
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA rotaxane-based platform for tailoring the pharmacokinetics of cancer-targeted radiotracers†
Faustine
d'Orchymont
 and
Jason P.
Holland
*
and
Jason P.
Holland
*
University of Zurich, Department of Chemistry, Winterthurerstrasse 190, CH-8057, Zurich, Switzerland. E-mail: jason.holland@chem.uzh.ch; Web: https://www.hollandlab.orgfaustine.dorchymont@chem.uzh.ch; Web: https://twitter.com/HollandLab Fax: +41-44-63-53990; Tel: +41-44-63-53990
First published on 11th October 2022
Abstract
Radiolabelled monoclonal antibodies (mAbs) are a cornerstone of molecular diagnostic imaging and targeted radioimmunotherapy in nuclear medicine, but one of the major challenges in the field is to identify ways of reducing the radiation burden to patients. We reasoned that a rotaxane-based platform featuring a non-covalent mechanical bond between the radionuclide complex and the biologically active mAb could offer new ways of controlling the biophysical properties of cancer-specific radiotracers for positron emission tomography (PET). Herein, we present the photoradiosynthesis and characterisation of [89Zr]ZrFe-[4]rotaxane-azepin-onartuzumab ([89Zr]ZrFe-2), a unique rotaxane-antibody conjugate for PET imaging and quantification of the human hepatocyte growth factor receptor (c-MET). Multiple component self-assembly reactions were combined with simultaneous 89Zr-radiolabelling and light-induced bioconjugation methods to give [89Zr]ZrFe-2 in 15 ± 1% (n = 3) decay-corrected radiochemical yield, with >90% radiochemical purity, and molar activities suitable for PET imaging studies (>6.1 MBq mg−1 of protein). Cellular assays confirmed the specificity of [89Zr]ZrFe-2 binding to the c-MET receptor. Temporal PET imaging in athymic nude mice bearing subcutaneous MKN-45 gastric adenocarcinoma xenografts demonstrated specific binding of [89Zr]ZrFe-2 toward c-MET in vivo, where tumour uptake reached 9.8 ± 1.3 %ID g−1 (72 h, n = 5) in a normal group and was reduced by ∼56% in a control (blocking) group. Head-to-head comparison of the biodistribution and excretion profile of [89Zr]ZrFe-2versus two control compounds, alongside characterisation of two potential metabolites, showed that the rotaxane-radiotracer has an improved clearance profile with higher tumour-to-tissue contrast ratios and reduced radiation exposure to critical (dose-limiting) organs including liver, spleen, and kidneys. Collectively, the experimental results suggested that non-covalent mechanical bonds between the radionuclide and mAb can be used to fine-tune the pharmacokinetic profile of supramolecular radiopharmaceuticals in ways that are simply not accessible when using traditional covalent design.
Introduction
Monoclonal antibodies (mAbs) are excellent tools for the delivery of drugs and radionuclides to tumours. Thus, a wide variety of radiolabelled mAbs that display high tumour uptake and specificity has been developed for diagnostic imaging and (radio) therapeutic applications.1,2 Despite their success in the clinic, mAb-based radiopharmaceuticals have encountered important vulnerabilities.3 For instance, the prolonged circulation of mAbs in the blood and their slow distribution, metabolism and excretion profile causes limited clearance of the radioactivity from the body and high radiation doses in healthy tissues.3 As the field of Nuclear Medicine progresses toward the utilisation of therapeutic radionuclides, which involve emission of high energy α- or β-particles and increased administered doses of radiolabelled mAbs, new methods that minimise undesirable radiation exposure to critical organs are essential.4,5Radiochemists have explored different approaches to address the dosimetry concerns of radiolabelled mAbs, which include the use of pretargeting strategies, chelation therapy, or mechanisms for controlling metabolic degradation.3,5–7 In the pretargeting approach (Fig. 1A), fast bioorthogonal reactions are used to ‘synthesise’ the radiolabelled mAb in vivo, whereby the radioactivity is administered several hours or days after mAb injection. Pre-administration of the functionalised mAb – which often display slow pharmacokinetics – provides time for these macromolecules to accumulate at tumour sites and clear from the blood pool and background tissues. Subsequent injection of the small-molecule radiolabelled reagent – which is designed to exhibit rapid tissue penetration and clearance – facilitates the use of shorter-lived radionuclides like 18F, thereby reducing patient exposure to ionising radiation and allowing clinicians to implement same-day imaging regimen. Chelation therapy takes a different approach which is based on the competitive complexation of the radioactive metal ion with a small ligand (Fig. 1B). Here, a radiolabelled mAb is administered, and after waiting for optimal distribution, peak tumour uptake, and blood pool or background tissue clearance, nuclear images are recorded before a small-molecule chelating ligand is administered. The chelating ligand must have a rapid pharmacokinetic profile and is selected to ensure that it possesses a strong thermodynamic and kinetic preference for complexation of the radiometal ion, and whereby the new radiometal ion complex that forms, is rapidly excreted from the body.
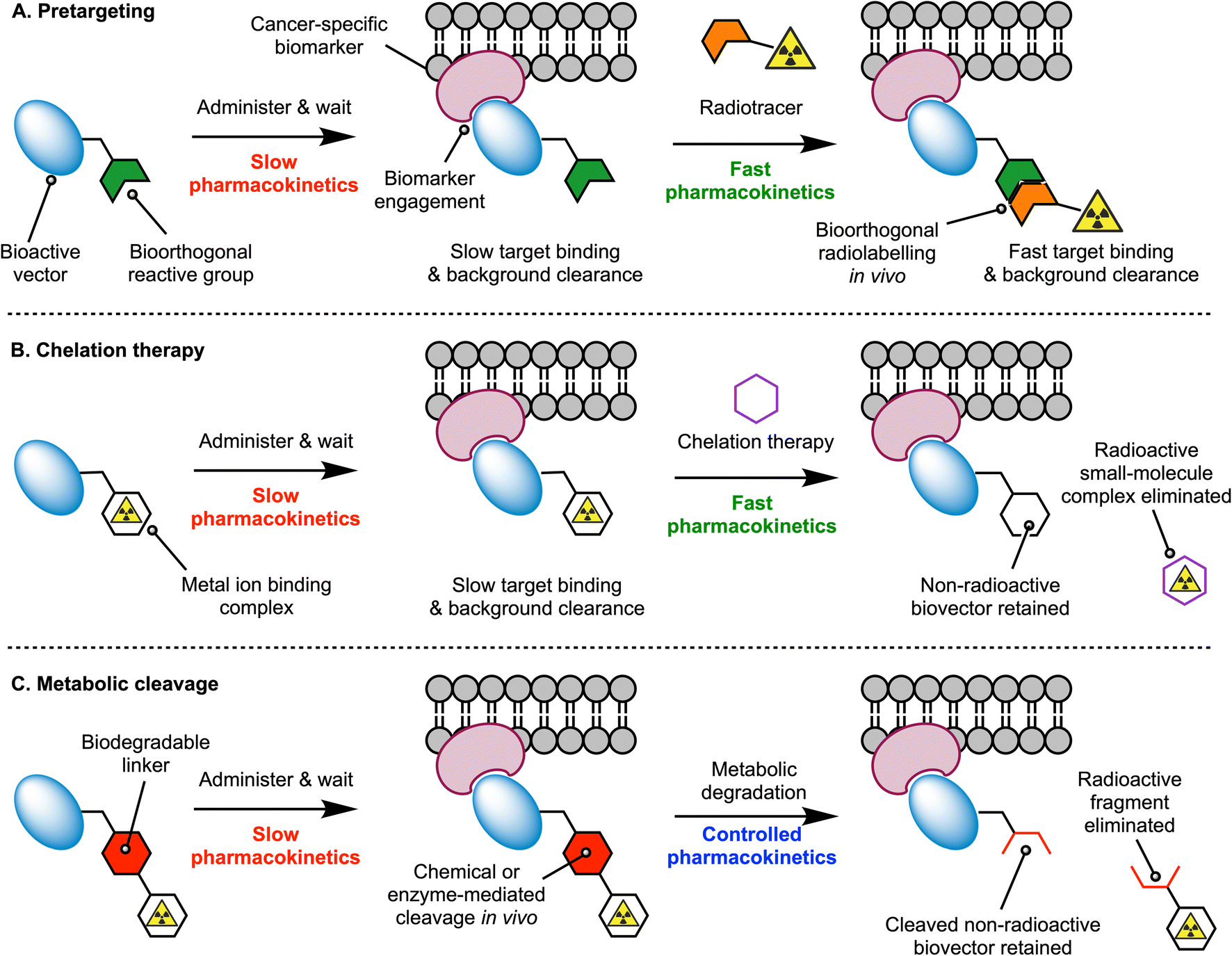 | ||
| Fig. 1 Three mechanistically distinct approaches for reducing the dosimetry burden of protein-based radiopharmaceuticals. Schemes illustrate the concepts of (A) pretargeting, (B) chelation therapy, and (C) controlled metabolic cleavage which harnesses catabolism in vivo to create a radiolabelled molecular fragment that is eliminated rapidly from the body. | ||
Experimental implementations of the multi-step pretargeting and chelation therapy strategies have demonstrated dosimetric advantages over conventional imaging with radiolabelled antibodies.7–11 However, these methods suffer from several drawbacks, including the need to produce, characterise, and administer two separate drug molecules, each having a distinct pharmacokinetic profile that complicates dosing regimen. The separate components can be difficult to synthesise, and expensive to characterise, potentially requiring separate absorption, distribution, metabolism, excretion and toxicological (ADME-tox) studies for each compound.7,12 Reliance on chelation therapy in vivo is also likely to encounter challenges regarding the kinetics of transchelation and competition from endogenous metal ions, the depletion of which may cause undesirable side-effects. The chelates themselves would also need to be administered at relatively high doses, and therefore, chemotoxic side-effects stemming directly from the administration of these drugs is also a potential problem.
To tackle the intrinsic limitations of the multi-step targeting strategies, an alternative approach is to re-engineer the radiolabelled antibody with functional groups that allow for controlled metabolism and disassembly in vivo, facilitating whole-body excretion (Fig. 1C). The installation of metabolically labile linkages that undergo cleavage in response to changes in pH, redox potential, exposure to light, or enzyme-mediated hydrolysis have been exploited in antibody-drug conjugate13,14 (ADC) and in radiotracer design.15–17 For ADC technologies, the objectives of using cleavable linkers are to ensure that the ADC-prodrug remains stable in vivo for sufficient time to allow tumour-specific localisation, and that release of active drug occurs selectively in the disease loci, thus reducing the chemotoxic burden to background tissues. In the context of using metabolically labile linkers in radiotracer design, the objectives are slightly different. First, the radiolabelled mAb should remain stable in the circulation to provide high, tumour-specific uptake. However, the main goal is to tailor the pharmacokinetic profile and reduce the effective half-life (t1/2(eff)/h) of the radiotracer in vivo by harnessing chemical or enzyme-mediated degradation processes. Ideally, the cleavage should operate in background tissues like the liver, spleen, and kidneys, which are prone to accumulate dose-limiting levels of activity when using protein-based radiotracers. Key to the success of this strategy is to ensure that the radioactive metabolites are eliminated rapidly from the body.
Supramolecular systems and mechanically interlocked molecules (MIMs) present interesting opportunities in the design of diagnostic imaging agents and therapeutic drugs.15,18–26 Supramolecular platforms have been shown to be viable routes for the synthesis of disease-targeted imaging probes, with key features including synthetic flexibility, rapid synthesis, facile labelling, water solubility, and biocompatibility of the construct. Here, we used mechanical bonding to make a supramolecular rotaxane-based 89Zr-radiolabelled mAb for PET imaging of the human hepatocyte growth factor receptor (c-MET) and evaluated the impact of the non-covalent design on the radiotracer pharmacokinetics in vivo.
Results and discussion
Synthesis and radiochemistry
Our rotaxane-based imaging agents are synthesised via a multi-component, cooperative capture strategy involving cucurbit[6]uril, the CB[6]-promoted ‘click’ reaction first introduced by Mock et al. and later developed by Stoddart and co-workers.27–29 With the objective of conjugating our radiolabelled rotaxane construct to mAbs, we introduced a photochemically active handle (aryl azide, ArN3) on to the β-cyclodextrin unit (Scheme 1, β-CD-ArN3).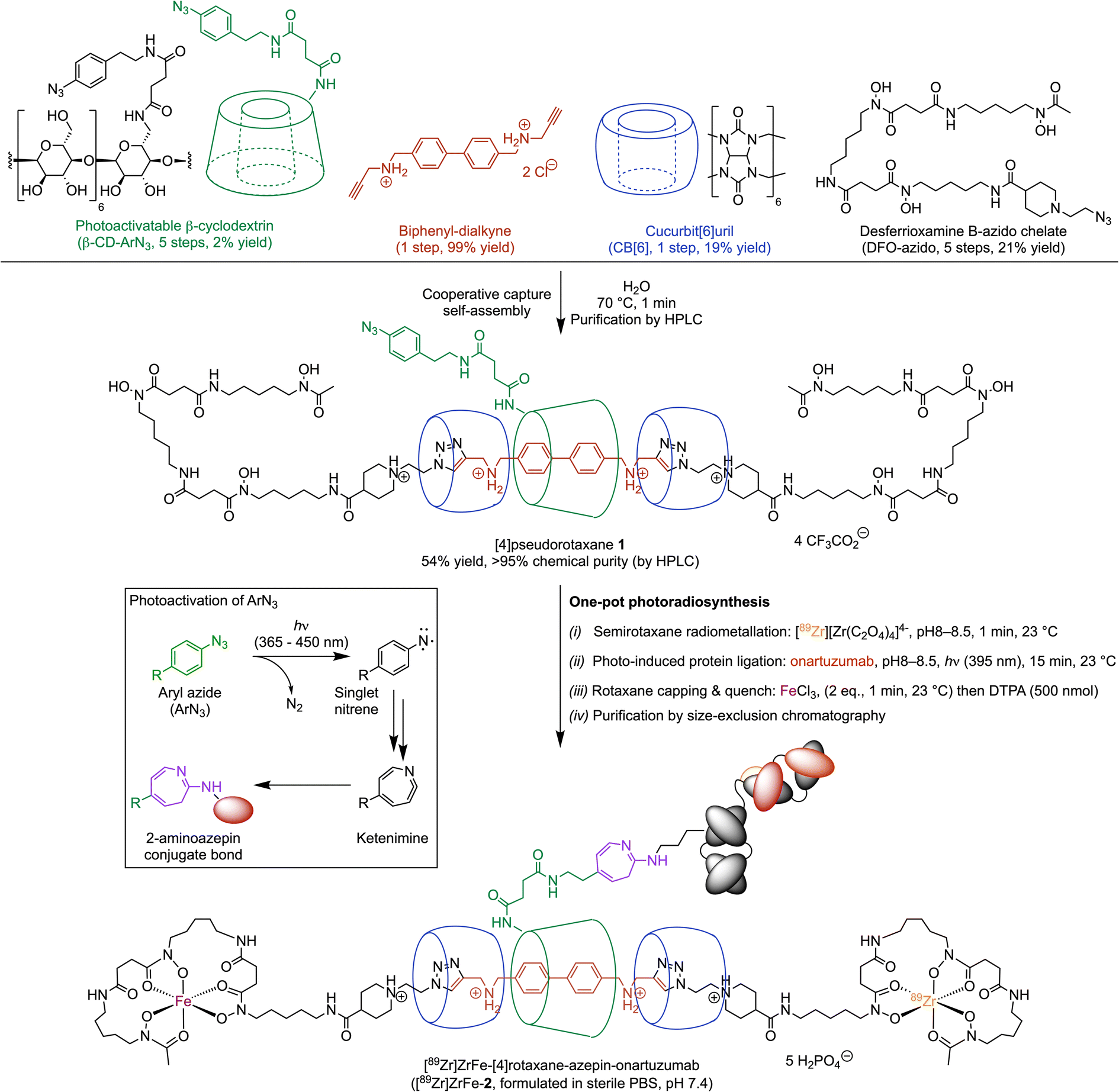 | ||
| Scheme 1 Cooperative capture self-assembly and the one-pot photoradiosynthesis of [89Zr]ZrFe-[4]rotaxane-azepin-onartuzumab ([89Zr]ZrFe-2). (Box inset) Mechanistic scheme of the photochemical activation of ArN3 showing formation of the open-shell singlet nitrene, multiple step rearrangement to the ketenimine heterocycle, and nucleophilic attack to give the 2-aminoazepin bioconjugate (based on reactivity with the ε-NH2 side-chain groups on surface-exposed lysine residues). | ||
The [4]pseudorotaxane 1 was synthesised in 54% yield after purification (Scheme 1). The reaction proceeds via a one-pot, four component self-assembly pathway after mixing the monofunctionalised photoactivatable β-CD-ArN3, a biphenyl-dialkyne guest molecule, the desferrioxamine B-azido chelate, and CB[6] in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2:2 mole ratio at 70 °C for 1 minute in aqueous media. Full details on the synthesis and characterisation of the precursors and [4]pseudorotaxane 1, the photochemical activation step, the radiochemistry and characterisation of both the 68Ga- and 89Zr-radiolabelled compounds were reported (see also ESI Fig. S1†).26 Simultaneous 89Zr-radiolabelling and light-induced bioconjugation to onartuzumab – an engineered monovalent antibody that binds the extracellular domain of the human hepatocyte growth factor receptor (c-MET)30,31 – afforded the [89Zr]Zr-[4]semirotaxane-azepin-onartuzumab intermediate. Subsequent capping with FeCl3 and reaction quenching with diethylenetriamine pentaacetic acid (DTPA) gave the bimetallic full rotaxane-mAb conjugate, [89Zr]ZrFe-[4]rotaxane-azepin-onartuzumab (Scheme 1, [89Zr]ZrFe-2). The light-induced activation of ArN3 used here to form the protein conjugate was demonstrated to be a viable route in the design of radiolabelled mAbs.32,33 Following light absorption (typically at photon wavelengths between 365 nm to 450 nm), dinitrogen N2(g) is released, producing a short-lived and highly reactive open-shell singlet aryl nitrene species.34,35 This aryl nitrene undergoes a series of extremely fast reactions where intramolecular rearrangements lead to the formation of a 7-membered electrophilic ketenimine ring, which reacts with nucleophiles (including the NH2 group of lysine residues) forming an azepine linkage between the protein and the chelate (Scheme 1, inset).32,33
:1:2:2 mole ratio at 70 °C for 1 minute in aqueous media. Full details on the synthesis and characterisation of the precursors and [4]pseudorotaxane 1, the photochemical activation step, the radiochemistry and characterisation of both the 68Ga- and 89Zr-radiolabelled compounds were reported (see also ESI Fig. S1†).26 Simultaneous 89Zr-radiolabelling and light-induced bioconjugation to onartuzumab – an engineered monovalent antibody that binds the extracellular domain of the human hepatocyte growth factor receptor (c-MET)30,31 – afforded the [89Zr]Zr-[4]semirotaxane-azepin-onartuzumab intermediate. Subsequent capping with FeCl3 and reaction quenching with diethylenetriamine pentaacetic acid (DTPA) gave the bimetallic full rotaxane-mAb conjugate, [89Zr]ZrFe-[4]rotaxane-azepin-onartuzumab (Scheme 1, [89Zr]ZrFe-2). The light-induced activation of ArN3 used here to form the protein conjugate was demonstrated to be a viable route in the design of radiolabelled mAbs.32,33 Following light absorption (typically at photon wavelengths between 365 nm to 450 nm), dinitrogen N2(g) is released, producing a short-lived and highly reactive open-shell singlet aryl nitrene species.34,35 This aryl nitrene undergoes a series of extremely fast reactions where intramolecular rearrangements lead to the formation of a 7-membered electrophilic ketenimine ring, which reacts with nucleophiles (including the NH2 group of lysine residues) forming an azepine linkage between the protein and the chelate (Scheme 1, inset).32,33
The one-pot, simultaneous 89Zr-radiolabelling and photochemical conjugation of 1 to onartuzumab was performed in accordance with the following general procedure. (i) The photoactivable [4]pseudorotaxane 1 and an aliquot of the neutralised stock solution of [89Zr][Zr(C2O4)4]4− (also known as 89Zr-oxalate) were added in a transparent glass vial containing H2O and a magnetic stir bar. The reaction pH was controlled to 8–8.5 and the mixture was stirred gently at 23 °C for 1 minute. (ii) An aliquot of onartuzumab (the antibody component of MetMab™, Genentech, South San Francisco, USA, prepurified by spin filtration methods and reconstituted in sterile PBS, pH7.4) was added at 23 °C to the reaction vial with an initial chelate-to-mAb ratio of 2-to-1. Then the reaction mixture was irradiated at 395 nm at 23 °C for 15 minutes to ensure that the light-induced activation step was complete. (iii) After irradiation, FeCl3 was added to cap the rotaxane with Fe3+ ions by formation of the bulky FeDFO complex at all non-radiolabelled positions. This capping process was motivated by previous reports, where full rotaxanes were demonstrated to be more stable in comparison with the pseudo- or semirotaxane species.36,37 Under the radiolabelling conditions used, it is statistically unlikely that the full Zr-rotaxane is formed whereby both ends of the axle are capped with a radioactive 89ZrDFO complex. The 89Zr-stock solution has an effective molar activity between 17.39–44.22 MBq nmol−1 which equates to 0.19–0.50 nmol of total material (radioactive and non-radioactive) used in the radiolabelling reactions. However, we note that ∼8.73 MBq of 89Zr was used which is equivalent to only 5.9 pmol of radioactive atoms and this gives an isotopic dilution factor of radioactive to non-radioactive atoms between 33–85. Since a total of 13.66 nmol of [4]pseudorotaxane 1 (27.32 nmol of DFO) was used in the reactions, then between 0.7%–1.8% of the available DFO chelate was labelled with the combined material present in the 89Zr-stock. The remaining chelate was quenched quantitatively by the Fe3+ ions. Overall, we estimate that in the reaction, the probability of dual radiolabelling one molecule of rotaxane with two radioactive 89Zr atoms lies between 0.014%–0.089%. Thus, after the radiolabelling step is complete, the [89Zr]Zr-[4]semirotaxane-azepin-onartuzumab species would be the most likely product formed. Capping with Fe3+ has the added benefit that the product can be detected due to the absorption of the FeDFO complex at 430 nm. After rotaxane capping, the reaction was quenched with excess DTPA (500 nmol) to ensure that only 89Zr4+ and Fe3+ ions that were specifically bound to the rotaxane-mAb remained. (iv) Finally, the crude reaction mixture was purified by using preparative Sephadex G-100 size-exclusion chromatography (G-100 SEC) to afford the bimetallic product [89Zr]ZrFe-2 (Scheme 1) formulated in sterile PBS (pH 7.4).
At this point we note that [89Zr]ZrFe-2 exists as two mechanical planar epimers with absolute assignments: (D, Rmp) and (D, Smp). These isomers arise from the fixed covalent stereochemistry of the functionalised β-[D]-CD (constructed from enantiomerically pure [D]-glucose) which gives an oriented macrocycle, in combination with the mechanically planar chirality introduced by the [4]rotaxane architecture (Fig. 2). For further details on the chirality and isomerism of molecularly interlocked molecules, as well as rules for stereochemical assignment, please refer to the reference works from Goldup and co-workers.38,39 It is unclear what impact (if any) isomerism of the rotaxane might have on the biological properties of large mAb-based radiotracers.
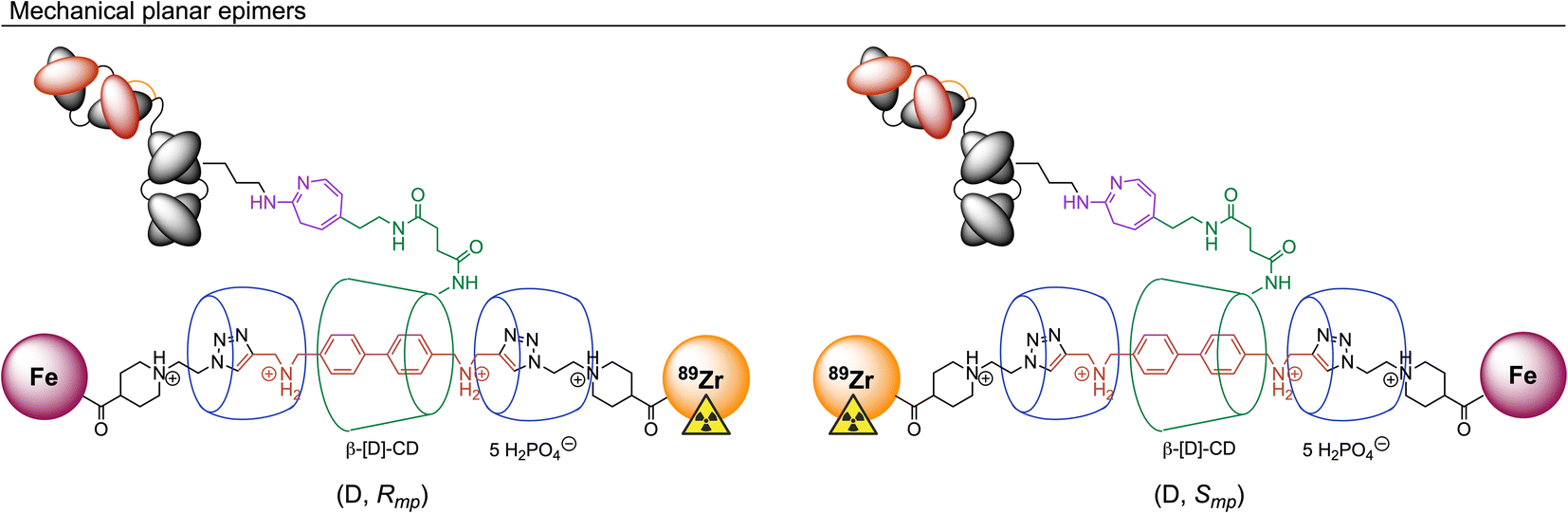 | ||
| Fig. 2 Illustration of the isomerism displayed by [89Zr]ZrFe-2. Stereochemical assignments for the two mechanical planar epimers were made in accordance with the Cahn–Ingold–Prelog rules and the methodology reported by Jamieson et al. where Zr has a higher priority than Fe, and clockwise rotation occurs for the primary face of the functionalised β-[D]-CD macrocycle.38,39 | ||
During the radiolabelling process, crude and purified aliquots of the reaction mixtures were retained for subsequent analysis by using radioactive instant thin-layer chromatography (radio-iTLC), manual size-exclusion chromatography (SEC; using custom-made columns containing a Sephadex G100 stationary phase eluted with sterile PBS, pH7.4), and automated high-performance liquid chromatography HPLC coupled to a SEC gel-filtration column (SEC-HPLC). Representative chromatographic data obtained during the photoradiosynthesis of [89Zr]ZrFe-2 are presented in Fig. 3.
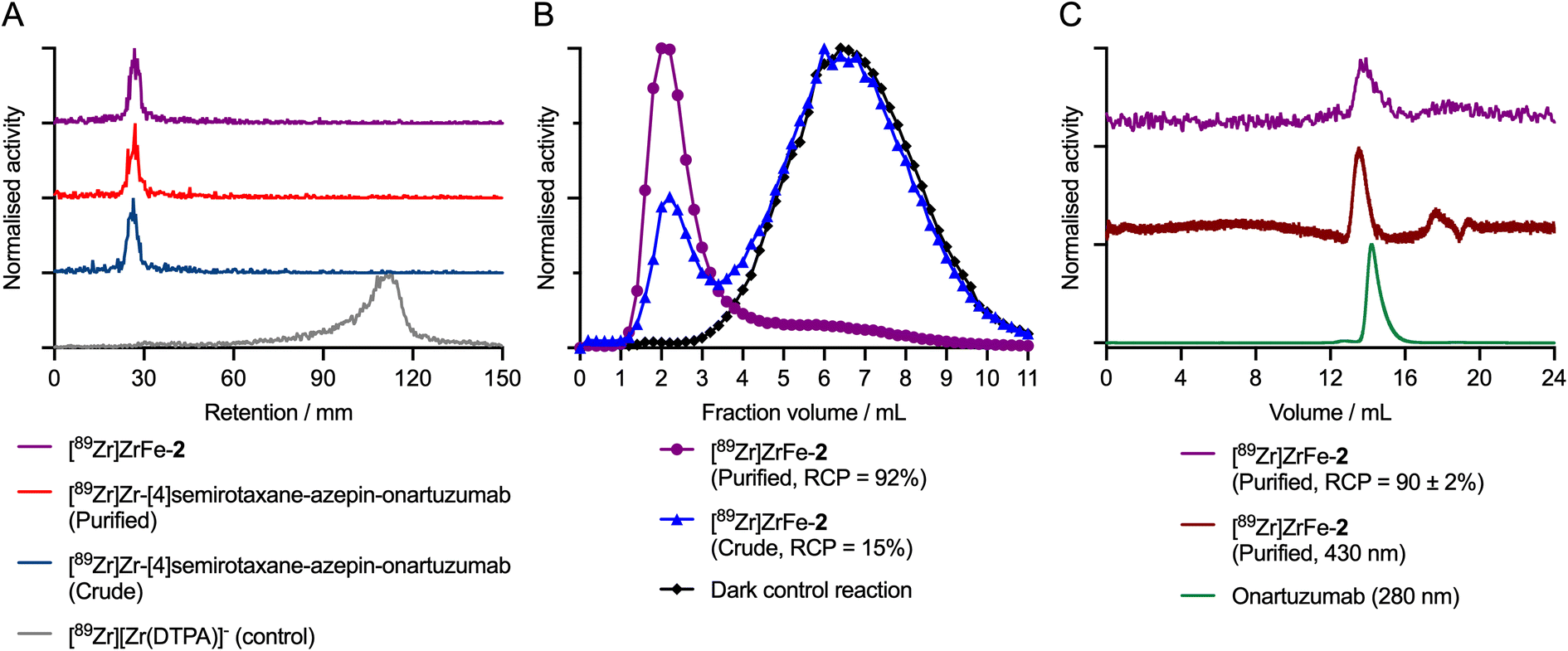 | ||
| Fig. 3 Characterisation data for the photoradiosynthesis of [89Zr]ZrFe-2. (A) Radio-iTLC chromatograms developed by using DTPA (50 mM, pH 7) as the mobile phase showing the quantitative complexation of 89Zr4+ ions for the crude (dark blue) and purified (red) samples of the [89Zr]Zr-[4]semirotaxane-onartuzumab intermediate, and the purified sample of the capped rotaxane-mAb [89Zr]ZrFe-2 (purple). The elution profile of the control compound [89Zr][Zr(DTPA)]− (grey) showing migration of ‘free’ or non-specifically complexed 89Zr4+ ions to the solvent front is also shown. (B) Manual analytical G100 SEC elution profiles of [89Zr]ZrFe-2 showing the purified (purple circles, RCP = 92%) and the crude samples (blue triangles, RCP = 15%), as well as the profile obtained from the dark control reaction (black diamonds). Activity eluting in the first ∼3.5 mL is associated with the high-molecular weight protein fraction, whereas the peak for small-molecule components occurs at ∼6.5 mL. (C) SEC-HPLC chromatograms of the purified sample of [89Zr]ZrFe-2 showing the radioactivity channel (purple trace) and the chromatogram produced by electron absorption at 430 nm corresponding to photon absorption by the FeDFO complex on the rotaxane axle (dark red trace). For comparison, the electronic absorption chromatogram of the non-modified protein (green trace, onartuzumab; 280 nm) is presented confirming co-elution with the radiolabelled product [89Zr]ZrFe-2. | ||
The radio-iTLC analysis confirmed that quantitative complexation of 89Zr4+ ions occurred rapidly. Under the radio-iTLC conditions employed, 89Zr-radiolabelled DFO samples and proteins are retained at the baseline (Rf = 0.0–0.1), whereas unbound or non-specifically bound 89Zr4+ ions immediately form the [89Zr][Zr(DTPA)]− complex which migrates to the solvent front (Rf = 0.8–1.0). Complexation of 89Zr4+ ions occurs immediately upon addition of [89Zr][Zr(C2O4)4]4− to the solution of [4]pseudorotaxane 1, giving the photoactivatable intermediate species [89Zr]Zr-[4]semirotaxane-ArN3 ([89Zr]Zr-1; see ESI Fig. S1†).26 Subsequent addition of protein to the reaction mixture (using an initial rotaxane-to-protein ratio of ∼2:1), followed by pH adjustment,32 and irradiation of the mixture with a powerful light emitting diode (LED, 395 nm, 15 min, 23 °C) leads to quantitative photochemical activation of the ArN3 group, and stable protein ligation to give the [89Zr]Zr-[4]semirotaxane-azepin-onartuzumab intermediate. Radio-iTLC chromatograms of the crude (Fig. 3A dark blue trace) and purified (Fig. 3A red trace) samples of [89Zr]Zr-[4]semirotaxane-azepin-onartuzumab confirmed that the activity remained bound to the DFO chelate and was also attached to the protein after irradiation. Subsequent addition of FeCl3 capped the rotaxane to give the desired product [89Zr]ZrFe-2, and importantly, did not induce any dissociation (via transmetallation) of the 89Zr4+ from the rotaxane-mAb (Fig. 3A purple trace).
Manual SEC analysis of the crude sample of [89Zr]ZrFe-2 (Fig. 3B blue trace) indicated that the photo-induced protein ligation step had a conjugation efficiency of ∼15%. After preparative isolation, the purified sample of [89Zr]ZrFe-2 (Fig. 3B purple trace) had a radiochemical purity of ∼92%. In a representative pilot experiment starting from 10.49 MBq of [89Zr][Zr(C2O4)4]4− and a total of 7.26 nmol (0.72 mg) of onartuzumab, the isolated product had a decay-corrected radiochemical yield (d. c. RCY) of 13.3%, an RCP of 92%, and a molar activity (Am) of 0.11 MBq nmol−1 of protein (equivalent to 1.108 MBq mg−1). The reaction was then repeated with three independent replicates, giving an average radiochemical conversion of 15 ± 1% (n = 3) of [89Zr]ZrFe-2 (see ESI Fig. S2†). Importantly, control reactions performed in the dark, which produce the [89Zr]Zr-1 intermediate, confirmed that no protein binding occurred in the absence of light (Fig. 3B black trace, where <0.5% 89Zr-activity was measured in the 1.0–3.5 mL fraction). Definitive confirmation of protein labelling was obtained from the SEC-HPLC analysis of [89Zr]ZrFe-2, which showed that for the purified sample, the radioactivity (Fig. 3C purple trace, peak retention time ∼14.1 min) coeluted with the peak observed from electronic absorption chromatogram measured at either 430 nm corresponding to absorption by the FeDFO complex (Fig. 3C dark red trace, peak retention time ∼13.9 min), or at 280 nm showing the elution profile of the protein component (Fig. 3C green trace, peak retention time ∼14.2 min). Interestingly, no increase in the proportion of aggregated protein, which elutes at an earlier time point (∼12.8 min) than the monomeric protein peak (∼14.2 min), was observed confirming that the light-induced protein ligation reaction is mild and does not compromise the structural integrity of the mAb.
After demonstrating that [89Zr]ZrFe-2 could be isolated, the photoradiosynthesis was scaled-up to provide sufficient activity for in vitro and in vivo experiments. In a scaled-up reaction, the initial amount of [89Zr][Zr(C2O4)4]4− activity was increased to 63.99 MBq but the total amount of protein (7.26 nmol), and the initial rotaxane-to-protein ratio (∼2:1) were kept constant. [89Zr]ZrFe-2 was isolated with a d. c. RCY of 10.6%, an RCP of 90%, and an approximate 6-fold higher molar activity, with Am = 0.61 MBq nmol−1 of protein (6.176 MBq mg−1 of protein). The scaled-up reaction confirmed that the photoradiochemistry can be adapted to produce useful quantities of rotaxane-based radiotracers.
Radiotracer stability and cellular binding experiments
To evaluate the stability of [89Zr]ZrFe-2, the purified and formulated sample was stored for 72 h in PBS under ambient conditions. Manual SEC analysis of the product reveal that ∼53% of the 89Zr-activity remained bound to the mAb (ESI Fig. S3†) confirming that the radiotracer remains intact for sufficient time to perform longitudinal studies in cells and animals. The potential chemical and metabolic degradation of the 89Zr-labelled rotaxane-onartuzumab conjugate is discussed in later sections.To confirm that conjugation and radiolabelling of onartuzumab did not compromise the binding specificity for c-MET, cellular binding (Lindmo-type)40 experiments were performed by using MKN-45 (c-MET positive) human gastric adenocarcinoma cells (ESI Fig. S4†). Cellular assays confirmed that [89Zr]ZrFe-2 remained biologically active and displayed specific binding to the c-MET receptor with an estimated immunoreactive fraction of ∼35%. We note that in our experiments, only an expired ex-clinical and pre-purified sample of onartuzumab (Genentech/Roche, South San Francisco, USA) was available, whereby the absence of formulation buffer is the likely cause of the lower immunoreactive fraction. Nevertheless, the observed specific binding provided confidence in pursuing further biological experiments with [89Zr]ZrFe-2 in animal models.
Small-animal PET imaging and biodistribution studies
Following the successful results obtained in vitro, PET imaging experiments were conducted in female athymic nude mice bearing subcutaneous MKN-45 tumours on the right flank (n = 5 mice/group). Mice were randomised into two groups and administered either a high molar activity formulation of [89Zr]ZrFe-2 (normal group, Am = 6.176 MBq mg−1, 0.192–0.198 MBq, 31–32 μg of protein, in 200 μL sterile PBS) or a low molar activity formulation of [89Zr]ZrFe-2 (blocking group, Am = 0.188 MBq mg−1, 0.219–0.229 MBq, ∼1.02 mg of protein, in 200 μL sterile PBS). For the standard competitive inhibition (blocking) group, the molar activity of [89Zr]ZrFe-2 was reduced by adding non-radiolabelled onartuzumab (MetMAb) to each dose of radiotracer to modulate the tumour uptake by saturating the available c-MET receptors. We note that the ∼97% reduction in molar activity of the blocking group (equivalent to ∼32-times increase in the administered protein mass) represents a standard difference when modulating radiotracer uptake and demonstrating specificity in vivo in tumours that express high levels of the target protein. Representative PET images recorded between 1 and 72 h post-intravenous teil-vein administration of [89Zr]ZrFe-2 are presented in Fig. 4, and maximum intensity projections are shown in ESI Fig. S5.†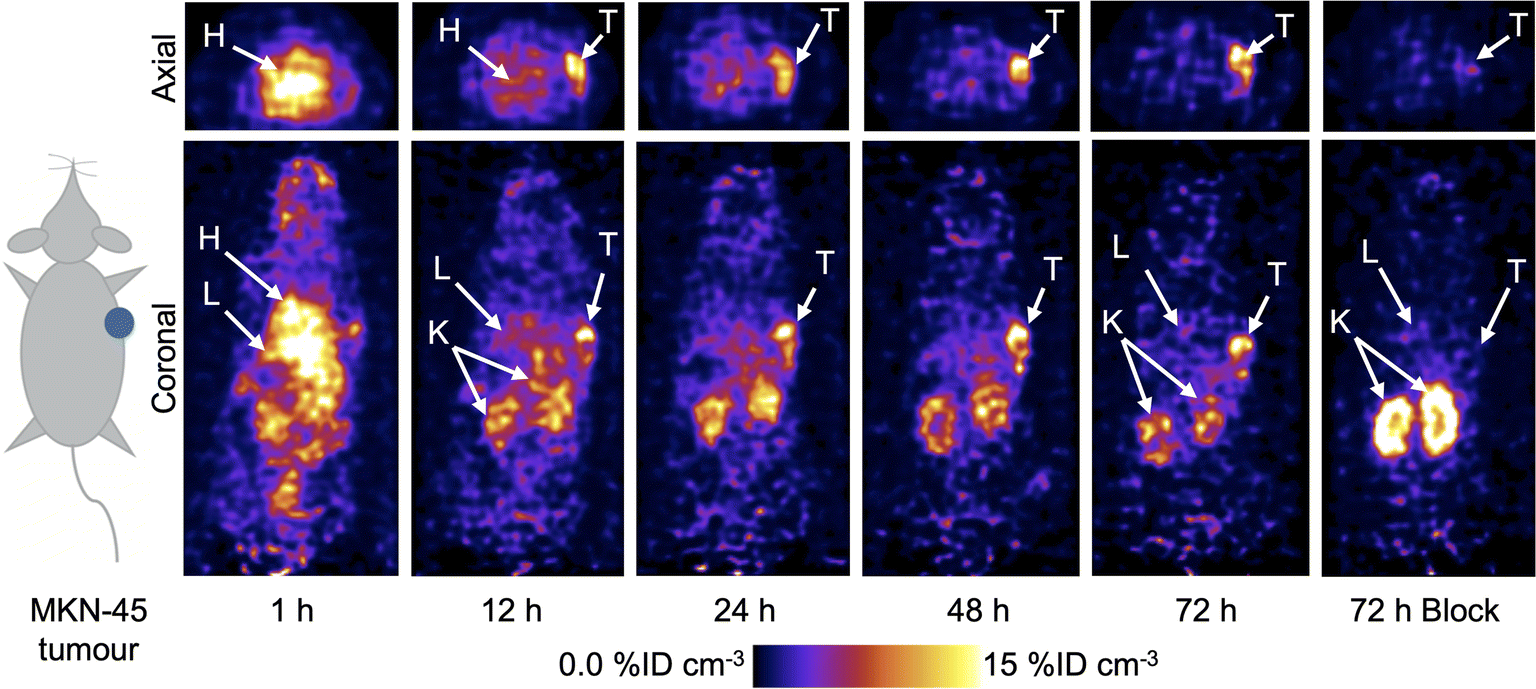 | ||
| Fig. 4 Representative PET images showing the distribution of [89Zr]ZrFe-2 in mice bearing subcutaneous MKN-45 tumours on the right flank. Images show coronal and axial tomographic views taken through the centre of the tumour for a mouse that was imaged at five time points between 1–72 h after radiotracer administration. T = tumour, H = heart, L = liver, B = bladder, K = kidneys. | ||
Volume of interest (VOI) analysis was used to quantify radiotracer distribution in different tissues based on the calibrated PET images (Fig. 5, ESI Fig. S6 and Table S1†). The mean tumour uptake of [89Zr]ZrFe-2 was significantly higher for the normal group (9.8 ± 1.3 %ID cm−3, 72 h, n = 5) when compared with the uptake observed in mice that received the blocking formulation (4.3 ± 0.3 %ID cm−3, 72 h, n = 5, 56% reduction, Student's t-test P-value < 0.001). These data indicate that [89Zr]ZrFe-2 displayed specific uptake in the MKN-45 tumours in vivo. The difference in kidney accumulation between groups was also shown to be statistically significant with a lower uptake of 9.7 ± 0.4 %ID cm−3 (n = 5, P-value < 0.001) observed for the normal group compared with of 17.6 ± 1.3 %ID cm−3 (n = 5) in the blocking group. This higher kidney accumulation with increasing administered protein dose is a known dose-dependent phenomenon for onartuzumab.41–43
 | ||
| Fig. 5 Bar chart showing time-activity profile data in various tissues obtained from the quantitative VOI analysis of [89Zr]ZrFe-2 PET images for the normal group (colour bars) and the blocking group (grey-to-white bars). Data are displayed in units of percentage injected dose per cubic centimetre (%ID cm−3) with data obtained from the calibrated PET images. Student's t-test: (*) P-value < 0.05, (**) P-value < 0.01. An equivalent representation of these VOI data as time-activity curves is presented in ESI Fig. S6.† | ||
To establish the effect of the rotaxane scaffold on the radiotracer pharmacokinetics, we monitored the whole-body excretion profile by using dose calibrator measurements to determine the effective (t1/2(eff)/h) and biological (t1/2(biol)/h) half-lives of our rotaxane-based imaging agent [89Zr]ZrFe-2. These data were compared to two separate onartuzumab-based radiotracers, which were synthetised previously by using either conventional or photochemical conjugation methods to create covalent linkages between the 89Zr-labelled DFO complex and the mAb (Fig. 6A). [89Zr]Zr-DFO-Bn-NCS-onartuzumab (orange) was obtained via standard (clinically used) methods involving thiourea formation,44 whereas [89Zr]Zr-DFO-azepin-onartuzumab (blue) was produced by our photoradiosynthesis method to create the equivalent 2-aminoazepin linker that also features in the structure of [89Zr]ZrFe-2 (Fig. 6A).42 It is important to emphasise that for [89Zr]Zr-DFO-Bn-NCS-onartuzumab and [89Zr]Zr-DFO-azepin-onartuzumab, a covalent linker connects the mAb directly to the 89Zr-labelled DFO complex, unlike our rotaxane-based agent where the radiolabelled rotaxane axle is connected to the mAb by a mechanical bond.
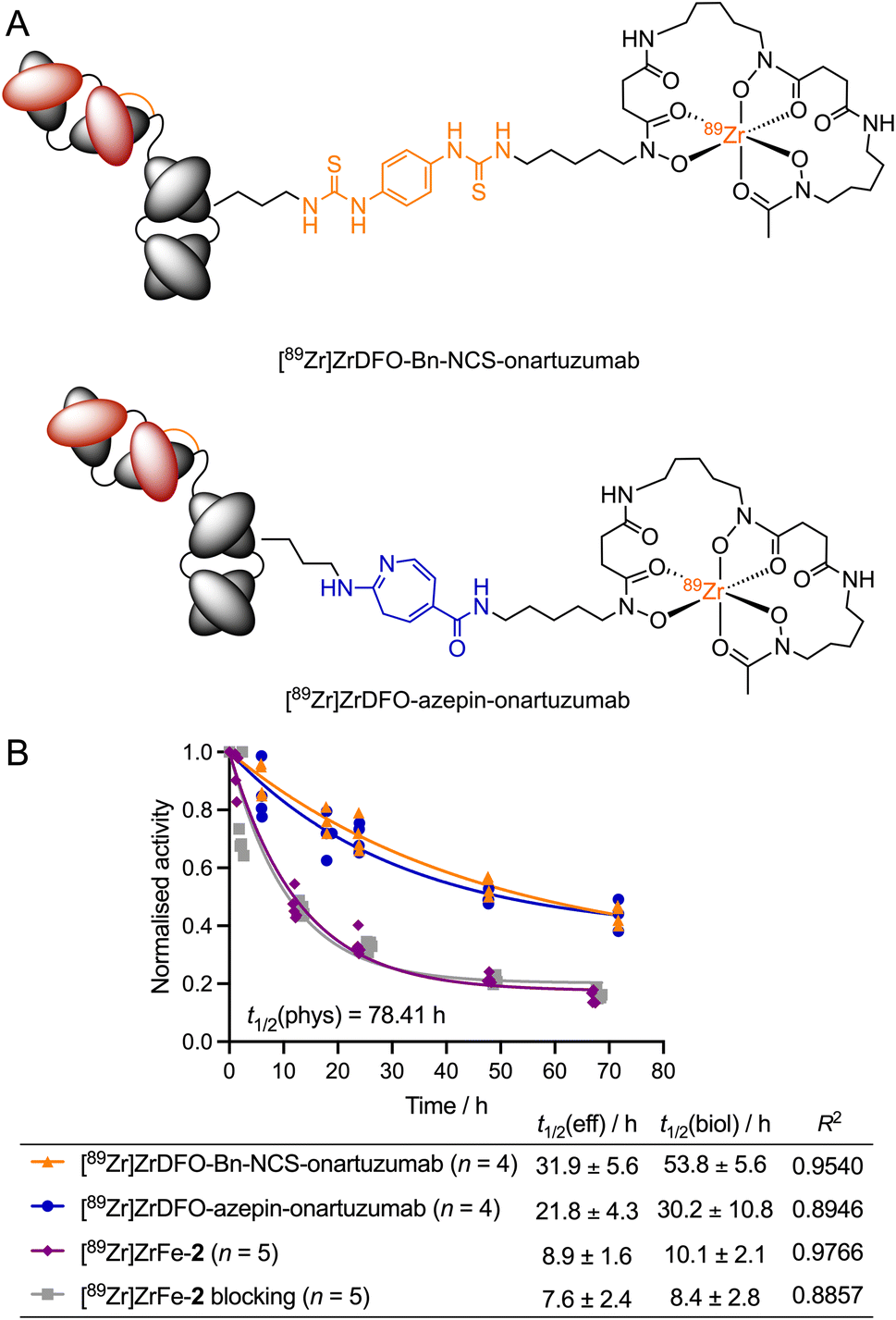 | ||
| Fig. 6 Influence of the non-covalent mechanical bond on the whole-body effective half-life of the rotaxane-based radiotracer [89Zr]ZrFe-2versus two covalently-modified radiotracers. (A) Structures of [89Zr]ZrDFO-Bn-NCS-onartuzumab and [89Zr]ZrDFO-azepin-onartuzumab. (B) Plot of the normalised whole-body activity retained in the animals versus time, after administration of [89Zr]ZrDFO-Bn-NCS-onartuzumab (orange), [89Zr]ZrDFO-azepin-onartuzumab (blue), or [89Zr]ZrFe-2 showing data obtained from both the normal group (purple) and the blocking group (grey). Curves show the one-phase exponential fit of the data with regression coefficients given in the table. | ||
The rotaxane-based radiotracer [89Zr]ZrFe-2 gave measured t1/2(eff) values of 8.9 ± 1.6 h (n = 5) for the normal group and 7.6 ± 2.4 h (n = 5) for the blocking group (no significant difference). In contrast, the measured t1/2(eff) values for the covalently-linked radiotracers [89Zr]ZrDFO-Bn-NCS-onartuzumab (31.9 ± 5.6 h, n = 4) and [89Zr]Zr-DFO-azepin-onartuzumab (21.8 ± 4.3 h, n = 4) indicated a prolonged retention of the activity in the whole-body of the animals. As anticipated, the same trend was also observed in the calculated biological half-lives of the different radiotracers. These data confirmed that 89Zr-labelled rotaxane-onartuzumab displays a favourable whole-body dosimetry profile whereby the 89Zr-activity localised specifically in the tumours generating high tumour-to-tissue contrast, but the radiotracer displayed faster clearance leading to reduced exposure of the background organs to the potentially damaging effects of ionising radiation.
After the final imaging time point at 72 h post-radiotracer injection, mice were euthanised and biodistribution studies were performed to quantify the radiotracer uptake across 15 different tissues and to determine the tumour-to-tissue ratios (Fig. 7 and ESI Table S2†). Ex vivo analysis confirmed that most of the activity remained in the tumour and kidneys for the normal group of mice injected with [89Zr]ZrFe-2 (Fig. 7A, purple bars), which was consistent with the PET image analysis. Accumulation of radioactivity in the MKN-45 tumours was 9.9 ± 2.8 %ID g−1 (n = 5). In contrast, tumour-associated uptake of [89Zr]ZrFe-2 decreased by ∼76% to 2.4 ± 0.3 %ID g−1 (n = 5, Student's t-test P-value < 0.01) in the blocking study (Fig. 7A, grey bars). Hence, the blocking study confirmed the specific targeting of c-MET by [89Zr]ZrFe-2in vivo. As expected from the PET images, the kidney accumulation was found to be statistically higher in the blocking group where an uptake of 25.4 ± 3.0 %ID g−1 (n = 5) was measured, compared with 13.1 ± 1.5 %ID g−1 (n = 5, P-value < 0.001) in the normal group. Except for the tumour and the kidneys, no other differences were observed in the biodistribution data sets for the normal and blocking groups that received [89Zr]ZrFe-2. We also compared the ex vivo data of our rotaxane-based imaging agent with equivalent data obtained for [89Zr]Zr-DFO-Bn-NCS-onartuzumab and [89Zr]Zr-DFO-azepin-onartuzumab (Fig. 7, orange and blue bars respectively).42 Comparison of the tumour-associated activity for the three radiotracers showed no statistically significant difference. However, we note that the tumour uptake of [89Zr]ZrFe-2 in the normal group tends toward a lower value than the two covalently-linked radiotracers with values of 9.9 ± 2.8 %ID g−1 for [89Zr]ZrFe-2versus 21.4 ± 11.6 %ID g−1 for [89Zr]Zr-DFO-Bn-NCS-onartuzumab, and 15.4 ± 5.2 %ID g−1 for [89Zr]Zr-DFO-azepin-onartuzumab. This observation is consistent with the lower effective and biological half-life values measured experimentally for [89Zr]ZrFe-2.
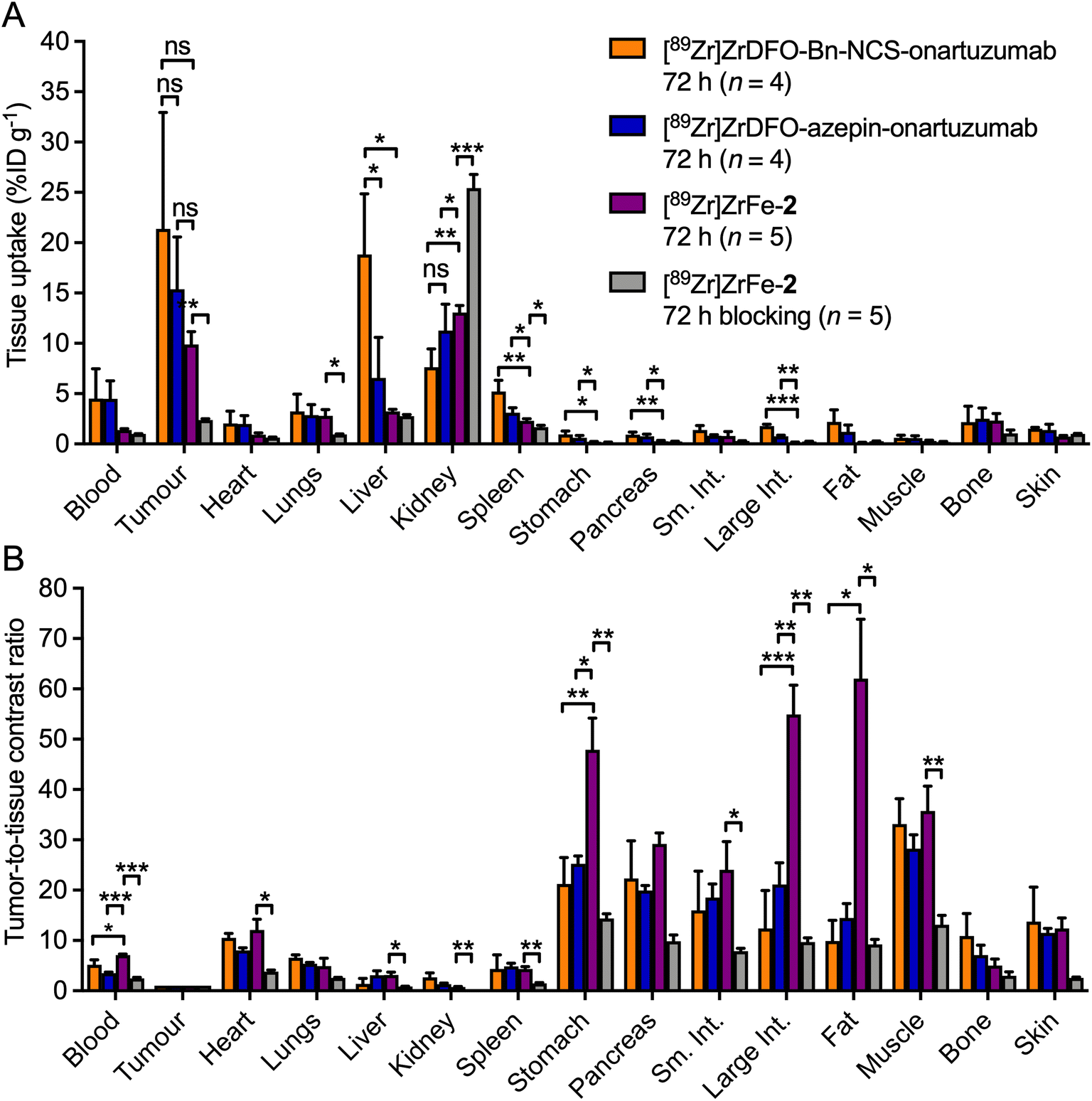 | ||
| Fig. 7 Bar charts showing (A) the full ex vivo biodistribution profile (in units of percentage injected dose per gram of tissue, %ID g−1), and (B) the tumour-to-tissue contrast ratios measured after the final imaging time point at 72 h post-injection in MKN-45 tumour bearing mice for [89Zr]ZrDFO-Bn-NCS-onartuzumab (orange), [89Zr]ZrDFO-azepin-onartuzumab (blue), and for [89Zr]ZrFe-2 in the normal (purple) and blocking groups (grey). Student's t-test: (*) P-value < 0.05, (**) P-value < 0.01. | ||
An overall decrease of activity was observed in background tissues for the normal group that received [89Zr]ZrFe-2 compared with mice that received either [89Zr]Zr-DFO-Bn-NCS-onartuzumab or [89Zr]Zr-DFO-azepin-onartuzumab. The lower retention of [89Zr]ZrFe-2 activity was statistically significant in the spleen, stomach, pancreas, and large intestine when compared against both the covalently-linked radiotracers. In addition, accumulation of activity in the liver – which is frequently the dose limiting organ for 89Zr-radiolabelled mAbs used in the clinic45 – was significantly lower for [89Zr]ZrFe-2versus [89Zr]Zr-DFO-Bn-NCS-onartuzumab (P-value < 0.05).
While no statistically significant differences were observed in the measured tumour-to-tissue contrast ratios between [89Zr]Zr-DFO-Bn-NCS-onartuzumab and [89Zr]Zr-DFO-azepin-onartuzumab, tumour uptake of [89Zr]ZrFe-2 showed higher contrast over background organs, particularly in the blood, stomach, large intestine, and fat (Fig. 7B, purple bars). These results suggest that the mechanical bond of the rotaxane-mAb construct has a positive influence over the radiotracer distribution and pharmacokinetics, where the data indicate that high target specificity, tumour uptake, and tumour-to-tissue contrast ratios can be retained whilst simultaneously enhancing the excretion rates and reducing the dose exposure of background organs.
Data from the PET imaging, biodistribution, and whole-body excretion measurements of [89Zr]ZrFe-2 suggest that this radiotracer undergoes metabolic degradation via mechanisms that are not accessible to the covalent radiotracers [89Zr]Zr-DFO-Bn-NCS-onartuzumab or [89Zr]Zr-DFO-azepin-onartuzumab. In addition, the data suggest that catabolic degradation does not release free 89Zr4+ ions – which would localise in the bone – but instead produces small, radiolabelled fragments that are eliminated rapidly from the animal (consistent with the mechanism shown in Fig. 1C).
To assess the potential mechanisms for chemical or metabolic degradation, we considered several plausible sites for bond disconnection/fragmentation in the structure of [89Zr]ZrFe-2 (Fig. 8, disconnections indicated by the dashed red lines at positions 1–6). As for all radiotracers bearing a 89Zr-radiolabelled DFO complex, a decrease in thermodynamic and kinetic stability can occur when the chemical structure of the coordination complex is compromised (Fig. 8, disconnections 1–4).46,47 The release of “free” Zr4+ ions by demetallation (or transmetallation, Fig. 8, disconnection 1) was demonstrated previously to induce higher retention in the bones and joints.48 Bond cleavage at disconnections 2, 3, or 4 break the mechanical bond and would produce semirotaxane species where dethreading of the macrocycles from the rotaxane axle is expected to release radioactive fragments that would likely be eliminated rapidly from the body.48
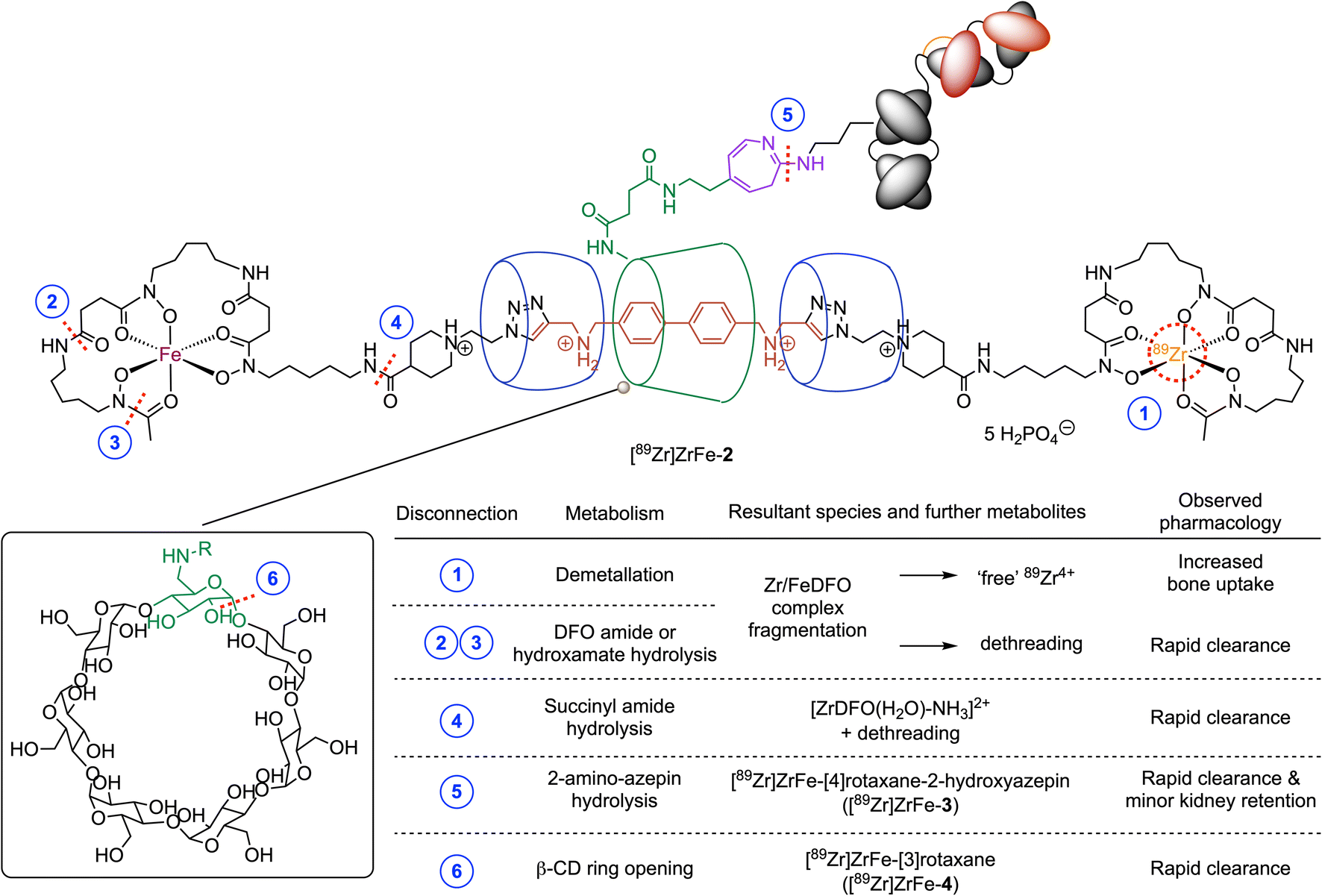 | ||
| Fig. 8 Chemical structure of the rotaxane-mAb construct [89Zr]ZrFe-2 showing the potential sites of chemical or catabolic degradation (dashed red lines positions 1–6). Resultant metabolites and the observed or anticipated pharmacology of the radiolabelled fragments are given in the table. | ||
Experimental data were obtained for both disconnections 5 and 6 (Fig. 9). Hydrolysis of the 2-aminoazepin linker leads to the formation of [89Zr]ZrFe-[4]rotaxane-2-hydroxyazepin ([89Zr]ZrFe-3, Fig. 9A), which is also an observed byproduct of the photoradiosynthesis of [89Zr]ZrFe-2 that was synthesised, characterised and found to exhibit rapid hepatobiliary and renal clearance with t1/2(eff) value 0.58 ± 0.13 h (Fig. 9B, green diamonds and curve).26 A small fraction (approximatively 6% of the total injectate) of [89Zr]ZrFe-3 was found to be retained in the kidneys as shown by the ex vivo biodistribution data collected at 24 h post-injection (Fig. 9C, green bars).
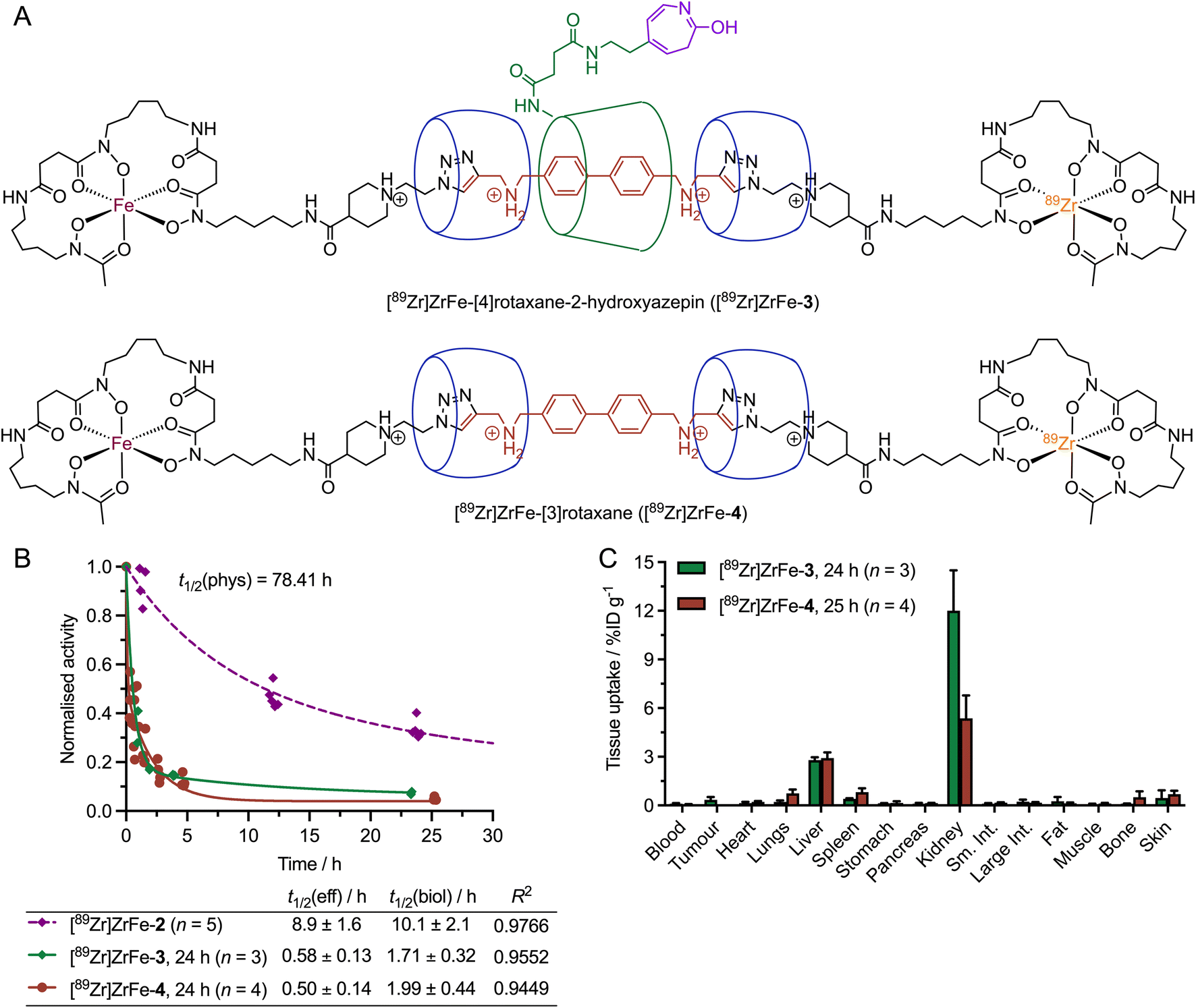 | ||
| Fig. 9 (A) Chemical structures of the metabolites [89Zr]ZrFe-[4]rotaxane-2-hydroxyazepin ([89Zr]ZrFe-3) formed by 2-aminoazepin hydrolysis and [89Zr]ZrFe-[3]rotaxane ([89Zr]ZrFe-4) formed by ring-opening hydrolysis of the β-CD macrocycle. (B) Plot of the measured whole-body excretion profiles of [89Zr]ZrFe-3 (green diamonds and curve) and [89Zr]ZrFe-4 (red circles and curve) versus the profile obtained for [89Zr]ZrFe-2 (purple diamonds and curve). (C) Bar chart showing the ex vivo biodistribution profiles obtained in mice for [89Zr]ZrFe-3 (green bars) and [89Zr]ZrFe-4 (red bars) in selected tissues (in units of percentage injected dose per gram of tissue, %ID g−1) at 24 h and 25 h post-injection, respectively. | ||
Another important bond fragmentation considered for [89Zr]ZrFe-2 is the hydrolytic ring-opening of β-CD (Fig. 8, disconnection 6). This degradation pathway is specific to the rotaxane design and leads to a dissociation of the radiolabelled [89Zr]ZrFe-[3]rotaxane ([89Zr]ZrFe-4) from the mAb. [89Zr]ZrFe-4 is a [3]rotaxane species composed of the DFO-guest-DFO axle and the two CB[6] macrocycles (Fig. 9A). The radiolabelling precursor, [3]pseudorotaxane 4, was synthesised and characterised by using 1-dimension and 2-dimensional multinuclear 1H- and 13C{1H}-NMR spectroscopy, high-resolution mass spectrometry (HRMS), and HPLC methods (ESI Fig. S7–S14†). In addition, the non-radiolabelled species natZr-4 was synthesised and characterised by HPLC and HRMS (ESI Fig. S15 and S16†), which confirmed that the [3]rotaxane can be synthesised by using standard metallation reactions. 89Zr-radiolabelling produced [89Zr]ZrFe-4 which was characterised by radio-iTLC (ESI Fig. S17†) and was administered to mice (n = 4) via intravenous teil-vein injections to measure the whole-body effective half-life and the biodistribution profile (Fig. 9B red circles, and Fig. 9C red bars, respectively). Whole-body excretion measurements of [89Zr]ZrFe-4 gave an t1/2(eff) value of 0.50 ± 0.1 h, confirming that this potential metabolite is rapidly eliminated from the mice. In addition, the biodistribution studies performed 25 h post-injection confirmed that [89Zr]ZrFe-4 displays similar clearance characteristics to [89Zr]ZrFe-3, with only minor retention of activity observed in the liver and kidneys, and <4% of the total injected activity retained in the whole animal (ESI Table S3†). We note that in both cases (Fig. 9B), a simple one-phase exponential model was used to fit the activity versus time data, giving a reasonably accurate estimation of the clearance profile for [89Zr]ZrFe-3 and [89Zr]ZrFe-4, and confirming that the behaviour of these ‘metabolites’ is completely different from that observed for the parent rotaxane-mAb, [89Zr]ZrFe-2. However, more detailed study of the elimination kinetics of these species is likely to require biphasic or multi-compartmental modelling in the future. As an example, a two-phase excretion model was applied to the data sets obtained for [89Zr]ZrFe-3 and [89Zr]ZrFe-4 (ESI Fig. S19†). In both cases, biphasic modelling also gave reasonable fits with the experimental data, resulting in correlation coefficient R2 values of 0.9831 for [89Zr]ZrFe-3 and 0.9449 for [89Zr]ZrFe-4. In this biphasic model, a fast elimination process is assigned to renal clearance which dominates, and a slower excretion pathway is tentatively assigned to a fraction of the activity (approximatively 15%) which is cleared via a hepatobiliary route. Further investigations are underway to explore the stability profile and metabolic fate of various rotaxane-based radiotracers.
Cyclodextrins (CDs) are cyclic oligosaccharides consisting of six or more α-1,4-linked glucose units (n ≥ 6), which can be degraded via enzyme-mediated hydrolysis to produce the corresponding maltooligosaccharides.49,50 Further hydrolysis results in smaller molecules, mainly maltotriose, maltose, and glucose, which are eventually absorbed and metabolised into CO2 and H2O.51–55 Known CD-hydrolysing enzymes include cyclomaltodextrinases and α-amylases.49,50 Chemical conditions, including low pH with Brønsted acids, or the use of Lewis acids, were also reported to catalyse the hydrolytic ring-opening of CDs.50 Cyclodextrins have found wide-spread application as excipients in pharmaceutical formulations of many drugs, and hence, the degradation of CDs in vivo has been a subject of long-standing interest.56–58 The three most common cyclodextrins, α-CD (n = 6 glucose monomers), β-CD (n = 7), and γ-CD (n = 8) were found to display different rates of degradation when exposed to gastric acid, or amylases from saliva or pancreatic tissue in vivo.55,59–62 While α-CD displays resistance and negligible degradation, and γ-CD is essentially completely digested in the small intestine, β-CD (as employed in our radiotracer design for [89Zr]ZrFe-2) follows an intermediate degradation pathway. Experiments reported that a fraction between 10% and 20% of administered β-CD is digested in the small intestine after 24 h.63,64 Major degradation of β-CD occurs in the microflora of the intestine or colon.55,64 β-CD also showed a trend towards renal enrichment,55 which could explain the higher accumulation of [89Zr]ZrFe-3 (the [4]rotaxane species that contains a β-CD unit) over [89Zr]ZrFe-4 (the [3]rotaxane model ‘metabolite’ without β-CD) in the kidneys (12.0 ± 2.5 %ID g−1 [n = 3] versus 5.4 ± 1.4 %ID g−1, [n = 4]). Additionally, we note that different biological behaviour and slower elimination rates were generally reported for the drug-β-CD inclusion complexes compared with free β-CD.55
Conclusion
Collectively, our experimental data from the radiosynthesis, PET imaging, biodistribution studies, and measurements of the whole-body excretion profiles of [89Zr]ZrFe-2, and several key ‘metabolites’ provide compelling evidence that mechanical bonding in the design of rotaxane-based radiotracers can be used to control the tumour-specificity, the organ distribution, and the mechanisms of metabolism and excretion. We believe that supramolecular chemistry harbours untapped potential in the development of molecular imaging agents and targeted therapeutics (particularly ADCs). Importantly, mechanical bonding offers new ways of tailoring the pharmacokinetics of radiotracers and drug molecules in vivo.Author contributions
FdO and JPH designed all experiments, analysed the data, and wrote the manuscript. FdO conducted all experiments including the synthesis, characterisation, radiochemistry, cellular studies, and imaging in vivo. JPH assisted with the radiochemistry and animal experiments and supervised the project.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Acknowledgements
JPH is supported by the Swiss National Science Foundation (SNSF Professorship PP00P2_163683 and PP00P2_190093) and the University of Zurich (UZH) for financial support. FdO received a Swiss Government Excellence Scholarship (ESKAS-Nr: 2017.0043). This project also received funding in part from the European Union's Horizon 2020 research and innovation programme/from the European Research Council (Grant No. 101001734, ERC-CoG-2020, PhotoPHARMA). We thank all members of the Holland group for helpful discussions and continuous support. We thank Prof. Chenfeng Ke, Prof. Michal Juricek and Prof. Oliver Zerbe for helpful discussions. We thank Prof. Steve Goldup for help and advice regarding the stereochemistry of rotaxanes.References
- M. Chomet, G. A. M. S. Van Dongen and D. J. Vugts, Bioconjugate Chem., 2021, 32, 1315–1330 CrossRef CAS PubMed.
- S. Parakh, S. T. Lee, H. K. Gan and A. M. Scott, Cancers, 2022, 14, 1454 CrossRef CAS PubMed.
- R. M. Sharkey, C. H. Chang, E. A. Rossi, W. J. McBride and D. M. Goldenberg, Tumor Biol., 2012, 33, 591–600 CrossRef CAS PubMed.
- R. M. Sharkey and D. M. Goldenberg, J. Nucl. Med., 2004, 46, 115S–127S Search PubMed.
- S. M. Larson, J. A. Carrasquillo, N. K. V. Cheung and O. W. Press, Nat. Rev. Cancer, 2015, 15, 347–360 CrossRef CAS PubMed.
- D. M. Goldenberg, C. H. Chang, E. A. Rossi, W. J. McBride and R. M. Sharkey, Theranostics, 2012, 2, 523–540 CrossRef CAS PubMed.
- M. Altai, R. Membreno, B. Cook, V. Tolmachev and B. M. Zeglis, J. Nucl. Med., 2017, 58, 1553–1559 CrossRef CAS PubMed.
- O. Keinänen, K. Fung, J. M. Brennan, N. Zia, M. Harris, E. Van Dam, C. Biggin, A. Hedt, J. Stoner, P. S. Donnelly, J. S. Lewis, B. M. Zeglis and J. K. Barton, PNAS, 2020, 117, 28316–28327 CrossRef PubMed.
- S. M. Cheal, H. Xu, H. F. Guo, P. B. Zanzonico, S. M. Larson and N. K. Cheung, Mol. Cancer Ther., 2014, 13, 1803–1812 CrossRef CAS PubMed.
- S. M. Cheal, H. Xu, H. fen Guo, S. gyu Lee, B. Punzalan, S. Chalasani, E. K. Fung, A. Jungbluth, P. B. Zanzonico, J. A. Carrasquillo, J. O'Donoghue, P. M. Smith-Jones, K. D. Wittrup, N. K. V. Cheung and S. M. Larson, Eur. J. Nucl. Med. Mol. Imaging, 2016, 43, 925–937 CrossRef CAS PubMed.
- S. M. Cheal, M. R. McDevitt, B. H. Santich, M. Patel, G. Yang, E. K. Fung, D. R. Veach, M. Bell, A. Ahad, D. B. Vargas, B. Punzalan, N. V. Kishore Pillarsetty, H. Xu, H. F. Guo, S. Monette, A. O. Michel, A. Piersigilli, D. A. Scheinberg, O. Ouerfelli, N. K. V. Cheung and S. M. Larson, Theranostics, 2020, 10, 11359–11375 CrossRef CAS PubMed.
- M. Patra, K. Zarschler, H. J. Pietzsch, H. Stephan and G. Gasser, Chem. Soc. Rev., 2016, 45, 6415–6431 RSC.
- A. M. Wu and P. D. Senter, Nat. Biotechnol., 2005, 23, 1137–1146 CrossRef CAS PubMed.
- E. L. Sievers and P. D. Senter, Annu. Rev. Med., 2013, 64, 15–29 CrossRef CAS PubMed.
- R. Alberto, G. Bergamaschi, H. Braband, T. Fox and V. Amendola, Angew. Chem., Int. Ed., 2012, 51, 9772–9776 CrossRef CAS PubMed.
- B. P. Burke, W. Grantham, M. J. Burke, G. S. Nichol, D. Roberts, I. Renard, R. Hargreaves, C. Cawthorne, S. J. Archibald and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 16877–16881 CrossRef CAS PubMed.
- B. Woods, R. D. M. Silva, C. Schmidt, D. Wragg, M. Cavaco, V. Neves, V. F. C. Ferreira, L. Gano, T. S. Morais, F. Mendes, J. D. G. Correia and A. Casini, Bioconjugate Chem., 2021, 32, 1399–1408 CrossRef CAS PubMed.
- E. Arunkumar, N. Fu and B. D. Smith, Chem. - Eur. J., 2006, 12, 4684–4690 CrossRef CAS PubMed.
- J. J. Gassensmith, J. M. Baumes and B. D. Smith, Chem. Commun., 2009, 6329–6338 RSC.
- C. Zhai, C. L. Schreiber, S. Padilla-coley, A. G. Oliver and B. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 23740–23747 CrossRef CAS PubMed.
- E. A. Katayev, G. V Kolesnikov and J. L. Sessler, Chem. Soc. Rev., 2009, 38, 1572–1586 RSC.
- J. W. Fredy, J. Scelle, A. Guenet, E. Morel, S. Adam De Beaumais, M. Ménand, V. Marvaud, C. S. Bonnet, E. Tõth, M. Sollogoub, G. Vives and B. Hasenknopf, Chem. - Eur. J., 2014, 20, 10915–10920 CrossRef CAS PubMed.
- J. W. Fredy, J. Scelle, G. Ramniceanu, B. T. Doan, C. S. Bonnet, É. Tóth, M. Ménand, M. Sollogoub, G. Vives and B. Hasenknopf, Org. Lett., 2017, 19, 1136–1139 CrossRef CAS PubMed.
- J. Riebe and J. Niemeyer, Eur. J. Org. Chem., 2021, 2021, 5106–5116 CrossRef CAS.
- H. V Schröder, Y. Zhang and A. J. Link, Nat. Chem., 2021, 13, 850–857 CrossRef PubMed.
- F. d'Orchymont and J. P. Holland, Angew. Chem., Int. Ed., 2022, 61, e2022040 CrossRef PubMed.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, J. Org. Chem., 1983, 48, 3619–3620 CrossRef CAS.
- C. Ke, R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 381–387 CrossRef CAS PubMed.
- X. Hou, C. Ke and J. Fraser Stoddart, Chem. Soc. Rev., 2016, 45, 3766–3780 RSC.
- M. Merchant, X. Ma, H. R. Maun, Z. Zheng, J. Peng, M. Romero, A. Huang, M. A. Starovasnik, R. A. Lazarus and D. G. Yansura, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(32), E2987–E2996 CrossRef CAS PubMed.
- H. Xiang, B. C. Bender, A. E. Reyes, M. Merchant, N. L. Shasha Jumbe, M. Romero, T. Davancaze, I. Nijem, E. Mai, J. Young, A. Peterson and L. A. Damico-Beyer, Clin. Cancer Res., 2013, 19, 5068–5078 CrossRef CAS PubMed.
- A. Guillou and D. F. Earley, Nat. Protoc., 2020, 15, 3579–3594 CrossRef CAS PubMed.
- D. F. Earley, A. Guillou, S. Klingler, R. Fay, M. Gut, F. d'Orchymont, S. Behmaneshfar, L. Reichert and J. P. Holland, JACS Au, 2022, 2(3), 646–664 CrossRef CAS PubMed.
- G. W. Preston and A. J. Wilson, Chem. Soc. Rev., 2013, 42, 3289–3301 RSC.
- N. P. Gritsan and M. S. Platz, Chem. Rev., 2006, 106, 3844–3867 CrossRef CAS PubMed.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- C. J. Bruns and J. F. Stoddart, The nature of the mechanical bond, 2016 Search PubMed.
- E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC.
- J. R. J. Maynard and S. M. Goldup, Chem, 2020, 6, 1914–1932 CAS.
- T. Lindmo, E. Boven, F. Cuttitta, J. Fedorko and P. A. Bunn, J. Immunol. Methods, 1984, 72, 77–89 CrossRef CAS PubMed.
- R. Fay, M. Gut and J. P. Holland, Bioconjugate Chem., 2019, 30, 1814–1820 CrossRef CAS PubMed.
- S. Klingler, R. Fay, J. P. Holland and S. Klingler, J. Nucl. Med., 2020, 61, 1072–1078 CrossRef CAS PubMed.
- A. Guillou, D. F. Earley and J. P. Holland, Chem. - Eur. J., 2020, 26, 7185–7189 CrossRef CAS PubMed.
- M. J. W. D. Vosjan, L. R. Perk, G. W. M. Visser, M. Budde, P. Jurek, G. E. Kiefer and G. A. M. S. Van Dongen, Nat. Protoc., 2010, 5, 739–743 CrossRef CAS PubMed.
- F. Bensch, M. M. Smeenk, S. C. Van Es, J. R. De Jong, C. P. Schröder, S. F. Oosting, M. N. L. Hooge and C. W. M. D. H. Van Oordt, Theranostics, 2018, 8, 4295–4304 CrossRef CAS PubMed.
- J. P. Holland and N. Vasdev, Dalton Trans., 2014, 43, 9872–9884 RSC.
- J. P. Holland, Inorg. Chem., 2020, 59, 2070–2082 CrossRef CAS PubMed.
- A. Guillou, D. F. Earley, S. Klingler, E. Nisli, L. J. Nüesch, R. Fay and J. P. Holland, Bioconjugate Chem., 2021, 32, 1263–1275 CrossRef CAS PubMed.
- B. C. Saha and J. G. Zeikus, Starch/Staerke, 1992, 44, 312–315 CrossRef CAS.
- M. Pélingre, D. S.-E. Koffi Teki, J. El-Abid, V. Chagnault, J. Kovensky and V. Bonnet, Organics, 2021, 2, 287–305 CrossRef.
- J. Szejtli, A. Gerlóczy and A. Fónagy, Arzneimittelforschung, 1980, 30, 808–810 CAS.
- A. Gerlóczy, A. Fónagy, P. Keresztes, L. Perlaky and J. Szejtli, Arzneimittelforschung, 1985, 35, 1042–1047 Search PubMed.
- G. H. Andersen, F. M. Robbins, F. J. Domingues, ∼ R. G. Moores and P. B. Brigham, Toxicol. Appl. Pharmacol., 1963, 5, 257–266 CrossRef CAS PubMed.
- M. Abdalla, B. Jiang, H. A. M. Hassanin, L. Zheng and J. Chen, Enzyme Microb. Technol., 2021, 149, 109847 CrossRef CAS PubMed.
- K. Mu, K. Jiang, Y. Wang, Z. Zhao, S. Cang, K. Bi, Q. Li and R. Liu, Molecules, 2022, 27(3), 1138 CrossRef CAS PubMed.
- T. Loftsson and M. E. Brewster, Am. J. Drug Delivery, 2004, 2, 261–275 CrossRef CAS.
- C. Muankaew and T. Loftsson, Basic Clin. Pharmacol. Toxicol., 2018, 122, 46–55 CrossRef CAS PubMed.
- M. E. Davis and M. E. Brewster, Nat. Rev. Drug Discovery, 2004, 3, 1023–1035 CrossRef CAS PubMed.
- I. Jodal, L. Kandra, J. Harangi, P. Nanasi, Debrecen and J. Szejtli, Starch/Staerke, 1984, 4, 140–143 Search PubMed.
- J. J. Marshall and I. Miwa, Biochim. Biophys. Acta, 1981, 661, 142–147 CrossRef CAS.
- H. Kondo, H. Nakatani and K. Hiromi, Carbohydr. Res., 1990, 204, 207–213 CrossRef CAS PubMed.
- A. T. H. J. De Bie, B. Van Ommen and A. Bär, Regul. Toxicol. Pharmacol., 1998, 27, 150–158 CrossRef CAS PubMed.
- R. N. Antenucci and J. K. Palmer, J. Agric. Food Chem., 1984, 32, 1316–1321 CrossRef CAS.
- B. Flourié, C. Molis, L. Achour, H. Dupas, C. Hatat and J. C. Rambaud, J. Nutr., 1993, 123, 676–680 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary information (PDF) is available and contains experimental details, high-resolution mass spectrometry data and HPLC chromatograms for all relevant compounds, as well as additional data from the radiochemistry and in vivo experiments. See: https://doi.org/10.1039/d2sc03928a |
| This journal is © The Royal Society of Chemistry 2022 |