
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photoinduced transient activating strategy for late-stage chemoselective C(sp3)–H trifluoromethylation of azines†
Mengjun
Huang
,
Jiawei
Ma
 ,
Zhenlei
Zou
,
Heyin
Li
,
Jiyang
Liu
,
Lingyu
Kong
,
Yi
Pan
,
Weigang
Zhang
*,
Yong
Liang
* and
Yi
Wang
*
,
Zhenlei
Zou
,
Heyin
Li
,
Jiyang
Liu
,
Lingyu
Kong
,
Yi
Pan
,
Weigang
Zhang
*,
Yong
Liang
* and
Yi
Wang
*
State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, Collaborative Innovation Center of Advanced Microstructures, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: yiwang@nju.edu.cn
First published on 1st September 2022
Abstract
The direct functionalization of C(sp3)–H bonds is an ultimately ideal synthetic strategy with high atom economy and step efficiency. However, the direct trifluoromethylation of electron-deficient heteroaryl adjacent C(sp3)–H bonds remains a formidable challenge. We have described a transient activating strategy involving a Tf-shift process and π–π stacking interaction for catalyst-free direct benzylic C(sp3)–H trifluoromethylation of azines, such as pyridine, pyrimidine, quinoline, dihydropyridinone, tetrahydroisoquinoline and tetrahydroquinazoline, with an air-stable crystalline imidazolium sulfonate reagent IMDN-Tf. This bench-stable cationic reagent offers a scalable and practical protocol for the late-stage modification of drug molecules with high site selectivity, which avoids the prefunctionalization and the use of stoichiometric metals and strong oxidants. Furthermore, comprehensive mechanistic studies revealed the determining effect of π–π stacking for the activation of azinylic C(sp3)–H bonds.
Introduction
N-Heterocycles as essential fragments exist in many bioactive molecules.1–4 Azines with six-membered ring systems containing one or several nitrogen atoms demonstrate various activities and applications in medicine and agrochemicals. The selective introduction of functionality at the heterobenzylic site of azines is of significant interest for modification of drugs.5–9 However, due to the strong basicity of most common azines, namely pyridine, pyrimidine, quinoline, etc., the late-stage functionalization of alkylated azines exhibits inevitable difficulties and often requires harsh conditions. Recent advances in the activation of C(sp3)–H bonds using pyridinium salts as precursors have found success in transformations such as fluoroalkylation7,8 and alkylation9 (Fig. 1a, left). Considering the significance of fluorine-containing moieties in medicine,10–17 the introduction of trifluoromethyl groups into organic molecules has attracted considerable attention, and significant progress has been achieved in this field in the past few decades.18–28 The fluoroalkyl modification of pyridylic C–H bonds to regulate the metabolism is particularly appealing,29–31 In order to introduce fluoroalkyl groups into heterocyclic scaffolds, the trifluoromethylation of pyridylic C–H bonds has been achieved using prefunctionalized N-oxides7 and N-amido pyridinium salts8via alkylidene dihydropyridine intermediates followed by nucleophilic or radical addition. However, the formation of the new C(sp3)–CF3 bond through azinylic C(sp3)–H fluoroalkylation was limited by tedious substrate preparation and hazardous procedures. In contrast, the direct functionalization of unactivated heterobenzylic C(sp3)–H bonds would be strategically appealing and highly desirable. Highly reactive alkylidene dihydropyridine intermediates which were generated from the deprotonation of heterobenzylic C(sp3)–H bonds have been effectively exploited in functionalization. Recent advances in Lewis acid or alkali metal base-promoted deprotonation of pyridylic C–H bonds have been reported.32–35 Transition-metal catalyzed C–H bond addition has also been reported.36–38 However, these methods usually require harsh conditions, which severely limits the range of functional groups and their practical applications (Fig. 1a, right). Meanwhile, no general protocol for direct azinylic C(sp3)–H fluoroalkylation and alkylation is available. In this context, several issues need to be addressed, including inevitable C(sp2)–H fluoroalkylation through a radical addition process39–41 and oxidation of azinylic C–H bonds.42–44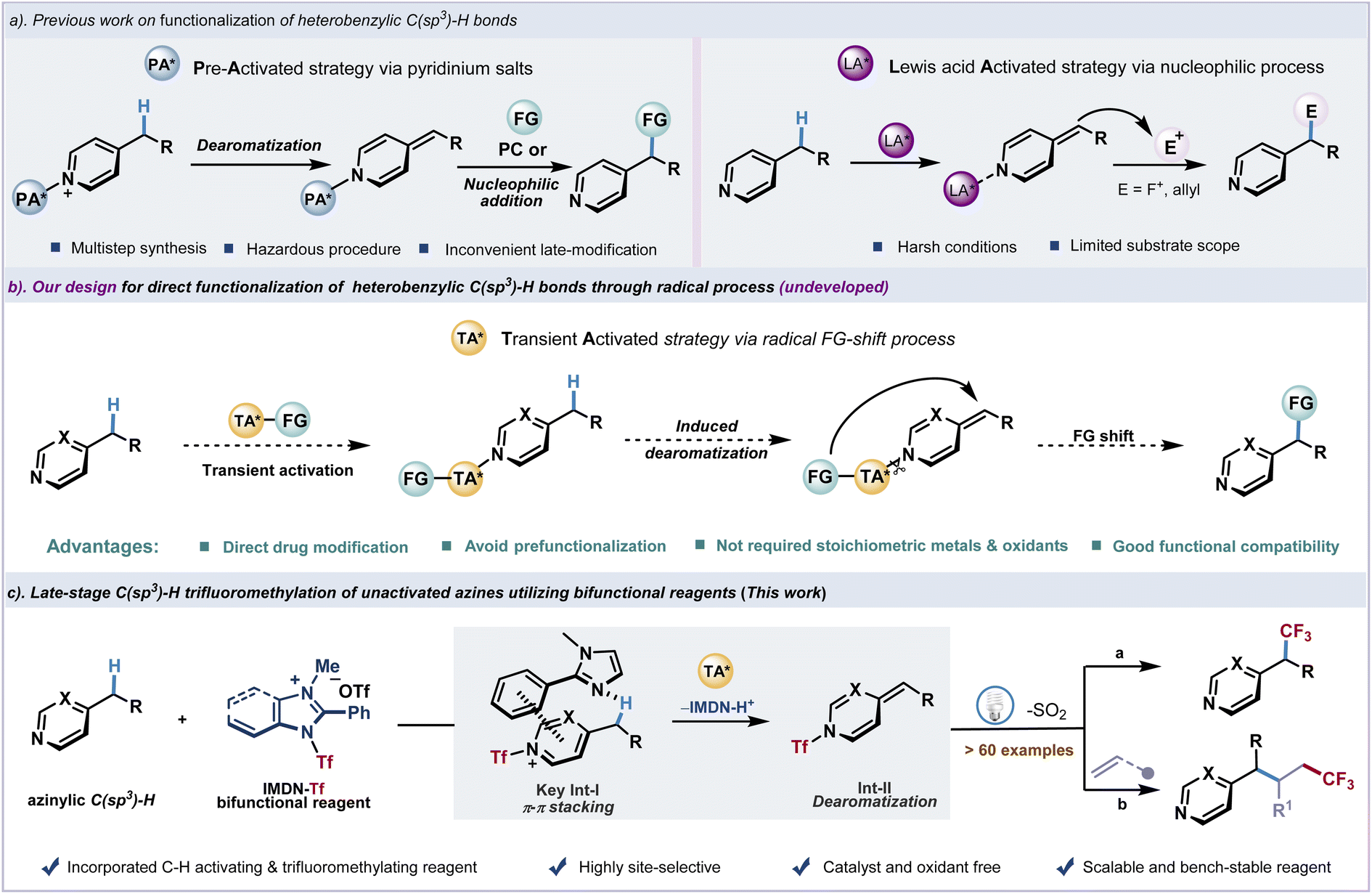 | ||
| Fig. 1 Synthetic strategies for direct C(sp3)–H bond trifluoromethylation. | ||
We envision that a transient activation strategy to rapidly introduce functional groups into azines could spontaneously generate the alkylidene dihydropyridine intermediate under mild photocatalytic conditions. After the dissociation of the transient activating group, functional groups would shift to the azinylic position to furnish the C(sp3)–H alkylation product (Fig. 1b). Recently, we have developed a bench-stable redox-active imidazolium sulfonate reagent (IMDN-Tf) from inexpensive Tf2O and imidazole.45 Owing to the electron deficiency and strong electrophilicity of this cationic reagent, IMDN-Tf could smoothly undergo an electrophilic Tf transfer process to generate Nu-Tf species through the cleavage of the weak N–S bond (BDE ≈ 70 kcal mol−1).46 Inspired by recent reports on Tf2O activation of pyridines,47 we design a Tf-shift process of IMDN-Tf to generate the azinylium salt in situ and imidazole residue, which also activates the azinylic C(sp3)–H bond via π–π stacking interaction (intermediate I) to produce the resonance-stabilized alkylidene dihydropyridine intermediate II. Finally, a rapid fragmentation and recombination event of II under photoexcitation affords the azinylic trifluoromethylation product and releases SO2 (Fig. 1c). This reasonable hypothesis could lead to an unprecedented late-stage azinylic C(sp3)–H functionalization protocol in a catalyst-free manner. Pre-activation of azine substrates and commonly observed C(sp2)–H alkylation could be avoided.
Results and discussion
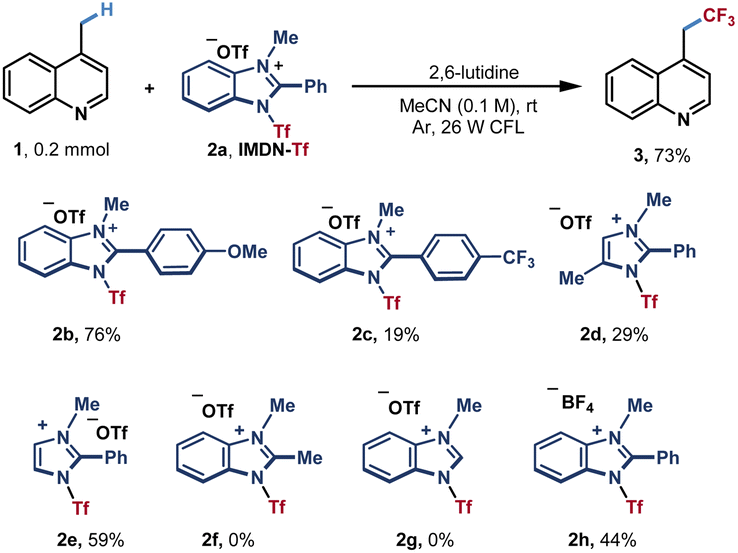
Based on our hypothesis, we selected lepidine (1) as a pilot substrate to test azinylic trifluoromethylation (Table 1). After extensive screening of conditions, we found that when using 1.8 equiv. of IMDN-Tf 2a and 1.75 equiv. of 2,6-lutidine in CH3CN at room temperature under the irradiation of 26 W CFL, the trifluoromethylation product 3 could be obtained in 73% NMR yield (65% isolated yield). Different IMDN-Tf salts 2b–2f were then examined. The imidazolium reagent 2b bearing a methoxy group afforded product 3 in a higher yield (76%), while trifluoromethyl-substituted phenyl benzimidazolium salt 2c furnished product 3 in a very low yield (19%). With imidazolium reagents 2d and 2e, the reaction could proceed in lower yields (29% and 59%, respectively). Furthermore, no product 3 was obtained with 2f and 2g, in which the methyl substituent or hydrogen was present at the 2-position of imidazole. These results indicated that the π–π stacking effect is significant for the activation of azinyl C(sp3)–H bonds. When highly corrosive Tf2O or CF3SO2Cl was used as a trifluoromethyl source, no desired product could be obtained, only the decomposition of the reagents was observed (entries 2 and 3). Other organic or inorganic bases could promote the reaction with lower efficiencies (23–52% yields, entry 4). The basicity of the reaction solution has a significant influence on the yield (see ESI Table S1†). A weak base such as pyridine could not promote the dissociation of imidazoles, while a strong base such as Na2CO3 can lead to the decomposition of IMDN-Tf reagents (see Table S1†). When using other solvents such as acetone, THF and ethyl acetate instead of MeCN, the yields of trifluoromethylated product 3 were significantly decreased (entry 5). This may be due to the poor solubility of IMDN-Tf 2a in those solvents. Basic solvents including DMF and DMSO were also attempted and the decomposition of IMDN-Tf was observed (Table S2†). A blue LED also led to a lower yield compared with that using CFL (entry 6 and Table S4†). In addition, control experiments proved that both light and lutidine were necessary for the reaction (entries 7 and 8). When 1.5 equiv. of 2a was used, the reaction occurred generating the product in a slightly lower yield with a small amount of unreacted starting material (52%, entry 9) When the amount of IMDN-Tf 2a was increased to 2.0 equiv., the yield of the product was also decreased with the generation of the bis-trifluoromethylated byproduct (Table S3†).|
|
||
|---|---|---|
| Entry | Variation from the above conditions | Yieldb/% |
| a Reaction conditions: 1 (0.2 mmol), 2a (1.8 equiv.), 2,6-lutidine (1.75 equiv.), MeCN (2 mL), Ar, irradiation with 26 W CFL at r.t. for 2–12 h. b Yield determined by 19F NMR spectroscopy using trifluoromethoxybenzene as an internal standard. c Isolated yield. | ||
| 1 | None | 73(65)c |
| 2 | Tf 2 O instead of 2a | 0 |
| 3 | CF 3 SO 2 Cl instead of 2a | 0 |
| 4 | Other bases instead of 2,6-lutidine | 23–55 |
| 5 | Other solvents instead of CH3CN | 5–33 |
| 6 | 60 W blue LED instead of CFL | 53 |
| 7 | Under darkness | 18 |
| 8 | W/o 2,6-lutidine | 42 |
| 9 | 1.5 equiv. of 2a was used | 52 |
With the optimized reaction conditions in hand, we next examined the generality of this transformation with different azines (Scheme 1). The primary C(sp3)–H substrates including methyl-substituted pyridine, pyrimidine and quinoline could all furnish the corresponding trifluoromethylated products with good yields (3–5). Pyridines and quinolines bearing electron withdrawing or electron-donating groups afforded products 6–14 in moderate to good yields (51–75%). Next, we investigated the direct trifluoromethylation of diversified heteroaromatic secondary C–H bonds. Pyridines and quinolines containing ethyl, propyl, isobutyl, heptyl, cyclohexyl and benzyl substituents afforded products 15–16 and 22–27 in moderate to good yields. Silyl-protected hydroxymethylpyridine reacted with cationic reagent 2a to afford product 17 in 21% yield. Dihydropyridinone, tetrahydroisoquinoline and tetrahydroquinazoline also furnished trifluoromethylated products in good yields (19–21). Pyrimidine and quinoline derivatives bearing ester and amide groups could be well tolerated under standard conditions (28–35). Naphthyl-, furanyl- and pyridyl-ester substituted quinolines also readily transformed into the corresponding products 36–38 in moderate yields. The reaction could also be applied to quinoline derivatives involving cycloalkane to produce the desired products in 61–64% yields (39–40, 42). Pyridine derived ester substrates also have good compatibility, and the corresponding products (43 and 44) could be obtained in moderate yields. An attempt using more challenging ether substrates resulted in the desired product 41 in moderate yield. To further investigate the reaction scope, this catalyst-free photosynthesis has been applied to late-stage modification of C(sp3)–H bonds of azine-containing natural products. Piperonylic acid (45), probenecid (46), gemfibrozil (47), camphanic acid (48), lauroyl chloride (49) and dehydroabietylamine (50) could furnish the corresponding trifluoromethylated products in good yields (42–67%). The insecticide flonicamid analogue (51) was also afforded in 76% yield.
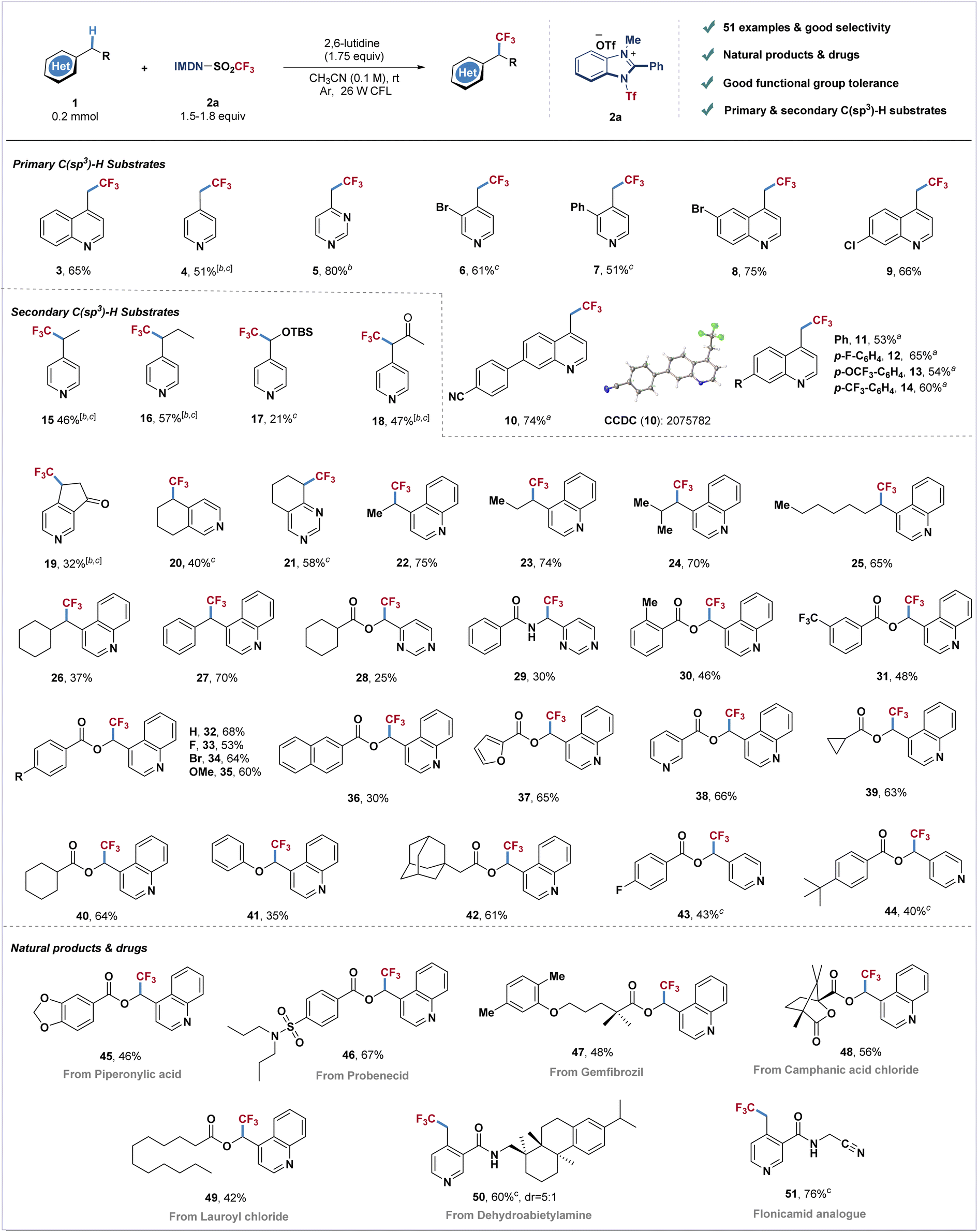 | ||
| Scheme 1 Substrate scope of azinylic C(sp3)–H bond trifluoromethylation. a1.5 equiv. of IMDN-Tf 2a was used. bYield determined by 19F NMR spectroscopy using trifluoromethoxybenzene as the internal standard. c50 °C. | ||
Difunctionalization of alkenes to incorporate two functional groups across a double bond has emerged as a powerful transformation to greatly increase molecular complexity in organic synthesis with improved efficiency. However, the Giese reaction involving the activation of electron-deficient heterobenzylic C(sp3)–H bonds and fluoroalkyl radical addition remains unexplored.48 By varying the reaction conditions, this practical strategy has been applied to difunctionalization of α,β-unsaturated olefins with the azinylic C(sp3)–H bond (Scheme 2). Several common electron-deficient α,β-unsaturated olefins can participate in the reaction, and three-component coupling products (52–61) were obtained in moderate yields (34–60%). Additionally, trifluoromethyl radicals were successfully attached to unactivated olefins to afford fluoroalkylated product 56. It is noteworthy that biorelevant molecules, such as diacetonefructose, paracetamol, picaridin, benzocaine, diacetone-D-glucose and epiandrosterone derived alkenes, afforded three-component coupled trifluoromethylated products in good yields (63–68).
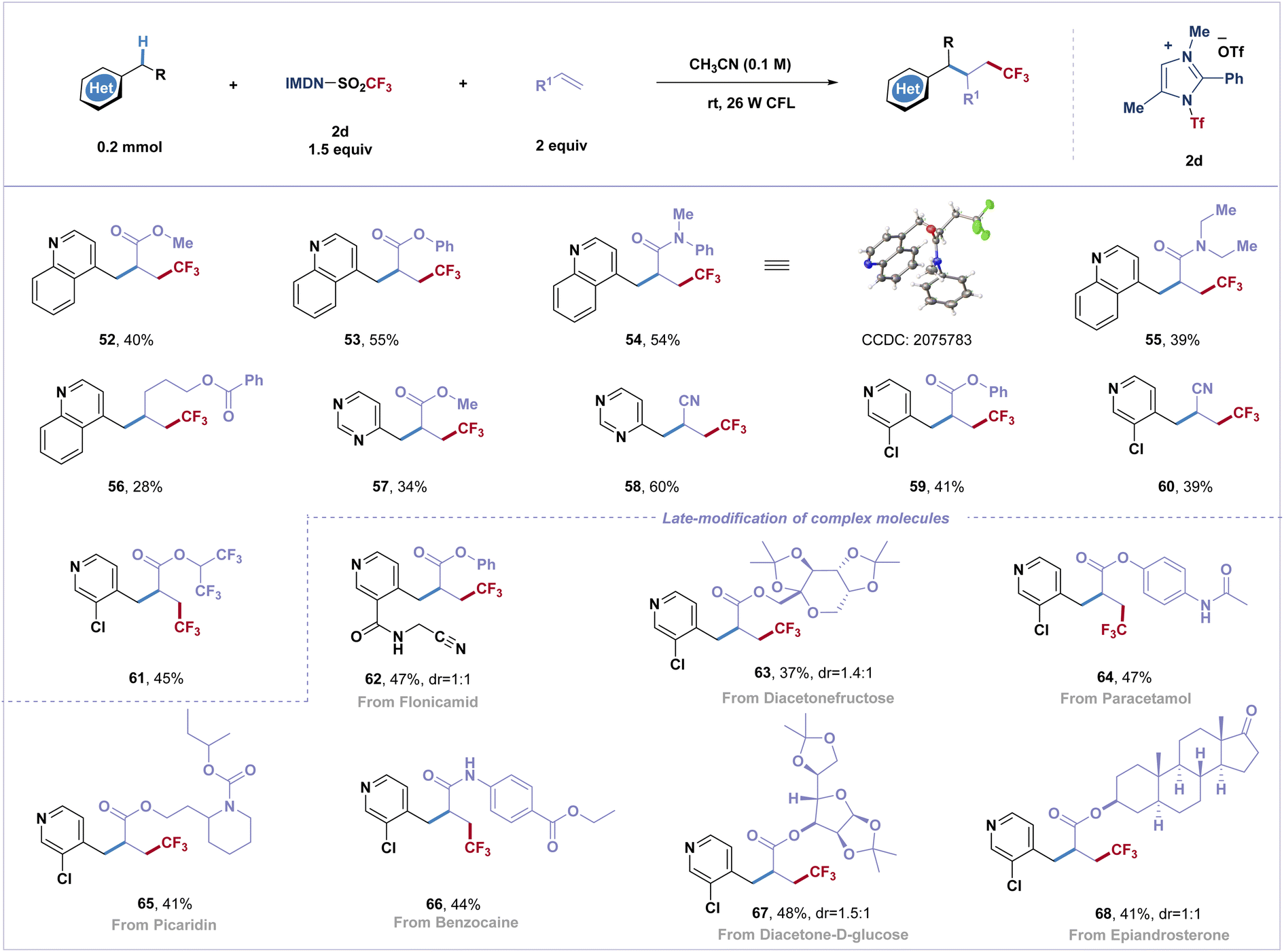 | ||
| Scheme 2 Substrate scope of the three-component direct azinylic C(sp3)–H bond functionalization. All reactions were carried out with azines (0.2 mmol), alkenes (0.4 mmol, 2.0 equiv.), IMDN-Tf 2d (0.3 mmol, 1.5 equiv.) in MeCN (2.0 mL) under Ar and 26 W CFL about 12 h. | ||
To gain further insight into the mechanism of this direct trifluoromethylation of the heteroaryl adjacent C(sp3)–H bond, a series of control experiments have been carried out. The formation of the proposed Tf-transfer process and π–π stacking effect between quinoline 1 and imidazolium reagent 2a was monitored by UV/vis absorption spectroscopy. Compared to quinoline 1 and IMDN-Tf 2a, a strong absorption peak of the mixture [1 + 2a] was observed at 280 nm (see ESI†). The treatment of imidazolium 2a with substrates 1 and 70 furnished the key intermediates 69 and 72, which were confirmed by GC-MS and X-ray crystallography (Scheme 3a and b). In addition, the optimization of the imidazolium sulfonate reagents 2a–2g indicates that the activation of the C(sp3)–H bond is the key step involving the π–π stacking interaction between intermediate I and 2-phenylimidazole. When three equivalents of TEMPO were added under standard conditions, the TEMPO adducts 73 and 74 were detected by 19F NMR and HRMS (Scheme 3c). Furthermore, the treatment of 2a with 4-benzylpyridine 75 afforded the trifluoromethylated product 76 in 17% yield and the dimerized byproduct 77 could be monitored by HRMS (Scheme 3d). These experimental results indicate that the intermediate II may result in a pair of CF3 and benzyl radicals through the homolytic cleavage of the weak N–S bond.
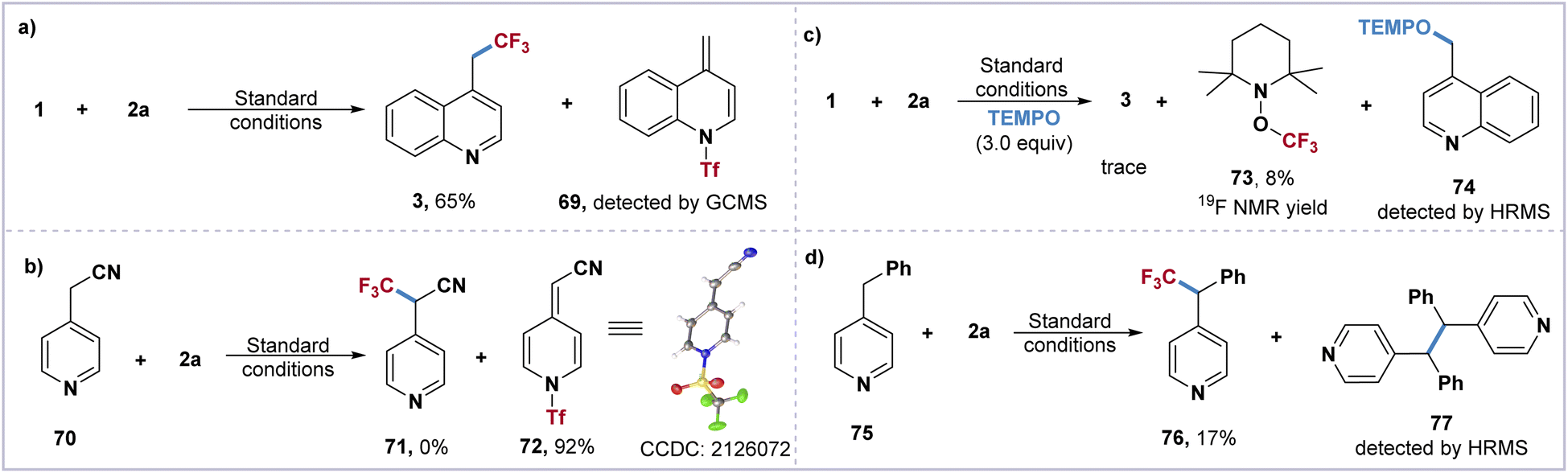 | ||
| Scheme 3 Mechanistic studies. (a and b) Tf-shift intermediate verification experiment. (c and d) Radical trapping experiment. | ||
DFT calculations have been conducted to better understand the mechanism of the overall reaction. The computed potential energy surface is shown in Scheme 4. Initially, the Tf group is readily transferred from 2a to 4-methylquinoline 1 through a SN2 process viaTS1, forming INT1 and benzimidazole INT2. Then INT1 is deprotonated by INT2viaTS2, with a barrier of 24.1 kcal mol−1, leading to protonated benzimidazole INT3 and the dearomatized intermediate 69. The deprotonation process strongly depends on the imidazolium sulfonate reagent. In TS2, the π–π stacking between the electron-deficient aromatic ring of the substrate and the phenyl on imidazole is clearly observed (highlighted in red). Combined with our experiments, when the electron-rich 4-methoxyphenyl-containing 2b gave a comparable yield with that using 2a but the electron-poor 4-(trifluoromethyl)phenyl-bearing 2c gave a much lower yield (Table 1), we believe that this non-covalent interaction is crucial to this step. For deprotonation using reagent 2f, in which such π–π stacking does not exist, it is found that an activation free energy as high as 27.3 kcal mol−1 is required, too high for a reaction occurring at room temperature (see ESI†), agreeing with the experiment. After the deprotonation, INT3 further undergoes an acid–base reaction with 2,6-lutidine, which regenerates INT2 and shifts the chemical equilibrium toward 69. Now as the formation of 69 is slightly exergonic (ΔG = −1.3 kcal mol−1), it could be detected by GC-MS (Scheme 3a).
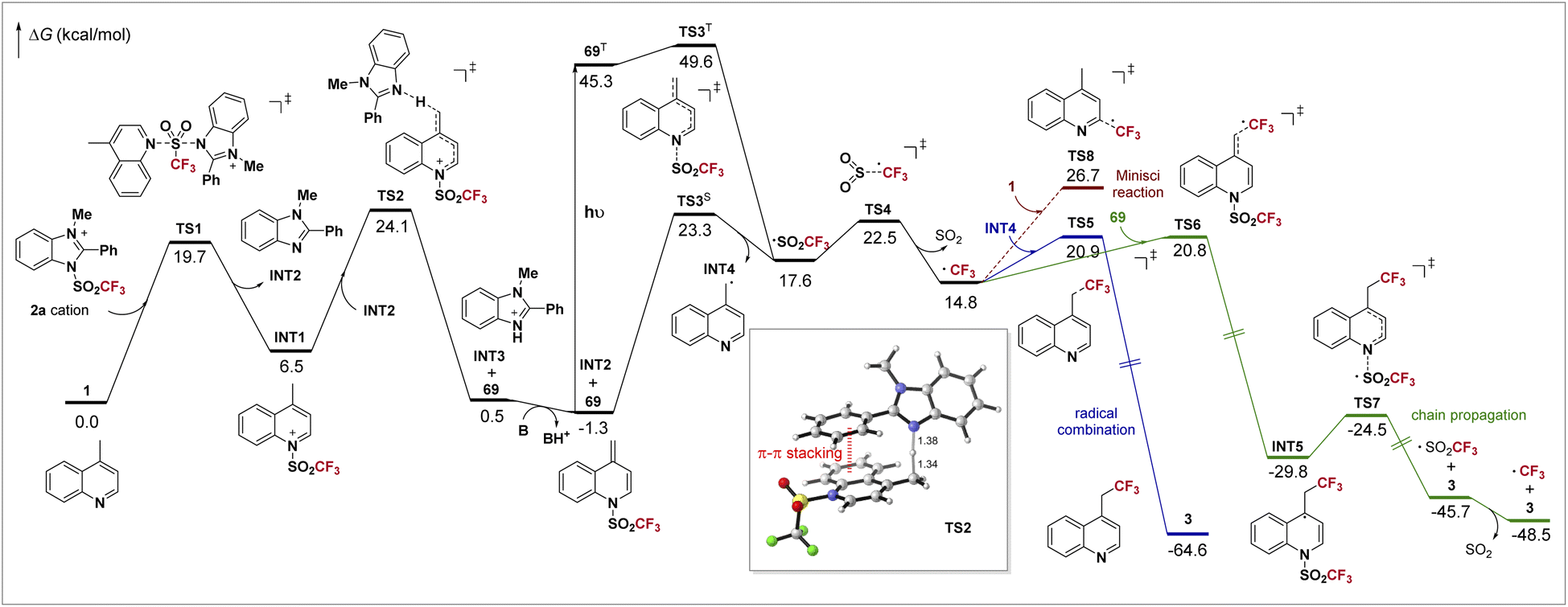 | ||
| Scheme 4 DFT study of the reaction mechanism. B = 2,6-lutidine. Relative Gibbs free energies (kcal mol−1) were computed with SMD(MeCN)-(U)M06-2X/6-311+G(2d,p)//SMD(MeCN)-(U)M06-2X/6-31G(d,p). S: singlet; T: triplet. All distances are in Å. | ||
For the radical formation step, as the singlet-triplet gap of 69 (46.6 kcal mol−1) is well in range of the energy of visible light (when λ = 400 nm, hv = 71.5 kcal mol−1), 69 can be excited to its triplet state 69T under irradiation of visible light, and then undergoes a facile homolysis viaTS3T, forming INT4 and Tf radicals. We find that the homolysis is also plausible under thermal conditions viaTS3S with a barrier of 24.6 kcal mol−1, which is in accordance with a bit lower yield in the dark (entry 7, Table 1). The unstable Tf radical quickly dissociates viaTS4 to form the CF3 radical. The involvement of these radical species agrees with our mechanistic experiments (Scheme 3c and d). The CF3 radical may undergo a combination with INT4viaTS5, yielding product 3 directly (blue line). Otherwise, a chain mechanism is also possible (green line). The CF3 radical may attack intermediate 69viaTS6 and forms INT5, which then undergoes N–S cleavage viaTS7 and gives 3 and the Tf radical. Finally, the decomposition of the Tf radical completes the chain propagation. Our calculations indicate that the barriers for both non-chain and chain mechanisms are almost identical. Furthermore, a quantum yield of 0.99 was observed (for details, see ESI†). Such results mean that both pathways contribute to the product formation in our system, as the quantum yield should be far below 1.0 if only the non-chain mechanism is present,49 and it should be far above 1.0 if the chain mechanism works exclusively.44,50 In addition, the Minisci reaction resulting in C(sp2)–H trifluoromethylation was also documented,51 which is possibly competitive in our system. However, the reaction between the CF3 radical and 1 (red line, viaTS8) has a barrier of 28.0 kcal mol−1, about 6.0 kcal mol−1 higher than that of the desired C(sp3)–H trifluoromethylation. The exclusion of such side reaction ensures excellent regioselectivity in our reaction.
The computational results can also explain why the reaction of 4-(cyanomethyl)pyridine 70 gives a stable trifluoromethanesulfonyl transfer intermediate 72. Energy profiles of 70 along with its analog, 4-methylpyridine, were calculated (Scheme 5). Both substrates have a faster deprotonation process than 1. For 4-methylpyridine, the dearomatized intermediate INT6 is slightly more stable than the substrate (−1.8 kcal mol−1). The subsequent homolysis viaTS3-bS requires a slightly higher activation free energy of 28.2 kcal mol−1, making it less reactive and need an elevated temperature (Scheme 1). On the other hand, owing to the conjugative effect of the cyano group, the intermediate 72 is especially stable (−8.3 kcal mol−1). The loss of such a favorable conjugation dramatically increases the difficulty in the next homolysis viaTS3-cS. The computed overall barrier is as high as 31.2 kcal mol−1, showing that the following radical reactions could not proceed, and thus 72 was eventually isolated (Scheme 3b).
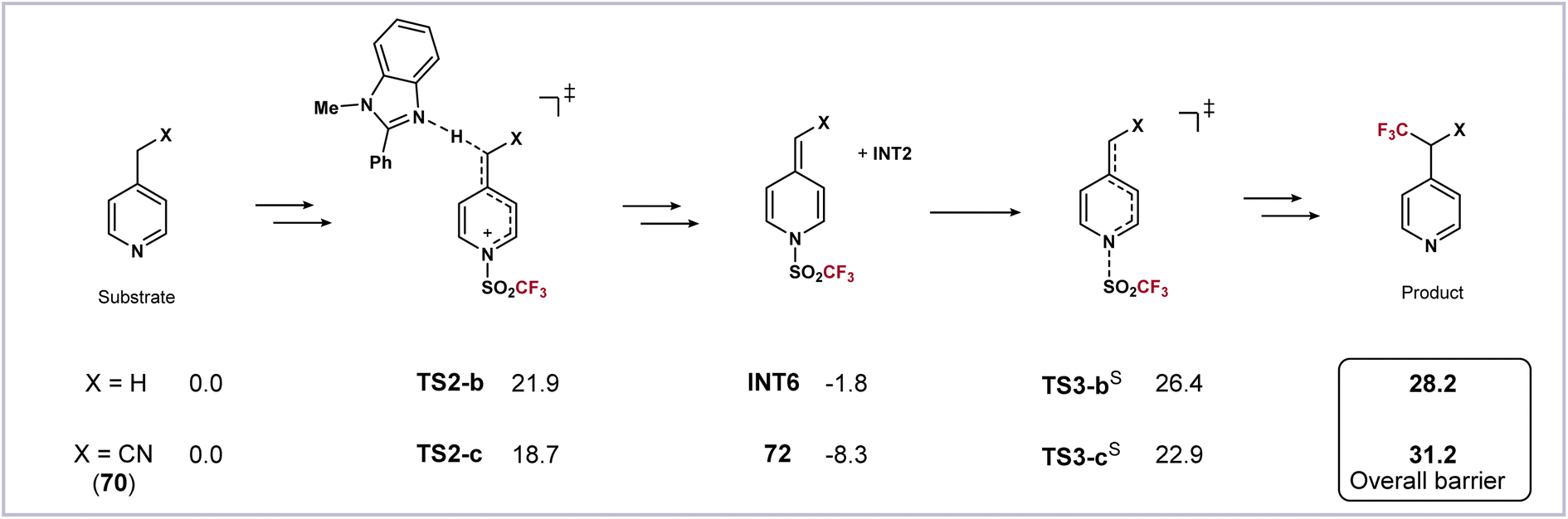 | ||
| Scheme 5 DFT study on the reactions using 4-methylpyridine and 4-(cyanomethyl)pyridine (70) as substrates. Relative Gibbs free energies (kcal mol−1) were computed with SMD(MeCN)-(U)M06-2X/6-311+G(2d,p)//SMD(MeCN)-(U)M06-2X/6-31G(d,p). S: singlet. | ||
Based on the above investigations and DFT calculations, a plausible mechanism was proposed for this reaction (Scheme 6). First, intermediate I was formed in the presence of azines 1 and imidazolium sulfonate reagent 2a through a Tf-shift process and dissociated out 2-phenylimidazole. Then the azinylic C(sp3)–H bond was activated by imidazole via π–π stacking interaction to generate the alkylidene dihydropyridine intermediate III. Under visible light irradiation, intermediate III produced the CF3 radical and benzyl radical through the homolytic cleavage of the weak N–S bond and released SO2. Finally, the CF3 radical and benzyl radical underwent a radical–radical coupling process to afford trifluoromethylated azines (Scheme 6, path a). Alternatively, the possibility of a chain reaction could not be excluded. The addition of CF3 radicals to intermediate III and fragmentation resulted in the desired product and trifluoromethyl radicals to complete the mechanistic cycle (Scheme 6, path b).
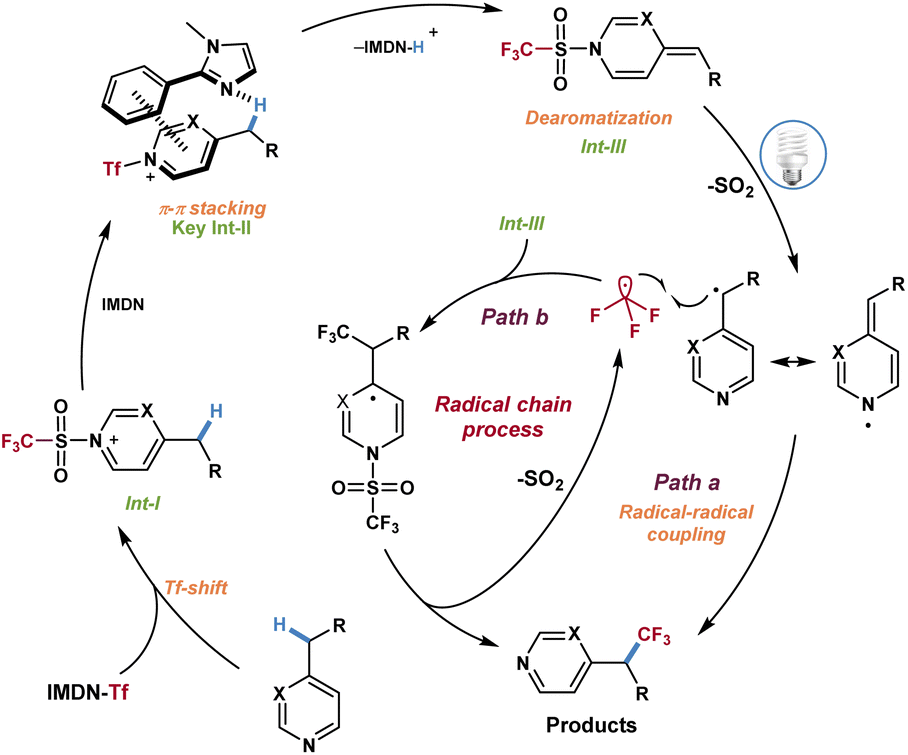 | ||
| Scheme 6 Proposed reaction mechanisms. | ||
Experimental
General procedure for the synthesis of products 3–51
Under argon, corresponding substrates (0.2 mmol) were added to a solution of 2a (0.36 mmol, 1.8 equiv.) and 2,6-lutidine (0.35 mmol, 1.75 equiv.) in CH3CN (2 mL) at room temperature. After that, the tube was exposed to a 26 W compact fluorescent light at room temperature for about 2–12 h until the reaction was complete as monitored by TLC analysis. The reaction mixture was evaporated in vacuo. The crude products were directly purified by flash chromatography on silica gel to give the desired product.General procedure for the synthesis of products 52–68
Under argon, corresponding substrates (0.2 mmol) were added to a solution of 2d (0.3 mmol, 1.5 equiv.) and alkenes (0.4 mmol, 2 equiv.) in CH3CN (2 mL) at room temperature. After that, the tube was exposed to a 26 W compact fluorescent light at room temperature for about 12 h until the reaction was complete as monitored by TLC analysis. The reaction mixture was evaporated in vacuo. The crude products were directly purified by flash chromatography on silica gel to give the desired product.Conclusions
In summary, we have developed a Tf-transfer strategy for photoinduced direct trifluoromethylation of azinylic C(sp3)–H bonds with the imidazolium sulfonate cationic reagent IMDN-Tf. The unique electronic character of this bench-stable reagent enables rapid trifluoromethanesulfonyl transfer toward azines. Meanwhile, phenyl-substituted imidazole can also promote the benzylic deprotonation of azines due to the favorable π–π stacking effect. This incorporated C–H activating & trifluoromethylating reagent provides a practical toolbox for site-selective late-stage C(sp3)–H functionalization in an unconventional catalyst-free manner. Further investigation of this reagent is underway in the laboratory.Data availability
The data that support the findings of this study are available in the ESI† or on request from the corresponding author.Author contributions
M. H. conducted all experiments and characterized the novel compounds. Y. W. designed the experiments. Y. L. and J. M. conducted the computational studies. W. Z., Y. L., Y. W. wrote the manuscript. Y. P. was responsible for funding application. Z. Z., H. L., J. L. and L. K. contributed to the analysis and interpretation of the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21971107 and 2201101), China Postdoctoral Science Foundation (2021T140309 and 2021M691511), the Fundamental Research Funds for the Central Universities (020514380253 and 020514380275), and the Natural Science Foundation of Jiangsu Province (BK20211555). We thank the High Performance Computing Center (HPCC) of Nanjing University for doing the numerical calculations in this paper on its blade cluster system.Notes and references
- A. R. Katritzky, Chem. Rev., 2004, 104, 2125–2126 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- B. Zhao, B. Prabagar and Z. Shi, Chem, 2021, 7, 2585–2634 CAS.
- M. Kaur and J. F. V. Humbeck, Org. Biomol. Chem., 2020, 18, 606–617 RSC.
- K. E. Danahy, J. C. Cooper and J. F. V. Humbeck, Angew. Chem., Int. Ed., 2018, 57, 5134–5138 CrossRef CAS PubMed.
- M. Meanwell, M. B. Nodwell, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2016, 55, 13244–13248 CrossRef CAS PubMed.
- Y. Kuninobu, M. Nagase and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 10263–10266 CrossRef CAS PubMed.
- During the preparation of this manuscript, a report of C(sp3)–H functionalization using N-amidopyridinium salts and sodium fluoroalkylsulfinate has been published, see: M. Kim, E. You, J. Kim and S. Hong, Angew. Chem., Int. Ed., 61, e202204217, DOI:10.1002/anie.202204217.
- Q. Liu, C. Zhang, H. Sheng, D. Enders, Z. Wang and X. Chen, Org. Lett., 2020, 22, 5617–5621 CrossRef CAS PubMed.
- B. Manteau, S. Pazenok and J.-P. Vors, J. Fluorine Chem., 2014, 131, 140–158 CrossRef.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- P. Jeschke, ChemBioChem, 2004, 5, 571–589 CrossRef PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- H. Yanai, S. Hoshikawa, Y. Moriiwa, A. Shoji, A. Yanagida and T. Matsumoto, Angew. Chem., Int. Ed., 2021, 60, 5168–5172 CrossRef CAS PubMed.
- E. J. McClain, T. M. Monos, M. Mori, J. W. Beatty and C. R. J. Stephenson, ACS Catal., 2020, 10, 12636–12641 CrossRef CAS.
- B. Yang, D. Yu, X.-H. Xu and F.-L. Qing, ACS Catal., 2018, 8, 2839–2843 CrossRef CAS.
- L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5809–5812 CrossRef CAS PubMed.
- D. J. W. Beatty, J. J. Douglas, R. Miller, R. C. McAtee, K. P. Cole and C. R. J. Stephenson, Chem, 2016, 1, 456–472 Search PubMed.
- J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6, 7919 CrossRef PubMed.
- G. Choi, G. S. Lee, B. Park, D. Kim and S. H. Hong, Angew. Chem., Int. Ed., 2021, 60, 5467–5474 CrossRef CAS PubMed.
- J. He, T. N. Nguyen, S. Guo and S. P. Cook, Org. Lett., 2021, 23, 702–705 CrossRef CAS.
- M. Paeth, W. Carson, J. Luo, D. Tierney, Z. Cao, M. Cheng and W. Liu, Chem. Eur. J., 2018, 24, 11559–11563 CrossRef CAS PubMed.
- S. Guo, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2018, 140, 12378–12382 CrossRef CAS PubMed.
- H. Xiao, Z. Liu, H. Shen, B. Zhang, L. Zhu and C. Li, Chem, 2019, 5, 940–949 CAS.
- P. J. Sarver, V. Bacauanu, D. M. Schultz, D. A. DiRocco, Y. Lam, E. C. Sherer and D. W. C. MacMillan, Nat. Chem., 2020, 12, 459–467 CrossRef CAS PubMed.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- J. Wang, M. Sanchez-Rosello, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- H. Komai, T. Yoshino, S. Matsunaga and M. Kanai, Org. Lett., 2011, 13, 1706–1709 CrossRef CAS.
- M. Rueping and N. Tolstoluzhsky, Org. Lett., 2011, 13, 1095–1097 CrossRef CAS PubMed.
- H. Suzuki, R. Igarashi, Y. Yamashita and S. Kobayashi, Angew. Chem., Int. Ed., 2017, 56, 4520–4524 CrossRef CAS PubMed.
- D. Zhai, X. Zhang, Y. Liu, L. Zheng and B. Guan, Angew. Chem., Int. Ed., 2018, 57, 1650–1653 CrossRef CAS PubMed.
- S. Duez, A. Steib, S. Manolikakes and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 7686–7690 CrossRef CAS PubMed.
- B. Qian, S. Guo, J. Shao, Q. Zhu, L. Yang, C. Xia and H. Huang, J. Am. Chem. Soc., 2010, 132, 3650–3651 CrossRef CAS PubMed.
- M. Joshi and F. Pigge, ACS Catal., 2016, 6, 4465–4469 CrossRef CAS.
- V. D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef PubMed.
- A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868–11871 CrossRef PubMed.
- Z. Zou, W. Zhang, Y. Wang and Y. Pan, Org. Chem. Front., 2021, 8, 2786–2798 RSC.
- R. Vanjari and K. N. Singh, Chem. Soc. Rev., 2015, 44, 8062–8096 RSC.
- E. W. Webb, J. B. Park, E. L. Cole, D. J. Donnelly, S. J. Bonacorsi, W. R. Ewing and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 9493–9500 CrossRef CAS PubMed.
- L. Yang, D. Tan, W. Fan, X. Liu, J. Wu, Z. Huang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 3454–3458 CrossRef CAS PubMed.
- W. Zhang, Z. Zou, W. Zhao, S. Lu, Z. Wu, M. Huang, X. Wang, Y. Wang, Y. Liang, Y. Zhu, Y. Zheng and Y. Pan, Nat. Commun., 2020, 11, 2572 CrossRef CAS PubMed.
- E. Kraka, D. Setiawan and D. Cremer, J. Comput. Chem., 2016, 37, 130–142 CrossRef CAS PubMed.
- Y. Ouyang, X.-H. Xu and F.-L. Qing, Angew. Chem., Int. Ed., 2018, 57, 6926–6929 CrossRef CAS PubMed.
- D. M. Kitcatt, S. Nicolle and A.-L. Lee, Chem. Soc. Rev., 2022, 51, 1415–1453 RSC.
- H.-H. Zhang and S. Yu, Org. Lett., 2019, 21, 3711–3715 CrossRef CAS PubMed.
- Y. Zhang, H. Liu, L. Tang, H.-J. Tang, L. Wang, C. Zhu and C. Feng, J. Am. Chem. Soc., 2018, 140, 10695–10699 CrossRef CAS PubMed.
- Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95–99 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2075782, 2075783 and 2126072. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03989c |
| This journal is © The Royal Society of Chemistry 2022 |