
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bis-NHC–CAAC dimer derived dicationic diradical†‡
Mithilesh Kumar
Nayak
a,
Pallavi
Sarkar
b,
Benedict J.
Elvers
 c,
Sakshi
Mehta
d,
Fangyuan
Zhang
e,
Nicolas
Chrysochos
a,
Ivo
Krummenacher
f,
Thangavel
Vijayakanth
g,
Ramakirushnan Suriya
Narayanan
a,
Ramapada
Dolai
a,
Biswarup
Roy
a,
Vishal
Malik
a,
Hemant
Rawat
a,
Abhishake
Mondal
*d,
Ramamoorthy
Boomishankar
*g,
Swapan K.
Pati
*b,
Holger
Braunschweig
*f,
Carola
Schulzke
*c,
Prince
Ravat
*e and
Anukul
Jana
*a
c,
Sakshi
Mehta
d,
Fangyuan
Zhang
e,
Nicolas
Chrysochos
a,
Ivo
Krummenacher
f,
Thangavel
Vijayakanth
g,
Ramakirushnan Suriya
Narayanan
a,
Ramapada
Dolai
a,
Biswarup
Roy
a,
Vishal
Malik
a,
Hemant
Rawat
a,
Abhishake
Mondal
*d,
Ramamoorthy
Boomishankar
*g,
Swapan K.
Pati
*b,
Holger
Braunschweig
*f,
Carola
Schulzke
*c,
Prince
Ravat
*e and
Anukul
Jana
*a
aTata Institute of Fundamental Research Hyderabad, Gopanpally, Hyderabad-500107, India. E-mail: ajana@tifrh.res.in
bTheoretical Sciences Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore-560064, India. E-mail: pati@jncasr.ac.in
cInstitut für Biochemie, Universität Greifswald, Felix-Hausdorff-Straße 4, D-17489, Greifswald, Germany. E-mail: carola.schulzke@uni-greifswald.de
dSolid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore, 560012, India. E-mail: mondal@iisc.ac.in
eInstitute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: princekumar.ravat@uni-wuerzburg.de
fInstitute of Inorganic Chemistry and Institute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
gDepartment of Chemistry, Indian Institute of Science Education and Research Pune, Dr Homi Bhabha Road, Pune 411008, India. E-mail: boomi@iiserpune.ac.in
First published on 21st September 2022
Abstract
The isolation of carbon-centered diradicals is always challenging due to synthetic difficulties and their limited stability. Herein we report the synthesis of a trans-1,4-cyclohexylene bridged bis-NHC–CAAC dimer derived thermally stable dicationic diradical. The diradical character of this compound was confirmed by EPR spectroscopy. The variable temperature EPR study suggests the singlet state to be marginally more stable than the triplet state (2J = −5.5 cm−1 (ΔEST = 0.065 kJ mol−1)). The presence of the trans-1,4-cyclohexylene bridge is instrumental for the successful isolation of this dicationic diradical. Notably, in the case of ethylene or propylene bridged bis-NHC–CAAC dimers, the corresponding dicationic diradicals are transient and rearrange to hydrogen abstracted products.
Introduction
Diradical compounds are of quite substantial scientific importance due to their versatile applications in various research fields, ranging from synthetic chemistry to modern chemical physics.1 In comparison to carbon-centered diradicals, the heteroatom-centered diradicals with unpaired electrons residing on nitrogen or oxygen centres are quite well studied.2 This is due to the fact that carbon-centered diradicals are generally much less stable.3,4 The design of synthetic pathways and the subsequent successful isolation of carbon-centered diradicals are therefore always challenging. Strategically, the synthesis of carbon-centered diradicals can be facilitated by applying electron transfer processes to redox-active organic molecules. In recent years, several such compounds have moved into the focus of interest: among them the electron donor tetrathiafulvalene (TTF) I5 and the electron acceptor N,N′-dimethyl-4,4′-bipyridinium dication (BIPY) II6 are of particular interest (Scheme 1). These comprehensively studied species I and II exhibit two reversible one-electron redox processes each and exist in three isolable oxidation states. Considering this kind of redox property, the synthesis of dicationic biradicals/diradicals involving two units of TTF as well as BIPY have been known such as III7 and IV.8 Moreover, compounds based on two or more TTF and/or BIPY units were intensely investigated for properties based on this notable redox-activity and for various applications including those as organic semiconductors, electrochromic devices, and molecular machines.9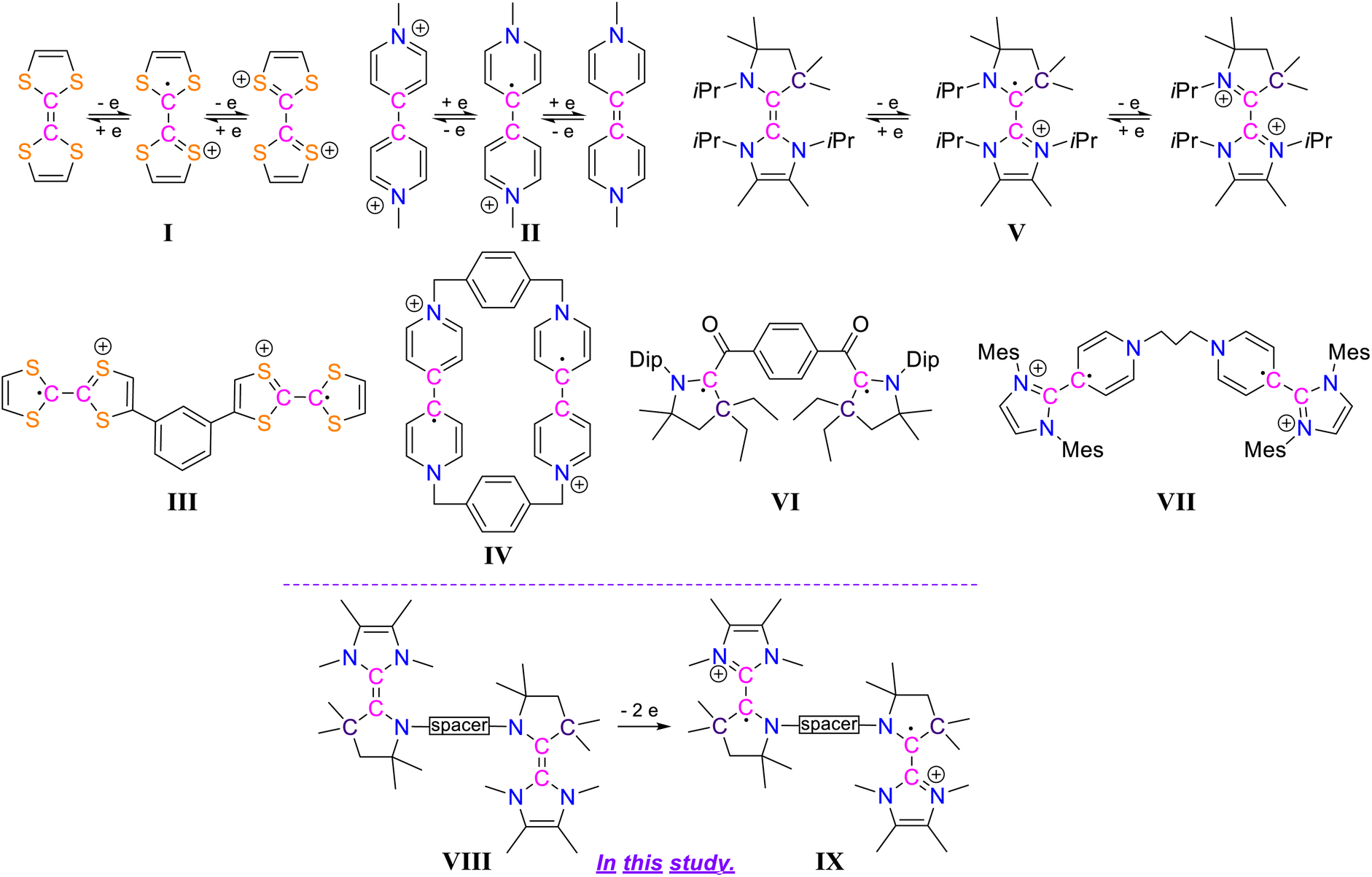 | ||
| Scheme 1 Chemical structures of I–IX. | ||
Electronically related to TTF, in recent years several carbene-based electron-rich alkenes have been synthesized,10–14 such as the NHC-CAAC dimer V11 (Scheme 1). These compounds exhibit two reversible one-electron redox processes and all three oxidation states can be isolated: neutral, radical cation, and dication, similar to I and II. In the case of the radical cation derived from NHC–CAAC dimers, the respective spin density resides mostly on the carbenic carbon of the CAAC-scaffolds.10 In recent years CAAC-scaffolds have become known to stabilize various carbon-centered based open-shell compounds.15 Among them the isolation of biradical VI16 (Scheme 1) demonstrates the potential of CAAC-scaffolds to act as paramagnetic building blocks. Also there was a report of pyridinium–NHC derived diradical VII (Scheme 1).13 Thinking strategically, the generation of radical cation based biradicals/diradicals should, hence, be possible by developing a new class of compounds bearing two NHC-CAAC dimer motifs, i.e. species VIII (Scheme 1).
Herein we report the synthesis of the trans-1,4-cyclohexylene bridged bis-NHC–CAAC dimer VIII derived dicationic diradical IX (Scheme 1). The presence of the trans-1,4-cyclohexylene bridge, which is tethered to the N-atoms of the two CAAC scaffolds, is crucial for the successful isolation of the dicationic diradical. In the case of an ethylene or a propylene bridge being used for the bis-NHC–CAAC dimers, their dicationic diradicals were only transiently formed and rearranged to the hydrogen abstracted products.
Results and discussion
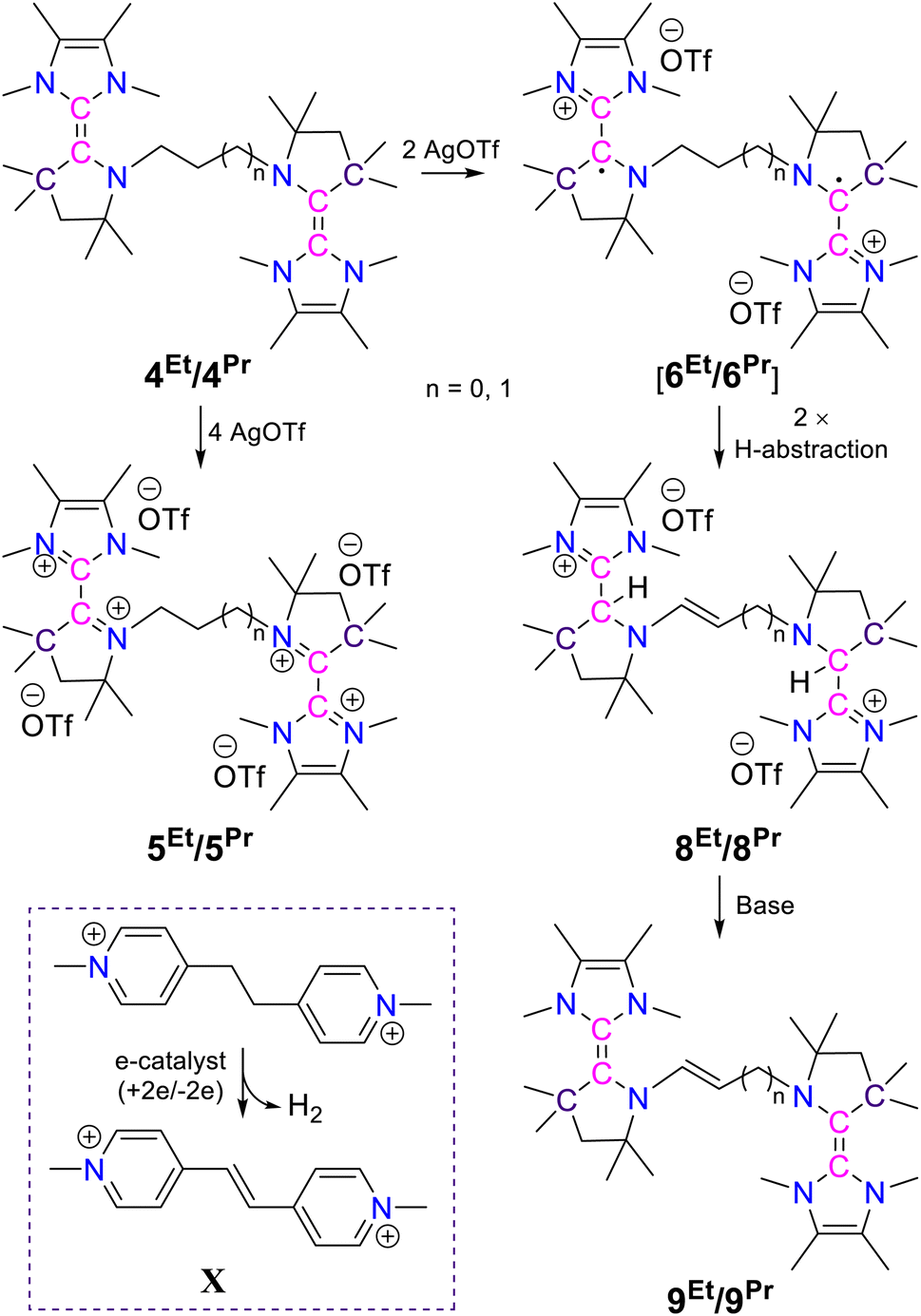
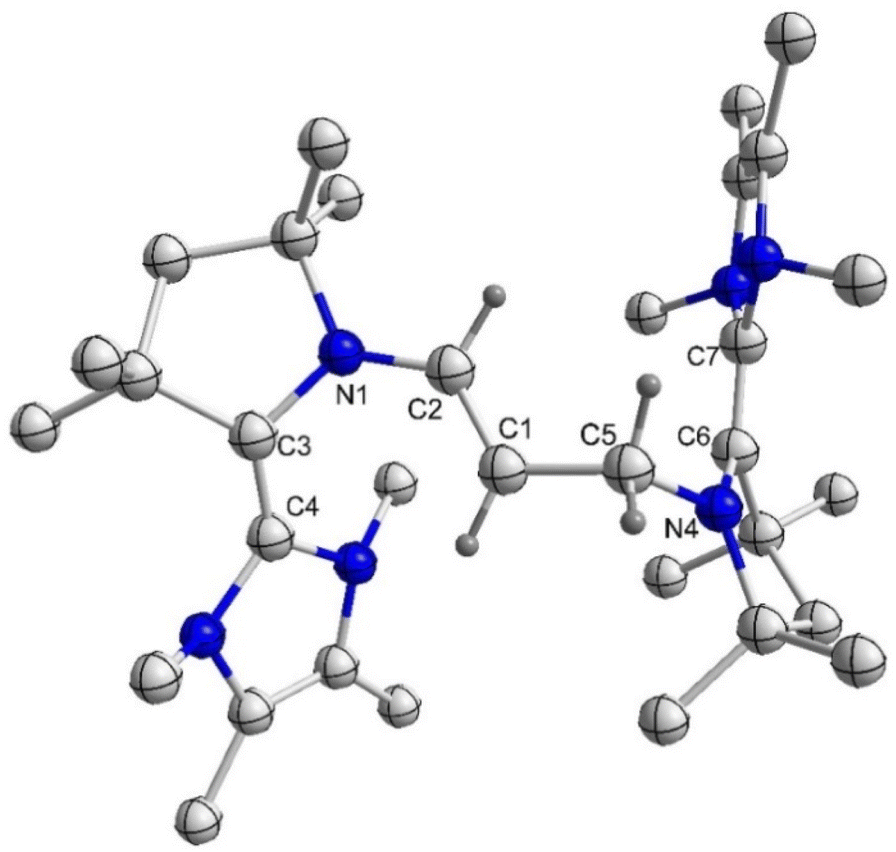
First, in order to obtain the trans-1,4-cyclohexylene bridged bis-NHC–CAAC dimer, 1Cy was reacted with 217 giving 3Cy in a 90% yield (Scheme 2).18 The reaction proceeds either through the nucleophilic addition of NHC to the pyrrolinium cation or acid–base reaction under the formation of bis-CAAC followed by C–H insertion.13,19 Recently, a related bis-CAAC with trans-1,4-cyclohexylene framework was reported.20 The subsequent reaction of 3Cy with two equivalents of LDA leads to the formation of the light yellow colored bis-NHC–CAAC dimer 4Cy in a 83% yield (Scheme 2).18 Compound 4Cy is inherently prone to undergo a four-electron oxidation, which was, consequently, tested. The reaction with four equivalents of AgOTf leads to the formation of the tetracation 5Cy in a 89% yield and this redox transition is reversible (Scheme 2).18 Subsequently, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of 4Cy with 5Cy yielded thermally stable 6Cy as a dark red crystalline solid almost quantitatively. We also observed the formation of 6Cy from the reaction of 4Cy with two equivalents of AgOTf, but the associated purification step is essentially unmanageable. Formation of 6Cy was confirmed by its solid-state molecular structure determination (Fig. 1). The C2–C3/C5–C6 bond lengths between the NHC and CAAC moieties of 6Cy are 1.439(7)/1.431(7) Å, i.e. close to the related radical cation of the NHC–CAAC dimer V (1.444 Å) and longer than that of the neutral species V (1.358 Å).11
:1 reaction of 4Cy with 5Cy yielded thermally stable 6Cy as a dark red crystalline solid almost quantitatively. We also observed the formation of 6Cy from the reaction of 4Cy with two equivalents of AgOTf, but the associated purification step is essentially unmanageable. Formation of 6Cy was confirmed by its solid-state molecular structure determination (Fig. 1). The C2–C3/C5–C6 bond lengths between the NHC and CAAC moieties of 6Cy are 1.439(7)/1.431(7) Å, i.e. close to the related radical cation of the NHC–CAAC dimer V (1.444 Å) and longer than that of the neutral species V (1.358 Å).11
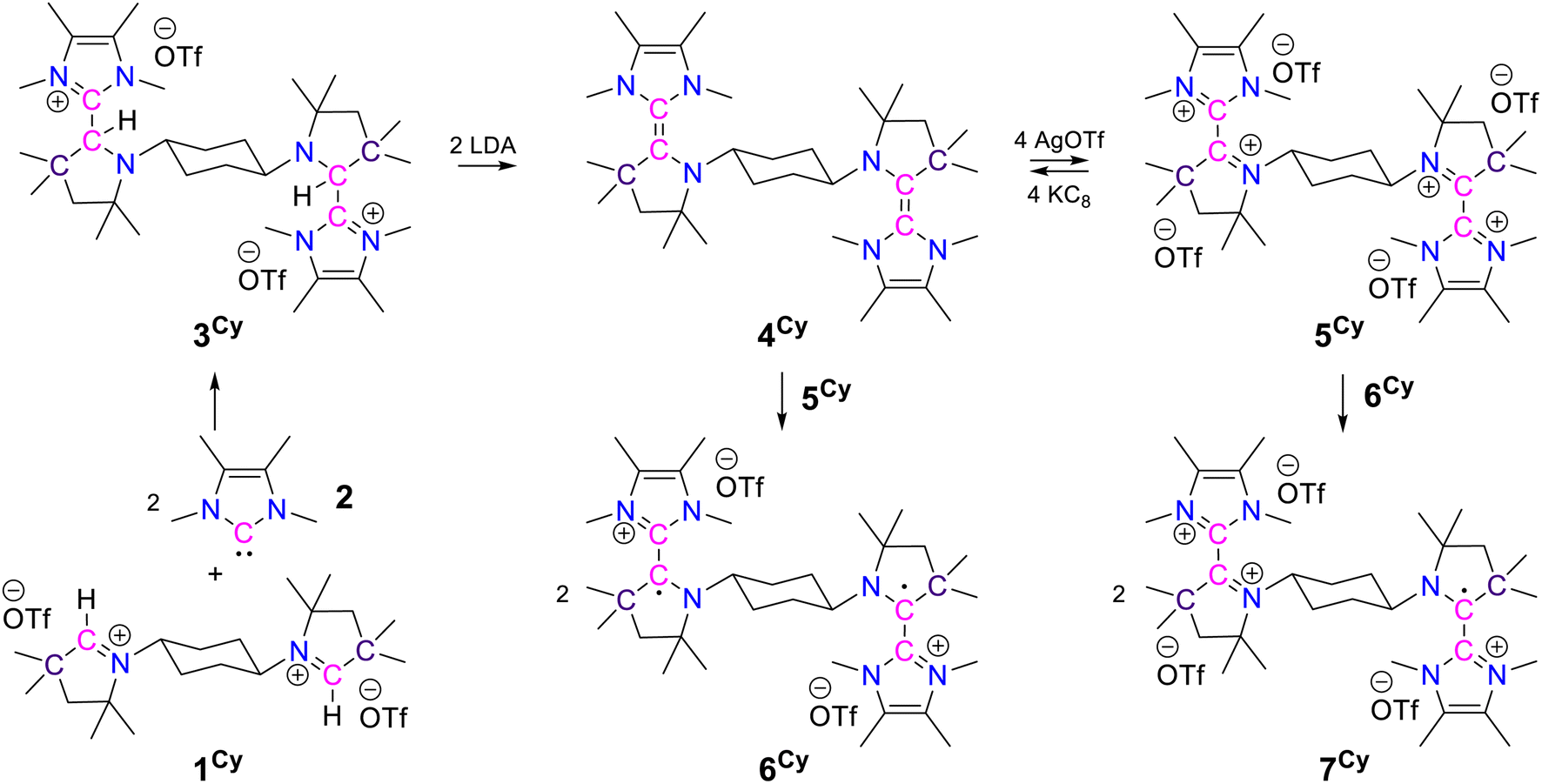 | ||
| Scheme 2 Synthesis of 3Cy, 4Cy, 5Cy, 6Cy, and 7Cy. | ||
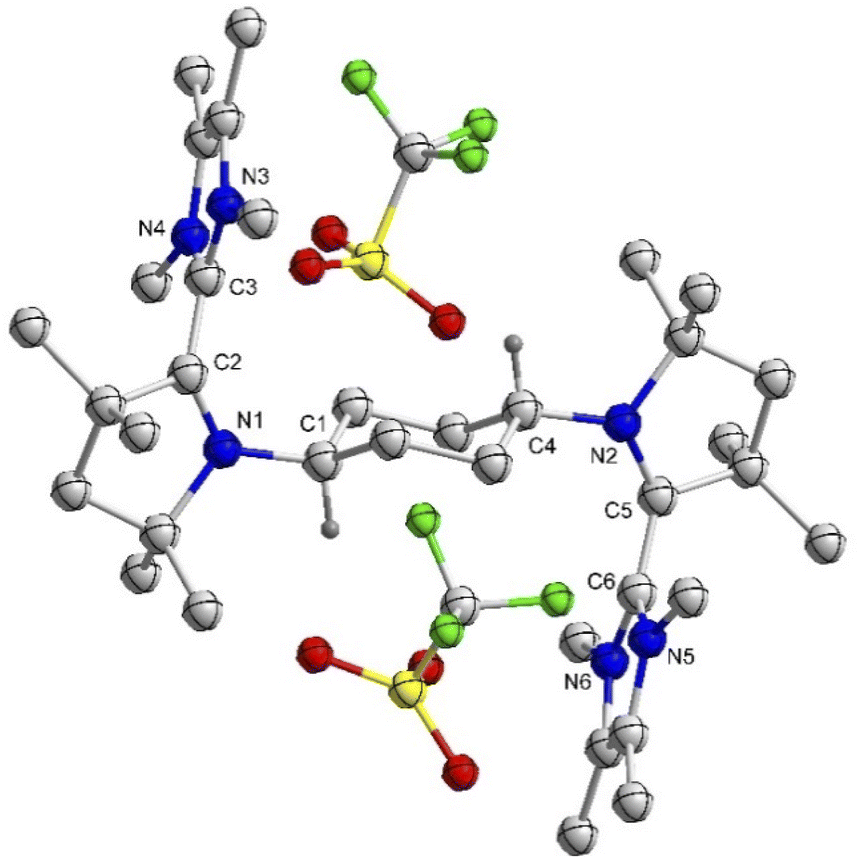 | ||
| Fig. 1 Molecular structure of 6Cy in the solid state with thermal ellipsoids at the 50% probability level. All hydrogen atoms except on C1 and C4 are omitted for clarity. Selected bond lengths (Å) and bond angles (°): N1–C2 1.380(6), C2–C3 1.439(7), N2–C5 1.366(6), C5–C6 1.431(7); N1–C2–C3 122.3(4), N2–C5–C6 122.5(4). | ||
The spin density plot of 6Cy places the maximum spin-density on the carbenic carbon atoms of the CAAC-scaffolds (Fig. S109‡). In the UV/Vis spectrum, compound 6Cy exhibits the longest wavelength absorbance at λmax = 455 nm. TD-DFT calculations suggest that the lowest-energy absorbance band arises mainly from the HOMO-α → LUMO-α + 1 transition, while the HOMO-α → LUMO-α and HOMO-β → LUMO-β transitions exhibit very weak oscillator strengths (Fig. S108 and Table S17‡).
EPR spectroscopy of 6Cy in frozen 1:1 toluene/acetonitrile solution confirmed its diradical nature with a weak half-field signal (Fig. 2 – left), which is readily detectable in the temperature range of 10 to 140 K (Fig. 2 – middle). Fitting of the temperature-dependent double-integral intensity to the Bleaney-Bowers model gives a small singlet-triplet gap of 2J = −5.5 cm−1 (ΔEST = 0.065 kJ mol−1) (Fig. S83‡). Similarly, weak exchange couplings were also observed for diradicals III,7VI,16 and VII.13 The small singlet-triplet energy gap was confirmed by a SQUID magnetometer measurement of 6Cy as the high-temperature Curie Weiss fitting provides a Weiss constant of θ = −3.065 K, corresponding to an energy gap of 0.025 kJ mol−1 (Fig. S88‡). Using the relative intensity of the half-field (ΔMS = ±2) to the allowed transitions (ΔMS = ±1), the distance r between the unpaired spins can be estimated to be ca. 8.8 Å,21 relatively similar, hence, to the distances between the two carbenic carbon atoms of CAAC-scaffolds in 6Cy (7.364 Å).
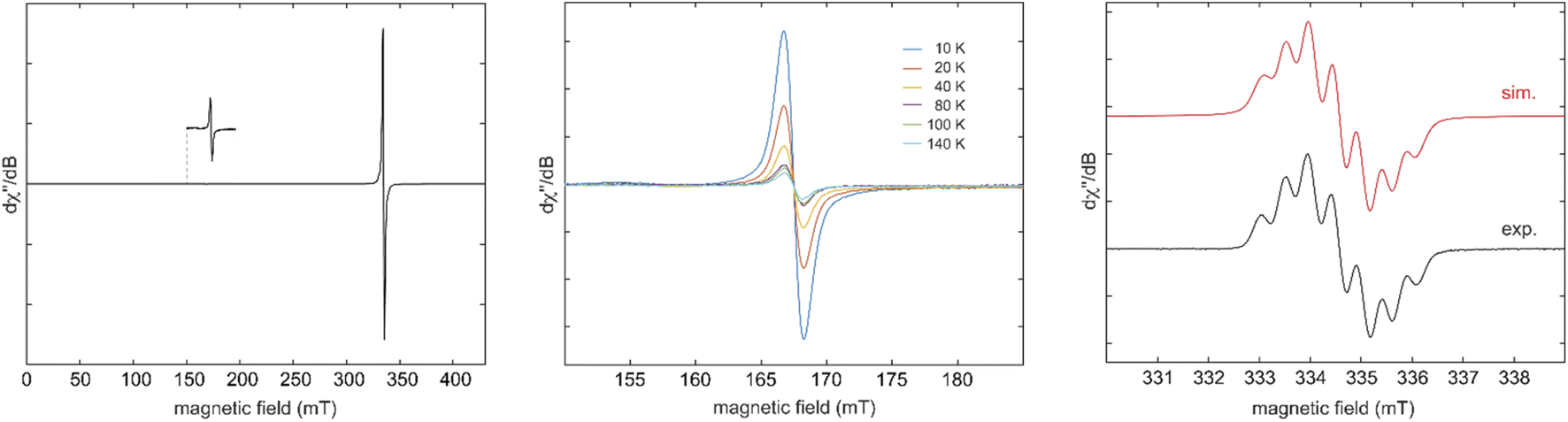 | ||
| Fig. 2 CW EPR spectrum of 6Cy in a 1:1 toluene/acetonitrile mixture at 40 K (left), temperature dependence of the half-field transitions between 10 and 140 K (middle), and experimental (black) and simulated (red) EPR spectra of 7Cy in acetonitrile (right). Best-fit simulation parameters: giso = 2.0029, a(14N) = 14.2 MHz (1N), 11.8 MHz (2N), and a(1H) = 3.2 MHz (6H). | ||
Quantum chemical calculations were performed to verify the nature of the ground state and estimate the energy gap between the singlet and triplet states of 6Cy. Initially, the energies of singlet and triplet states were calculated by DFT using four different functionals ωB97XD, MN12SX, B3LYP-D3, and PBE0 with the Def2SVP basis-set (Table S16‡). Calculations provided values of ΔEST in the range of −0.015 to 0.10 kJ mol−1, which is in accordance with the small ΔEST obtained from EPR experiments. The negative value of the exchange coupling constant J from EPR studies indicates antiferromagnetic coupling between two-spin centers and, hence, a singlet ground state. Interestingly, while the ωB97XD functional gave a positive value for J, MN12SX, B3LYP-D3, and PBE0 yielded negative values corroborating the EPR results. This may be based on the fact that the ΔEST is very small. Multi-reference theoretical methods are known to precisely describe the energies of singlet and triplet states. Indeed the multi-reference CASSCF(2,2)/NEVPT2/def2-TZVP approach22 verified the singlet ground states and provided a much more accurate adiabatic ΔEST of −0.037 kJ mol−1.
Subsequently, we have synthesized the radical-trication 7Cy by the 1:1 reaction of 5Cy with 6Cy in a 88% yield (Scheme 2).18 The comproportionation reaction between 6Cy and 5Cy gives rise to an EPR signal centered around g = 2.0029, indicating the formation of radical-trication 7Cy (Fig. 2 – right). The single crystal X-ray structural analysis (Fig. 3) identifies two different C–C bond lengths between the NHC and CAAC moieties C2–C3 1.459(4)/C5–C6 1.488(4) Å, which suggests that one NHC–CAAC unit is oxidized twice and the other one is oxidized only once. This is further supported by the C–N bond distances of the two CAAC-scaffolds (C2–N1 1.361(4)/C5–N2 1.277(3) Å).
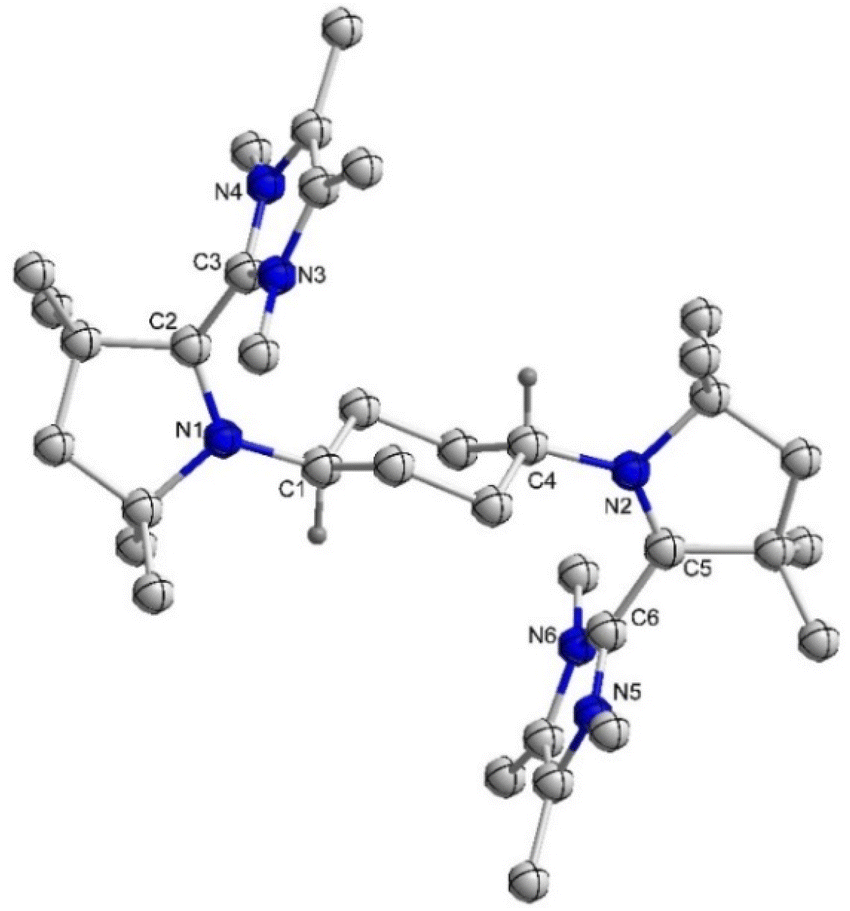 | ||
| Fig. 3 Molecular structure of 7Cy in the solid state with thermal ellipsoids at the 50% probability level. All hydrogen atoms except on C1 and C4 and three triflate anions are omitted for clarity. Selected bond lengths (Å) and bond angles (°): N1–C2 1.361(4), C2–C3 1.459(4), N2–C5 1.277(3), C5–C6 1.488(4); N1–C2–C3 122.0(3), N2–C5–C6 125.2(2). | ||
To address the influence of the alkylene bridge, which is tethered to the N-atoms of the CAAC scaffolds, we have also synthesized and studied the ethylene and the propylene bridged bis-NHC–CAAC dimers 4Et/4Pr. Both of these bis-NHC–CAAC dimers 4Et/4Pr undergo four-electron oxidation with AgOTf under the formation of 5Et/5Pr in good yields (5Et = 76% and 5Pr = 89%) (Scheme 3).18 However, in contrast to trans-1,4-cyclohexylene bridged 4Cy, the ethylene/propylene bridged bis-NHC–CAAC dimers 4Et/4Pr upon two-electron oxidation do not facilitate the isolation of bis-radical cations 6Et/6Pr. Instead these transient species proceed to double H-abstraction23 under the formation of ethenylene/propenylene-bridged dications 8Et/8Pr (Scheme 3).18 The formation of 8Et/8Pr was also realized by the 1:1 reaction of 4Et/4Pr and 5Et/5Pr.Moreover, the formation of 8Et has been confirmed by the solid-state molecular structure determination (Fig. 4). Compound 8Et can be classified as disubstituted E-diamino alkene and exhibits electron-rich nature. Its oxidation with AgOTf leads to the partial formation of corresponding radical trication, which has been confirmed by solid-state structure determination (Fig. S77‡) and EPR spectroscopy (Fig. S87‡). Quantitative EPR measurement indicates a radical concentration of ca. 54% in the crystalline crude products considering TEMPO as standard.
 | ||
| Scheme 3 Synthesis of 5Et/5Pr, 8Et/8Pr, and 9Et/9Pr (Insert: Schematic Presentation of X). | ||
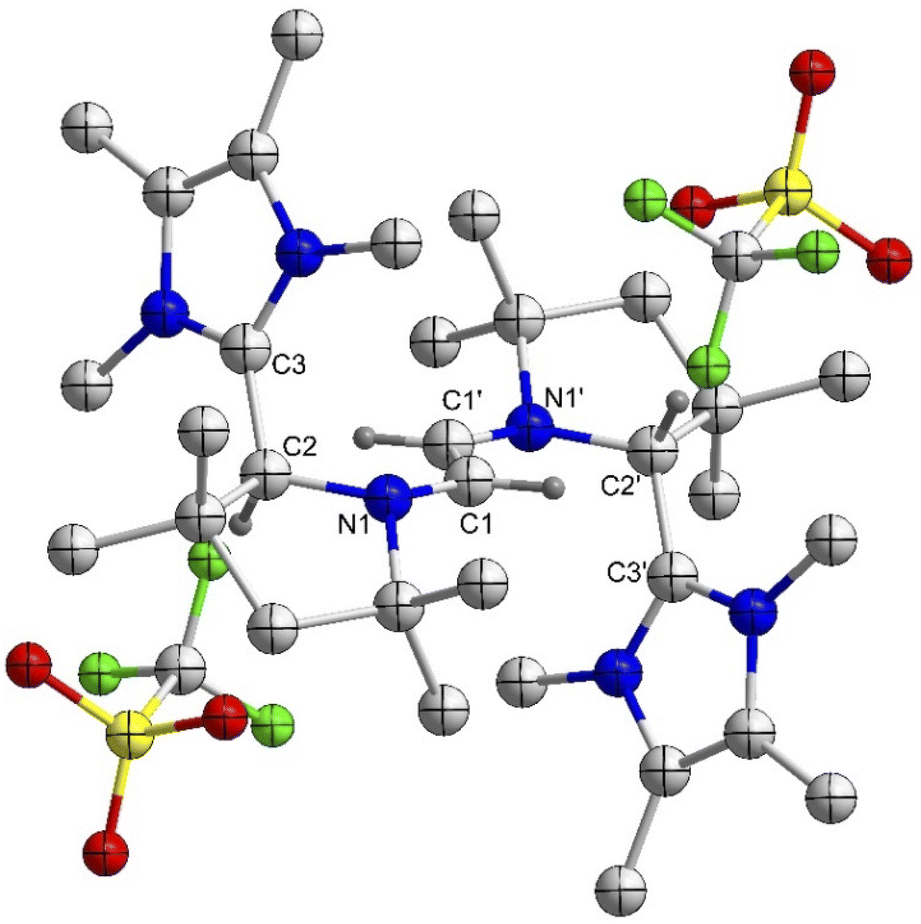 | ||
| Fig. 4 Molecular structure of 8Et in the solid state with thermal ellipsoids at the 50% probability level. All hydrogen atoms except on C1, C1′, C2, and C2′ are omitted for clarity. Selected bond lengths (Å) and bond angles (°): N1–C2 1.457(2), C2–C3 1.505(3), C1–C1′ 1.324 (4); N1–C2–C3 111.82(15), N1–C1–C1′ 127.4(3). | ||
These observations confirm the formation of bis-radical cation intermediates 6Et/6Pr which have very limited stability compared to 6Cy, emphasizing the critically important influence of the alkylene bridge. In case of the ethylene/propylene bridge, most likely the facile C–C double bond formation after the double H-abstraction is responsible for the limited stability of their dicationic diradicals. Such type of facile C–C double bond formation at the trans-1,4-cyclohexylene bridge is impaired by conformational restriction. As a result, the trans-1,4-cyclohexylene bridge is instrumental in stabilizing the dicationic diradical.
These newly synthesized bis-NHC–CAAC dimers 4Cy, 4Et, and 4Pr react with ([nBu4N][PF6]) (Fig. S6‡) and therefore we were not able to obtain cyclic voltammetric results. Subsequently, we carried out the cyclic voltammetric studies of the corresponding tetracations 5Cy, 5Et, and 5Pr along with dicationic diradical 6Cy and radical tri-cation 7Cy in acetonitrile (0.1 M [nBu4N][PF6]). Compound 5Cy exhibits two cathodic reversible waves at Epk = −1.07 V (with a shoulder at Epk = −0.95 V) and Epk = −1.66 V (at 100 mV s−1 scan rate) likely associated with the formation of dicationic diradical 6Cy and bis-NHC–CAAC dimer 7Cy (Fig. 5). The first cathodic wave is closely associated with two one-electron reduction processes and the second cathodic wave is for a two-electron reduction process, which was confirmed by a differential pulse voltammetry (DPV) study (Fig. S93‡). Considering the relative current height of the backward oxidation process from the measurements at 100 mV s−1 and 1000 mV s−1 scan rates, confirms the instability of the generated bis-alkene towards the electrolyte [nBu4N][PF6]. The cyclic voltammetric studies of 6Cy and 7Cy exhibit the redox processes as expected and along the line of 5Cy (Fig. S94–S99‡). On the other hand, the scan rate dependent cyclic voltammetry along with differential pulse voltammetry (DPV) studies of 5Et and 5Pr indicate that follow up chemical reactions occurred after the two-electron reduction (Fig. S100–S107‡) which is in accordance with the chemical reduction experiments. The first reduction event in both cases 5Et and 5Pr is reversible, though (Fig. S102‡).
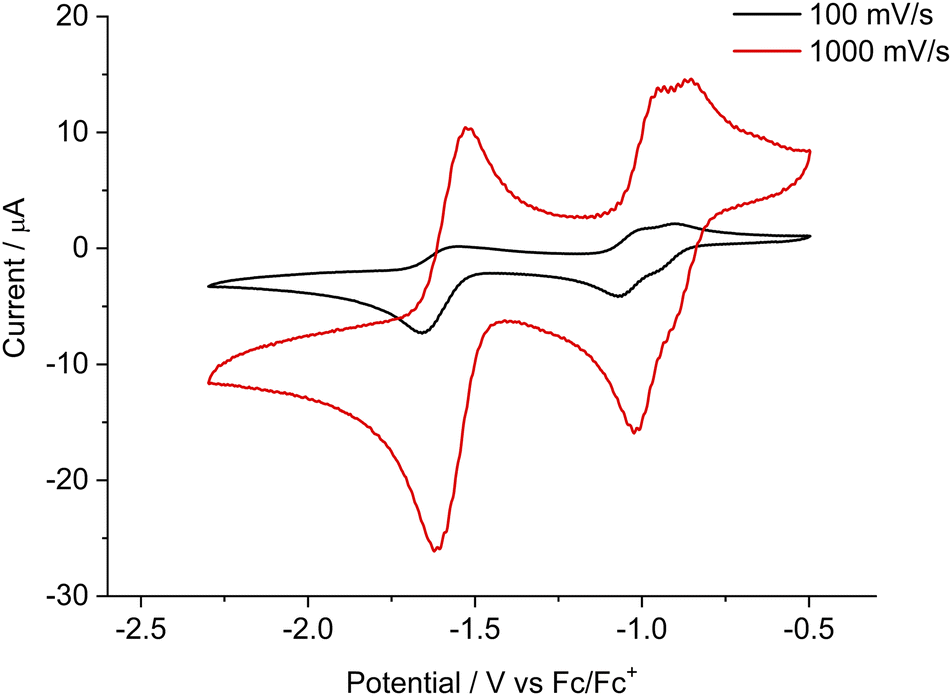 | ||
| Fig. 5 Cyclic voltammetry plots for 5Cy at a scan rate of 100 mV s−1 and 1000 mV s−1 in acetonitrile (0.1 M [nBu4N][PF6]). | ||
Lastly it was shown that 8Et/8Pr can be converted to the crystalline N,N′-ethenylene/propenylene-bridged bis-NHC–CAAC dimers 9Et/9Pr by double deprotonation reactions (Scheme 3).18 The formation of N,N′-ethenylene/propenylene-bridged bis-NHC–CAAC dimers 9Et/9Pr have been unambiguously confirmed by their solid state molecular structure determination (Fig. S76‡ and 6). The overall transformation of the N,N′-ethylene/propylene-bridged bis-NHC–CAAC dimers 4Et/4Pr into the N,N′-ethenylene/propenylene-bridged bis-NHC–CAAC dimers 9Et/9Pr is notably reminiscent of the recently reported electron-catalyzed dehydrogenation of a saturated bipyridinium-ethane backbone to a conjugated bipyridinium-ethene backbone X by Stoddart et al. (Scheme 3 – insert).23
 | ||
| Fig. 6 Molecular structure of 9Pr in the solid state with thermal ellipsoids at the 50% probability level. All hydrogen atoms except on C2, C1, and C5 are omitted for clarity. Selected bond lengths (Å) and bond angles (°):N1–C3 1.4200(18), C3–C4 1.352(2), C1–C2 1.330(2); N1–C3–C4 122.66(12), N1–C2–C1 129.63(14). | ||
Conclusions
In conclusion, we have synthesized a trans-1,4-cyclohexylene-bridged bis-NHC–CAAC dimer derived radical-cation based crystalline and thermally stable diradical. Its singlet state is only marginally more stable than the triplet state (2J = −5.5 cm−1 (ΔEST = 0.065 kJ mol−1)). The related radical-trication and the tetra-cation were also isolated and characterized. Importantly, this study points to the choice of the alkylene bridge being crucial for the stabilization and successful isolation of the dicationic diradical. With ethylene/propylene bridged bis-NHC–CAAC dimers, the corresponding dicationic diradicals are transient and rearrange to the hydrogen abstracted products. The latter can be converted to ethenylene and propenylene-bridged bis-NHC–CAAC dimers; these transformations constitute dehydrogenations of the alkylene bridges. Notably, with this synthetic route an extensive substrate scope can be realized both in the NHC and CAAC scaffolds as well as in the bridging unit.Data availability
Full experimental and computational details are provided as part of the ESI.‡Author contributions
M. K. N. lead all the synthesis and characterization along with initial contribution from R. D., B. R. and V. M., P. S., S. K. P. and P. R. performed the theoretical calculations. T. V., R. S. N., H. R., R. B. and C. S. for the single crystal XRD study. I. K. and H. B. for EPR study. S. M. and A. M. for SQUID study. B. J. E., F. Z., N. C., C. S. and P. R. for electrochemistry study. M. K. N. and A. J. designed the research work. M. K. N. and A. J. prepared the ESI.‡ A. J. wrote the manuscript and all authors assisted in writing and editing the manuscript. A. J. supervised the study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge generous support of the Department of Atomic Energy, Government of India, under Project Identification No. RTI 4007 and SERB (CRG/2019/003415), India. The National Facility for High-Field NMR, TIFR-Hyderabad, is highly acknowledged. AM thanks the Council of Scientific and Industrial Research (CSIR), Govt. of India (Project No: 01(3031)/21/EMR-II), and the Solid State and Structural Chemistry Unit at the Indian Institute of Science (IISc) Bangalore, India, for providing the SQUID Magnetometer facility. We thank the reviewers for their insightful comments helping to improve the quality of the manuscript. AJ is very much grateful to the Alexander von Humboldt Foundation for the sponsoring of a renewed research stay.Notes and references
- (a) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed; (b) T. Stuyver, B. Chen, T. Zeng, P. Geerlings, F. D. Proft and R. Hoffmann, Chem. Rev., 2019, 119, 11291–11351 CrossRef CAS PubMed.
- (a) S. Zhang, M. Pink, T. Junghoefer, W. Zhao, S. N. Hsu, S. Rajca, A. Calzolari, B. W. Boudouris, M. B. Casu and A. Rajca, J. Am. Chem. Soc., 2022, 144, 6059–6070 CrossRef CAS PubMed; (b) N. Gallagher, H. Zhang, T. Junghoefer, E. Giangrisostomi, R. Ovsyannikov, M. Pink, S. Rajca, M. B. Casu and A. Rajca, J. Am. Chem. Soc., 2019, 141, 4764–4774 CrossRef CAS PubMed; (c) A. Rajca, A. Olankitwanit and S. Rajca, J. Am. Chem. Soc., 2011, 133, 4750–4753 CrossRef CAS PubMed; (d) W. T. Borden, R. Hoffmann, T. Stuyver and B. Chen, J. Am. Chem. Soc., 2017, 139, 9010–9018 CrossRef CAS PubMed; (e) H. Han, D. Zhang, Z. Zhu, R. Wei, X. Xiao, X. Wang, Y. Liu, Y. Ma and D. Zhao, J. Am. Chem. Soc., 2021, 143, 17690–17700 CrossRef CAS PubMed; (f) S. Arikawa, A. Shimizu, D. Shiomi, K. Sato and R. Shintani, J. Am. Chem. Soc., 2021, 143, 19599–19605 CrossRef CAS PubMed.
- (a) A. Rajca, S. Utamapanya and J. Xu, J. Am. Chem. Soc., 1991, 113, 9235–9241 CrossRef CAS; (b) C. Shu, H. Zhang, A. Olankitwanit, S. Rajca and A. Rajca, J. Am. Chem. Soc., 2019, 141, 17287–17294 CrossRef CAS PubMed; (c) D. Schmidt, M. Son, J. M. Lim, M. J. Lin, I. Krummenacher, H. Braunschweig, D. Kim and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 13980–13984 CrossRef CAS PubMed; (d) A. Rajca, K. Shiraishia and S. Rajcaa, Chem. Commun., 2009, 4372–4374 RSC.
- (a) A. Jana, M. Ishida, J. S. Park, S. Bähring, J. O. Jeppesen and J. L. Sessler, Chem. Rev., 2017, 117, 2641–2710 CrossRef CAS PubMed; (b) H. V. Schröder and C. A. Schalley, Beilstein J. Org. Chem., 2018, 14, 2163–2185 CrossRef PubMed; (c) N. Duvva, U. Chilakamarthi and L. Giribabu, Sustainable Energy Fuels, 2017, 1, 678–688 RSC.
- (a) L. Striepe and T. Baumgartner, Chem.–Eur. J., 2017, 23, 16924–16940 CrossRef CAS PubMed; (b) M. Berville, J. Richard, M. Stolar, S. Choua, N. Le Breton, C. Gourlaouen, C. Boudon, L. Ruhlmann, T. Baumgartner, J. A. Wytko and J. Weiss, Org. Lett., 2018, 20, 8004–8008 CrossRef CAS PubMed; (c) T. Nakazato, H. Takekoshi, T. Sakurai, H. Shinokubo and Y. Miyake, Angew. Chem., Int. Ed., 2021, 60, 13877–13881 CrossRef CAS PubMed.
- F. Riobé, N. Avarvari, P. Grosshans, H. Sidorenkova, T. Berclazb and M. Geoffroy, Phys. Chem. Chem. Phys., 2010, 12, 9650–9660 RSC.
- M. Frasconi, I. R. Fernando, Y. Wu, Z. Liu, W.-G. Liu, S. M. Dyar, G. Barin, M. R. Wasielewski, W. A. Goddard III and J. Fraser Stoddart, J. Am. Chem. Soc., 2015, 137, 11057–11068 CrossRef CAS PubMed.
- (a) J. Jung, W. Liu, S. Kim and D. Lee, J. Org. Chem., 2019, 84, 6258–6269 CrossRef CAS PubMed; (b) A. Jana, S. Bähring, M. Ishida, S. Goeb, D. Canevet, M. Salle, J. O. Jeppesen and J. L. Sessler, Chem. Soc. Rev., 2018, 47, 5614–5645 RSC; (c) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed; (d) Y. Wang, M. Frasconi and J. F. Stoddart, ACS Cent. Sci., 2017, 3, 927–935 CrossRef CAS PubMed.
- D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886–12890 CrossRef CAS PubMed.
- (a) D. Mandal, R. Dolai, R. Kumar, S. Suhr, N. Chrysochos, P. Kalita, R. S. Narayanan, G. Rajaraman, C. Schulzke, B. Sarkar, V. Chandrasekhar and A. Jana, Org. Lett., 2017, 19, 5605–5608 CrossRef CAS PubMed; (b) D. Mandal, R. Dolai, N. Chrysochos, P. Kalita, R. Kumar, D. Dhara, A. Maiti, R. S. Narayanan, G. Rajaraman, C. Schulzke, V. Chandrasekhar and A. Jana, J. Org. Chem., 2019, 84, 8899–8909 CrossRef CAS PubMed; (c) D. Mandal, F. Stein, S. Chandra, N. I. Neuman, P. Sarkar, S. Das, A. Kundu, A. Sarkar, H. Rawat, S. K. Pati, V. Chandrasekhar, B. Sarkar and A. Jana, Chem. Commun., 2020, 56, 8233–8236 RSC; (d) M. K. Nayak, S. Suhr, N. Chrysochos, H. Rawat, C. Schulzke, V. Chandrasekhar, B. Sarkar and A. Jana, Chem. Commun., 2021, 57, 1210–1213 ( Chem. Commun. , 2021 , 57 , 4979–4980 ) RSC.
- U. S. D. Paul and U. Radius, Chem.–Eur. J., 2017, 23, 3993–4009 CrossRef CAS PubMed.
- (a) P. W. Antoni and M. M. Hansmann, J. Am. Chem. Soc., 2018, 140, 14823–14835 CrossRef PubMed; (b) P. W. Antoni, T. Bruckhoff and M. M. Hansmann, J. Am. Chem. Soc., 2019, 141, 9701–9711 CrossRef CAS PubMed; (c) P. W. Antoni, C. Golz and M. M. Hansmann, Angew. Chem., Int. Ed., 2022, 61, e202203064 CrossRef CAS PubMed.
- J. Messelberger, A. Grünwald, S. J. Goodner, F. Zeilinger, P. Pinter, M. E. Miehlich, F. W. Heinemann, M. M. Hansmann and D. Munz, Chem. Sci., 2020, 11, 4138–4149 RSC.
- (a) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed; (b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; (c) S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Chem. Sci., 2019, 10, 4727–4741 RSC; (d) J. K. Mahoney, D. Martin, F. Thomas, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519–7525 CrossRef CAS PubMed; (e) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 15620–15623 CrossRef CAS PubMed; (f) D. Mandal, S. Sobottka, R. Dolai, A. Maiti, D. Dhara, P. Kalita, R. S. Narayanan, V. Chandrasekhar, B. Sarkar and A. Jana, Chem. Sci., 2019, 10, 4077–4081 RSC; (g) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2206–2213 CrossRef CAS PubMed; (h) M. M. Hansmann, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2546–2554 CrossRef CAS PubMed.
- J. K. Mahoney, D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 18766–18769 CrossRef CAS PubMed.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS.
- See the ESI† for the experimental details, analytical data, NMR spectra, UV/Vis spectra, X-ray crystallographic details, and details of quantum chemical calculation.
- (a) D. Mandal, R. Dolai, P. Kalita, R. S. Narayanan, R. Kumar, S. Sobottka, B. Sarkar, G. Rajaraman, V. Chandrasekhar and A. Jana, Chem.–Eur. J., 2018, 24, 12722–12727 CrossRef CAS PubMed; (b) U. S. D. Paul and U. Radius, Chem.–Eur. J, 2017, 23, 3993–4009 CrossRef CAS PubMed.
- B. M. P. Lombardi, E. R. Pezoulas, R. A. Suvinen, A. Harrison, Z. S. Dubrawski, B. S. Gelfand, H. M. Tuononen and R. Roesler, Chem. Commun., 2022, 58, 6482–6485 RSC.
- S. S. Eaton, K. M. More, B. M. Sawant and G. R. Eaton, J. Am. Chem. Soc., 1983, 105, 6560–6567 CrossRef CAS.
- Y. Guo, K. Sivalingam, E. F. Valeev and F. Neese, J. Chem. Phys., 2017, 147, 064110 CrossRef PubMed.
- (a) M. K. Nayak, J. Stubbe, N. I. Neuman, R. S. Narayanan, S. Maji, C. Schulzke, V. Chandrasekhar, B. Sarkar and A. Jana, Chem.–Eur. J., 2020, 26, 4425–4431 CrossRef CAS PubMed; (b) R. Kumar, S. Chandra, M. K. Nayak, A. S. Hazari, B. J. Elvers, C. Schulzke, B. Sarkar and A. Jana, ACS Omega, 2022, 7, 837–843 CrossRef CAS PubMed.
- H. Chen, F. Jiang, C. Hu, Y. Jiao, S. Chen, Y. Qiu, P. Zhou, L. Zhang, K. Cai, B. Song, X. Y. Chen, X. Zhao, M. R. Wasielewski, H. Guo, W. Hong and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 8476–8487 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Sriram Ramaswamy on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, analytical data, NMR spectra, UV/Vis spectra, X-ray crystallographic details and details of theoretical calculation. CCDC 2047110 (3Et), 2047111 (4Et), 2047113 (10Et), 2047114 (5Et), 2047112 (8Et), 2047115 (9Et), 2161952 (3Pr), 2161953 (4Pr), 2161955 (9Pr), 2161956 (3Cy), 2161957 (5Cy), 2161958 (6Cy), 2161959 (7Cy), 2204009 (5Pr). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03937k |
| This journal is © The Royal Society of Chemistry 2022 |