 60115, USA. E-mail: ycheng@niu.edu
First published on 25th July 2022
Abstract
Electrochemical nitrogen fixation under ambient conditions is promising for sustainable ammonia production but is hampered by high reaction barrier and strong competition from hydrogen evolution, leading to low specificity and faradaic efficiency with existing catalysts. Here we describe the activation of MoS2 in molten sodium that leads to simultaneous formation of a sulfur vacancy-rich heterostructured 1T/2H-MoSx monolayer via reduction and phase transformation. The resultant catalyst exhibits intrinsic activities for electrocatalytic N2-to-NH3 conversion, delivering a faradaic efficiency of 20.5% and an average NH3 rate of 93.2 μg h−1 mgcat−1. The interfacial heterojunctions with sulfur vacancies function synergistically to increase electron localization for locking up nitrogen and suppressing proton recombination. The 1T phase facilitates H–OH dissociation, with S serving as H-shuttling sites and to stabilize 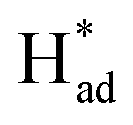 . The
. The  subsequently couple with nearby N2 and NHx intermediates bound at Mo sites, thus greatly promoting the activity of the catalyst. First-principles calculations revealed that the heterojunction with sulfur vacancies effectively lowered the energy barrier in the potential-determining step for nitrogen reduction, and, in combination with operando spectroscopic analysis, validated the associative electrochemical nitrogen reduction pathway. This work provides new insights on manipulating chalcogenide vacancies and phase junctions for preparing monolayered MoS2 with unique catalytic properties.
subsequently couple with nearby N2 and NHx intermediates bound at Mo sites, thus greatly promoting the activity of the catalyst. First-principles calculations revealed that the heterojunction with sulfur vacancies effectively lowered the energy barrier in the potential-determining step for nitrogen reduction, and, in combination with operando spectroscopic analysis, validated the associative electrochemical nitrogen reduction pathway. This work provides new insights on manipulating chalcogenide vacancies and phase junctions for preparing monolayered MoS2 with unique catalytic properties.
1. Introduction
Ammonia, a vital chemical feedstock with high energy density (e.g., 3 kWh kg−1, 4.3 kWh h−1), is of great importance to the agricultural industry and clean energy exploration. Commercial industrial NH3 synthesis is dominated by the Haber–Bosch process with harsh synthetic conditions, which consumes 1–2% of global energy production and results in carbon dioxide emissions.1,2 This can be potentially replaced by direct electrochemical nitrogen fixation to ammonia using renewable electricity as an attractive ‘carbon-neutral’ alternative strategy.3,4 However, current heterogeneous electrocatalysts have very limited activity to activate nitrogen molecules due to their chemical inertness (dissociation energy of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond: 940.95 kJ mol−1). As a result, electrocatalytic nitrogen conversion typically operates via high overpotentials and under potentials that are well below the hydrogen evolution reaction (HER). Unfortunately, the HER typically exhibits fast kinetics in aqueous solutions and the selectivity towards NH3 is still far from being competitive with that of the Haber–Bosch process. In addition, ambient ammonia contamination during catalysis is significant and inevitable, thus leading to it also being difficult to detect ammonia generated by the nitrogen reduction reaction (NRR) accurately. So, the effect of ambient ammonia contamination also needs to be identified in as much detail as possible.5,6 Further advances on novel catalyst design and fundamental understanding regarding the multi-step transfer of electrons and protons during nitrogen hydrogenation are required to overcome the limitations of existing nitrogen reduction electrocatalysts such as slow kinetics and high energy barrier.7–10
N triple bond: 940.95 kJ mol−1). As a result, electrocatalytic nitrogen conversion typically operates via high overpotentials and under potentials that are well below the hydrogen evolution reaction (HER). Unfortunately, the HER typically exhibits fast kinetics in aqueous solutions and the selectivity towards NH3 is still far from being competitive with that of the Haber–Bosch process. In addition, ambient ammonia contamination during catalysis is significant and inevitable, thus leading to it also being difficult to detect ammonia generated by the nitrogen reduction reaction (NRR) accurately. So, the effect of ambient ammonia contamination also needs to be identified in as much detail as possible.5,6 Further advances on novel catalyst design and fundamental understanding regarding the multi-step transfer of electrons and protons during nitrogen hydrogenation are required to overcome the limitations of existing nitrogen reduction electrocatalysts such as slow kinetics and high energy barrier.7–10
Transition metal dichalcogenides with tunable electronic properties have attracted wide attention and have been extensively exploited in electrocatalysis.11–14 Of them, inspired by natural metalloenzymes, such as Mo-dependent nitrogenase, MoS2 exhibits very similar constituent elements and is viewed as a very promising nitrogen functionalization catalyst.15–19 In this regard, the low-coordinated Mo atoms at the edge sites are generally accepted as the nitrogen reduction catalytic centers for activating N2 molecules. The abundant saturated coordinative atoms in the base planes, on the other hand, are inactive and the overall catalytic efficiency is severely slowed by a deficiency of active sites on the basical plane.20 To this end, defect, phase and strain engineering have been explored to boost the intrinsic activity of MoS2 catalysts.21–24 However, their poor conductivity and inert basal planes result in NH3 yield rates that are generally less than 50 μg h−1 mgcat−1, such as 13.09 μg h−1 mgcat−1 for MoS2 nanosheets and 29.28 μg h−1 mgcat−1 for defect-rich 2H-MoS2 nanoflowers.25 By far, 2H-MoS2 is the most studied material, but its trigonal prismatic Mo coordination leads to fully occupied dz2 and empty dxy, dx2–y2 orbitals, making them intrinsically less active. On the other hand, the metastable 1T phase MoS2 has octahedral Mo coordination that splits the 4d2 orbitals of Mo4+ into eg and t2g orbitals and could be more attractive as partially occupied t2g orbitals provide mechanisms to overlap with N2 and form strong σ bonds and transfer electrons to the anti-bonding molecular orbital of N2 for lowering the barrier associated with initial activation.25,26 In addition, the Mo–Mo distance, potentially adjustable via doping or crystal defects, could be further employed to modulate Mo–N interaction for nitrogen conversion. Multi-functional heterojunctions that strengthen nitrogen adsorption and promote its activation while suppressing hydrogen evolution represent a promising direction for catalyst design.26 Notably, various research endeavors have been devoted to the phase engineering of MoS2, especially focusing on the transformation from the 2H to 1T phase (such as the n-butyllithium intercalation approach) for realizing metallic features with strong hydrophilicity. Besides this, defect engineering is also an effective strategy to improve the activity of MoS2 base planes. The formation of sulfur vacancies can become interstitials by lodging in nearby locations, which will greatly promote the charge distributions of monolayer MoS2 base planes.24–26 However, facile effective experimental strategies by which to synthesize MoS2-based catalysts with a tunable 1T/2H phase and sulfur vacancies have not yet been identified.
Herein, we report a facile chemical conversion technique to transform bulk MoS2via an amalgamated MoS2-Na method to 1T/2H-MoSx with engineered sulfur vacancies that exhibits high intrinsic activity. The strong chemical interactions between metallic Na and MoS2 sites under molten Na states generate ∼10% sulfur vacancies and the conversion of stabilized 1T phases, realizing the activation of the basal plane. The final heterostructured 1T/2H–MoS1.79 monolayer exhibits intrinsic selectivity and yields excellent nitrogen fixation performance, with an average ammonia formation rate of 93.2 μg h−1 mgcat−1 at −0.4 V versus the reversible hydrogen electrode (RHE) and a faradaic efficiency of 20.5% with remarkable long-term operation stability over 30 h. Specifically, it was identified that the undercoordinated Mo regions and the intimate 1T/2H interfacial heterojunctions with sulfur vacancies can lock up nitrogen at localized hotspots, and phase transformation promotes H–OH dissociation in mild electrolyte, whereas S sites serve as a H-provider and are conducive to 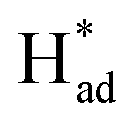 stabilization, thus both processes work synergistically to lower the nitrogen reduction energy barrier in the potential-determining step and assist selective N2 transformation, further supported by density functional theory (DFT) calculations.
stabilization, thus both processes work synergistically to lower the nitrogen reduction energy barrier in the potential-determining step and assist selective N2 transformation, further supported by density functional theory (DFT) calculations.
2. Results and discussion
Fig. 1a illustrates our molten-Na-assisted intercalation and subsequent chemical conversion approach for generating abundant 1T/2H interfacial heterojunctions with sulfur vacancies (SVs) starting from bulk MoS2 (MoS2-H). Briefly, bulk MoS2 powder (Fig. S1†) was infiltrated with molten Na metal and then calcined under an Ar atmosphere (optimized at 120 °C). During the blending process, the difference between the electron affinity of MoS2 (4.45) and the ionization potential of Na (5.12 eV) results the intercalation of Na into MoS2 interlayers (MoS2 accepts electrons due to high electron affinity) to realize reduction and phase control, producing an exfoliated nanosheet morphology.27–29 In addition, the intercalated Na atoms generate Na–S bonds, and the large electronegativity difference between the Na and S atoms facilitates the transfer of electrons and induces the formation of SVs and a 1T phase (Table S1†).30 Therefore, junction type MoSx-H/T monolayers could be synthesized through a highly-scalable molten-Na-assisted approach.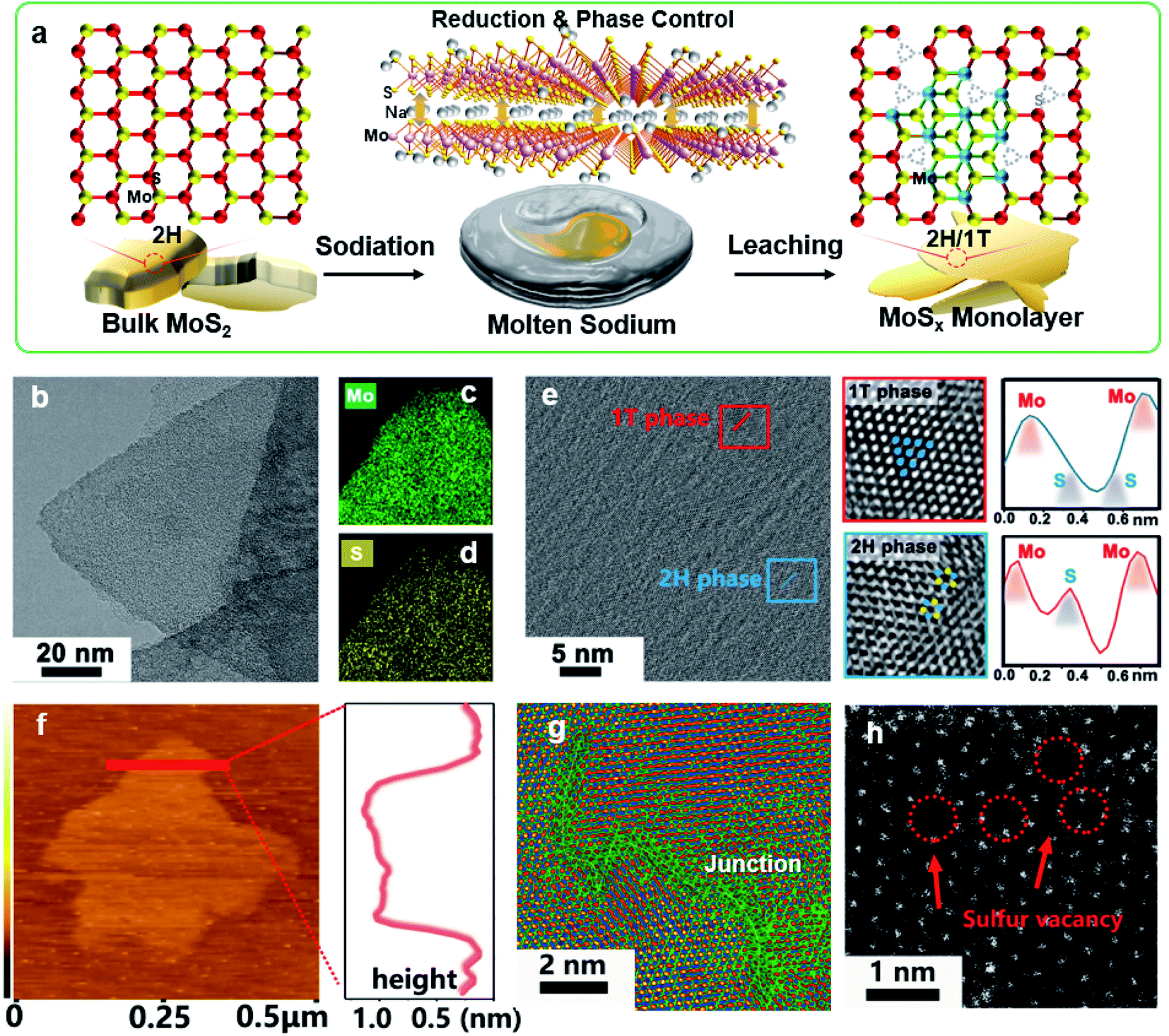 | ||
| Fig. 1 Schematic illustration of the catalyst synthesis and representative electron microscopy morphology. (a) Schematic illustration of the preparation of MoSx-H/T. (b) TEM and the corresponding (c, d) EDS mapping analysis of MoSx-H/T sample, confirming the homogeneous distributions of Mo and S. (e) TEM and HRTEM images of MoS1.79-H/T, showing the hybrid 1T-phase and 2H-phase domains. Zoomed-in images of the 1T (red frame) and 2H (blue frame) phase regions in (e), showing the octahedral (1T) and trigonal-prismatic (2H) coordinated lattice structure with corresponding atomic profiles, respectively. (f) Tapping-mode AFM image and the corresponding line scan of the MoSx-H/T film. (g) HRTEM image of MoS1.79-H/T with pseudo color labelling. (h) Atomic-resolution HAADF-STEM images of MoSx-H/T with vacancies. | ||
To confirm the phase transformation of the exfoliated MoSx-H/T nanoplatelets, the morphology and lattice properties were investigated by high resolution transmission electron microscopy (HRTEM). Fig. 1b–d show the microscopy images and energy-dispersive X-ray spectroscopy (EDS) mapping of the MoS1.79-H/T prepared under the optimal synthesis conditions of 120 °C and 10 min. The Mo and S are homogeneously distributed on the basal plane of the MoSx nanosheets. Fig. 1e presents the junction where the 1T- and 2H-phases meet. The characteristic crystalline lattice structures of the 1T- and 2H-phases were clarified from the zoomed-in images (red and blue frames) and the atomic arragenement profiles. The 1T-phase region shows characteristic Mo–S octahedral coordination, corresponding to the (100) plane of MoS2 with two sulfur atoms between neighboring molybdenum atoms (Mo–S–S–Mo), whereas the 2H-phase region exhibits typical Mo–S trigonalprismatic coordination (Mo–S–Mo) (red line).31,32 The monolayer nanosheet nature of MoSx-H/T was verified, with a height of 1.05 nm in the atomic force microscopy (AFM) image (Fig. 1f). In addition, the HRTEM image in Fig. 1g highlights the 1T- and 2H-phase heterojunctions within the basal plane of MoSx-H/T. Moreover, aberration-corrected scanning transmission electron microscopy (AC-STEM) was carried out to disclose the atomic configurations of MoSx-H/T. Prominent defect sites were observed in MoSx-H/T, as highlighted by the red arrows in Fig. 1h. This evidence indicates that the SVs were clearly generated after the molten-Na treatment.12,17,21
Due to the formation of SVs, the atomic ratio of MoSx-H/T (x = 1.59–2) with different molten-Na-assisted intercalation reaction times was quantified by inductively coupled plasma atomic emission spectroscopy (see Table S1†). The molten-Na-assisted intercalation and subsequent chemical conversion and its effect on the MoSx phase were further verified by X-ray diffraction (XRD) analysis. Fig. 2a presents the XRD peaks of Na-intercalated MoS2. The (002) plane of pristine MoS2 can be observed at ∼14°, which gradually widened (the full width at half maximum was significantly increased from 0.2° to 1.1°) during Na intercalation and hence suggests increased MoS2 interlayer spacing after Na intercalation. Meanwhile, the diffraction signals of the (002) plane were blueshifted (from 14.1° to 13.2°) for MoSx-H/T relative to MoS2-H, indicating the effective intercalation and phase transformation from 2H to 1T in MoSx.32,33 The phase control and molten-Na-assisted generation of SVs were investigated by conducting X-ray photoelectron spectroscopy (XPS) measurements on the as-prepared MoSx-H/T (Fig. 2b). Mo 3d5/2 and 3d3/2 peaks can be observed at ∼229.8 and ∼232.9 eV, respectively, in the spectra of 2H-MoS2 (red areas), whereas for 1T-MoS2 (green areas), these two peaks appear at ∼229.0 and ∼232.1 eV (Fig. 2b), respectively. A similar downshift in the binding energies of the 1T phase were identified from the S 2p3/2 and S 2p1/2 signals (Fig. 2c), which arise at ∼163.0 and ∼164.1 eV in the spectra of the 2H phase, and at ∼162.1 and ∼163.4 eV for the 1T phase, respectively. These downshifts in the 1T phase are attributable to the change in the Fermi level induced by electron filling in the Mo d orbitals.32–34 Remarkably, the signals from the 1T phase (green areas) gradually dominate the main composition with an increase in the reaction time from 0 to 30 min, which comfirms that the molten-Na-assisted process effectively transformed from 2H to 1T. Furthermore, in the XPS results, the phase compositions of the 1T phase were calculated from 0 to 72%, indicating that the proposed molten-Na-assisted intercalation approach successfully controls the 2H and 1T phases (Fig. 2d). As shown in Fig. 2e, after molten-Na-assisted intercalation treatment, MoSx-H/T shows different vibration modes in its Raman spectra. For 2H-MoS2 (black line) two characteristic Raman peaks at 383 cm−1 (E12g mode, in-plane stretching) and 401 cm−1 (A1g mode, out-of-plane vibration) arise from S–Mo–S. In addition, during the molten-Na-assisted intercalation treatment, the intensities of the E12g and A1g modes decrease dramatically, meanwhile, the new Raman peaks at 280 cm−1 (E1g mode, attributable to the octahedral coordination of Mo), 152 cm−1 (J1 mode), 201 cm−1 (J2 mode) and 337 cm−1 (J3 mode) indicate the formation of the 1T phase. In particular, the amplification of the Raman peaks at 383 cm−1 (E12g mode) and 401 cm−1 (A1g mode), and the gaps (Δk) between the two Raman peaks of A1g to E12g progressively decrease from 25 to 22, further demonstrating that the Na intercalation process weakens the interactions between neighboring MoS2 layers along with the exfoliation of the monolayers.32,35
 | ||
| Fig. 2 Physical characterization of MoS2 samples before and after molten-Na-assisted intercalation. (a) Selected XRD spectra of pristine and treated MoS2 samples. The clearly widened and blue shifted 2θ peaks indicate that the Na intercalation expands the interlayer spacing of 2H- and 1T-MoS2. Comparison of the high-resolution (b) Mo 3d and (c) S2p XPS spectra of the different MoSx samples. (d) Relative fractions of the 1T phase in the original and treated MoS2 samples. (e) Raman spectra of the different MoSx samples as noted. | ||
Electron spin resonance (ESR) was utilized to quantify the paramagnetic signals of MoSx-H/T and the formation of SVs (Fig. 3a), in which the ∼337 mT (g = 2.009) intensity reveals the concentration of unsaturated sites with unpaired electrons.25 Notably, the pristine 2H-MoS2 demonstrates a relatively low signal intensity, which can be attributed to the inherent in-plane and edge SVs. Remarkably, the MoSx-H/T exhibits approximately 10 times higher signal intensity than pristine 2H-MoS2, corroborating the formation of abundant SVs, which also demonstrates that the SV concentration correlates well with the 1T phase. Temperature-programmed desorption (TPD) of nitrogen was used to investigate the N2 adsorption properties (Fig. 3b). Pristine 2H-MoS2 exhibits a Brunauer–Emmett–Teller (BET) specific surface area of 33.2 m2 g−1 (Fig. S2†) and inferior N2 physisorption and chemisorption. In particular, strong correlation between physisorption/chemisorption and SV density was observed. All of the MoSx-H/T groups display enhanced chemisorption and physisorption capability with increased BET surface areas from 85.1 to 174.2 m2 g−1 due to their high SV concentration in the hybrid 2H/1T heterojunction system. Thus, the density of interfacial heterojunctions plays a vital role in strengthening the chemisorption. Among them, MoS1.79-H/T exhibits the highest chemisorption capacity, thus the heterojunction and SVs can act as strong acitve sites to “lock up” the nitrogen reactant for subsequent triple-bond dissociation. In addition, the Fourier-transform infrared (FTIR) spectra exhibits an obvious blueshift for the resonance peak from adsorbed nitrogen species from 2365 to 2358 cm−1 after Na-assisted intercalation (Fig. 3c), suggesting the enlongation of the NN triple bond length when adsorbed on the MoSx-H/T catalyst.36 Therefore, we conclude that the abundant localized electron induced by heterojunction and SVs in MoSx-H/T could better activate the NN triple bond to accelerate the catalysis.
 | ||
| Fig. 3 Structural and primary electrochemical characterization of the different catalysts. (a) ESR spectra. (b) N2 TPD curves of the different MoSx samples as noted. (c) FTIR spectra of N2 adsorption on the different MoSx samples. (d) LSV curves and (e) the corresponding Tafel slopes of the different MoSx in Ar-saturated electrolyte. (f) Comparison of the electrochemical surface areas of the different MoSx samples. | ||
Fig. 3d and e compare the hydrogen evolution reaction (HER) activities and kinetics of the pristine MoS2 and MoSx-H/T. As expected, pristine MoS2 is a poor HER catalyst with an onset potential of ∼−0.5 V vs. RHE and a high Tafel slope of 190 mV dec−1 due to its non-defective basal planes featuring few active sites.31–35 The MoSx-H/T catalysts, on the other hand, exhibited enhanced HER activities, and in particular, MoS1.59-H/T exhibits the best performance (with an onset overpotential of ∼−0.3 V vs. RHE and a Tafel slope of 93 mV dec−1), highlighting the advantages of the 1T phase. Meanwhile, the electrochemically active surface areas (ECSA, Fig. S3†) of MoSx-H/T were analysed and compared to reveal the electrochemical properties of the different electrocatalysts. MoS1.59-H/T exhibited the highest ECSA value (58.1 cm2), suggesting that it contains abundant active sites, owing to its higher 1T-phase content, providing a favorable platform for electrocatalytic activity. MoS1.59-H/T displays the lowest charge transfer resistance, which should be attributed to its high metallic 1T phase content (Fig. S4†). In addition, we can see from Fig. 3f that the monolayer catalyst shows a very similar ECSA value and that the 1T/2H interfacial junctions could have a positive effect on promoting both the NRR and HER kinetics.17,20,26
The resultant MoSx-H/T catalysts were deposited on carbon paper working electrodes and employed for the NRR in a three-compartment cell under ambient conditions (Fig. S5†). The NRR performance was evaluated by quantifying the generated NH3via the indophenol blue method (Fig. S6†), and the possible formation of byproduct (N2H4) was also quantified using the Watt and Chrisp method.37Fig. 4a shows the faradaic efficiencies (FEs) of the different MoSx-H/T catalysts at various potentials, of which MoS1.79-H/T exhibits the highest average NH3 yield rate and FE of 93.2 μg h−1 mgcat−1 and 20.5% at −0.4 V vs. RHE, respectively, much higher than that of pristine 2H-MoS2 (FE: 0.5%) and the other MoS2-H/T catalysts (FEs from 3.2 to 11.5%), implying that the 1T phases and SVs mainly promote the electrocatalytic NRR performance. It is worth mentioning that, due to the competing HER, the FEs of the catalysts significantly decreased when the applied potential was lower than −0.5 V. Moreover, as illustrated in Fig. 4b, the linear sweep voltammograms (LSVs) show a gas-dependent response for the MoS1.79-H/T catalyst (the electrolyte was purged with N2 or Ar for 30 min before measurements). Compared to the Ar measurements, the N2 LSVs displayed evidently higher cathodic currents, especially after −0.26 V vs. RHE, indicating the catalytic reduction of N2. In addition, the MoS1.59-H/T catalyst exhibits a relatively poor HER activity under an Ar atmosphere (with a Tafel slope of 128 mV dec−1) and could be beneficial for efficient N2 reduction. In comparison, a decreased Tafel slope of 114 mV dec−1 was achieved in a N2 saturated environment, indicating that the N2 electrochemical processes accelerate the interfacial  generation and/or removal. Considering the competitive relationship between the HER and NRR, the difference in the Tafel slopes under Ar and N2 diminished when the applied potential was lower than −0.77 V, indicating dominant *H adsorption and suppressed *N2 adsorption under high overpotentials. It should be noted that at the highest potential, more protons occupied the active sites and dominate proton reduction, seriously impeding the yield of the NRR. Further, the ammonia yields were 4.96, 26.2, 93.2, 84.9, and 36.7 μg h−1 mgcat−1, with FEs of 9.7%, 17.6%, 20.5%, 8.1%, and 2.2% at −0.20, −0.30, −0.40, −0.50, and −0.60 V vs. RHE (Fig. 4d, S7–S8,† average experimental values), respectively. In addition, none of the N2H4 by-products were observed in the cycled electrolyte (Fig. S9†), thus further confirming the high selectivity of the monolayer catalyst. The highest NH3 yield rate and FE reached 93.2 μg h−1 mgcat−1 and 20.5% at −0.4 V vs. RHE, respectively. The normalized LSV results (Fig. S10†) again confirmed the synergistic function of the 1T phase and SVs for facilitating nitrogen activation and conversion, especially in comparison with the pristine MoS2. The MoS1.79-H/T monolayer catalyst possesses the most catalytic active sites for the NRR. And the highest activity of MoS1.79-H/T catalyst under our testing conditions, when normalized to the electroactive surface area, was 0.491 μg h−1·cmECSA−2. Importantly, the catalyst exhibited an appreciable FE of 0.43% at an almost thermodynamic equilibrium potential of 0 V vs. RHE, indicating highly active catalysts in comparison with previously reported work.
generation and/or removal. Considering the competitive relationship between the HER and NRR, the difference in the Tafel slopes under Ar and N2 diminished when the applied potential was lower than −0.77 V, indicating dominant *H adsorption and suppressed *N2 adsorption under high overpotentials. It should be noted that at the highest potential, more protons occupied the active sites and dominate proton reduction, seriously impeding the yield of the NRR. Further, the ammonia yields were 4.96, 26.2, 93.2, 84.9, and 36.7 μg h−1 mgcat−1, with FEs of 9.7%, 17.6%, 20.5%, 8.1%, and 2.2% at −0.20, −0.30, −0.40, −0.50, and −0.60 V vs. RHE (Fig. 4d, S7–S8,† average experimental values), respectively. In addition, none of the N2H4 by-products were observed in the cycled electrolyte (Fig. S9†), thus further confirming the high selectivity of the monolayer catalyst. The highest NH3 yield rate and FE reached 93.2 μg h−1 mgcat−1 and 20.5% at −0.4 V vs. RHE, respectively. The normalized LSV results (Fig. S10†) again confirmed the synergistic function of the 1T phase and SVs for facilitating nitrogen activation and conversion, especially in comparison with the pristine MoS2. The MoS1.79-H/T monolayer catalyst possesses the most catalytic active sites for the NRR. And the highest activity of MoS1.79-H/T catalyst under our testing conditions, when normalized to the electroactive surface area, was 0.491 μg h−1·cmECSA−2. Importantly, the catalyst exhibited an appreciable FE of 0.43% at an almost thermodynamic equilibrium potential of 0 V vs. RHE, indicating highly active catalysts in comparison with previously reported work.
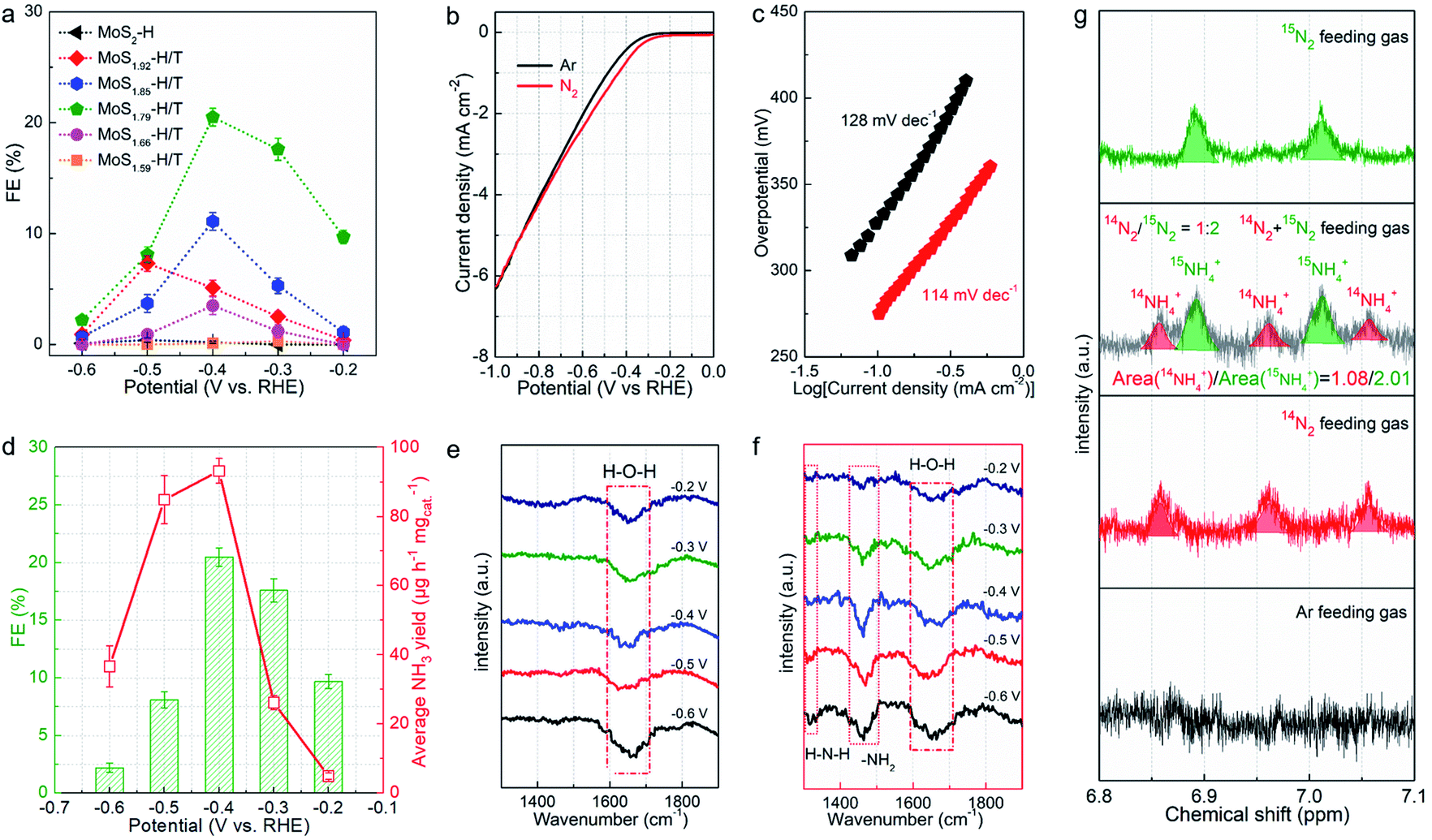 | ||
| Fig. 4 Ambient electrochemical NRR activity of the MoS2-based monolayer catalyst. (a) Average FEs of the different MoSx samples at different applied potentials. (b) LSV curves of MoS1.79-H/T in Ar- and N2-saturated electrolyte at a scan rate of 10 mV s−1, and (c) the corresponding Tafel slopes of the MoS1.79-H/T electrode. (d) Average NH3 yields and FEs of MoS1.79-H/T at different applied potentials. (e, f) FTIR spectra from −0.2 to −0.6 V vs. RHE for MoS1.79-H/T using Ar or N2 as the feed gas. (g) 15N isotope labeling experiment. The integrated peak areas associated with 14NH4+ and 15NH4+ are proportional to the initial ratio of the 14N2/15N2 feed gas. | ||
Fig. 4e and f exhibit the FTIR spectra of MoS1.59-H/T in Ar- and N2-saturated 0.1 M Na2SO4 solution at room temperature and −0.2 to −0.6 V vs. RHE. Different from the spectra under Ar, the spectra under N2 have additional peaks at 1317 and 1461 cm−1 corresponding to H–N–H and –NH2 vibrations. These results show that −NHx intermediates were indeed generated on MoS1.59-H/T during the NRR. An isotopic 15N labeling experiment was conducted to confirm the origin of the nitrogen resources (Fig. 4g). With 14N and an Ar feed gas, the obtained doublet pattern in the 1H NMR spectra can be attributed to 15NH4+. The ratio of the 14NH4+/15NH4+ products is proportional to the initial feed gas ratio of 14N2/15N2 (1.86 vs. 2.00), showing that the product originates electrocatalytic N2 gas reduction rather than the contamination and/or decomposition of the catalysts.37–39 In addition, the almost linear correlation between the NH3 concentration and electrolysis duration further demonstrate that MoS1.59-H/T is a stable catalyst for electrochemical nitrogen fixation (Fig. S11†). The slightly decreased ammonia formation rate can be ascribed to the changes in pH (the pH change of the electrolyte was from 6.36 to 9.11 within 30 h) or the accumulation of formed ammonia that slightly modifies the thermodynamic equilibrium potential for NH3 formation, thus making it impossible to apply a constant driving force for nitrogen fixation.38
Electrochemical in situ Fourier-transform infrared (FTIR) spectroscopy was employed to monitor the complex nitrogen reduction pathway. Fig. 5a shows the catalyst interfacial FTIR spectrum at −0.4 V vs. RHE for 4000 s in N2-saturated electrolyte. The resonance peak at 1110 cm−1 can be assigned to the N–N stretching of adsorbed N2Hx (1 ≤ x ≤ 4) species, with the peak intensity increasing with increasing reaction time.40,41 This clearly reflects that the NN triple bond is cleaved into a single bond within the catalyst/electrolyte interface. Notably, extra peaks that increased with the reaction time were observed, located at 1320 and 1465 cm−1, corresponding to H–N–H bending and –NH2 wagging,38 respectively. These results indicate the formation of N2Hx (1 ≤ x ≤4) species on the MoS1.79-H/T interface during the associative nitrogen reduction process. In detail, the molecular N2 reactant is firstly locked on the surface of the heterogeneous electrocatalyst, and then a proton is attached to dinitrogen to form a –NN–H bond, which initially activates the stable triple bond. In the subsequent step, the further hydrogenation process directs the formation of the –H–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–H intermediates. Thereafter, along with the continuous electron transfer and hydrogenation, the –H–NN–H intermediates were further weakened and the double bond was cleaved to form –H–N–NH2 species, and finally NH3 was formed on the surface of MoS1.79-H/T under ambient conditions.38,39 The MoS1.79-H/T catalyst exhibits negligible changes in FEs and ammonia yielding rates even after 30 continuous electrolysis cycles (Fig. 5b), indicating remarkably stable catalytic activity. After 30 h of electrolysis, MoS1.79-H/T still maintains good electrochemical durability without apparent current fluctuation (Fig. 5c and d). The high stability of the MoS1.79-H/T monolayer catalyst was also further confirmed by TEM (structure stability) and XPS (phase stability) after stability measurements, from which it is apparent that the catalyst still retains its initial monolayer structure without aggregation and the phase content does not change, as shown in Fig. 5e. Also, as compared in Fig. 5f, apart from the robustness of MoS1.79-H/T towards nitrogen fixation, the performance of our monolayer catalyst still surpasses those of previously reported MoS2-based materials, especially when considering the FEs and ammonia yield rates.24,31–35,42–54
N–H intermediates. Thereafter, along with the continuous electron transfer and hydrogenation, the –H–NN–H intermediates were further weakened and the double bond was cleaved to form –H–N–NH2 species, and finally NH3 was formed on the surface of MoS1.79-H/T under ambient conditions.38,39 The MoS1.79-H/T catalyst exhibits negligible changes in FEs and ammonia yielding rates even after 30 continuous electrolysis cycles (Fig. 5b), indicating remarkably stable catalytic activity. After 30 h of electrolysis, MoS1.79-H/T still maintains good electrochemical durability without apparent current fluctuation (Fig. 5c and d). The high stability of the MoS1.79-H/T monolayer catalyst was also further confirmed by TEM (structure stability) and XPS (phase stability) after stability measurements, from which it is apparent that the catalyst still retains its initial monolayer structure without aggregation and the phase content does not change, as shown in Fig. 5e. Also, as compared in Fig. 5f, apart from the robustness of MoS1.79-H/T towards nitrogen fixation, the performance of our monolayer catalyst still surpasses those of previously reported MoS2-based materials, especially when considering the FEs and ammonia yield rates.24,31–35,42–54
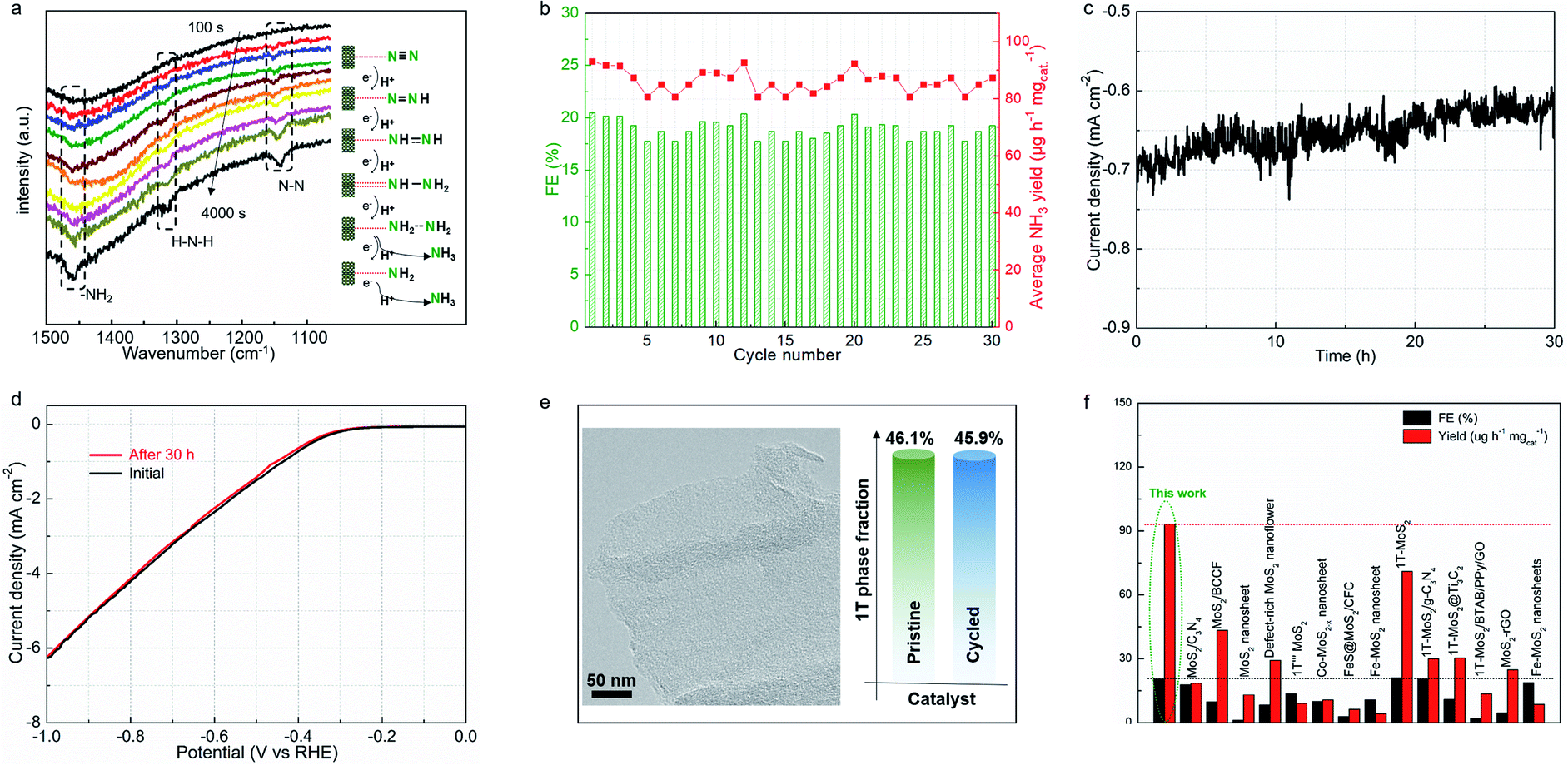 | ||
| Fig. 5 Operando spectroscopy analysis and stability studies. (a) Electrochemical in situ FTIR spectra of the N2 reduction on the MoS1.79-H/T electrode. (b) Ammonia formation rate and FE of cycles at −0.4 V versus RHE. (c) Evolution of current density for nitrogen reduction in N2-saturated electrolyte for 30 h at −0.4 V. (d) LSV curves before and after constant potential tests at −0.4 V vs. RHE. (e) High-resolution TEM image and evolution of the 1T content of MoS1.79-H/T after 30 h of stability testing. (f) NH3 yield rates and the corresponding FEs of MoS1.79-H/T compared with the reported state-of-the-art MoS2-based catalysts. | ||
To gain deeper mechanistic insights into the nitrogen adsorption and conversion on the SV-rich heterojunctions of 2H/1T-MoS2, first principle calculations based on DFT were carried out to identify a feasible NRR pathway (bridge enzymatic-alternating-hybrid pathway). The results of pristine 2H-MoS2 and 1T-MoS2 were also used for comparision (Fig. S12 and S13†). As shown in Fig. 6a and b, the computed Gibbs free energy diagrams demonstrate that 1T-MoS2 adsorbs inert N2 more strongly than the 2H- and heterojunctions of the 2H/1T-phase at the Mo atom (which existed in the heterojunctions and was neighbored by SVs) with the energy for N–N → *N–N being −0.37 eV, −1.46 eV and −1.10 eV for 2H/1T-MoS2, 1T-MoS2 and 2H-MoS2, respectively. The potential-determining step (PDS) was identified as the second hydrogenation of *N–NH to produce *NH–NH for the 2H/1T-MoS2 catalyst, with the effective energy barrier (Ueff) values of −0.69 eV, while the NH3 desorption was the PDS for the 2H-MoS2 catalyst, and a related high PDS (−1.22 eV) was calculated for the 1T-MoS2 system. Besides this, the computed Gibbs free energy for N–N → *N–N were −0.37 eV and −0.04 eV for Mo edges adjacent to SVs and Mo-edges adjacent to sulfur atoms in 2H/1T-MoS2, respectively, which demonstrated that Mo edges adjacent to SVs are preferential to adsorb nitrogen molecules. The results illustrate that the SVs and heterojunctions in 2H/1T-phase play a key role in enhancing NRR activity, with a lower barrier in PDS toward NRR than that of the 2H or 1T phases.12,31
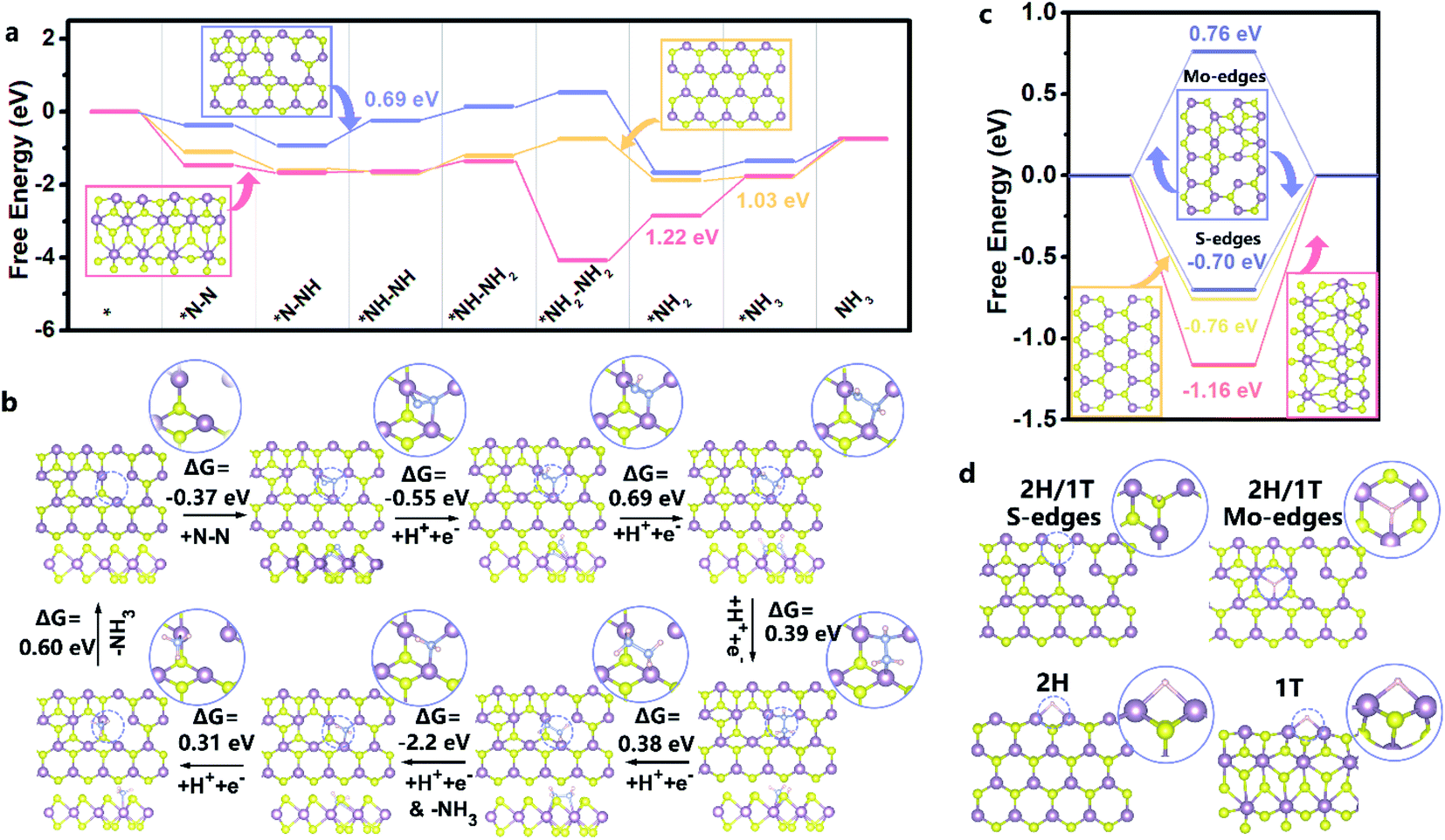 | ||
| Fig. 6 Density functional theory results and the likely overall NRR mechanism. (a) Gibbs free energy diagrams of the 1T, 2H and SVs-rich heterojunctions of the 2H/1T-phases of MoS2 with the optimum NRR pathway. The PDS and original structures for each reaction are labeled. (b) The front and top views of the 1T, 2H and SV-rich heterojunctions of the 2H/1T-phases of MoS2 along the bridge enzymatic-alternating-hybrid pathway. (c) HER Gibbs free energy diagrams of the 1T, 2H and SV-rich heterojunctions of the 2H/1T-phases in MoS2. (d) The front view of the 1T, 2H and SV-rich heterojunctions of 2H/1T-phases (with Mo and S-edges) in MoS2. | ||
The HER activity on the 1T, 2H and vacancy-rich heterojunctions of 2H/1T-phases were also explored as it is the major competition to the NRR. As exhibited in Fig. 6c and d, the H atom bound stronger to the Mo atom at the edge of the 1T and 2H phases with hydrogen desorption or combination as the PDS, calculated as −1.16 eV and −0.76 eV, respectively. In addition, the more negative energy of ΔG (N–N → *N–N) in both 1T- and 2H-MoS2 systems demonstrates that the Mo atom at the edge of the 1T and 2H phases are mainly covered by nitrogen adsorbate. However, in the 1T and 2H phases, the higher PDS (|Ueff (NRR-2H-MoS2) = −1.22| > |Ueff (HER-2H-MoS2) = −1.16| and |Ueff (NRR-2H-MoS2) = −1.03| > |Ueff (HER-2H-MoS2) = −0.76|) inhibit the further NRR selectivity. In sharp contrast, the first step and energy barrier of the Mo edges of the 2H/1T-phases of MoS2 (−0.37 and 0.69 eV) during the NRR is much lower than that of the HER process, which suggests that the Mo sites at the heterojunctions of the 2H/1T-phases prefer the NRR. Significantly, in S sites at heterojunctions of the 2H/1T-phase reaction, the ΔG (H+ → H*) = −0.70 eV is more negative than that of Mo, the hydrogenated S sites at heterojunctions act as a H provider to stabilize 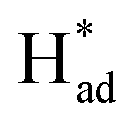 and promote the transformation of
and promote the transformation of 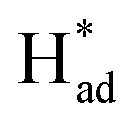 from S sites to adsorbed N2 or nitrogen reduction intermediates on nearby Mo.31,44,51,52 Therefore, SV-rich heterostructured 1T/2H-MoS2 can synchronously lower the energy barrier of the PDS and stabilize
from S sites to adsorbed N2 or nitrogen reduction intermediates on nearby Mo.31,44,51,52 Therefore, SV-rich heterostructured 1T/2H-MoS2 can synchronously lower the energy barrier of the PDS and stabilize 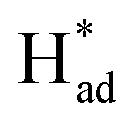 , resulting in a reduction of the competition between the HER and NRR, thus amplifying the selectivity and activity of the NRR.
, resulting in a reduction of the competition between the HER and NRR, thus amplifying the selectivity and activity of the NRR.
3. Conclusions
In this contribution, based on molten-sodium-assisted intercalation, a new kind of vacancy- and phase-engineering strategy was developed to prepare a multisite SV-rich heterostructured 1T/2H-MoSx monolayer catalyst. The formation of SVs and a heterojunction induced by efficient transition from the 2H to 1T phase of MoS2 change the local electron density and lower the nitrogen reduction energy barrier in PDS associated with the MoSx electrocatalyst. The resultant monolayer catalyst exhibits intrinsic selectivities and achieves a faradaic efficiency of 20.5% at −0.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) V vs. RHE and an average rate of 93.2 μg h−1 mgcat−1 for electrocatalytic NH3 generation. The monolayer catalyst operated stably over 30h. This study shows fundamental insight into the structure–property relationships of a SV-rich heterostructured MoSx electrocatalyst for practical application in the conversion of nitrogen.
V vs. RHE and an average rate of 93.2 μg h−1 mgcat−1 for electrocatalytic NH3 generation. The monolayer catalyst operated stably over 30h. This study shows fundamental insight into the structure–property relationships of a SV-rich heterostructured MoSx electrocatalyst for practical application in the conversion of nitrogen.
Data availability
Data available on request.Author contributions
K. L. conceived and designed the project. H. Z. and B. S. performed all experimental and mechanism studies and wrote the required scripts. All authors were involved in the analysis of the results and further editing and reviewing process.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was financially supported by the Natural Scientific Foundation of China (22109001), the Anhui Provincial Natural Science Foundation (2108085QB58), the Hefei National Laboratory for Physical Sciences at the Microscale (KF2020106), the China Postdoctoral Science Foundation (2022M713039), and the startup funds provided to K. L. from Anhui University.References
- J. Choi, H. Du, M. Chatti, B. H. R. Suryanto, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2022, 5, 382 CrossRef CAS.
- M. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896 CrossRef PubMed.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490 CrossRef.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294 CrossRef CAS PubMed.
- C. Tang and S. Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166 RSC.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. Mcenaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504 CrossRef CAS PubMed.
- G. Soloveichik, Nat. Catal., 2019, 2, 377 CrossRef CAS.
- M. Ravi and J. W. Makepeace, Chem. Sci., 2022, 13, 890–908 RSC.
- B. H. R. Suryanto, H. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290 CrossRef CAS.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh and T. W. Hamann, Chem. Rev., 2020, 120, 5437 CrossRef CAS PubMed.
- S. J. K. Forrest, B. Schluscha, E. Y. Yuzik-Klimova and S. Schneider, Chem. Rev., 2021, 121, 6522 CrossRef CAS PubMed.
- X. Chia, A. Y. S. Eng, A. Ambrosi, S. M. Tan and M. Pumera, Chem. Rev., 2015, 115, 11941 CrossRef CAS PubMed.
- J. Y. Tsai, J. Pan, H. Lin, A. Bansil and Q. Yan, Nat. Commun., 2022, 13, 492 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef PubMed.
- Y. Yu, F. Yang, X. F. Lu, Y. J. Yan, Y. Cho, L. Ma, X. Niu, S. Kim, Y. Son, D. Feng, S. Li, S. Cheong, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2015, 10, 270 CrossRef CAS PubMed.
- Z. Zheng, L. Yu, M. Gao, X. Chen, W. Zhou, C. Ma, L. Wu, J. Zhu, X. Meng, J. Hu, Y. Tu, S. Wu, J. Mao, Z. Tian and D. Deng, Nat. Commun., 2020, 11, 3315 CrossRef CAS PubMed.
- Y. Zhou, J. Zhang, E. Song, J. Lin, J. Zhou, K. Suenaga, W. Zhou, Z. Liu, J. Liu, J. Lou and H. J. Fan, Nat. Commun., 2020, 11, 2253 CrossRef CAS PubMed.
- J. Xu, G. Shao, X. Tang, F. Lv, H. Xiang, C. Jing, S. Liu, S. Dai, Y. Li, J. Luo and Z. Zhou, Nat. Commun., 2022, 13, 2193 CrossRef CAS PubMed.
- S. Li, Y. Liu, X. Zhao, K. Cui, Q. Shen, P. Li, X. Qu and L. Jiao, Angew. Chem., Int. Ed., 2021, 133, 20286 CrossRef PubMed.
- Y. Wu, J. Wang, Y. Li, J. Zhou, B. Y. Wang, A. Yang, L. Wang, H. Y. Hwang and Y. Cui, Nat. Commun., 2022, 13, 3008 CrossRef CAS PubMed.
- K. F. Mak, K. He, C. Lee, G. H. Lee, J. Hone, T. F. Heinz and J. Shan, Nat. Mater., 2021, 12, nmat3505-211 Search PubMed.
- J. Hong, Z. Hu, M. Probert, K. Li, D. Lv, X. Yang, L. Gu, N. Mao, Q. Feng, L. Xie, J. Zhang, D. Wu, Z. Zhang, C. Jin, W. Ji, X. Zhang, J. Yuan and Z. Zhang, Nat. Commun., 2015, 6, 197 Search PubMed.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- J. M. Lu, O. Zheliuk, I. Leermakers, N. F. Q. Yuan, U. Zeitler, K. T. Law and J. T. Ye, Science, 2015, 350, 1353 CrossRef CAS PubMed.
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Energy Mater., 2018, 8, 1801357 CrossRef.
- J. Peng, Y. Liu, X. Luo, J. Wu, Y. Lin, Y. Guo, J. Zhao, X. Wu, C. Wu and Y. Xie, Adv. Mater., 2019, 31, 1900568 CrossRef PubMed.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313 CrossRef CAS PubMed.
- J. Zou, F. Li, M. A. Bissett, F. Kim and L. J. Hardwick, Electrochim. Acta, 2020, 331, 135284 CrossRef CAS.
- K. Lu, Y. Liu, F. Lin, I. A. Cordova, S. Gao, B. Li, B. Peng, H. Xu, J. Kaelin, D. Coliz, C. Wang, Y. Shao and Y. Cheng, J. Am. Chem. Soc., 2020, 142, 12613 CrossRef CAS PubMed.
- S. Park, C. Kim, S. O. Park, N. K. Oh, U. Kim, J. Lee, J. Seo, Y. Yang, H. Y. Lim, S. K. Kwak, G. Kim and H. Park, Adv. Mater., 2020, 32, 2001889 CrossRef CAS PubMed.
- J. Zhang, A. Yang, X. Wu, J. Groep, P. Tang, S. Li, B. Liu, F. Shi, J. Wan, Q. Li, Y. Sun, Z. Lu, X. Zheng, G. Zhou, C. Wu, S. Zhang, M. L. Brongersma, J. Li and Y. Cui, Nat. Commun., 2018, 9, 5289 CrossRef CAS PubMed.
- R. Liu, T. Guo, H. Fei, Z. Wu, D. Wang and F. Liu, Adv. Sci., 2021, 2103583 Search PubMed.
- G. Lin, Q. Ju, X. Guo, W. Zhao, S. Adimi, J. Ye, Q. Bi, J. Wang, M. Yang and F. Huang, Adv. Mater., 2021, 2007509 CrossRef CAS PubMed.
- X. Xua, X. Tian, B. Sun, Z. Liang, H. Cui, J. Tian and M. Shao, Appl. Catal. B, 2020, 272, 118984 CrossRef.
- H. Mao, Y. Fu, H. Yang, Z. Deng, Y. Sun, D. Liu, Q. Wu, T. Ma and X. Song, ACS Appl. Mater. Interfaces, 2020, 12, 25189 CrossRef CAS PubMed.
- B. Pattengale, Y. Huang, X. Yan, S. Yang, S. Younan, W. Hu, Z. Li, S. Lee, X. Pan, J. Gu and J. Huang, Nat. Commun., 2020, 11, 4114 CrossRef CAS PubMed.
- Z. Shang, B. Song, H. Li, H. Zhang, F. Feng, J. Kaelin, W. Zhang, B. Xie, Y. Cheng, K. Lu and Q. Chen, CCS Chem., 2021, 3, 2104 CrossRef.
- K. Lu, F. Xia, B. Li, Y. Liu, I. B. A. Razak, S. Gao, J. Kaelin, D. E. Brown and Y. Cheng, ACS Nano, 2021, 15, 16887 CrossRef CAS PubMed.
- H. Cheng, L. Ding, G. Chen, L. Zhang, J. Xue and H. Wang, Adv. Mater., 2018, 30, 1803694 CrossRef PubMed.
- M. Wang, S. Liu, H. Ji, T. Yang, T. Qian and C. Yan, Nat. Commun., 2021, 12, 3198 CrossRef CAS PubMed.
- Z. Shang, B. Song, H. Li, H. Zhang, F. Feng, J. Kaelin, W. Zhang, B. Xie, Y. Cheng, K. Lu and Q. Chen, CCS Chem., 2022, 4, 2115 CrossRef CAS.
- Y. Yao, H. Wang, X. Yuan, H. Li and M. Shao, ACS Energy Lett., 2019, 4, 1336 CrossRef CAS.
- P. Song, H. Wang, L. Kang, B. Ran, H. Song and R. Wang, Chem. Commun., 2019, 55, 687 RSC.
- K. Chu, Y. Liu, Y. Li, Y. Guo and Y. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 7081 CrossRef CAS PubMed.
- Y. Liu, M. Han, Q. Xiong, S. Zhang, C. Zhao, W. Gong, G. Wang, H. Zhang and H. Zhao, Adv. Energy Mater., 2018, 9, 1803935 CrossRef.
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Mater., 2018, 30, 1800191 CrossRef PubMed.
- B. H. R. Suryanto, D. Wang, L. M. Azofra, M. Harb, L. Cavallo, R. Jalili, D. R. G. Mitchell, M. Chatti and D. R. MacFarlane, ACS Energy Lett., 2018, 4, 430 CrossRef.
- J. Zhang, X. Tian, M. Liu, H. Guo, J. Zhou, Q. Fang, Z. Liu, Q. Wu and J. Lou, J. Am. Chem. Soc., 2019, 141, 19269 CrossRef CAS PubMed.
- Y. Guo, Z. Yao, B. J. J. T. X. Sheng, L. Fan, Y. Li, F. Zhang and L. Sun, Nano Energy, 2019, 62, 282 CrossRef CAS.
- X. Zhao, X. Zhang, Z. Xue, W. Chen, Z. Zhou and T. Mu, J. Mater. Chem. A, 2019, 7, 27417 RSC.
- X. Xu, B. Sun, Z. Liang, H. Cui and J. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 26060 CrossRef CAS PubMed.
- X. Li, X. Ren, X. Liu, J. Zhao, X. Sun, Y. Zhang, X. Kuang, T. Yan, Q. Wei and D. Wu, J. Mater. Chem. A, 2019, 7, 2524 RSC.
- H. Su, L. Chen, Y. Chen, R. Si, Y. Wu, X. Wu, Z. Geng, W. Zhang and J. Zeng, Angew. Chem., Int. Ed., 2020, 132, 20591 CrossRef.
- Y. Ma, T. Yang, H. Zou, W. Zang, Z. Kou, L. Mao, Y. Feng, L. Shen, S. J. Pennycook, L. Duan, X. Li and J. Wang, Adv. Mater., 2020, 32, 200217 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03804h |
| This journal is © The Royal Society of Chemistry 2022 |