
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structure and reactivity of μ3-SnH capped trinuclear nickel cluster†
Nicole A.
Torquato
a,
Joseph M.
Palasz
a,
Quentin C.
Bertrand
b,
Felix M.
Brunner
a,
Thomas
Chan
a,
Milan
Gembicky
 a,
Anthony A.
Mrse
a and
Clifford P.
Kubiak
*a
a,
Anthony A.
Mrse
a and
Clifford P.
Kubiak
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr, La Jolla, CA 92093, USA. E-mail: ckubiak@ucsd.edu
bDepartment of Chemistry, Yale University, New HavenCT 06520USA
First published on 5th September 2022
Abstract
Treatment of the trichlorotin-capped trinuclear nickel cluster, [Ni3(dppm)3(μ3-Cl)(μ3-SnCl3)], 1, with 4 eq. NaHB(Et)3 yields a μ3-SnH capped trinuclear nickel cluster, [Ni3(dppm)3(μ3-H)(μ3-SnH)], 2 [dppm = bis(diphenylphosphino)methane]. Single-crystal X-ray diffraction, nuclear magnetic resonance (NMR) spectroscopy, and computational studies together support that cluster 2 is a divalent tin hydride. Complex 2 displays a wide range of reactivity including oxidative addition of bromoethane across the Sn center. Addition of 1 eq. iodoethane to complex 2 releases H2 (g) and generates an ethyltin-capped nickel cluster with a μ3-iodide, [Ni3(dppm)3(μ3-I)(μ3-Sn(CH2CH3))], 4. Notably, insertion of alkynes into the Sn–H bond of 2 can be achieved via addition of 1 eq. 1-hexyne to generate the 1-hexen-2-yl-tin-capped nickel cluster, [Ni3(dppm)3(μ3H)(μ3-Sn(C6H11))], 5. Addition of H2 (g) to 5 regenerates the starting material, 2, and hexane. The formally 44-electron cluster 2 also displays significant redox chemistry with two reversible one-electron oxidations (E = −1.3 V, −0.8 V vs. Fc0/+) and one-electron reduction process (E = −2.7 V vs. Fc0/+) observed by cyclic voltammetry.
Introduction
Tin hydrides have been widely employed in stoichiometric and catalytic hydrogen atom transfer to a diverse range of substrates.1,2 Industrially relevant tin hydrides predominately feature tin in the +4 oxidation state. However, recent advances in group 14 hydride chemistry have introduced a range of tin hydride species formally in the +2 oxidation state that enable new reactivity profiles and pathways.1,3 Over the past 20 years, seminal studies by Power and co-workers have demonstrated the synthesis, isolation, and structural characterization of low-valent Sn, Ge, and Pb hydrides.4–8 These studies have led to multiple advances in main group reactivity, such as aromatic C–F activation, C–H activation, hydrostannylation chemistry, and catalytic hydroboration.9–13 Furthermore, recent studies have revealed new functionalities for tin(II) species including reversible coordination of H2, coordination of NH3 and N–H activation, and oxidative addition of H2, H2O, and silyl and boryl SnIV species.14,15 In an effort to change the electronics and structural platform for tin hydride complexes, there have been reports of tin hydride complexes coordinated to transition metals, featuring tin in the +2 oxidation state, utilizing Lewis base/Lewis acid pairs for stabilization.16–21 Overall, these advancements in the tin chemistry platform specifically regarding the production of low valent and highly reduced tin species have led to the observation of transition-metal like reactivity of tin, including activation of small and unsaturated molecules.22 In an effort to expand on the known structural and reaction chemistry of tin, we sought to investigate the impacts of supporting different tin functional groups on polynuclear transition metal clusters.23–25 Herein, we describe the synthesis, structure, and reactivity of a divalent Sn hydride capped trinuclear nickel cluster, [Ni3(dppm)3(μ3-H)(μ3-SnH)], 2 [dppm = bis(diphenylphosphino)methane]. Complex 2, displays a wide range of reactivity, including oxidative addition of alkyl halides and alkyne insertion with subsequent hydrogenation.Results and discussion
The synthesis and structural determination of a trichlorostannyl-capped cluster [Ni3(dppm)3(μ3-Cl)(μ3-SnCl3)] (1) has been reported previously.26 Treatment of 1 with 4 eq. NaB(Et)3H at room temperature liberates H2 and affords 2 in 88% yield (Scheme 1). This complex was characterized by X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cyclic voltammetry. DFT calculations were also used to investigate the electronic structure.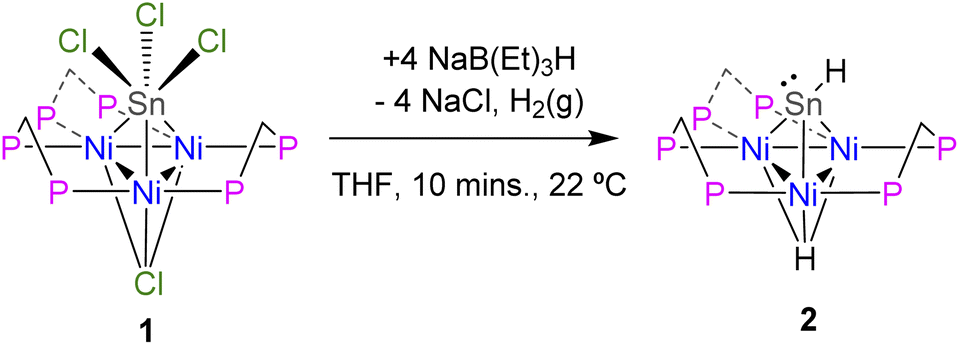 | ||
| Scheme 1 Synthesis of [Ni3(dppm)3(μ3-H)(μ3-SnH)], 2. | ||
Vapor diffusion of diethyl ether into a THF solution of 2 at −35 °C resulted in brownish-red X-ray quality crystals. X-ray diffraction revealed that cluster 2 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and features a μ3-Sn atom capping a triangular face consisting of three nickel atoms supported by three bridging dppm ligands (Fig. 1). Although the Sn–H hydrogen atom in 2 could not be precisely located in the difference electron-density map, the Sn–H linkage was unequivocally confirmed by 1H and 119Sn NMR spectroscopy (vide infra). Density functional theory (DFT) calculations and natural bond orbital (NBO) analysis also support this structural assignment (see ESI† for details).
and features a μ3-Sn atom capping a triangular face consisting of three nickel atoms supported by three bridging dppm ligands (Fig. 1). Although the Sn–H hydrogen atom in 2 could not be precisely located in the difference electron-density map, the Sn–H linkage was unequivocally confirmed by 1H and 119Sn NMR spectroscopy (vide infra). Density functional theory (DFT) calculations and natural bond orbital (NBO) analysis also support this structural assignment (see ESI† for details).
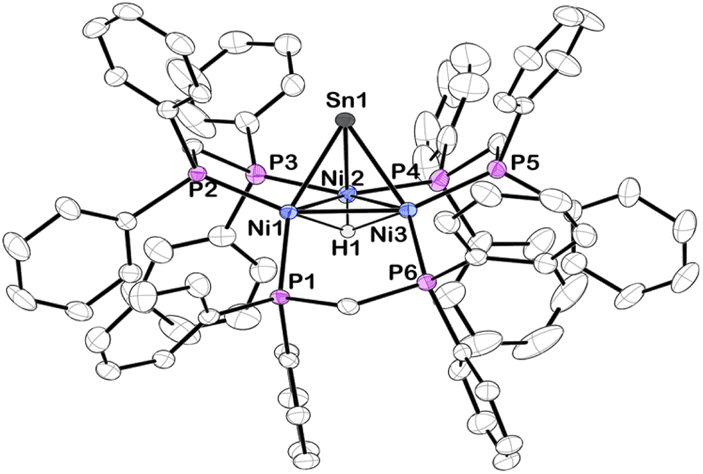 | ||
| Fig. 1 Solid-state structure of 2 as determined by single-crystal X-ray diffraction. Thermal ellipsoids were set at the 50% probability level. A diethyl ether molecule and carbon bound hydrogens were omitted for clarity. The hydride attached to Sn was not precisely located. | ||
Compared to 1, 2 exhibits substantially shorter Sn–Ni bond distances and considerably longer Ni–Ni bond distances (Table 1). While many low-valent Sn-hydrides form dimers,3,4,92 does not, likely as a result of the trinuclear Ni3(dppm)3 framework providing considerable steric bulk, an effect seen in other monomeric Sn–H complexes.27,28 The structure of 2 shows slight variations in Ni–Sn bond distances. Additionally, the Ni1–Ni3 bond distance is significantly shortened in comparison to the other Ni–Ni bonds (Fig. 1 and Table 1). This implies that, in the solid-state, the μ3 binding of the Sn–H is not entirely symmetric, and the interactions of Sn–H may be more localized on one nickel center. Similar variations in Ni–Sn bond distances are seen in the starting material, 1.25,26 These structural features are also observed in the results of DFT calculations. DFT and natural bond order (NBO) analysis for complex 2 (B3LYP/LANL2DZ) reveal that the Sn–H interacts principally with one nickel atom (Fig. 2, see also ESI†).
| Bond length [Å]/angle [°] | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Ni1–Ni2 | 2.4593(18) | 2.7445(6) | 2.4104(9) | 2.6840(5) | 2.5011(10) |
| Ni1–Ni3 | 2.4829(18) | 2.4449(5) | 2.4020(9) | 2.8886(5) | 2.5285(10) |
| Ni2–Ni3 | 2.4825(16) | 2.7315(6) | 2.3858(9) | 2.8227(5) | 2.6655(11) |
| Sn1–Ni1 | 2.6118(14) | 2.5187(5) | 2.7261(8) | 2.4423(4) | 2.4324(8) |
| Sn1–Ni2 | 2.5960(16) | 2.5502(5) | 2.5716(7) | 2.4419(4) | 2.5282(8) |
| Sn1–Ni3 | 2.6185(16) | 2.5348(5) | 2.7447(7) | 2.4607(4) | 2.4724(8) |
| ∠C–Sn–Ni1 | 143.57(8) | 162.02(16) | |||
| ∠C–Sn–Ni2 | 140.24(8) | 136.59(17) | |||
| ∠C–Sn–Ni3 | 132.11(8) | 125.91(16) |
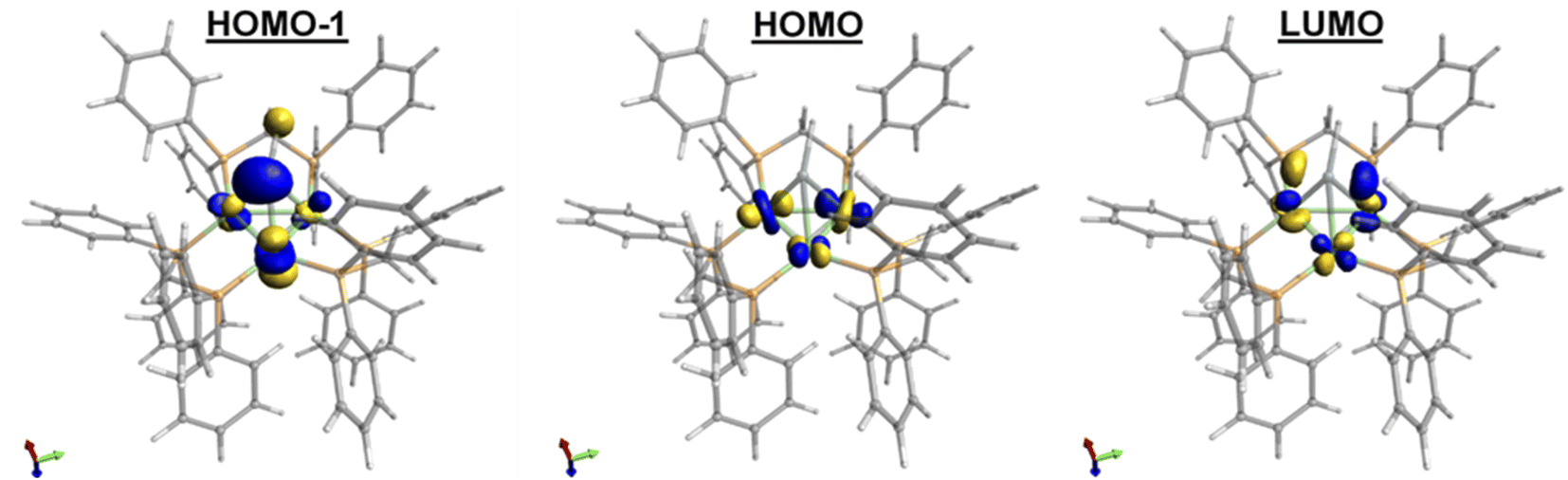 | ||
| Fig. 2 Computed molecular orbitals of 2, utilizing a B3LYP basis set and LANL2DZ functional showing the two highest energy occupied orbitals (HOMO−1 and HOMO) and the lowest energy unoccupied orbital (LUMO). | ||
Further details of the molecular and electronic structure were elucidated by 31P, 1H, and 119Sn NMR spectroscopy. The 31P NMR spectroscopy of 2 in benzene-d6 revealed symmetrical binding of the μ3-SnH and μ3-H capping groups on the NMR timescale as demonstrated by an isolated sharp singlet resonance at 23.4 ppm. The Ni–H resonance was located in the 1H NMR at −0.86 ppm and it appears as a heptet, featuring significant two-bond coupling (2JP−H) to the six phosphorus atoms coordinated to the nickel atoms. In addition, the Sn–H 1H NMR resonance was located at −0.42 ppm and it appears as a singlet. The assignment of this resonance as that belonging to the hydrogen-bound Sn was evidenced by the proton-coupled 119Sn NMR spectrum which, at room temperature, features a doublet at 2939.4 ppm (see ESI: S4†). The 1H decoupled 119Sn NMR spectrum contained a singlet resonance, indicating that there is one hydrogen atom bound to Sn, which is also further confirmed by integration of the peaks in the 1H NMR spectrum.
Satellite peaks arising from coupling of hydrogen to the 117Sn and 119Sn nuclei with 1J(Sn–H) values near 1900 Hz are normally observed for tetravalent Sn(IV) hydrides.3,29,30 In contrast, divalent Sn(II) hydride complexes typically exhibit much smaller coupling constants (1J(Sn–H)ca. 100 Hz) because in these Sn(II) hydrides most of the s-orbital electron density is localized to the lone pair on Sn.4,31–33 The 1J(117/119Sn–1H) coupling constant of 2 was found to be 360 Hz, consistent with a divalent Sn center. This assignment is corroborated by NBO analysis, which predicts that the Sn hybridization used to form the Sn–H linkage in 2 has about 20% s-character at Sn. Furthermore, DFT analysis indicates the presence of a lone pair on Sn in the HOMO−1 (Fig. 2). In addition, preliminary Mössbauer results on 2 depict a +2 oxidation state Sn center. Overall, NMR, computational studies, and preliminary Mössbauer results suggest that the Sn can be considered as sp2 hybridized with most of the s-orbital density localized on a lone pair on Sn.
To support experimentally the assignment of 2 as a Sn(II) hydride with a lone pair, we performed reactivity studies. Alkyl halides are known to undergo oxidative addition to Sn(II) complexes that feature a lone pair.34 Addition of 1 eq. BrCH2CH3 to 2 in THF at −20 °C, results in the oxidative addition of BrCH2CH3 at the Sn center and formation of [Ni3(dppm)3(μ3-H)(μ3-Sn((Br)(H)(CH2CH3)))], 3 in 75% yield (Scheme 2).
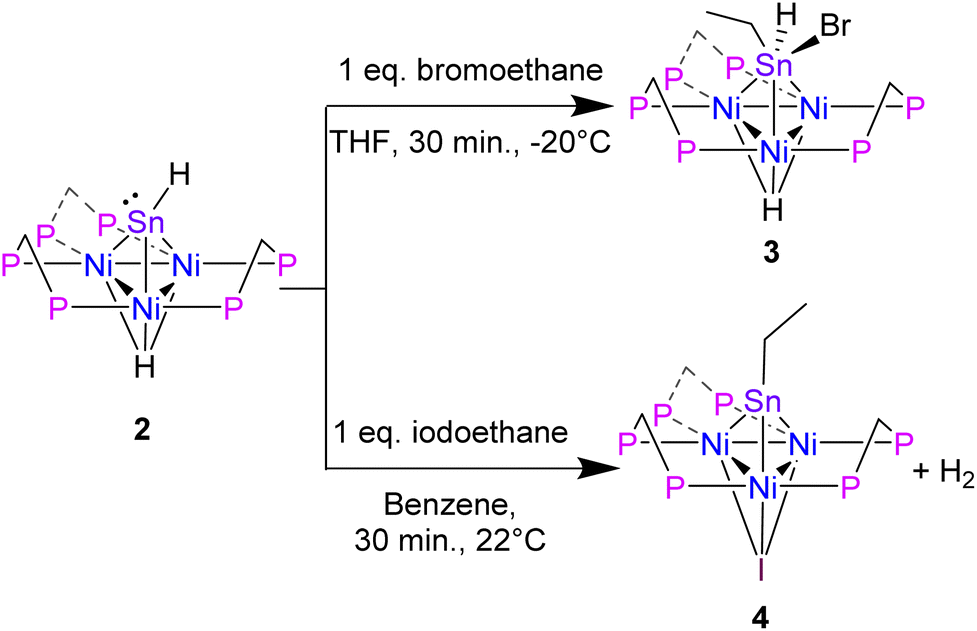 | ||
| Scheme 2 Synthesis of 3 and 4. | ||
The formation of 3 is characterized by a significant upfield shift in the 31P NMR spectrum compared to 2, with a resonance located at 4.2 ppm in benzene-d6. As for the 1H NMR signals, the Ni–H resonance shifts upfield to −4.70 appearing as a heptet, and the Sn–H resonance shifts downfield to 8.78 ppm, presumably a result of the close proximity to the Sn–Br bond. There is significant broadening in the 31P and 1H NMR spectra due to the break in symmetry from the three different functional groups attached to Sn. In comparison to 2, the proton-coupled 119Sn NMR of 3 depicts an upshifted doublet with a resonance located at 247.8 ppm and a 1J(Sn–H) = 993.6 Hz, suggesting a larger s orbital contribution from the Sn into the H atom bond (Table 2, also see ESI: S9†). The 1J(Sn–H) value of 3 is comparable to that reported by the Rivard group for a Sn(II) dihydride transition metal coordinated complex, IPr–SnH2–W(CO)5 (IPr = [(HCNAr′)2C:]; Ar′ = 2,6-iPr2C6H3).16
| 31P, δ (ppm) | Ni–H, δ (ppm) | Sn–H, δ (ppm) | 119Sn, δ (ppm) | 1 J (Sn–H) (Hz) | |
|---|---|---|---|---|---|
| 2 | 23.4 | −0.86 | −0.42 | 2939.4 | 346.7 |
| 3 | 4.2 | −4.80 | 8.77 | 248.0 | 993.6 |
| 4 | 34.1 | 2115.1 | |||
| 5 | 31.0 | 3.54–3.62 | 1837.8 |
Vapor diffusion of pentane into a THF solution of 3 at −20 °C resulted in red colored X-ray quality crystals (Fig. 3). The hydride attached to Sn was not precisely located. There appears to be significant asymmetry of the μ3-SnHBr(CH2CH3) binding to the trinuclear nickel core, suggested by the significant elongation of the Sn–Ni1 and Sn–Ni3 bonds versus the Sn–Ni2 bond (Table 1). Furthermore, the data indicates an overall shortening of the Ni–Ni bond distances more closely resembling that of cluster 1 than 2 (Table 1).
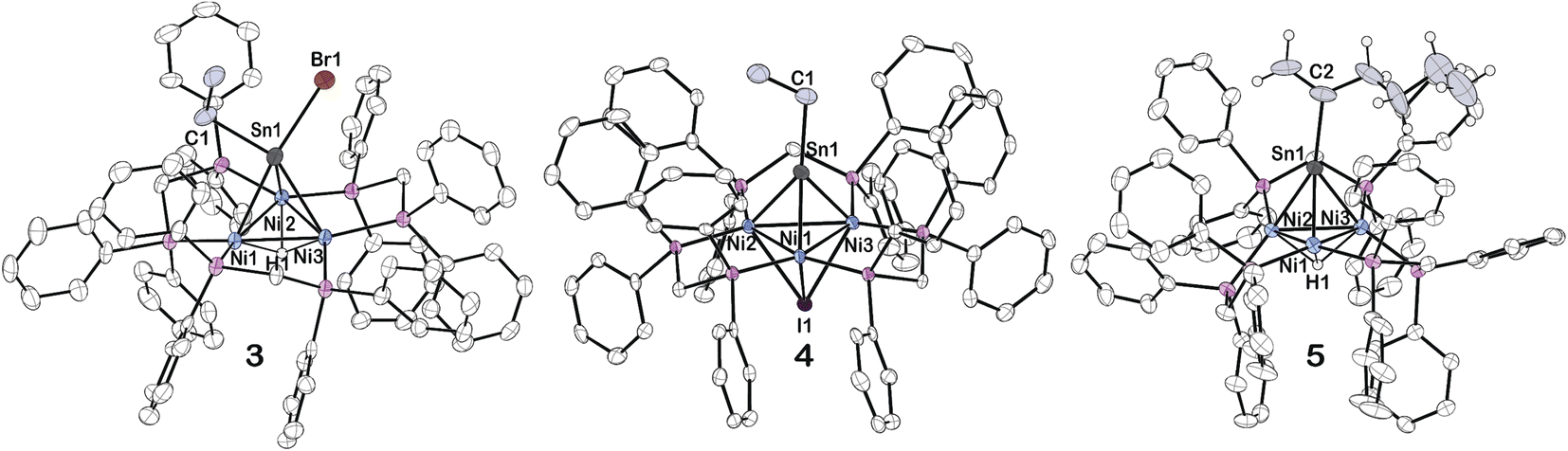 | ||
| Fig. 3 Solid-state structure of 3, 4 and 5 as determined by single-crystal X-ray diffraction. Thermal ellipsoids were set at the 50% probability level. A solvent molecule and carbon bound hydrogens were omitted for clarity. The hydride attached to tin was not precisely located in complex 3. Carbons atoms on bis(diphenylphosphino)methane ligands are colored white, while those on Sn are colored grey. | ||
Surprisingly, in an analogous reaction to forming cluster 3, treatment of 2 with 1 eq. ICH2CH3, releases H2 and generates an CH3CH2Sn-capped, nickel cluster with a μ3 iodide [Ni3(dppm)3(μ3-I)(μ3-Sn(CH2CH3))], 4, in 94% yield (Scheme 2). The formation of 4, is characterized by a significant downfield shift from the starting material in the 31P NMR to 34.1 ppm in benzene-d6, which is flanked by satellites arising from 2-bond coupling to the 119Sn nuclei (2J(Sn,P) = 98.8 Hz), while the 119Sn NMR shifts upfield to 2115.14 ppm (Table 2, see ESI: S14†).
Layering of pentane over a benzene solution of 4 at 22 °C resulted in brownish-red X-ray quality crystals (Fig. 3). The Sn–Ni bond distances and the C–Sn–Ni bond angles reveal an almost linear binding orientation of the μ3-SnCH2CH3 to the center of the trinuclear nickel core. Furthermore, the shortened Sn–Ni bond distances: 2.4423(4), 2.4419(4), and 2.4607(4), are of similar length to other transition metal coordinated stannylyne complexes in the literature, which primarily exhibit a linear arrangement unless there are considerable steric demands due to the ligand environment.35–40 We will report separately results of Mössbauer spectroscopy and computational studies to support this assignment.
Hydrostannylation is a well-known transformation in organic chemistry.41 Hydrostannylation utilizing tin(II) species has been demonstrated. These reactions feature transition metal and radical catalyst-free cis-hydrostannylation of the alkynes under mild conditions.42–44 Therefore, it was of interest to evaluate the reactivity of complex 2 with alkynes. 1 eq. of 1-hexyne was added to 2 in a benzene solution, which resulted in insertion to yield 1-hexen-2-yl-Sn-capped nickel cluster [Ni3(dppm)3(μ3-H)(μ3-Sn(C6H11))], 5, in 98% yield (Scheme 3). A study from the Power group demonstrated Sn(II) hydrostannylation of primary alkynes resulted in Sn binding to the terminal position.42 However, complex 5 exhibits a distinct preference to bind to the secondary position.
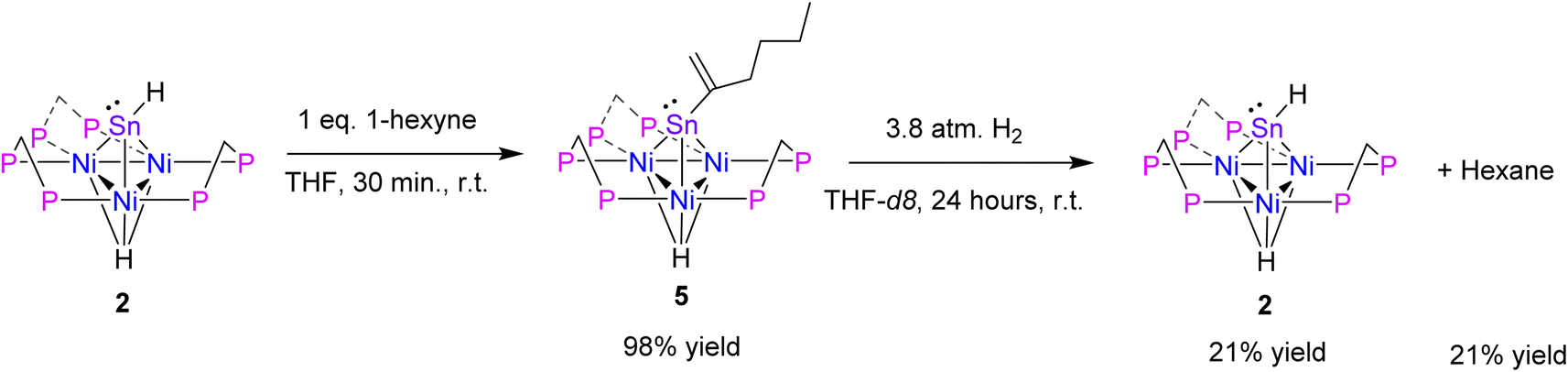 | ||
| Scheme 3 Synthesis of [Ni3(dppm)3(μ3-H)(μ3-Sn)(C6H11)], 5, and hydrogenation to [Ni3(dppm)3(μ3-H)(μ3-SnH)], 2. | ||
The formation of 5, is characterized by an upfield shift in the 31P NMR to 31.0 ppm from the starting material, 2. In comparison to 2, an upshifted Ni–H resonance in the 1H NMR was observed at −3.54 to −3.62 ppm, while the 119Sn NMR resonance shifted upfield to 1837.8 ppm. Vapor diffusion of pentane into a diethyl ether solution of 5 at −20 °C results in brown X-ray quality crystals (Fig. 3). The μ3-Sn(C6H11) features a significant bend from a 180-degree angle to the center of the three nickel clusters, suggesting the presence of a lone pair on the Sn, similar to cluster 2 (Table 1). This is supported by variations in the Sn–Ni bond distances in complex 5, in contrast to 4. Additionally, the Sn–Ni and Ni–Ni bond distances in 5 is similar to those of complex 2 (Table 1).
After isolating complex 5, hydrogenation of the 1-hexen-2-yl fragment and regeneration of 2 was attempted. Upon addition of H2 (g) to complex 5, complex 2 and hexane are formed in 21% yield by 1H NMR. In this process, an intermediate species is formed at −4.5 ppm via31P NMR and there is a shift in the Ni–H resonance to −6.2 ppm via1H NMR, before the regeneration of complex 2 (see ESI: S19–S21†). Thus far, this intermediate has proved elusive due to the chemical instability in solution. NMR stability studies of 5 in THF-d8 at 22 °C found that 92% of the initial integration of 1H NMR features were lost over 24 hours, leading to a variety of unidentified species (see ESI: S22 and S23†). Repeat studies in benzene-d6 yielded similar conclusions. This is an unexpected result, as previous studies demonstrating tin(II) hydrostannylation of alkynes only studied the formation of stable tin alkene species and have not shown the hydrogenation chemistry of these products.42–44 The literature indicates that, Sn has primarily been utilized as a stoichiometric reagent, however, this study demonstrates a platform that may lead to catalytic application of alkyne reduction by Sn–H utilizing H2 (g).41
Finally, the oxidative addition chemistry revealed by the novel low valent μ3-SnH capped trinuclear nickel cluster 2 raises the question of electronics pertaining to this species. Cyclic voltammetry (CV) of a solution of 2 in THF revealed two reversible, one-electron oxidations (at E = −0.8, −1.3 V vs. [Cp2Fe]0/+) and one partially-reversible one electron reduction process (E = −2.7 V vs. [Cp2Fe]0/+) (see ESI for CV: S27–S31†). Complex 2 is a formally 44-electron trinuclear cluster of the closo structural class and is therefore electronically deficient in accordance with Wade's rules.45,46 To elucidate the nature of these oxidation events, we computed molecular orbitals of 2 (Fig. 2). Analysis of the computed MOs suggests that the oxidation events involve depleting electron density from the d-orbital based Ni–Ni antibonding interactions, thereby providing relief from antibonding instability. The reduction process is best described as the introduction of electron density into the LUMO, which features electron density shared between the Ni3 core and Sn and d-orbital based Ni–Ni antibonding interactions. This is expected to increase Ni–Ni antibonding instability in the complex, which is consistent with the partial irreversibility observed for the redox process observed in the CV. Together, the electrochemical and DFT results of 2 suggest frontier d-orbitals interactions centered at the Ni3 core. Given that the coordination environment around the Ni3 cluster does not allow for activation by the Ni3 center, we propose these charge states may be mediated through the Sn center, potentially enabling the oxidative addition and unique hydrogenation chemistry displayed here.
Conclusion
In summary, this report describes the synthesis and structural analysis of a divalent Sn–H capped trinuclear nickel cluster. This cluster features three chemically reversible redox processes and may represent an example of a general and useful platform for isolating Sn–H species to investigate their electronic properties and chemical reactivity in greater detail. Cluster 2 displays a breadth of reactivity, including oxidative addition of alkyl halides and insertion of alkynes. Furthermore, the alkyne insertion product, 5, was found to undergo hydrogenation chemistry with H2 (g) to reform the starting material, 2, and hexane in 21% yield. While these reactivity studies are comparable to other tin(II) species that feature insertion of alkynes and oxidative addition at Sn; to our knowledge, we report the first example of tin(II) hydrogenation of the hydrostannylated alkyne.34,42–44 This demonstrates a platform that may lead to catalytic applications for the Sn–H mediated hydrogenation of unsaturated organic molecules. Ongoing studies of 2, 3, 4 and 5 are focused on Mössbauer characterization, DFT modeling and further chemical reactivity experiments. Results will be reported separately.Data availability
Crystallographic data for 2, 3, 4, and 5 have been deposited at the CCDC under 2102266, 2173131, 2169137, 2169160, respectively.†Author contributions
N. A. T. and C. P. K. conceived of the idea. N. A. T. developed the methodology. N. A. T. and Q. C. B. acquired the data for synthesis and characterization. N. A. T. and M. G. acquired and analysed the crystallographic data for compounds 2, 3, 4, and 5. J. M. P. and N. A. T. performed DFT studies for complex 2. A. A. M. facilitated with NMR spectroscopic studies. N. A. T., T. C., and F. M. B. acquired and analysed electrochemical data. N. A. T. and C. P. K. wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. A. T. acknowledges Micah Ziegler for invaluable discussions. N. A. T. acknowledges support from NSF for a Graduate Research Fellowship. Financial support from NSF is also gratefully acknowledged (CHE-1853908). The authors thank Theresa Block, Aylin Koldemir, and Rainer Pöttgen for Mössbauer investigations.Notes and references
- A. G. Davies, Organotin chemistry, John Wiley & Sons, 2006 Search PubMed.
- M. Pereyre, J.-P. Quintard and A. Rahm, Tin in organic synthesis, Butterworth-Heinemann, 2013 Search PubMed.
- E. Rivard, R. C. Fischer, R. Wolf, Y. Peng, W. A. Merrill, N. D. Schley, Z. Zhu, L. Pu, J. C. Fettinger, S. J. Teat, I. Nowik, R. H. Herber, N. Takagi, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2007, 129, 16197–16208 CrossRef CAS PubMed.
- B. E. Eichler and P. P. Power, J. Am. Chem. Soc., 2000, 122, 8785–8786 CrossRef CAS.
- Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef CAS PubMed.
- S. Wang, M. L. McCrea-Hendrick, C. M. Weinstein, C. A. Caputo, E. Hoppe, J. C. Fettinger, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2017, 139, 6586–6595 CrossRef CAS PubMed.
- S. Wang, M. L. McCrea-Hendrick, C. M. Weinstein, C. A. Caputo, E. Hoppe, J. C. Fettinger, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2017, 139, 6596–6604 CrossRef CAS PubMed.
- J. Schneider, C. P. Sindlinger, K. Eichele, H. Schubert and L. Wesemann, J. Am. Chem. Soc., 2017, 139, 6542–6545 CrossRef CAS PubMed.
- E. Rivard and P. P. Power, Dalton Trans., 2008, 4336–4343 RSC.
- A. Jana, H. W. Roesky, C. Schulzke and P. P. Samuel, Organometallics, 2010, 29, 4837–4841 CrossRef CAS.
- A. Jana, H. W. Roesky, C. Schulzke and A. Döring, Angew. Chem., Int. Ed., 2009, 48, 1106–1109 CrossRef CAS PubMed.
- T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028–3031 CrossRef CAS PubMed.
- O. T. Summerscales, C. A. Caputo, C. E. Knapp, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2012, 134, 14595–14603 CrossRef CAS.
- S. Wang, T. J. Sherbow, L. A. Berben and P. P. Power, J. Am. Chem. Soc., 2018, 140, 590–593 CrossRef CAS PubMed.
- A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed.
- S. M. I. Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson and E. Rivard, J. Am. Chem. Soc., 2011, 133, 777–779 CrossRef CAS PubMed.
- S. M. I. Al-Rafia, O. Shynkaruk, S. M. McDonald, S. K. Liew, M. J. Ferguson, R. McDonald, R. H. Herber and E. Rivard, Inorg. Chem., 2013, 52, 5581–5589 CrossRef CAS.
- J.-J. Maudrich, M. Widemann, F. Diab, R. H. Kern, P. Sirsch, C. P. Sindlinger, H. Schubert and L. Wesemann, Eur. J. Chem., 2019, 25, 16081–16087 CrossRef CAS PubMed.
- Q. Zhu, J. C. Fettinger and P. P. Power, Dalton Trans., 2021, 50, 12555–12562 RSC.
- M. M. D. Roy, A. A. Omaña, A. S. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Chem. Rev., 2021, 121, 12784–12965 CrossRef PubMed.
- S. M. I. Al-Rafia, A. C. Malcolm, R. McDonald, M. J. Ferguson and E. Rivard, Angew. Chem., Int. Ed., 2011, 50, 8354–8357 CrossRef CAS PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS.
- Q. Zhao, T. D. Harris and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 8293–8306 CrossRef CAS PubMed.
- E. V. Eames and T. A. Betley, Inorg. Chem., 2012, 51, 10274–10278 CrossRef CAS PubMed.
- B. K. Breedlove, P. E. Fanwick and C. P. Kubiak, Inorg. Chem., 2002, 41, 4306–4308 CrossRef CAS PubMed.
- E. Simón-Manso and C. P. Kubiak, Angew. Chem., Int. Ed., 2005, 44, 1125–1128 CrossRef.
- A. R. Leverett, V. Diachenko, M. L. Cole and A. I. McKay, Dalton Trans., 2019, 48, 13197–13204 RSC.
- T. J. Hadlington, M. Hermann, J. Li, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2013, 52, 10199–10203 CrossRef CAS PubMed.
- V. M. S. Gil and W. Von Philipsborn, Magn. Reson. Chem., 1989, 27, 409–430 CrossRef CAS.
- B. Wrackmeyer, Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, 1999, vol. 38, pp. 203–264 Search PubMed.
- R. E. Wasylishen and N. Burford, Can. J. Chem., 1987, 65, 2707–2712 CrossRef CAS.
- C. P. Sindlinger and L. Wesemann, Chem. Sci., 2014, 5, 2739–2746 RSC.
- L. W. Pineda, V. Jancik, K. Starke, R. B. Oswald and H. W. Roesky, Angew. Chem., Int. Ed., 2006, 45, 2602–2605 CrossRef CAS PubMed.
- M. J. S. Gynane, M. F. Lappert, S. J. Miles and P. P. Power, J. Chem. Soc., Chem. Commun., 1976, 256–257 RSC.
- A. C. Filippou, A. I. Philippopoulos and G. Schnakenburg, Organometallics, 2003, 22, 3339–3341 CrossRef CAS.
- A. C. Filippou, D. Hoffmann and G. Schnakenburg, Chem. Sci., 2017, 8, 6290–6299 RSC.
- J. D. Queen, A. C. Phung, C. A. Caputo, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2020, 142, 2233–2237 CrossRef CAS PubMed.
- M. Widemann, K. Eichele, H. Schubert, C. P. Sindlinger, S. Klenner, R. Pöttgen and L. Wesemann, Angew. Chem., Int. Ed., 2021, 60, 5882–5889 CrossRef CAS PubMed.
- A. C. Filippou, P. Ghana, U. Chakraborty and G. Schnakenburg, J. Am. Chem. Soc., 2013, 135, 11525–11528 CrossRef CAS PubMed.
- A. C. Filippou, P. Portius, A. I. Philippopoulos and H. Rohde, Angew. Chem., Int. Ed., 2003, 42, 445–447 CrossRef CAS PubMed.
- M. Alami, A. Hamze and O. Provot, ACS Catal., 2019, 9, 3437–3466 CrossRef CAS.
- M. L. McCrea-Hendrick, S. Wang, K. L. Gullett, J. C. Fettinger and P. P. Power, Organometallics, 2017, 36, 3799–3805 CrossRef CAS.
- T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, Chem. Sci., 2015, 6, 7249–7257 RSC.
- S. K. Mandal and H. W. Roesky, Acc. Chem. Res., 2012, 45, 298–307 CrossRef CAS PubMed.
- K. Wade, Chem. Commun., 1971, 792–793 RSC.
- D. M. P. Mingos, Nat. Phys. Sci., 1972, 236, 99–102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2173131, 2169137, 2169160 and 2102266. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04042e |
| This journal is © The Royal Society of Chemistry 2022 |