
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-free nickel catalyzed perfluoroalkylation of arenes and heteroarenes†
Shubham
Deolka
,
Ramadoss
Govindarajan
,
Serhii
Vasylevskyi
 ,
Michael C.
Roy
,
Julia R.
Khusnutdinova
and
Eugene
Khaskin
*
,
Michael C.
Roy
,
Julia R.
Khusnutdinova
and
Eugene
Khaskin
*
Coordination Chemistry and Catalysis Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna-son, 904-0495, Okinawa, Japan. E-mail: eugene.khaskin@oist.jp
First published on 17th October 2022
Abstract
We describe a “ligand-free” Ni-catalyzed perfluoroalkylation of heteroarenes to produce a diverse array of trfiluoromethyl, pentafluoroethyl and heptafluoropropyl adducts. Catalysis proceeds at room temperature via a radical pathway. The catalytic protocol is distinguished by its simplicity, and its wide scope demonstrates the potential in the late-stage functionalization of drug analogues and peptides.
Introduction
Over the past decade, nickel has emerged as a cheaper alternative,1–6 to noble metals for a wide variety of carbon–carbon,6–11 and carbon–heteroatom,3,12,13 bond formation reactions traditionally mediated by second and third-row metals,.14,15 First-row catalytic systems often require specialized ligands16,17 to stabilize the catalyst to enable reactivity similar to that of more expensive Pt group metals. For industrial-scale reactions however, specialized ligands can be the most expensive part of the protocol and can often raise the cost of chemical processes at an industrial scale to prohibitive levels.The incorporation of a fluoroalkyl group,18–32 into drug molecules has recently become a powerful strategy to modulate a known drug's properties,33 as fluoroalkyl modification is known to help with permeation through the blood–brain barrier,34,35 and to increase clearance half-life by inhibiting metabolic degradation. Other properties that are often significantly altered include hydrophobicity,36 and a compound's pH. So far, the trifluoromethyl group has attracted much more attention from medicinal chemists37,38 compared to the longer fluoroalkyl chains, including even the two-carbon chain pentafluoroethyl (C2F5) group. However, adding an extra CF2 spacer would cause significant changes in the above-listed properties,39 as well as the sterics and dipole moment of the molecule when compared to a CF3 group. For example, recently the incorporation of the C2F5 group in a bioactive molecule such as the anti-breast cancer drug fulvestrant,40 demonstrated the potential utility of this strategy and a research interest to move beyond the CF3 group in modulating properties of bioactive compounds.
Trifluoromethyl incorporation41–45 is often used as a ‘proof of concept’ for wider fluoroalkyl incorporation, but the properties of the longer fluoroalkyl radicals such as their stability under the reaction conditions, as well as the stability of intermediate nickel complexes change drastically when moving beyond one carbon. It is thus necessary to develop a functional and simple protocol for the general incorporation of larger fluoroalkyls into drug-like compounds.
Examples of inexpensive first-row transition metals being used as catalysts for perfluoroalkylation of arenes, in particular C2F5/C3F7 groups, are rare. One approach for the incorporation of the C2F5 group into organic molecules reported by Macmillan and coworker,46 involves the cross-coupling of organobromides using Ruppert–Prakash type reagents as perfluoroalkyl precursors. The protocol uses a copper catalyst in the presence of photosensitizer along with aminosilane, and proceeds via a silyl-radical-mediated halogen atom abstraction pathway (Fig. 1A). While the conditions are mild, the system requires several inputs (photosensitizer, light, silane reagent) and the substrate has to be pre-functionalized (i.e. a C–Br bond has to be present). Beller has demonstrated the perfluoroalkylation of heteroarenes using C10F20I as a perfluoroalkyl source (Fig. 1B). Interestingly, the reaction proceeds via direct C–H bond functionalization of the substrate. However, the reaction is not selective, and it takes place at 130 °C using an expensive ((dppf)Ni(o-tol)Cl) catalyst,47 and cesium carbonate as a stoichiometric base. These are quite harsh conditions that may be incompatible with a number of bioactive molecules. Further the reaction was expanded to terminal olefins at milder temperatures, with the concomitant difficulty of imperfect selectivity for the E/Z isomers.48
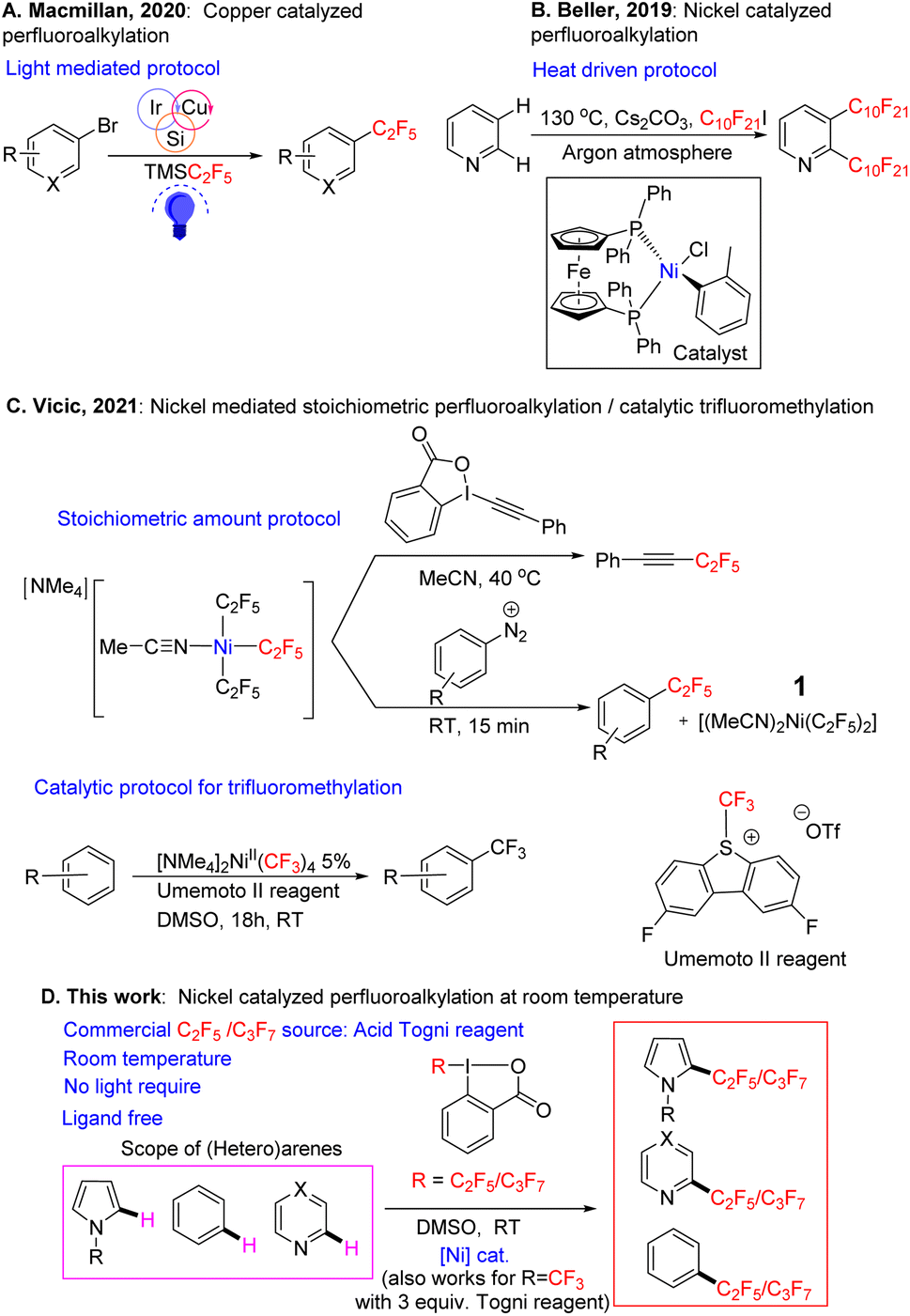 | ||
| Fig. 1 Protocols for the perfluoroalkylation of (hetero)arenes. | ||
Recently Vicic and coworkers reported perfluoroalkylation of iodonium salts and diazonium salts using ligand-free (Fig. 1C), solvated tris perfluoroalkyl Ni complexes.49 The reaction proceeded stoichiometrically in nickel to yield the desired fluoroalkylated arenes in good yield. In addition, the reaction was catalytic for trifluoromethylation in the presence of the Umemoto reagent (see also recent reports by Vicic on catalytic trifluoromethylation of brucine by a similar [NMe4]2Ni(II)(CF3)4 catalyst,50 or of trimethoxybenzene by [NMe4]Ni(III)(C4F8)(CF3) supported by an interesting, chelating C4F8 ligand51). In the stoichiometric reactions, the yields of the organic perfluorinated substrates were often comparable to those of the trifluoromethylated substrates, but the yield of the bis-C2F5/bis-CF3–Ni byproduct, that forms from the Ni tris-fluoroalkyl intermediate concurrently with the organic product, was much lower in the C2F5 case, and catalysis with C2F5 was not attempted. The stoichiometric substrates used for perfluoroalkylation were also limited to activated hydrocarbons.
In our laboratory, we began to examine nickel in several coupling reactions, and recently we reported that NiIII(CF3)2 complexes supported by simple naphthyridine ligands could form under aerobic conditions, with air serving as a NiII oxidant.52 These high valent nickel complexes were capable of trifluoromethylating arenes under air using blue LED light. During a further evaluation of the system, we found that a stoichiometric amount of simple nickel solvated complex Ni(C2F5)2(MeCN)21, first reported as a precursor by Vicic in 2013,53 in the presence of an oxidant can functionalize arenes and heteroarenes at room temperature in the absence of base. This complex however, is the byproduct that is formed in small or negligible yields after the stoichiometric perfluoroalkylation with the tris-perfluoroalkyl Ni complex in the latter 2021 Vicic report.49 Observing that 1 was capable of reacting with organic substrates, gave us hope that we could develop a simple, ligand-free catalytic protocol for higher perfluorocarbon CH functionalization.
After the publication of the above stoichiometric ligand-free Vicic protocol (Fig. 1C), we decided to focus on developing our perfluoroalkylation reaction as it would fill a valuable niche,54–58 since catalytic trifluoromethylation has been better developed both in the case of the ligand-free system,49,59 and for more challenging substrates with a ligand supported system.19,60
In the current paper we report a catalytic protocol based on the solvated bis perfluoroalkyl nickel complex, which is able to CH functionalize a variety of (hetero)arenes and natural products, demonstrating the reaction's potential in the late-stage functionalization of drugs (Fig. 1D). The catalysis proceeds with a cheap and easy-to-prepare nickel precursor and a C2F5/C3F7 reagent set; it works under mild conditions and does not require photosensitizer/light. Our preliminary radical trap studies suggest that catalysis proceeds via a radical pathway. To the best of our knowledge, this is the first example of a ligand-free, room temperature, Ni-catalyzed, catalytic C(sp2)–H perfluoroalkylation (C2F5/C3F7 groups) protocol. The yields obtained are moderate to almost quantitative, with the reaction requiring equimolar amounts of substrate and perfluoroalkylating Togni oxidant.
Results and discussion
Stoichiometric perfluoroalkylation using solvated nickel precursors
During initial screening of a stoichiometric protocol, we found that 1 did not react with 1,3,5-trimethoxybenzene substrate when simply mixing the two reactants in DMSO (Table 1, entry 1) suggesting that Ni–C bond homolysis and perfluoroalkyl transfer do not occur at the NiII oxidation state.2,9,52,61 We then added an oxidant to enable the formation of radical perfluorocarbons via homolysis of the Ni–C bond from a putative higher oxidation state nickel complex. As we expected, addition of potassium persulfate (K2S2O8) immediately gave 75% mono-pentafluoroethylated product (Table 1, entry 2).| Entry | Substrate | Conditions | Yield [%] |
|---|---|---|---|
| a Reactions were performed for 24 hours using 1 equiv. of the substrate, and 1 equiv. of 1 in DMSO at RT under N2 unless indicated otherwise. The yields were determined by 19F NMR based on integration against α,α,α-trifluorotoluene as an internal standard. | |||
| 1 |

|
— | 0 |
| 2 |

|
K2S2O8 (1 equiv.) | 75 |
| 3 |
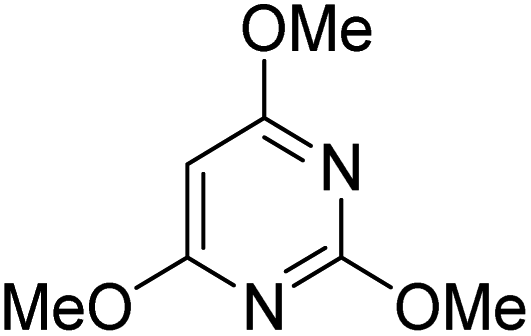
|
K2S2O8 (1 equiv.) | 80 |
| 4 |
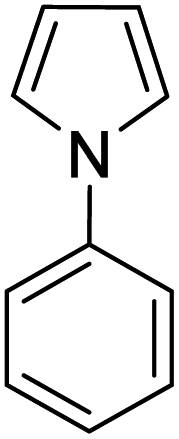
|
K2S2O8 (1 equiv.) | 38 |
| 5 |
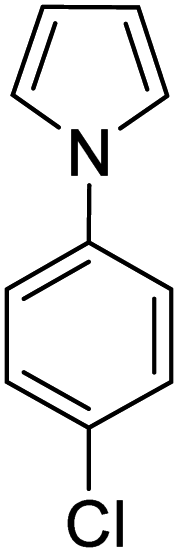
|
K2S2O8 (1 equiv.) | 50 |
| 6 |
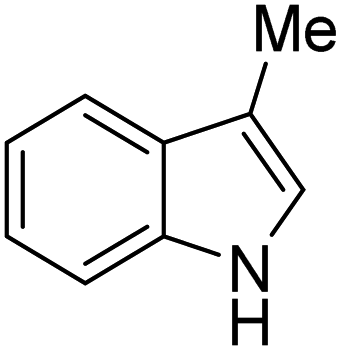
|
K2S2O8 (1 equiv.) | 37 |
A number of other substrates were also found to react stoichiometrically with this simple Ni(II)/oxidant system. While electron-rich six-member rings (1,3,5-trimethoxybenzene and 2,4,6-trimethoxypyrimidine) gave high yields of 75–80%, pyrroles (Table 1, entry 4 and 5) resulted in moderate yields of 38–50%. A relatively low yield of 37% was observed for 3-methylindole (Table 1, entry 6). Overall, these results showed that adding an oxidant to a solvated NiII bis-perfluoroalkyl complex does lead to a stoichiometric reaction and may eventually lead to a catalytic protocol.
We generalized the stoichiometric results to longer chain fluoroalkyls by preparing a new solvated nickel precursor Ni(MeCN)2(C3F7)2 (2) (Scheme 1). The addition of an equivalent amount of K2S2O8 to 2 results in an effective functionalization of 1,3,5-trimethoxybenzene and 2,4,6-trimethoxypyrimidine in moderate yields of 71–63% (see ESI, Fig. S7 and S9†). We also found that 2 reacts more effectively than 1 with 1-(4-chlorophenyl)-1H-pyrrole affording the product in a yield of 84% as opposed to 50% with 1 (see ESI, Fig. S11†).
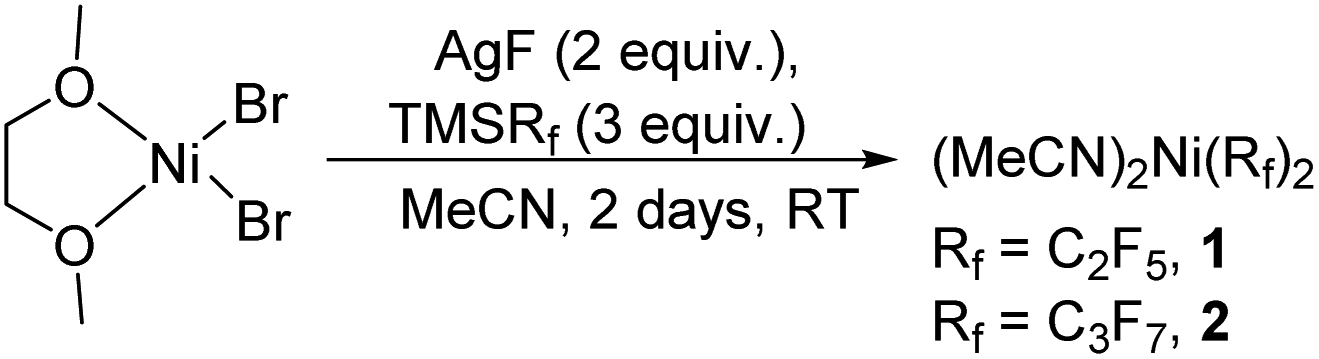 | ||
| Scheme 1 Synthesis of NiII–Rf precursors. | ||
To confirm that radical formation is involved in stoichiometric reactivity, we carried out (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and 1,1-diphenylethylene (DPE) radical trap experiments (Scheme 2). In the presence of 2 equiv. of TEMPO or DPE, no product was observed with 1 (Scheme 2a and b). We could not however, detect the TEMPO–C2F5 adduct, which may be due to its direct reaction with the K2S2O8 oxidant (see also Scheme 7 with a different oxidant below). However, the DPE–C2F5 adduct was confirmed by GC-MS (see ESI, Fig. S51†). We further confirmed the importance of oxidant by carrying out the DPE radical trap experiment in the absence of K2S2O8. In this case (Scheme 2c), we could not detect the DPE–C2F5 adduct by GC-MS. Overall, these results confirm that 1 reacts to generate a C2F5 radical in the presence of K2S2O8 oxidant.
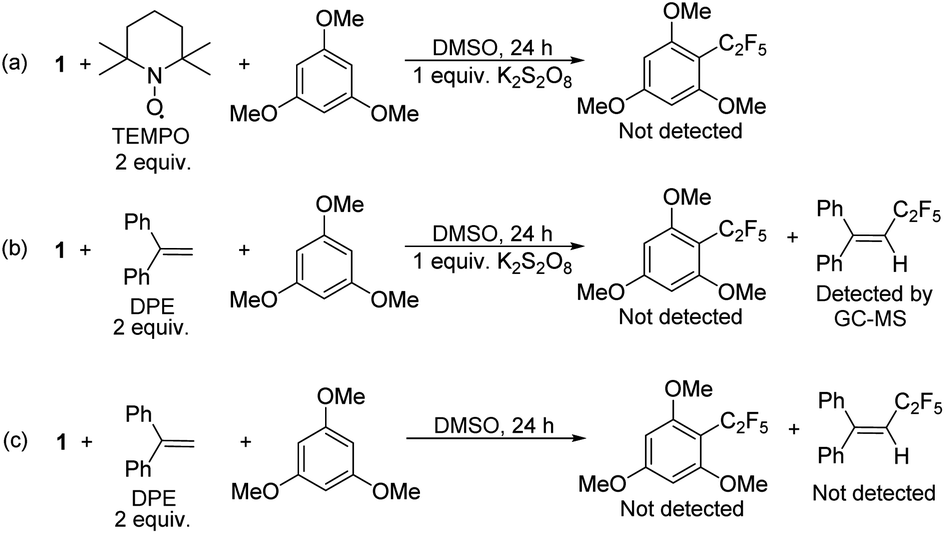 | ||
| Scheme 2 Radical trap experiments under stoichiometric conditions. | ||
Catalytic protocol
To develop a catalytic protocol based on the stoichiometric reaction, we looked for an accessible and commercially available reagent, which can act both as an oxidant and Ni perfluoroalkylation agent. We found that 1-pentafluoroethyl-1,2-benziodoxol-3(1H)-one (acid C2F5–Togni reagent; Fig. 1D) in the presence of 10 mol% of 1 can functionalize 1,3,5-trimethoxybenzene in 17% yield (Table 2, entry 1) under air in DMSO. Performing the reaction under nitrogen atmosphere however, we obtained the mono-pentafluoroethylated product in 97% yield (entry 2) after 24 h at RT. A similar alcohol C2F5–Togni reagent gave a lower yield. The yield did not change in the presence of pyridine acting as a possible ligand (95% yield) (entry 3). This reaction can be performed in different solvents, such as MeCN and MeOH, affording the product in 78 and 58% yields (entries 4 and 5) respectively. While the acid Togni reagent is commercially available, it can also be easily prepared with the desired perfluoroalkyl. Either commercially or synthetically, it is cheaper than the modified Umemoto reagents used in previous reports.62|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent, conditions | Yield [%] |
| a The reaction was performed under nitrogen unless indicated otherwise. b NMR yield was based on integration against α,α,α-trifluorotoluene as an internal standard. c 100 equiv. of silver salt used. d Para-chloro-N-phenylpyrrole used as substrate (*) indicates the average yield of three reactions. | |||
| 1 | 1 | DMSO, air | 17 |
| 2 | 1 | DMSO | 97 |
| 3 | 1 | DMSO, pyridine | 95 |
| 4 | 1 | MeCN | 78 |
| 5 | 1 | MeOH | 58 |
| 6 | NiCl2(glyme) | DMSO | 0 |
| 7 | NiBr2(glyme) | DMSO | 0 |
| 8 | AgF | DMSO | 26 |
| 9c | AgF | DMSO | 56 |
| 10c,d | AgF | DMSO | 13 |
| 11 | Ag(OSO2CF3) | DMSO | 0 |
| 12 | AgBr | DMSO | 0 |
| 13c | AgBr | DMSO | 0 |
| 14 | None | DMSO | 6* |

To investigate the role of metal precursor, we carried out the reaction in the presence of simple nickel salts such as NiCl2-glyme and NiBr2(glyme) (entries 6 and 7; glyme = dimethoxyethane) in catalytic amounts. No detectable pentafluoroethylated product was observed, suggesting that presence of 1 is important. Since the synthesis of 1 involves the use of AgF, minute residue of silver may remain in the nickel complex product; thus silver salts were evaluated as possible contributors to catalysis.63 However, silver fluoride and silver triflate (entry 8–11) under catalytic conditions afforded 26% and 0% yields respectively for 10 mol% silver, showing only a 56% yield of product if 100 mol% of silver was used.
The control reaction in the absence of 1 afforded only 6% of pentafluoroethylated product (entry 14), indicating that 1 is a necessary catalyst, and that some nickel and silver species (entries 6, 7, 11–13) effectively shut down this background reaction.
Adding more equivalents of the Togni reagent did not improve yield for a more difficult substrate, tryptophan (vide infra); the NMR yield increased from 52% for one equivalent of acid-C2F5–Togni to 54% with three equivalents.
Interestingly, using the same conditions (Table 2, entry 2) with the (MeCN)2Ni(CF3)2 complex and the associated acid Togni–CF3 reagent to attempt ligand-free trifluoromethylation gave only 30% of product. However, increasing the amount of the Togni reagent in this case significantly improved yields and 73% of trimethoxybenzene–CF3 was obtained with three equivalents of the Togni–CF3 reagent, while the control reaction with three equivalents and without the catalyst gave 8% of product. While a full study of ligand free fluoroalkylation is beyond the scope and purpose of this paper, we have included data in the ESI† on the trifluoromethylation of five representative substrates with three equivalents of the Togni reagent. The yields obtained ranged from 44–84% (see below; also ESI Section 9; p. S82).
Substrate scope
We applied the optimized protocol for trimethoxybenzene (Table 2, entry 2) to a broad number of other heteroarene substrates (Scheme 3). Expectedly, related substrates based on the original electron-rich trimethoxybenzene (a–f) resulted in pentafluoroethylated product in good to high 32–86% isolated yields, with exclusive selectivity for the mono-functionalized product.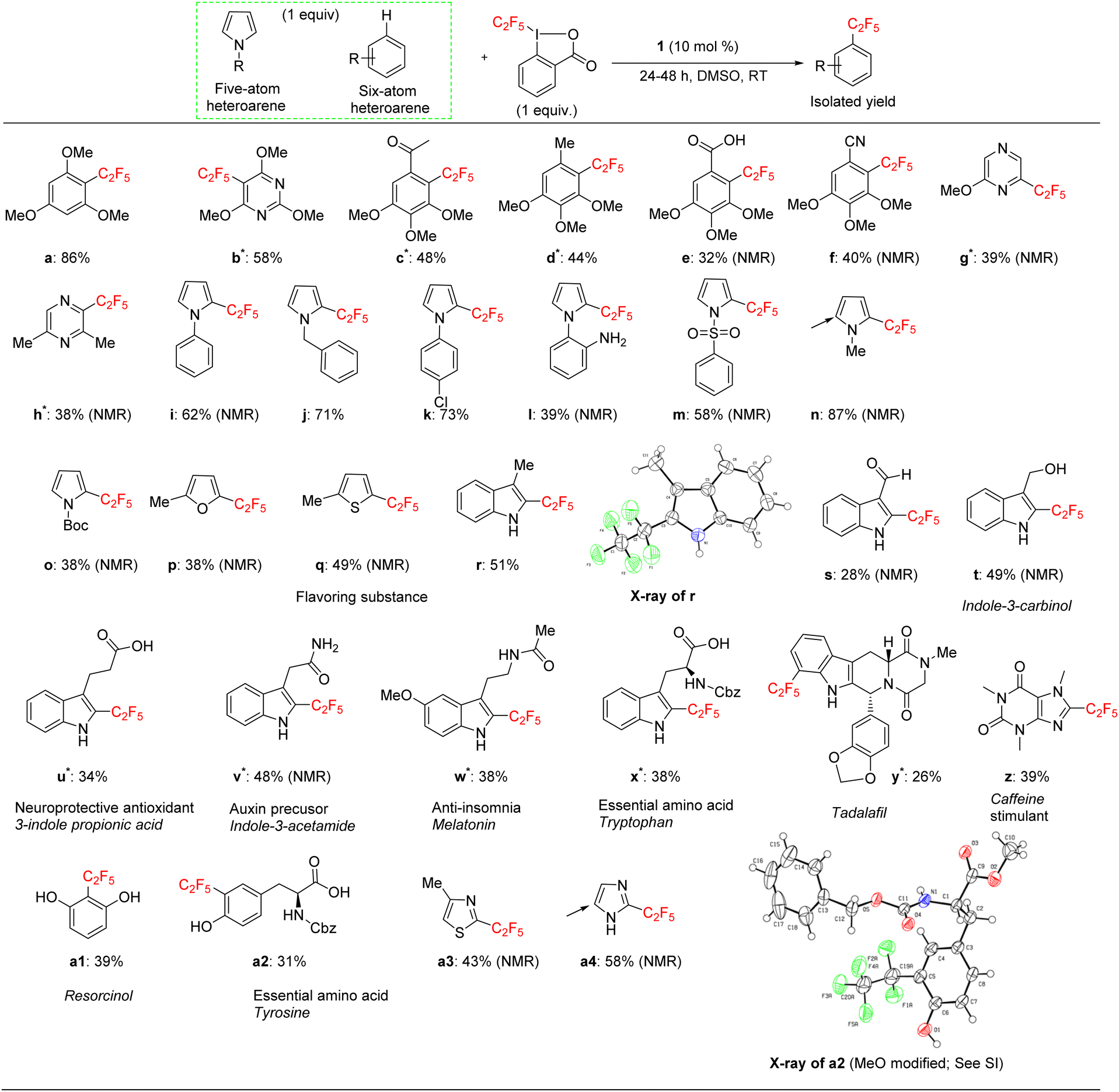 | ||
| Scheme 3 Substrate scope of pentafluoroethylation catalyzed by 1 using acid-C2F5–Togni reagent. General conditions: 1 equiv. of 1-pentafluoroethyl-1,2-benziodoxol-3(1H)-one, 1 equiv. of (hetero)arene, 10 mol% 1 in DMSO for 24 h. Isolated yield is shown here. Asterisk symbol signifies the reaction was performed for 48 hours. Arrows signify the second site of polyfluoroalkylation in the minor isomer. | ||
Other five to six membered heterocycles, such as pyrazines, pyrimidine, pyrroles, furan, and thiophene (b, g–q) gave the fluoroalkyl product with remarkable selectivity in 38–87% yields. Only N-methyl pyrrole (n) gave the bis-fluoromethylated species as a minor side product. N-Phenyl pyrrole (i) and N-benzyl pyrrole (j) have similar isolated yields (62% and 71%) with functionalization occurring at the sp2 carbon position for j. N-Parachlorophenyl pyrrole (k), 73% isolated yield, demonstrated the reaction's tolerance to halogenated substrates. However, the sterically demanding substrate l gave only a 39% NMR yield. The less bulky tosyl substituted entry m resulted in a 58% NMR yield. The Boc protected pyrrole, as well as a furan and a thiophene substrate were also shown to participate in the reaction (entries o–q), albeit at lower 38–49% NMR yields. The thiophene starting material for product q is a commercially important compound used as a flavouring substance by the food industry. The indole motif, which is found in many natural products and drugs was explored in seven varied substrates, giving the desired product (r–x) in up to 51% yield. Indoles substituted in the 3 position are of interest as possible drugs that are neuroactive. As fluorination helps with permeation of the blood–brain barrier, such substrates are natural candidates to explore under our protocol. The seven indoles we looked at had divergent reactivity based on the nature of the substituent. While the aldehyde (s) gave only a 28% NMR yield, the natural product indole-3-carbinol substituted species (entry t) was fluoroalkylated in 49% yield. The latter substance is found in high amounts in green vegetables and cabbage related plants and has been investigated for its anticarcinogenic and antioxidant properties.
3-Indolepropionic acid (u) gave a 34% isolated yield after 48 hours of reaction time. The parent compound is being studied as a possible therapeutic in Alzheimer's disease and is a potent hydroxyl radical inhibitor. Indole-3-acetamide is a plant metabolite and a precursor of the plant hormone auxin that is widely studied in plant science. We obtained its perfluoroalkylated product in 48% NMR yield after 48 hours of reaction time (entry v). We also investigated the reactivity of melatonin, a plant hormone which is used to treat insomnia (w), and tryptophan, an essential amino acid (x). Isolated yields for the two important natural products were 38% and 42% respectively after 48 hours of reaction time.
For the last four examples we investigated non-indole natural products with industrial applicability. A drug containing multiple functional groups, tadalafil, was found to undergo selective mono-pentafluoroethylation in a specific position on the phenyl ring that is part of the indole moiety; NMR did not show the presence of other isomers. The isolated yield was 26% after 48 hours of reaction time (entry y). Caffeine (z) and essential amino acid tyrosine (a2) are examples of a drug and a natural product that are significantly smaller than tadalafil and also do not contain an indole moiety. The pentafluoroethylation was found to occur at a specific sp2 carbon with isolated yields of 39% and 32% respectively. Finally, resorcinol is a fairly simple molecule (1,3 bis-hydroxy alcohol, entry a1) that is expensive to produce and has medical uses as an acne treatment and an anaesthetic. Although less electron rich than the tris-methoxy benzene substrates, resorcinol was amenable to our protocol and an isolated yield of 43% was obtained. The reaction is highly selective for the 2 position on the phenol ring.
The indole and drug/natural product results highlight the utility of our new perfluoroalkylation method in the late-stage functionalization of pharmaceuticals and other bioactive molecules. However, the protocol was not always successful in the case of relatively electron-deficient arenes. A list of these substrates that worked poorly is given in the ESI.†
Since we wanted to demonstrate that the reaction acts as a general method for perfluoroalkylation, we applied the catalytic protocol to heptafluoropropylation (the C3F7 group) in some heteroarenes (Scheme 4).
 | ||
| Scheme 4 Substrate scope of heptafluoropropylation catalyzed by 2. | ||
Under the same catalytic conditions, catalyst 2 can catalyze the functionalization of 1,3,5-trimethoxybenzene in the presence of the reaction specific 1-heptafluoropropyl-1,2-benziodoxol-3(1H)-one (acid C3F7–Togni) reagent in 74% yield (A1) (Scheme 4). As in Scheme 4, we found that this protocol was able to functionalize pyrimidine, pyrrole, and indole derivatives (A2–A6), including melatonin, in moderate to good yields (38–64%). Thus, the catalytic results of Scheme 3 could be generalized to a longer-chain C3F7 substituent.
For trifluoromethylation, 3 equiv. of the CF3–acid Togni reagent had to be used to obtain yields similar to those obtained for perfluoroalkylated products; the reaction is not as competitive with other methods published for catalytic nickel trifluoromethylation. Nevertheless, it may be preferred when considering the cost of the Umemoto against the Togni reagents. The five substrates that were modified are given below (Scheme 5).
 | ||
| Scheme 5 Substrate scope of trifluoromethylation. | ||
Finally, we targeted functionalization of short peptides for the installation of the C2F5 group. While a C2F5 substituted protein or polypeptide could be generated by solid state synthesis by including a C2F5 substituted amino acid (see Scheme 3, substrates, x and a2), the functionalization of polypeptides shows off the versatility of the method and its possible applicability to the modification of larger proteins, including difficult to access heme and metalloproteins. Such transformations can enhance the development of methods that advance therapeutics and diagnostics. To model such late-stage functionalization, we explored the functionalization of 2–4 amino acid long peptides. Under catalytic conditions, 1 can functionalize the sequences tyrosine–alanine, valine–tyrosine–valine, glycine–leucine–tyrosine, and glycine–glycine–tyrosine–arginine (P1–P4) (Scheme 6), as confirmed by an LC-MS analysis, hinting that late-stage functionalization of large natural products and proteins may be possible. By analogy with the tyrosine functionalization result (Scheme 3, entry a2), the functionalization is here assumed to also occur at the tyrosine, with the selectivity being for the ortho position next to the hydroxyphenyl moiety. The reaction is selective for mono-functionalization.
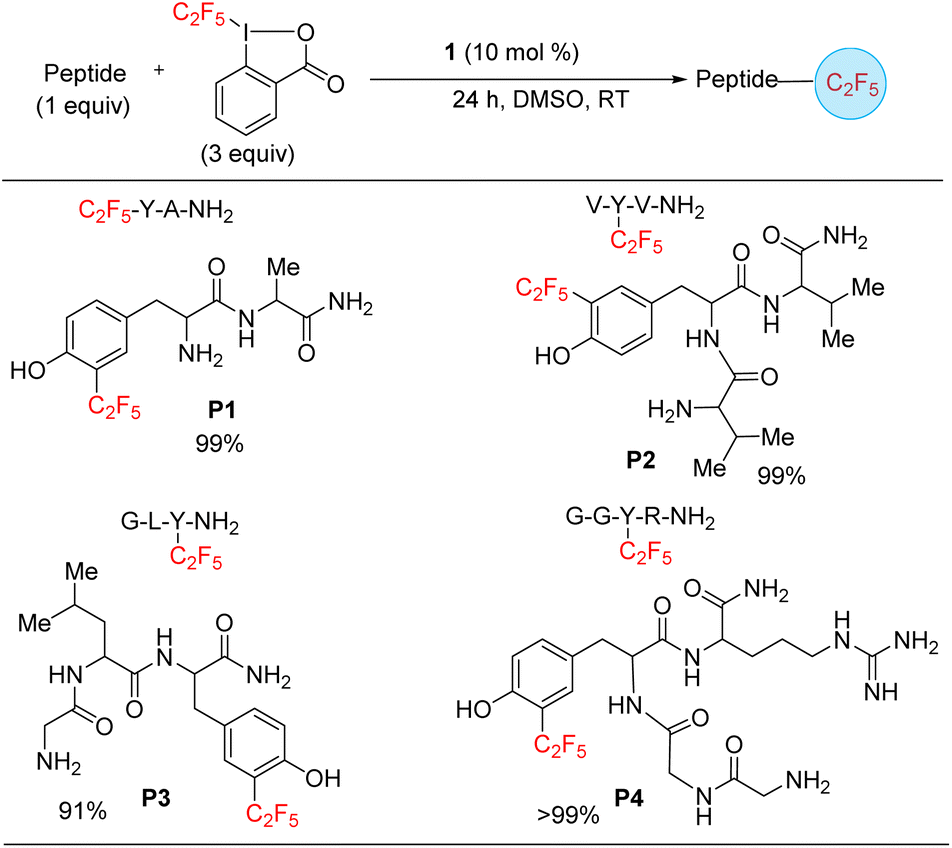 | ||
| Scheme 6 Substrate scope of peptides. The yields were approximated as the ratio of area of product/area total determined by LC-MS spectrum. | ||
Mechanistic experiments
To prove the homogeneity of the reaction, a mercury drop test was carried out. As the yields were not affected, the reaction is assumed to be homogeneous (see ESI Fig. S50†). We investigated whether radical formation occurs under typical catalytic conditions for 1 catalyzed perfluoroalkylation in the presence of substrate and acid C2F5–Togni reagent, by using TEMPO and 1,1-diphenylethylene (DPE) radical traps (Scheme 7).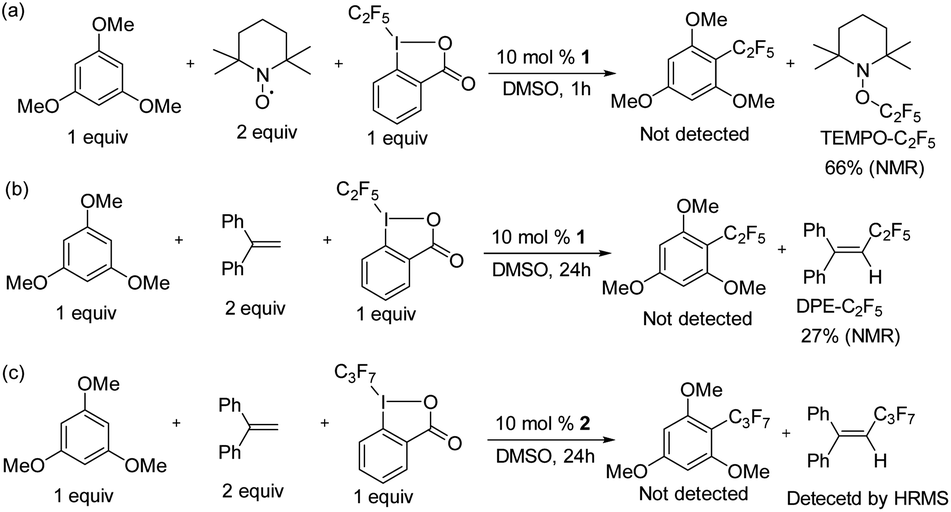 | ||
| Scheme 7 Radical trap experiments under catalytic conditions. | ||
In all cases the formation of TEMPO–C2F5 and DPE–C2F5 adducts was observed (Scheme 7a and b), while product formation was inhibited.
The quantification of the radical trap products was determined via19F NMR spectroscopy for TEMPO–C2F5 and DPE–C2F5, which formed in 66% and 27% yield respectively. Similarly, the reaction between 1,3,5-trimethoxybenzene and acid C3F7–Togni reagent catalyzed by 2 at room temperature was performed in the presence of DPE to form a DPE–C3F7 adduct as confirmed by HRMS, while product formation was inhibited (Scheme 7c). In a control reaction between the acid C2F5–Togni reagent and 1,3,5-trimethoxybenzene in the presence of TEMPO but in the absence of catalyst, no TEMPO–C2F5 adduct was observed, indicating that nickel is necessary for radical formation (see ESI, Fig. S55†).
Next, the reaction of 1, acid C2F5–Togni reagent, and substrate was followed by UV/vis (Fig. 2). Reacting an equivalent amount of yellow 1 with the acid C2F5–Togni reagent results in a color change to green. A new intense absorption band appears at λ 624 nm; this band is not present in the absence of 1, and it slowly disappears after two hours. The same band at λ 624 nm could be observed under catalytic conditions five minutes after mixing the reagents (see ESI Fig. S70†). These experiments confirm the formation of a new complex resulting from the interaction of 1 with the acid C2F5–Togni reagent.
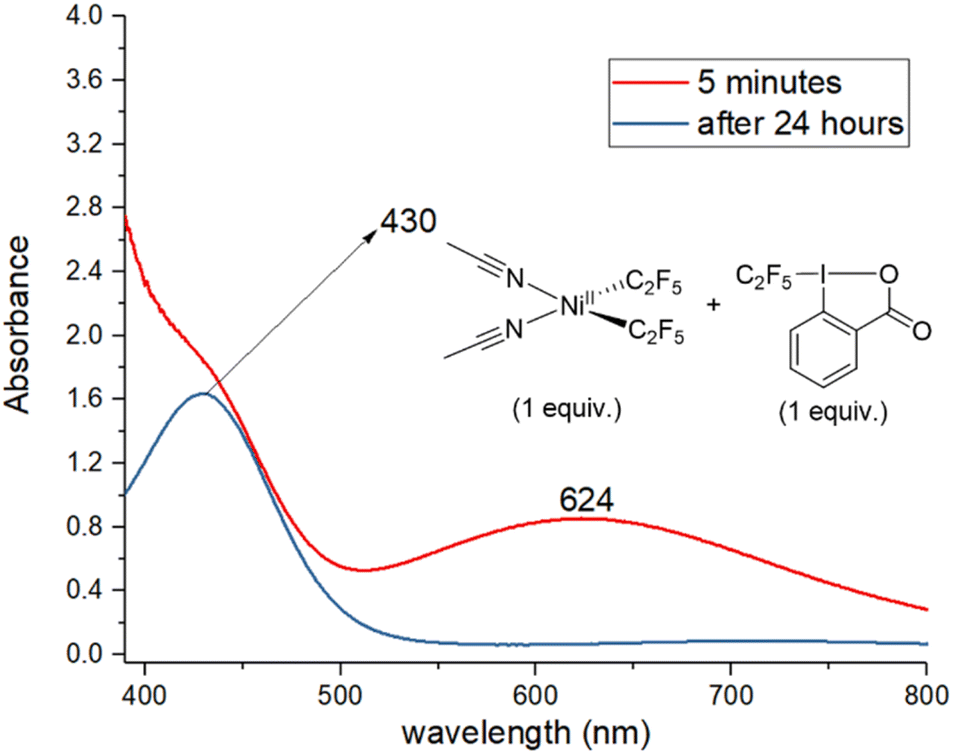 | ||
| Fig. 2 UV/vis spectra of the reaction mixture containing 1 and acid C2F5–Togni reagent in equivalent amount to form a new absorption band at 624 nm. | ||
The reaction between 1 and acid C2F5–Togni was followed by mass spectrometry (ESI-MS) to detect any high valent Ni intermediates formed during the oxidation process. Mixing 1 and acid C2F5–Togni at room temperature in an acetonitrile solvent generates the green color instantly.
The green color species gives an intense signal corresponding to complex 3 (m/z = 583.8720), where two C2F5 groups are bound to the nickel along with carboxylate group of the reacted Togni reagent and an acetonitrile ligand. It is likely this complex that is responsible for the λ 624 nm band. We were unsuccessful in isolating this unstable complex, precluding its structural determination. In a cross experiment, the reaction between 1 and acid C3F7–Togni also results in green color to form high valent nickel species 4. The reverse reaction with 2 also gives complex 4 (m/z = 633.8625), where one C2F5 and C3F7 molecules are bound to nickel along with iodo benzoate (Scheme 8). In addition, the reaction between (MeCN)2Ni(CF3)2 and the CF3–Togni reagent results in a similar NiIII intermediate that could be detected by ESI-MS. These experiments suggests that the acid–Togni reagent acts as both a perfluoroalkyl group source and an oxidant.
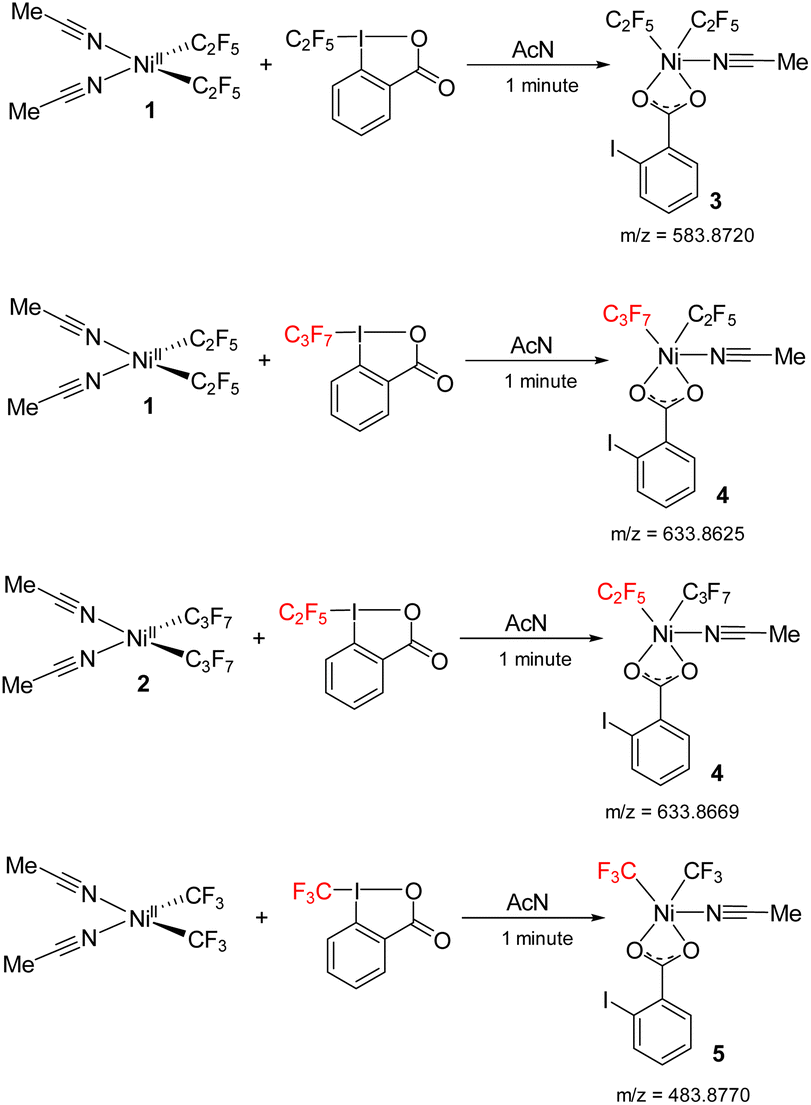 | ||
| Scheme 8 Reaction between 1 and acid C2F5/C3F7–Togni to form high valent nickel intermediates 3, 4 and reaction between Ni(CF3)2(MeCN)2 and acid-CF3 to form 5 detected by ESI-MS. | ||
Based on these observations and previous studies,19,52,59 we propose that the initial step involves addition of the Togni reagent to NiII complex 1 to give either an unobserved intermediate from which homolysis occurs, or concurrent homolysis with the addition of the Togni reagent to generate the NiIII complex 3, observed by UV/vis and mass spectroscopy, and a C2F5 radical (Scheme 9). The organic radical reacts with an aromatic substrate A to form a functionalized intermediate B. Oxidation/deprotonation of B by 3 gives the perfluoroalkyl product C, free 2-iodobenzoic acid, and regenerates complex 1.
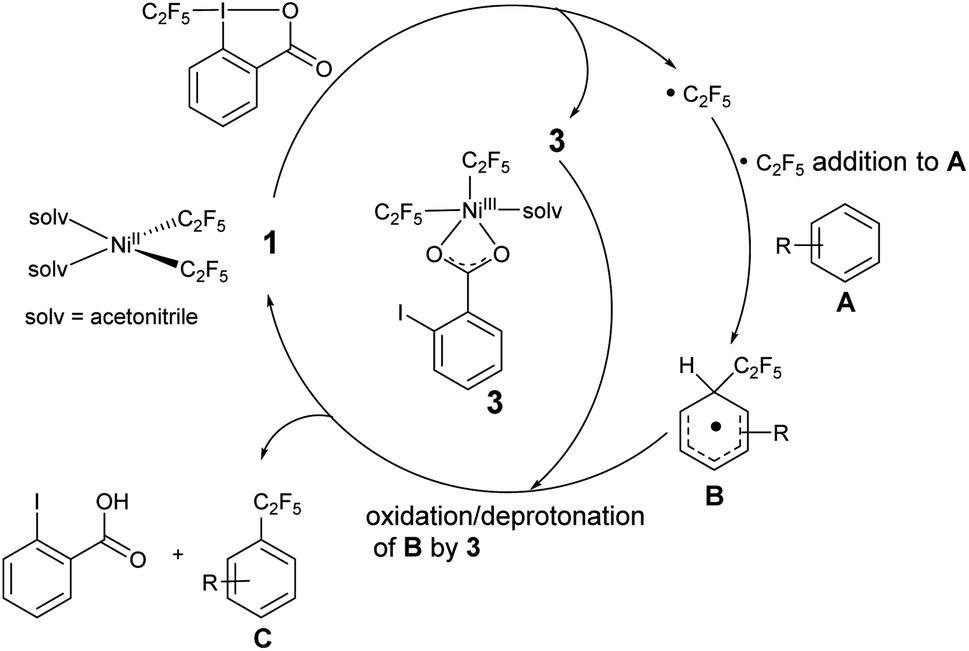 | ||
| Scheme 9 Proposed radical pathway for perfluoroalkylation. | ||
The mechanism does not rule out the intermediacy of an elusive short-lived NiIV(C2F5)3 intermediate formed from the reaction of 1 and the Togni reagent that quickly decays to a Ni(III) species 3 and a C2F5 radical. We were not able to detect an elusive Ni(IV) intermediate by low temperature NMR. We also observed an anisotropic signal in EPR spectrum obtained after mixing 1 and acid-C2F5–Togni reagent and freezing the reaction mixture at 94 K (Fig. 3). The anisotropic EPR signal with g values significantly shifted from 2.0023 (g1 = 2.230, g2 = 2.185, g3 = 2.026; gave 2.147) is consistent with the presence of a d7 Ni(III) center (see ESI† for details).
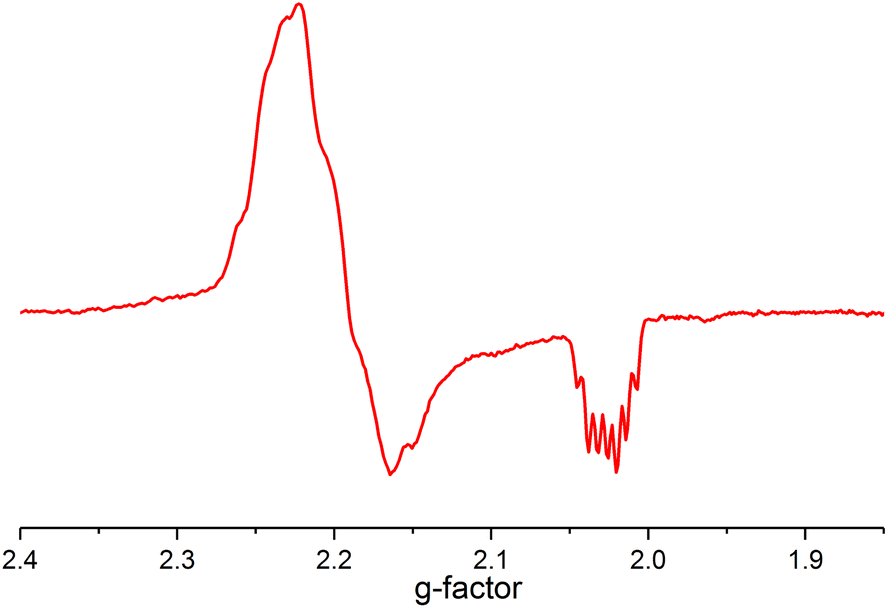 | ||
Fig. 3 EPR spectrum of the sample of the reaction mixture containing 1 and acid-C2F5–Togni reagent (PrCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtCN glass, 94 K, 9.07 GHz). :EtCN glass, 94 K, 9.07 GHz). | ||
The proposed mechanism indicates that acidification of the reaction mixture occurs as the reaction progresses. We tested the stability of 1 towards acidic conditions by reacting it with acid C2F5–Togni in the presence of 10 equivalents of 2-iodobenzoic acid, the reaction byproduct. In this experiment we were still able to observe the UV/vis band at 624 nm (see ESI, Fig. S76†).
Conclusions
In conclusion, we demonstrated that a “ligand-free” [(MeCN)2Ni(C2F5)2] and [(MeCN)2Ni(C3F7)2] system can successfully functionalize hetero(arenes) to incorporate fluoroalkyl groups in the presence of an oxidant. We showed that simple, inexpensive nickel precursors can catalytically functionalize natural products, drug analogues, and peptides containing an aromatic moiety, using the acid-C2F5–Togni reagent. This method is easy to set up, highly efficient, and is performed under mild conditions without special equipment such as a light setup. Preliminary mechanistic studies suggest that catalysis proceeds via a radical mechanism. In the future, we plan to investigate the mechanism of this reaction in more detail and attempt to develop a protocol/system that is capable of functionalizing sp3 C–H bonds.Data availability
All original data is deposited in the OIST Institutional Repository (OISTir) within one year of publication and is available to the public free of charge.Author contributions
SD and EK designed the study, interpreted the data, and wrote the manuscript; SD and RG carried out the experiments; SV collected and refined crystallographic data; MR helped to carry out the peptide experiments and mass spectrometry; JR helped in designing and carrying out the mechanistic EPR experiments, and helped in writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge CF Plus Chemicals for synthesizing acid-Togni–C3F7 on special order. All funding was provided by an OIST internal grant.Notes and references
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- F. Le Vaillant, E. J. Reijerse, M. Leutzsch and J. Cornella, J. Am. Chem. Soc., 2020, 142, 19540–19550 CrossRef CAS.
- N. M. Camasso and M. S. Sanford, Science, 2015, 347, 1218–1220 CrossRef CAS.
- E. A. Standley, S. Z. Tasker, K. L. Jensen and T. F. Jamison, Acc. Chem. Res., 2015, 48, 1503–1514 CrossRef CAS PubMed.
- C. M. Lavoie and M. Stradiotto, ACS Catal., 2018, 8, 7228–7250 CrossRef CAS.
- H. Xu, J. B. Diccianni, J. Katigbak, C. Hu, Y. Zhang and T. Diao, J. Am. Chem. Soc., 2016, 138, 4779–4786 CrossRef CAS PubMed.
- N. Nebra, Molecules, 2020, 25, 1141 CrossRef CAS.
- L. K. G. Ackerman, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS.
- J. R. Bour, N. M. Camasso, E. A. Meucci, J. W. Kampf, A. J. Canty and M. S. Sanford, J. Am. Chem. Soc., 2016, 138, 16105–16111 CrossRef CAS PubMed.
- M. B. Watson, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2017, 139, 35–38 CrossRef CAS PubMed.
- J. W. Schultz, K. Fuchigami, B. Zheng, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2016, 138, 12928–12934 CrossRef CAS.
- M. Yuan, Z. Song, S. O. Badir, G. A. Molander and O. Gutierrez, J. Am. Chem. Soc., 2020, 142, 7225–7234 CrossRef CAS.
- S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 624–627 CrossRef CAS.
- X. Ji, H. Huang, Y. Li, H. Chen and H. Jiang, Angew. Chem., Int. Ed., 2012, 51, 7292–7296 CrossRef CAS PubMed.
- R. Martinez, M.-O. Simon, R. Chevalier, C. Pautigny, J.-P. Genet and S. Darses, J. Am. Chem. Soc., 2009, 131, 7887–7895 CrossRef CAS PubMed.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- M. P. Doyle, Angew. Chem., Int. Ed., 2009, 48, 850–852 CrossRef CAS.
- F. D'Accriscio, P. Borja, N. Saffon-Merceron, M. Fustier-Boutignon, N. Mézailles and N. Nebra, Angew. Chem., Int. Ed., 2017, 56, 12898–12902 CrossRef PubMed.
- E. A. Meucci, S. N. Nguyen, N. M. Camasso, E. Chong, A. Ariafard, A. J. Canty and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 12872–12879 CrossRef CAS.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS.
- L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5809–5812 CrossRef CAS PubMed.
- K. Niedermann, N. Früh, R. Senn, B. Czarniecki, R. Verel and A. Togni, Angew. Chem., Int. Ed., 2012, 51, 6511–6515 CrossRef CAS.
- L. An, F.-F. Tong, S. Zhang and X. Zhang, J. Am. Chem. Soc., 2020, 142, 11884–11892 CrossRef CAS.
- H.-Y. Zhao, X. Gao, S. Zhang and X. Zhang, Org. Lett., 2019, 21, 1031–1036 CrossRef CAS.
- M. Zhou, H.-Y. Zhao, S. Zhang, Y. Zhang and X. Zhang, J. Am. Chem. Soc., 2020, 142, 18191–18199 CrossRef CAS.
- Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef CAS PubMed.
- L. Xu and D. A. Vicic, J. Am. Chem. Soc., 2016, 138, 2536–2539 CrossRef CAS.
- L. An, C. Xu and X. Zhang, Nat. Commun., 2017, 8, 1460 CrossRef PubMed.
- C. Xu, W.-H. Guo, X. He, Y.-L. Guo, X.-Y. Zhang and X. Zhang, Nat. Commun., 2018, 9, 1170 CrossRef.
- W.-Q. Hu, S. Pan, X.-H. Xu, D. A. Vicic and F.-L. Qing, Angew. Chem., Int. Ed., 2020, 59, 16076–16082 CrossRef CAS PubMed.
- C. F. Harris, C. S. Kuehner, J. Bacsa and J. D. Soper, Angew. Chem., Int. Ed., 2018, 57, 1311–1315 CrossRef CAS PubMed.
- M. Paeth, W. Carson, J.-H. Luo, D. Tierney, Z. Cao, M.-J. Cheng and W. Liu, Chem.–Eur. J., 2018, 24, 11559–11563 CrossRef CAS.
- L. Amini-Rentsch, E. Vanoli, S. Richard-Bildstein, R. Marti and G. Vilé, Ind. Eng. Chem. Res., 2019, 58, 10164–10171 CrossRef CAS.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef.
- E. Prchalová, O. Štěpánek, S. Smrček and M. Kotora, Future Med. Chem., 2014, 6, 1201–1229 CrossRef PubMed.
- M. Jagodzinska, F. Huguenot, G. Candiani and M. Zanda, ChemMedChem, 2009, 4, 49–51 CrossRef CAS.
- L. A. McAllister, C. R. Butler, S. Mente, S. V. O'Neil, K. R. Fonseca, J. R. Piro, J. A. Cianfrogna, T. L. Foley, A. M. Gilbert, A. R. Harris, C. J. Helal, D. S. Johnson, J. I. Montgomery, D. M. Nason, S. Noell, J. Pandit, B. N. Rogers, T. A. Samad, C. L. Shaffer, R. G. da Silva, D. P. Uccello, D. Webb and M. A. Brodney, J. Med. Chem., 2018, 61, 3008–3026 CrossRef CAS PubMed.
- B. Jeffries, Z. Wang, J. Graton, S. D. Holland, T. Brind, R. D. R. Greenwood, J.-Y. Le Questel, J. S. Scott, E. Chiarparin and B. Linclau, J. Med. Chem., 2018, 61, 10602–10618 CrossRef CAS.
- I. Vergote and P. Abram, Ann. Oncol., 2006, 17, 200–204 CrossRef CAS PubMed.
- N. Früh and A. Togni, Angew. Chem., Int. Ed., 2014, 53, 10813–10816 CrossRef.
- H. Jia, A. P. Häring, F. Berger, L. Zhang and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7623–7628 CrossRef CAS.
- Y. Wu, H.-R. Zhang, R.-X. Jin, Q. Lan and X.-S. Wang, Adv. Synth. Catal., 2016, 358, 3528–3533 CrossRef CAS.
- S. Martínez de Salinas, Á. L. Mudarra, C. Odena, M. Martínez Belmonte, J. Benet-Buchholz, F. Maseras and M. H. Pérez-Temprano, Chem.–Eur. J., 2019, 25, 9390–9394 CrossRef.
- S. Martínez-Salvador, J. Forniés, A. Martín and B. Menjón, Chem.–Eur. J., 2011, 17, 8085–8097 CrossRef.
- X. Zhao and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 19480–19486 CrossRef CAS.
- S. Zhang, N. Rotta-Loria, F. Weniger, J. Rabeah, H. Neumann, C. Taeschler and M. Beller, Chem. Commun., 2019, 55, 6723–6726 RSC.
- S. Zhang, F. Weniger, F. Ye, J. Rabeah, S. Ellinger, F. Zaragoza, C. Taeschler, H. Neumann, A. Brueckner and M. Beller, Chem. Commun., 2020, 56, 15157–15160 RSC.
- S. T. Shreiber and D. A. Vicic, Angew. Chem., Int. Ed., 2021, 60, 18162–18167 CrossRef CAS PubMed.
- S. T. Shreiber, G. I. Puchall and D. A. Vicic, Tetrahedron Lett., 2022, 97, 153795 CrossRef CAS.
- S. T. Shreiber, F. Amin, S. A. Schafer, R. E. Cramer, A. Klein and D. A. Vicic, Dalton Trans., 2022, 51, 5515–5523 RSC.
- S. Deolka, R. Govindarajan, E. Khaskin, R. R. Fayzullin, M. C. Roy and J. R. Khusnutdinova, Angew. Chem., Int. Ed., 2021, 60, 24620–24629 CrossRef CAS PubMed.
- C.-P. Zhang, H. Wang, A. Klein, C. Biewer, K. Stirnat, Y. Yamaguchi, L. Xu, V. Gomez-Benitez and D. A. Vicic, J. Am. Chem. Soc., 2013, 135, 8141–8144 CrossRef CAS.
- B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 79–81 CrossRef CAS.
- S. Fantasia, J. Windisch and M. Scalone, Adv. Synth. Catal., 2013, 355, 627–631 CrossRef CAS.
- D. R. Williams and S. A. Bawel, Org. Lett., 2017, 19, 1730–1733 CrossRef CAS PubMed.
- S. L. Wiskur, A. Korte and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 82–83 CrossRef CAS.
- J. Yin and C. J. T. Hyland, J. Org. Chem., 2015, 80, 6529–6536 CrossRef CAS PubMed.
- X. Gao, Y. Geng, S. Han, A. Liang, J. Li, D. Zou, Y. Wu and Y. Wu, Org. Lett., 2018, 20, 3732–3735 CrossRef CAS PubMed.
- X. Liu, G. Mao, J. Qiao, C. Xu, H. Liu, J. Ma, Z. Sun and W. Chu, Org. Chem. Front., 2019, 6, 1189–1193 RSC.
- J. R. Bour, P. Roy, A. J. Canty, J. W. Kampf and M. S. Sanford, Organometallics, 2020, 39, 3–7 CrossRef CAS.
- N. Shibata, A. Matsnev and D. Cahard, Beilstein J. Org. Chem., 2010, 6, 65 Search PubMed.
- L. Demonti, N. Saffon-Merceron, N. Mezailles and N. Nebra, Chem.–Eur. J., 2021, 27, 15396–15405 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc03879j |
| This journal is © The Royal Society of Chemistry 2022 |