
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA versatile o-aminoanilide linker for native chemical ligation†
Iván
Sánchez-Campillo
 a,
Judit
Miguel-Gracia
a,
Periklis
Karamanis‡
b and
Juan B.
Blanco-Canosa
*a
a,
Judit
Miguel-Gracia
a,
Periklis
Karamanis‡
b and
Juan B.
Blanco-Canosa
*a
aInstitute for Advanced Chemistry of Catalonia (IQAC-CSIC), Jordi Girona 18-26, 08034 Barcelona, Spain. E-mail: juanbautista.blanco@iqac.csic.es
bDept. of Chemistry “G. Ciamician”, University of Bologna, Via Selmi 2, 40126, Bologna, Italy
First published on 26th August 2022
Abstract
Chemical protein synthesis (CPS) is a consolidated field founded on the high chemospecificity of amide-forming reactions, most notably the native chemical ligation (NCL), but also on new technologies such as the Ser/Thr ligation of C-terminal salicylaldehyde esters and the α-ketoacid-hydroxylamine (KAHA) condensation. NCL was conceptually devised for the ligation of peptides having a C-terminal thioester and an N-terminal cysteine. The synthesis of C-terminal peptide thioesters has attracted a lot of interest, resulting in the invention of a wide diversity of different methods for their preparation. The N-acylurea (Nbz) approach relies on the use of the 3,4-diaminobenzoic (Dbz–COOH) and the 3-amino-(4-methylamino)benzoic (MeDbz–COOH) acids; the latter disclosed to eliminate the formation of branching peptides. Dbz–COOH has been also used for the development of the benzotriazole (Bt)-mediated NCL, in which the peptide–Dbz–CONH2 precursor is oxidized to a highly acylating peptide–Bt–CONH2 species. Here, we have brought together the Nbz and Bt approaches in a versatile linker, the 1,2-diaminobenzene (Dbz). The Dbz combines the robustness of MeDbz–COOH and the flexibility of Dbz–COOH: it can be converted into the Nbz or Bt C-terminal peptides. Both are ligated in high yields, and the reaction intermediates can be conveniently characterized. Our results show that the Bt precursors have faster NCL kinetics that is reflected by a rapid transthioesterification (<5 min). Taking advantage of this major acylating capacity, peptide–Bt can be transselenoesterified in the presence of selenols to afford peptide selenoesters which hold enormous potential in NCL.
Introduction
Native chemical ligation (NCL) forms with exclusive selectivity an amide bond between a C-terminal peptide α-thioester (peptide–COSR) and an N-terminal cysteinyl-peptide (Cys-peptide) under reaction conditions resembling the physiological ones.1,2 The chemospecific nature of this reaction prevents the need for the side chain protection of peptides, and thus it can be used just as effectively in recombinant protein–COSR obtained by splicing of protein-intein fusions.3 Further elaborations of the NCL extended the reaction to other N-terminal residues,4–7 peptide selenoesters8 and selenoester-diselenide ligations.9 Since Fmoc-solid phase peptide synthesis (Fmoc-SPPS) is currently the most adopted methodology for peptide synthesis, the direct preparation of peptide thioesters is poorly efficient, necessitating other approaches that overcome the piperidine-mediated aminolysis of the peptide–COSR. Some of these methods include the Kenner's sulfonamide,10,11 pyroglutamyl imides,12 cyclic urethane at Ser side chain-backbone sites,13 acyl hydrazides,14 SEAlide15 and other intramolecular O or N-to-S acyl scaffolds,16–19 benzotriazoles (Bt)20,21 and the N-acylurea (Nbz).22–24 These approaches rely on the use of thioester precursors (also called crypto-thioesters) that, upon activation under specific conditions, can be trapped in the presence of thiols that form the active peptide–COSR. For instance, the SEAlide is masked off through an intramolecular disulfide bond, and the addition of TCEP initiates the reduction of the disulfide bridge that allows the N-to-S acyl shift and subsequent transthioesterification.15 On the other hand, acyl hydrazides are popular due to their straightforward synthesis and activation, either by nitrite oxidation to yield acyl azides14 or acetyl acetone condensation to afford acyl pyrazoles.25The Nbz method has been also used for the chemical synthesis of a broad range of proteins. Disclosed in 2008,22 it is based on the 3,4-diaminobenzoic (Dbz–COOH) scaffold (Scheme 1). Monoacylation of the Dbz–CONH–resin, preferentially at the m-amine, followed by stepwise chain elongation, results in a peptide1–Dbz–CONH–resin (1a). Then, the free amine of the o-aminoanilide 1a is acylated with p-nitrophenyl chloroformate, and the resulting 3-peptidyl-4-(pNO2-phenyloxycarbonyl)Dbz–CONH–resin undergoes intramolecular cyclization under basic conditions to yield the peptide1–Nbz–CONH–resin product (Scheme 1 route A). After TFA-mediated acidolytic cleavage, the unprotected peptide1–Nbz–CONH2 (2a) is obtained in solution. This acyl-Nbz–CONH2 activated peptide 2a can react with Cys-peptide2 at neutral pH, although the slow ligation rates rather reduce the synthetic applicability of the direct condensation due to a faster hydrolysis. However, in the presence of exogenous thiols, the rapid thioesterification of 2a streamlines the NCL in high conversions (>95%).22
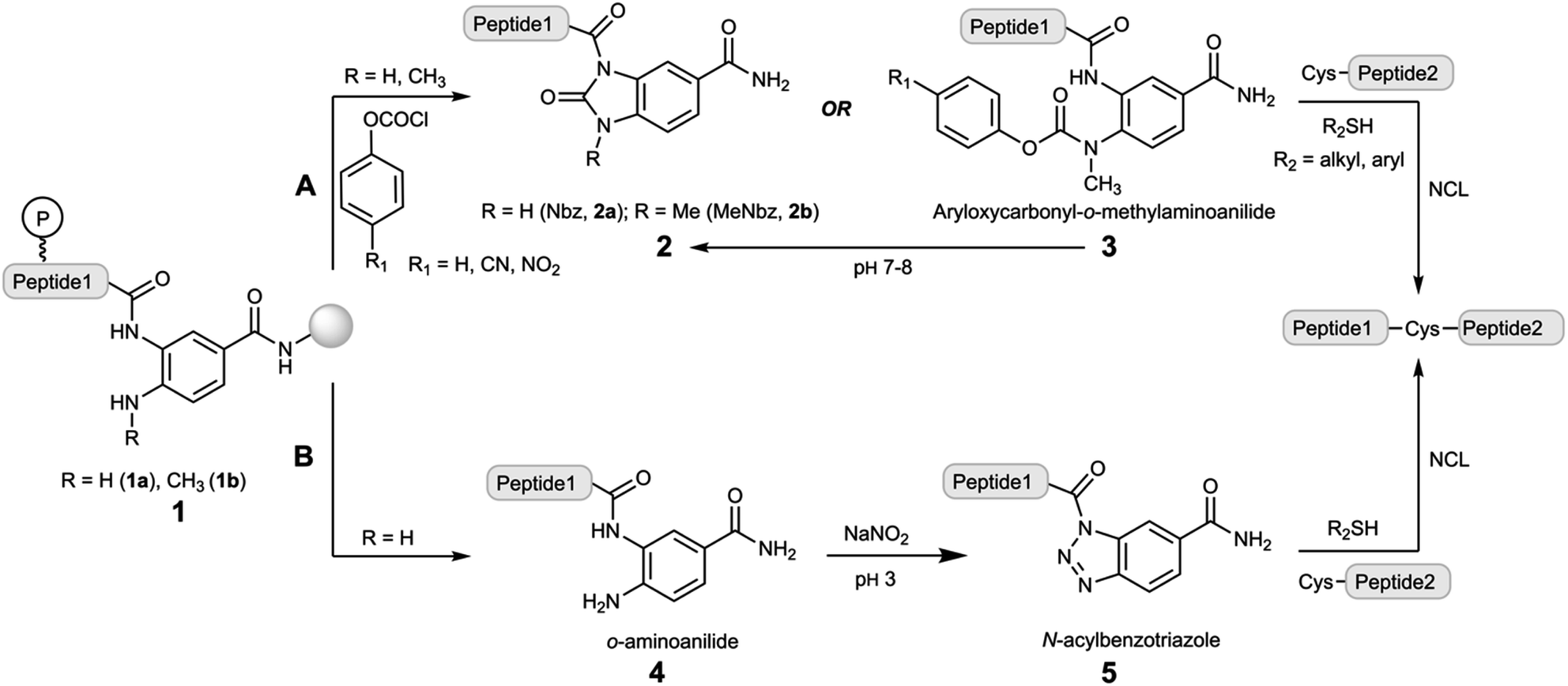 | ||
| Scheme 1 Previous work: synthetic avenues of 1,2-diaminobenzoyl derivatives for chemical protein synthesis. Following the route A, the peptide1–Dbz/MeDbz–CONH–resin is converted in the peptide1–Nbz/MeNbz–CONH2 on resin or in solution through a phosgenation-type process. Next, this peptide is ligated through NCL to another Cys-peptide2. In the route B, the peptide1–Dbz–CONH2 is transformed in peptide1–Bt–CONH2 and then ligated to a Cys-peptide2. P = protecting group. | ||
It is worth mentioning that during the Fmoc-SPPS, the o-aminoanilide of 1a remains mostly unmodified although unprotected. However, under specific conditions, i.e. microwave-assisted Fmoc-SPPS, use of basic coupling protocols and, especially, fragments containing Gly-rich sequences, the o-aminoanilide is acylated precluding further conversion to 2a.23,26 Therefore, apart from considering other strategies that involve the orthogonal protection of the o-aminoanilide,26,27 the methylation of the p-amino group provided a new linker, 3-amino-4-(methyl)aminobenzoic acid (MeDbz–COOH), that overcame the overacylation while it still undergoes MeNbz formation on resin23 (2b) or in aqueous solution (3, Scheme 1 route A).24 Several proteins have been synthesized using the Nbz/MeNbz methodology including ubiquitin chains,28 cyclotides,29 α-synuclein,30 the human selenoproteins SELW31 and F,32 and the transactivation domain of p53.33
In addition, the o-aminoanilide peptide1–Dbz–CONH2 (4) can undergo a chemoselective oxidation by sodium nitrite at acidic pH to give a peptide1–Bt–CONH2 (5) species, that is highly acylating and can be also intercepted by thiols to form a peptide–COSR (Scheme 1 route B).20,21,34 Although this route B has not received similar attention as for route A, the synthetic potential has been underestimated, particularly its use in sequential NCL.35
Besides CPS, the Nbz/MeNbz and Bt linkers have found other important applications, enabling the access to posttranslational modifications at the C-terminal peptide side of high structural and biomedical relevance, including C-terminal peptide esters and N-alkylamides and36–38 head-to-tail and side chain-to-tail cyclic peptides.39–41
From a synthetic point of view, it would be very attractive to use a single 1,2-diaminobenzene-type moiety for the preparation of peptides featuring C-terminal Nbz and Bt-type functionalities that combines the advantages of Dbz–COOH and MeDbz–COOH. Here, we show that C-terminal peptides amidated with 1,2-diaminobenzene (Dbz) can be efficiently converted in peptide–Nbz or peptide–Bt derivatives from the same initial peptide–Dbz–PAL–resin (PAL = 4-(4-(aminomethyl)-3,5-dimethoxyphenoxy)butanoic/pentanoic acid) precursor.
Results and discussion
Fmoc-SPPS of peptide–Dbz–PAL
Despite the Dbz–COOH compatibility with the Nbz and Bt approaches, the undesired side-acylations of the o-aminoanilide required the use of orthogonal protection for the p-amino group.26,27 Nevertheless, a facile and straightforward strategy based on a robust o-aminoanilide derivative would increase the final yield and purity. Thus, the Dbz group has been previously incorporated into different TFA-labile handles including Wang,42 2-Cl-trityl43 and Rink,43 to synthesize peptide–Bt derivatives. Unfortunately, when the synthesis was carried out, the steric hindrance led to incomplete acylations with p-nitrophenyl chloroformate in the peptide–Dbz–2-Cl-trityl–resin and peptide–Dbz–Rink–resin, while due to the nature of the carbamate bond, is not permitted in the peptide–Dbz–Wang–resin.42Then, we surmised that a labile aminomethyl-type linker could protect the Dbz from secondary acylations while still enabling the carbamoylation with chloroformate derivatives. The PAL linker is commercially available and widely used in Fmoc-SPPS.44 Its acid lability is in between the 2-Cl-trityl and the Rink, and importantly, it can provide a similar selectivity like MeDbz–COOH, though sufficiently reactive towards chloroformates and alike compounds.
Following a similar synthetic route as for the synthesis of Fmoc–MeDbz–COOH,23 2-fluoronitrobenzene was added to the PAL–resin to give the (2-PAL)nitrobenzene–resin (6, Scheme 2A). The reduction of the nitro group of 6 was efficiently achieved in PEG and PEG–polystyrene resins using Na2S2O4 as a reducing agent under basic (K2CO3) aqueous conditions (80–90%). However, due to the hydrophobic characteristics and low swelling properties of the polystyrene in H2O, this route gave poor yields. The reduction was significantly improved in a DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1) mixture using a transfer agent such as the tetrabutylammonium hydrogen sulfate, achieving conversions around 75% on the different types of polystyrene resins tested (see the ESI† for the resin manufacturers).45
:H2O (1:1) mixture using a transfer agent such as the tetrabutylammonium hydrogen sulfate, achieving conversions around 75% on the different types of polystyrene resins tested (see the ESI† for the resin manufacturers).45
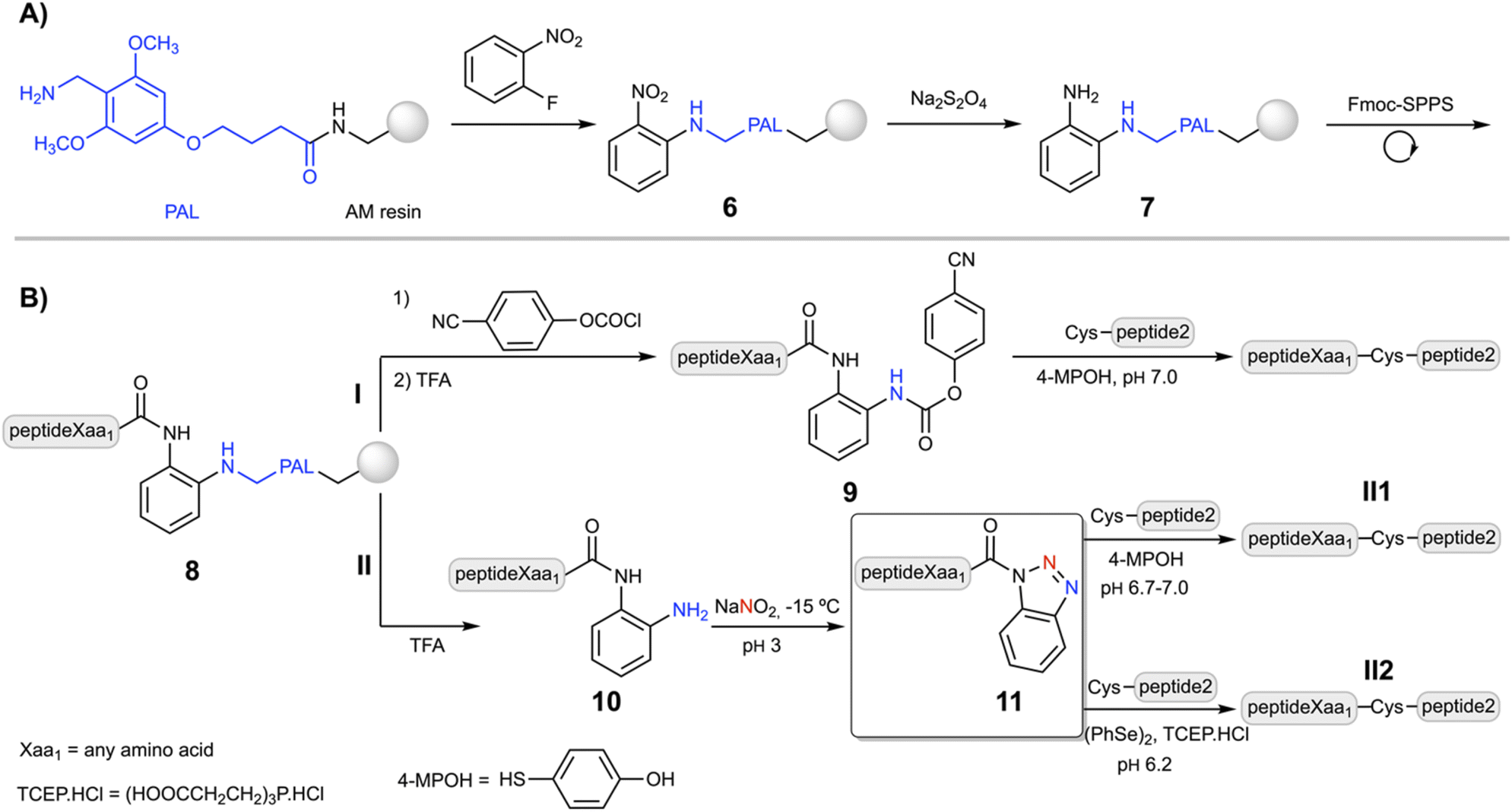 | ||
| Scheme 2 This work: (A) the 1,2-diaminobenzene–PAL strategy developed in this work for the synthesis of o-aminoanilide–PAL peptides. (B) Route I: synthesis of the peptideXaa1–(pCN-Phoc)Dbz and NCL. Route II: preparation of the peptideXaa1–Dbz precursor of the peptideXaa1–Bt and NCL mediated by peptideXaa1–(4-MPOH) thioesters (II1) and peptideXaa1–COSePh selenoesters (II2). | ||
The coupling of the first amino acid was accomplished in high yield (>99%) using HATU, although it is not strictly necessary for those amino acids with less bulky side chains. Fmoc-SPPS and chain elongation of 7 was carried out under standard conditions, using HBTU/DIEA or DIC/oxyma couplings (5 eq. of amino acid and the coupling agent), without noticeable branching products, confirmed by the synthesis of the LYRAG5–Dbz peptide (Fig. S1†). Acylation of peptideXaa1–Dbz–PAL–resin (8, Xaa1 = any amino acid, Scheme 2B) with p-nitrophenyl chloroformate followed by DIEA-induced cyclization gave the peptideXaa1–Nbz–PAL–resin peptide, confirmed by the yellow color of the p-nitrophenolate anion. Cleavage of the resin with TFA/scavengers (95%, v/v) yielded the expected peptideXaa1–Nbz product (LYRAF–Nbz, Fig. S2†). However, based on our own experience and the results of others in the preparation of C-terminal Nbz–CONH2 and MeNbz–CONH2 peptides (usually >30-mer), which can be slightly more problematic due to the intrinsic nature of the sequence,46 we decided to adopt a more reliable strategy. This approach is based on the C-terminal (pCN-Phoc)MeDbz–CONH2 peptides that undergo intramolecular cyclization to form the N-acylurea under aqueous conditions (Scheme 1 route A, conversion of 3 to 2).24
Thus, after acylation of 8 with p-cyanophenyl chloroformate, the peptideXaa1–(pCN-Phoc)Dbz–PAL–resin (peptideXaa1 = LYR(A/I)Xaa1; pCN-Phoc = pCN-phenyloxycarbonyl) was cleaved and simultaneously side chain deprotected using TFA/TIS/H2O (95:2.5:2.5%, v/v/v) during two and a half hours to give the peptideXaa1–(pCN-Phoc)Dbz (9) in good yields (>85%) independently of the C-terminal amino acid, which indicates a wide scope of the method (92% for LYRAG–(pCN-Phoc)Dbz, Fig. S3†). Alternatively, cleavage of 8 (Scheme 2B route II) afforded the desired peptideXaa1–Dbz (10) in similar good overall yields (>90%) and crude purities, as shown for LYRAG–Dbz (95% crude recovery, Fig. S14†).
Chemical ligations with peptideXaa1–(pCN-Phoc)Dbz and peptideXaa1–Dbz
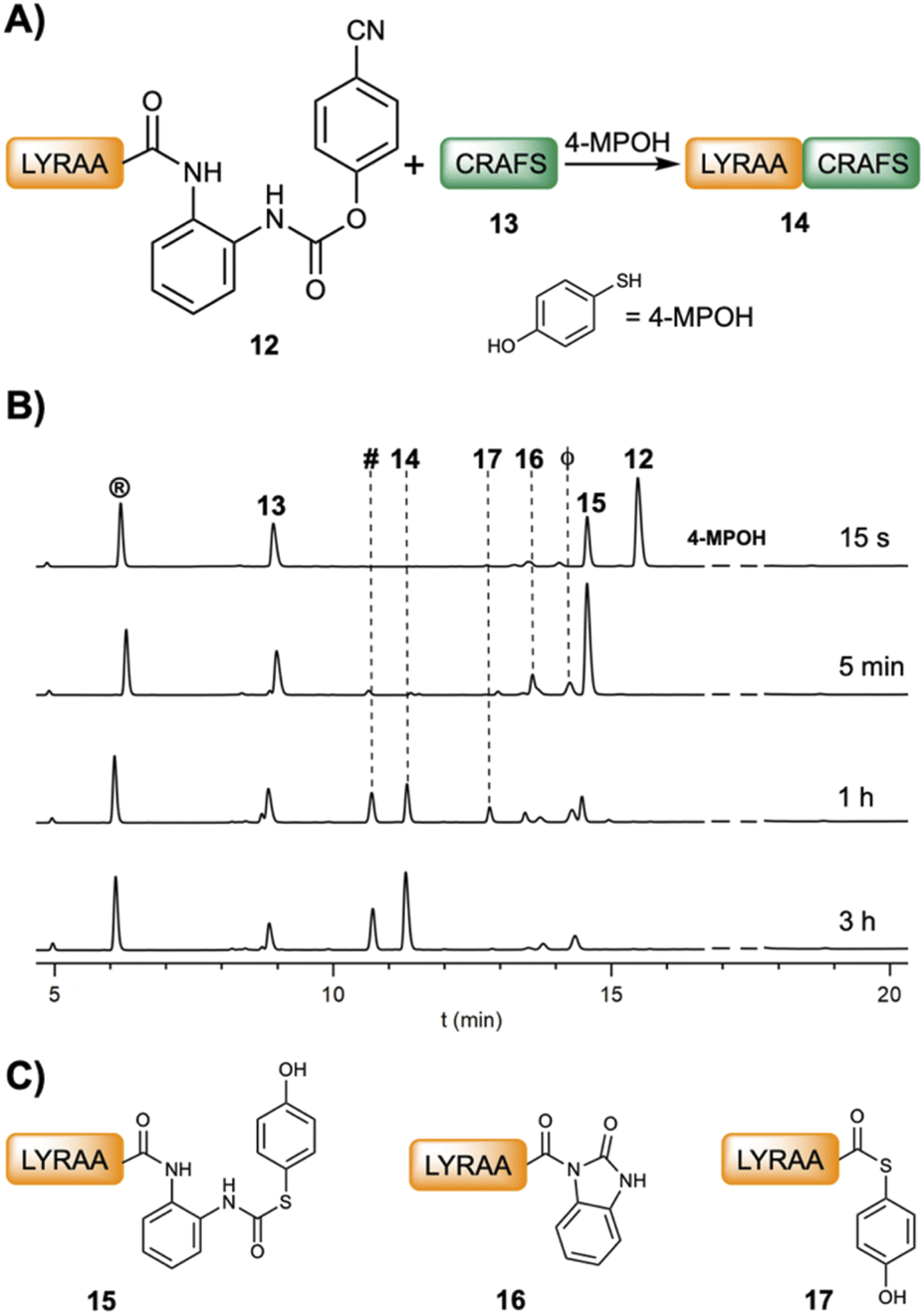 | ||
| Fig. 1 (A) Scheme of the NCL between LYRAA–(pCN-Phoc)Dbz (12, 2 mM) and CRAFS (13, 3.0 mM) in 6 M Gdm·HCl, 0.2 M NaPhos, 0.2 M 4-MPOH, and 50 mM TCEP·HCl at pH 7.0. (B) HPLC traces at 220 nm of the ligation at different times. ® = L-Tyr, # = 2-benzimidazolinone (Nbz); ϕ = p-cyanophenol. (C) Chemical structures of the intermediates LYRAA–(pOH-phenylthiocarbonyl)Dbz (15), LYRAA–Nbz (16) and LYRAA–(4-MPOH) (17). | ||
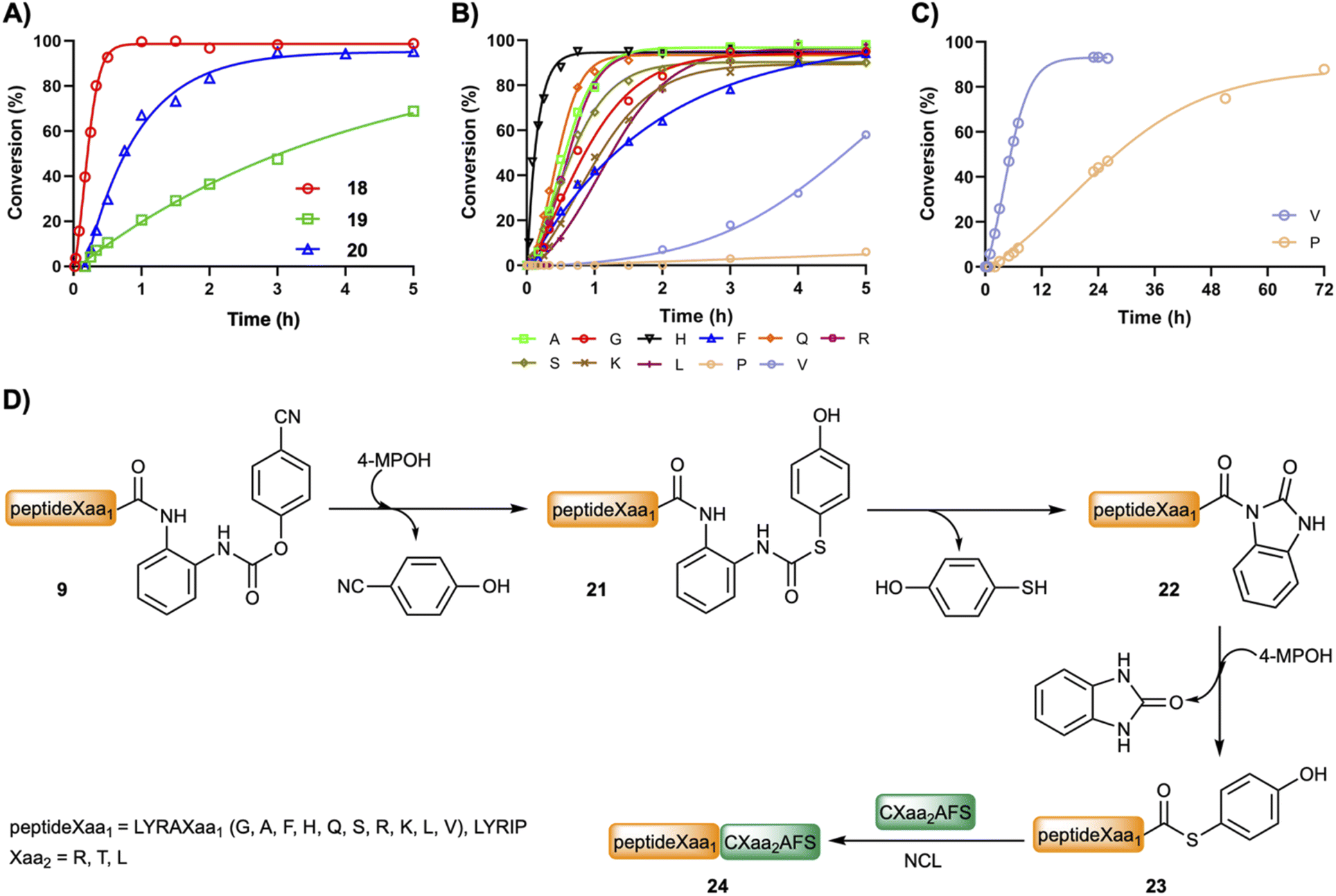 | ||
| Fig. 2 (A) Kinetics of the ligation between CLAFS and LYRAG–(pCN-Phoc)Dbz–CONH–G (18) and LYRAG–(pCN-Phoc)MeDbz–CONH–G (19) and LYRAG–(pCN-Phoc)Dbz (20). (B) Plot of the fraction ligated of LYRAXaa1CXaa2AFS. (C) Formation of LYRAVCTAFS and LYRIPCTAFS. (D) Plausible mechanism for the ligation. Reactions were carried out at pH 6.8–7.0 in 6 M Gdm·HCl and 0.2 M NaPhos at 22 °C. Concentrations of the peptides were: [LYRAXaa1–(pCN-Phoc)Dbz, LYRIP–(pCN-Phoc)Dbz] = 2 mM; [CXaa2AFS] = 3–3.5 mM. | ||
Fig. 2B and C show the product formation for the NCL between model peptides featuring different C-terminal amino acids (peptideXaa1–(pCN-Phoc)Dbz, 2 mM LYRAXaa1, Xaa1 = G, A, F, H, Q, S, K, L, V and LYRIP–(pCN-Phoc)Dbz) and CXaa2AFS (3–3.5 mM; Xaa2 = R, T, L), representative of model ligations displaying diverse kinetics, under aqueous conditions (6 M Gdm·HCl, 0.2 M NaPhos, 0.2 M 4-MPOH, 50 mM TCEP·HCl, pH = 6.8–7.0, and 22 °C). In all reactions, the conversion reached over 90% or close (88% for Pro) with only limited hydrolysis product as the major side product. Inspection of the kinetics showcased a sigmoidal plot-type behavior in which the ligated product started to form at around 5 min (Xaa1 = G, A, F, Q, S, L, H at 1 min) or even later (V at 1 h and P at 3 h). All the intermediate species formed during the ligation of 12 (Fig. 1B and C) were also observed (Fig. S28–S38†). Cumulatively, the C-terminal amino acid does not have a strong influence in the p-cyanophenol-to-(4-MPOH) (21) exchange (<10 min, Fig. 2D). Then, the cyclization follows up to give the corresponding Nbz derivative (22) at a speed that, although it depends on Xaa1, is not rate limiting (Table S1†). Interestingly, in the absence of 4-MPOH, the cyclization in LYRAG–(pCN-Phoc)Dbz at pH 7.0 displayed a similar rate to LYRAG–(pOH-phenylthiocarbonyl)Dbz (Fig. S40†). However, we hypothesize that although it is conceivable that the formation of the N-acylurea (22) can occur through both species (9 and 21), the several-fold excess of 4-MPOH predominantly leads the cyclization via the thiocarbamate peptideXaa1–(pOH-phenylthiocarbonyl)Dbz derivative (21, Fig. 2D). Next, the peptideXaa1–Nbz (22) is intercepted by 4-MPOH yielding the thioester peptideXaa1–(4-MPOH) (23). This step is primarily rate limiting, as is clearly seen from the accumulation of LYRAV–Nbz and LYRIP–Nbz products during the reaction (Fig. 2C, S37 and S38†). Once the thioester is formed, it is ligated to yield the final peptideXaa1–CXaa2AFS (24). The slow transthioesterification step and the low effective concentration of LYRAV–(4-MPOH) and LYRIP–(4-MPOH) explain the longer reaction times, particularly significative in the case of LYRIP–(4-MPOH) (72 h), since this ligation achieves similar conversions in 20 h when the concentration of LYRIP–(4-MPOH) is in the millimolar range (Table 1).
| Entry | C-Terminal Xaa1 | Time (h) | Conversion (%) |
|---|---|---|---|
| a NCL was carried out at pH 6.7–7.0 in 6 M Gdm·HCl and 0.2 M NaPhos at 22 °C. Concentrations of the peptides were: [LYRAXaa1–Dbz, LYRIP–Dbz] = 2.2–2.5 mM; [CXaa2AFS] = 4.3–4.7 mM. | |||
| 1 | G | 0.5 | 93 |
| 2 | A | 1 | 99 |
| 3 | F | 1 | 90 |
| 4 | H | 0.5 | 92 |
| 5 | Q | 1 | 85 |
| 6 | K | 1 | 95 |
| 7 | R | 1 | 91 |
| 8 | S | 1 | 94 |
| 9 | L | 1 | 88 |
| 10 | V | 8 | 86 |
| 11 | P | 20 | 84 |
Finally, thioesterification in solution using the peptideXaa1–(pCN-Phoc)Dbz appears to be a better strategy for the preparation of peptideXaa1–COSR because it prevents the formation of the peptide–Nbz on resin followed by the cleavage, which seems slightly problematic in the preparation of some peptide–Nbz/MeNbz–CONH–G derivatives.46
The ligation could be carried out stepwise (Fig. 3) or in a two-step one-pot fashion (Table 1). For instance, in a stepwise manner, after the oxidation of LYRAA–Dbz (25, 30 min, Fig. 3) to afford LYRAA–Bt (26), the reaction is diluted in a larger volume of a 4-MPOH solution, which also quenches the excess of NaNO2, and the pH is adjusted to 6.7–7.0. Remarkably, the LYRAA–Bt fragment can be characterized by HPLC and mass spectrometry (MALDI-TOF). Then, CTAFS (27) is added to the resulting LYRAA–(4-MPOH) (17) to initiate the ligation, which is complete in under 1 h.
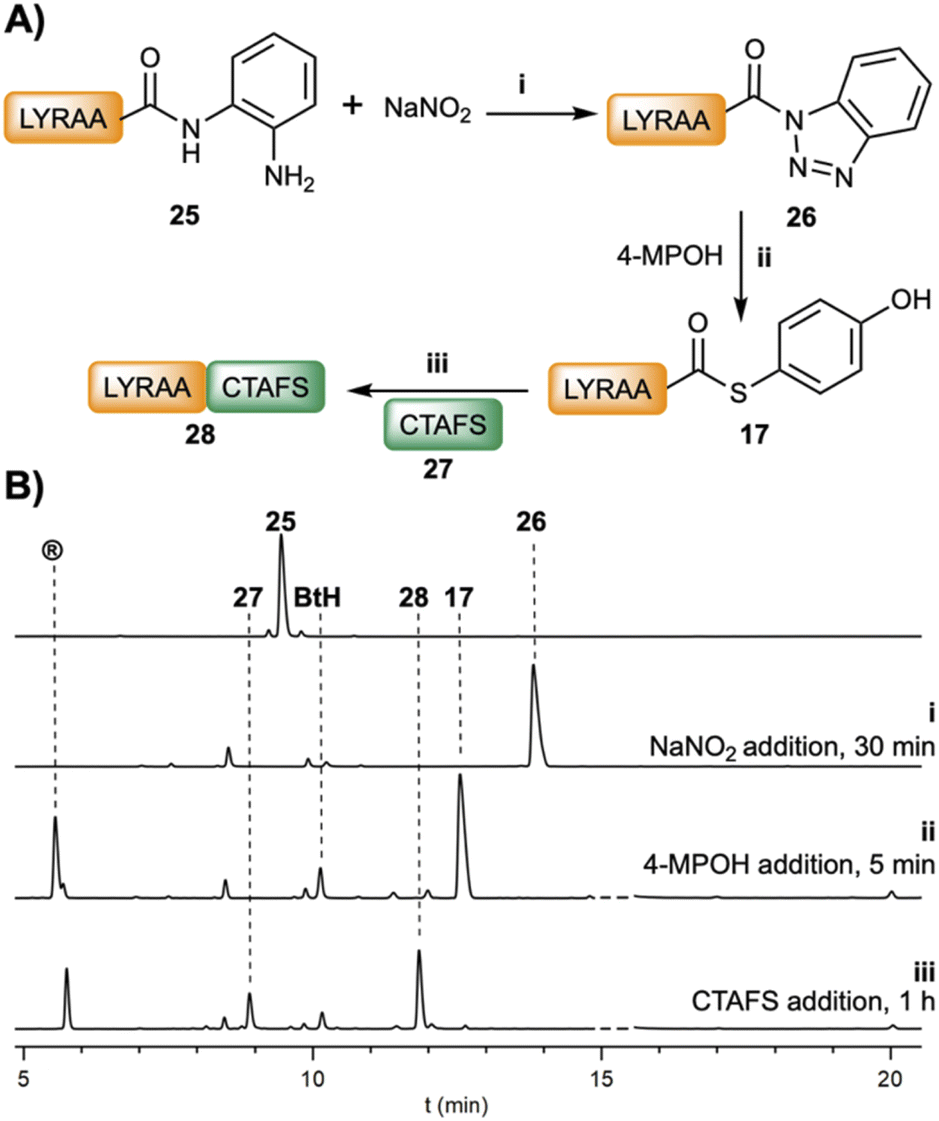 | ||
| Fig. 3 (A) Scheme of the stepwise ligation between LYRAA–Dbz (25, 2.5 mM) and CTAFS (27, 4.7 mM): (i) NaNO2 (3.5 eq.) oxidation at −15 °C, pH = 3, and 30 min; (ii) addition of 4-MPOH (200 mM) and TCEP·HCl (50 mM) at pH = 6.7; (iii) NCL, addition of CTAFS (27). (B) HPLC traces at 220 nm of the reaction after (i) (NaNO2 addition), (ii) (4-MPOH addition) and (iii) (CTAFS addition) steps. -- = 4-MPOH. | ||
Studies on the stability of LYRAG–Bt and LYRAG–Bt–CONH–G at pH 7 (Fig. S53†) revealed a higher stability of the former (t1/2 ≈ 16 min, 28% remaining peptide after 0.5 h vs. ∼2%), although much lower compared to LYRAG–Nbz (Fig. S40†). Nevertheless, bearing in mind that the transacylation reaction occurs on the time scale of seconds, we consider that the Bt approach is superior to the Bt–CONH–G strategy for the preparation of peptide–COSR. Indeed, the direct ligation of LYRIP–Bt and CTAFS gave an interesting 33% of the ligated LYRIPCTAFS peptide (Fig. S54†).48
When the reaction is performed in one pot (Table 1), the peptideXaa1–Dbz (2.2–2.5 mM) and CXaa2AFS (4.3–4.7 mM) were dissolved in buffer (6 M Gdm·HCl, 0.2 M NaPhos, pH = 3.0, ∼−15 °C). Then, NaNO2 (3.5 eq. pertaining to peptideXaa1–Dbz) was added, and the oxidation was carried out for 30 min. Of note, in the case of Xaa1 = P, for the Bt formation, it was necessary to leave it at 0 °C to achieve complete conversion. Following up, the reaction was diluted in a larger volume of 4-MPOH buffer (200 mM; final pH of the ligation is around 6.7–7.0) that started the ligation. It is noticeable that, under these ligation conditions, the N-terminal cysteine was not modified due to either the low pH of the oxidation or because the 4-MPOH can revert the S-nitrosylation of the β-mercapto group.14 The conversions achieved were over 90% for almost all peptides (except when Xaa1 = Q, L, V, P), and significantly faster than the Nbz-mediated ligations: 1 h for all peptides tested, except when Xaa1 was V (8 h) and P (20 h).
We have noticed that, although the Bt approach is compatible with stepwise or two-step one-pot reactions, in our experience, the chemical ligation of large peptide–COSR fragments (>20-mer) is more reliable by using a stepwise methodology. Thus, it is recommended that the peptide–Bt is first converted into a more stable peptide–COSR (R = alkyl) such as peptide–MESNa (MESNa = 2-mercaptoethanesulfonic acid sodium salt) and then ligated.
Recently, an approach based on unprotected C-terminal peptide pyrazoles prepared from peptide hydrazides yielded the corresponding peptide–COSeR,49 a methodology that has been further applied to a novel expressed protein–COSeR ligation.50 Accordingly, the synthesis of C-terminal peptide–COSeR would be greatly benefited by general methods that, starting from readily available selenoester precursors, facilitate an efficient conversion under smooth conditions. Following this premise, we investigated if the peptideXaa1–Nbz or the peptideXaa1–Bt could undergo acyl exchange in the presence of benzeneselenol. Thus PhSeH, generated in situ from the reduction of diphenyldiselenide (50 mM) using TCEP (100 mM), was incubated in the presence of LYRAF–Nbz (2 mM). However, only trace amounts of LYRAF–COSePh were detected after 2 h of reaction, confirming the previous results.8 In sharp contrast, LYRAF–Bt readily exchanged to give LYRAF–COSePh (<1 min, 30, Fig. 4B). This result was not surprising and reassures the superior acyl transfer characteristics of the Bt group. Stepwise NCL of LYRAF–Dbz (29, 2.2 mM, Fig. 4A), activated with NaNO2 (3.5 eq.), and CTAFS (27, 4.7 mM) yielded the ligated LYRAFCTAFS (32) product in 15 min (95% conversion). Very interestingly, under these NCL conditions (pH 6.0–6.2), a product (31) with a shorter HPLC retention time that had a mass identical to the ligated peptide (32), was detected during the first seconds of the reaction. We tentatively hypothesize and assign this compound to the S-acyl LYRAF(S)CTAFS (Fig. S55†) intermediate formed by the transacylation of LYRAF–COSePh (30) and CTAFS (27). Studies to determine the correct structure of this intermediate are underway.
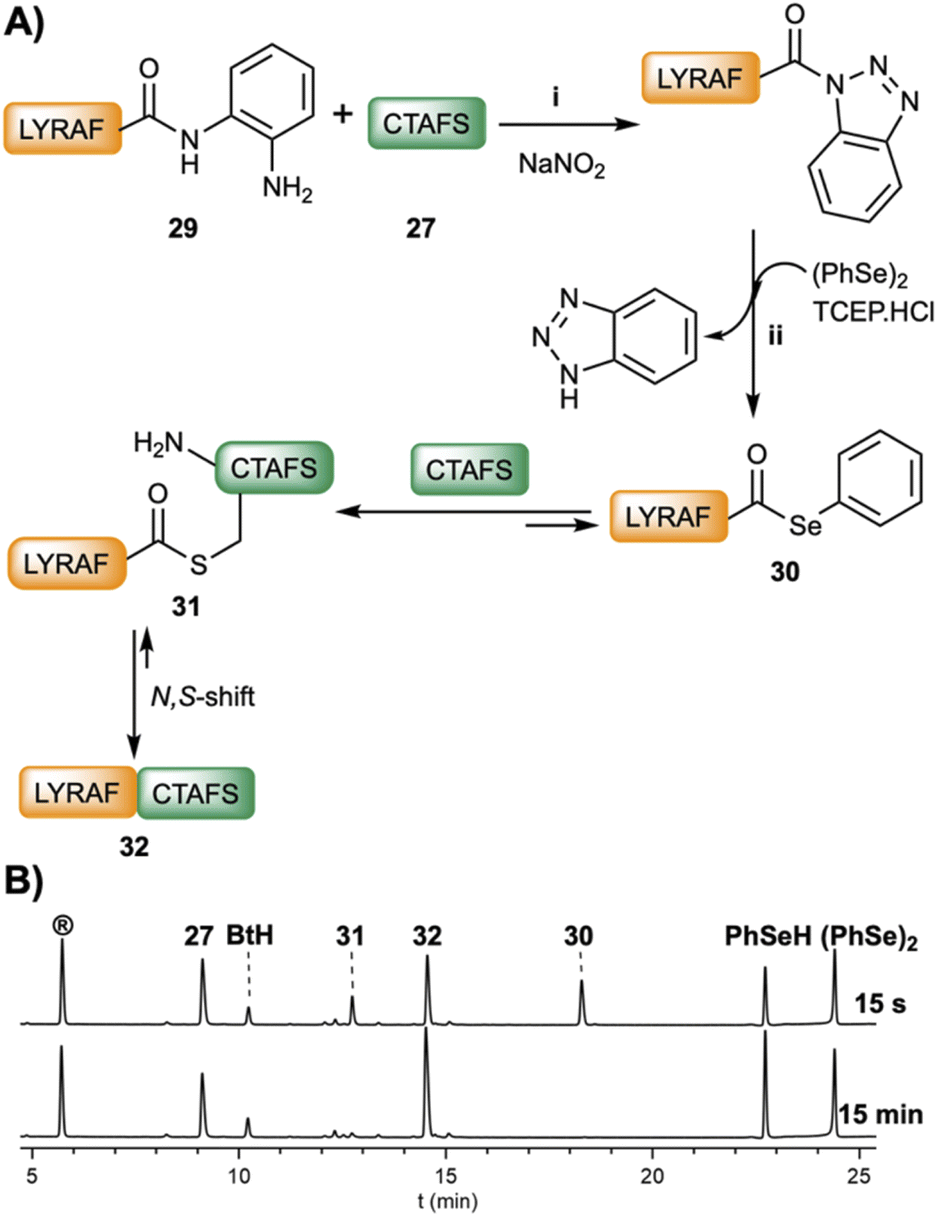 | ||
| Fig. 4 (A) Scheme of the stepwise ligation between LYRAF–Dbz (29, 2.2 mM) and CTAFS (27, 4.7 mM): (i) NaNO2 (3.5 eq.) oxidation at −15 °C; pH = 3, 30 min; (ii) addition of (PhSe)2 (50 mM) and TCEP·HCl (100 mM) at pH = 6.2. (B) HPLC traces at 220 nm of the reaction after addition of (PhSe)2 and TCEP·HCl at 15 s and 15 min. | ||
The fast and clean selenoester conversion imparted to the peptide–Bt intermediates is a very promising approach for selenoester-based ligations, especially at those sites in which the kinetics of the reaction is prohibitively slow or the concentration of the reaction partners is in the micromolar range.
Synthesis of the glucocorticoid receptor DNA binding domain
The glucocorticoid receptor (GR) is a transcription factor (TF) belonging to the zinc finger family. The GR is involved in the inflammatory response and cell growth, proliferation and survival.51 The DNA binding domain of the GR (GR DBD) spans a region of about 75 amino acids (418–493) and contains two zinc atoms tetracoordinate by four Cys residues, each Zn atom adopting a tetrahedral conformation (Fig. 5A and B). The GR proteins bind the GRE response elements (AGAACAaaaTGTTCT) as homodimers (Fig. 5A), activating or repressing the transcription of the target genes depending on the cell state.52–54 The GR has been and continue to be an important target for drug development.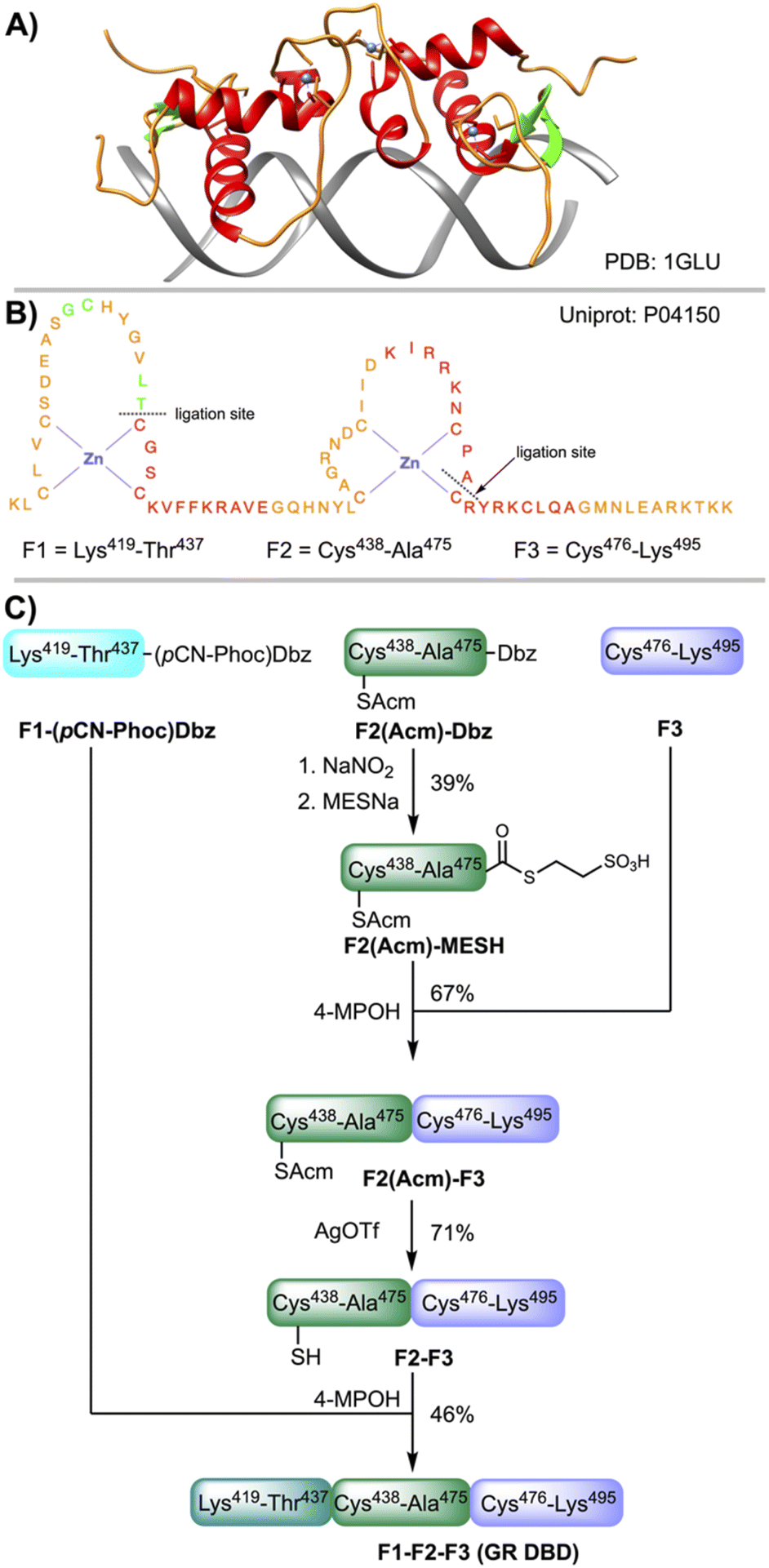 | ||
| Fig. 5 (A) X-Ray structure of the homodimer DNA binding domain (DBD) of the glucocorticoid receptor (GR) bound to the GRE site (AGAACAaaaTGTTCT).53 The red color denotes the α-helix, green the β-strand, orange the interconnecting loops and blue the Zn atoms. (B) Sequence of the Zn finger domain of the human GR and fragments F1, F2 and F3 used for CPS. (C) Synthetic scheme of the GR assembled linearly from three fragments: first F2 + F3; second F1 + F2–F3. | ||
For the CPS of the GR DBD (amino acids from 419 to 495), a linear synthesis comprising three fragments (Fig. 5B and C) was designed: F1 (419–437), F2 (438–475) and F3 (476–495). F1 and F2 were assembled on a ChemMatrix resin (Fig. S56 and S57†) and F3 in a 2-Cl-trityl functionalized resin (Fig. S58†). For F1 and F2, following the coupling of PAL and 2-fluoronitrobenzene, the nitro group was reduced to give the corresponding Dbz–PAL–resin. After chain elongation, F1 was acylated with p-cyanophenyl chloroformate. Next, the peptide F1–(pCN-Phoc)Dbz was cleaved from the resin and isolated in a good crude purity (∼60% by analytical HPLC) and recovery yield (70%). F2(Acm)–Dbz was prepared following a similar strategy (∼70% crude purity, 70% recovery yield), using the Acm instead of the thiazolidine protection for the sulfhydryl of the N-terminal Cys in order to prevent the nitrosylation of the α-amino group.
For the ligation of F2(Acm) and F3, although it could be carried out in a two-step one-pot approach, we found that the conversion to F2(Acm)–MESH enables a more reliable and scalable synthesis. Thus, F2(Acm)–Dbz was first transformed into the more stable F2(Acm)–MESH thioester (39% based on the F2(Acm)–Dbz crude) and then assembled with F3 in a good yield (67%, Fig. 5C, 6A(i and ii)). Remarkably, it is important to point out that despite the presence of the multiple Cys residues in the fragment F2, none of these were nitrosylated under the reaction conditions: (i) 10 eq. NaNO2, pH 3, and −15 °C and (ii) 100 mM MESNa, highlighting the exclusive chemospecificity of the oxidation–thioesterification process.
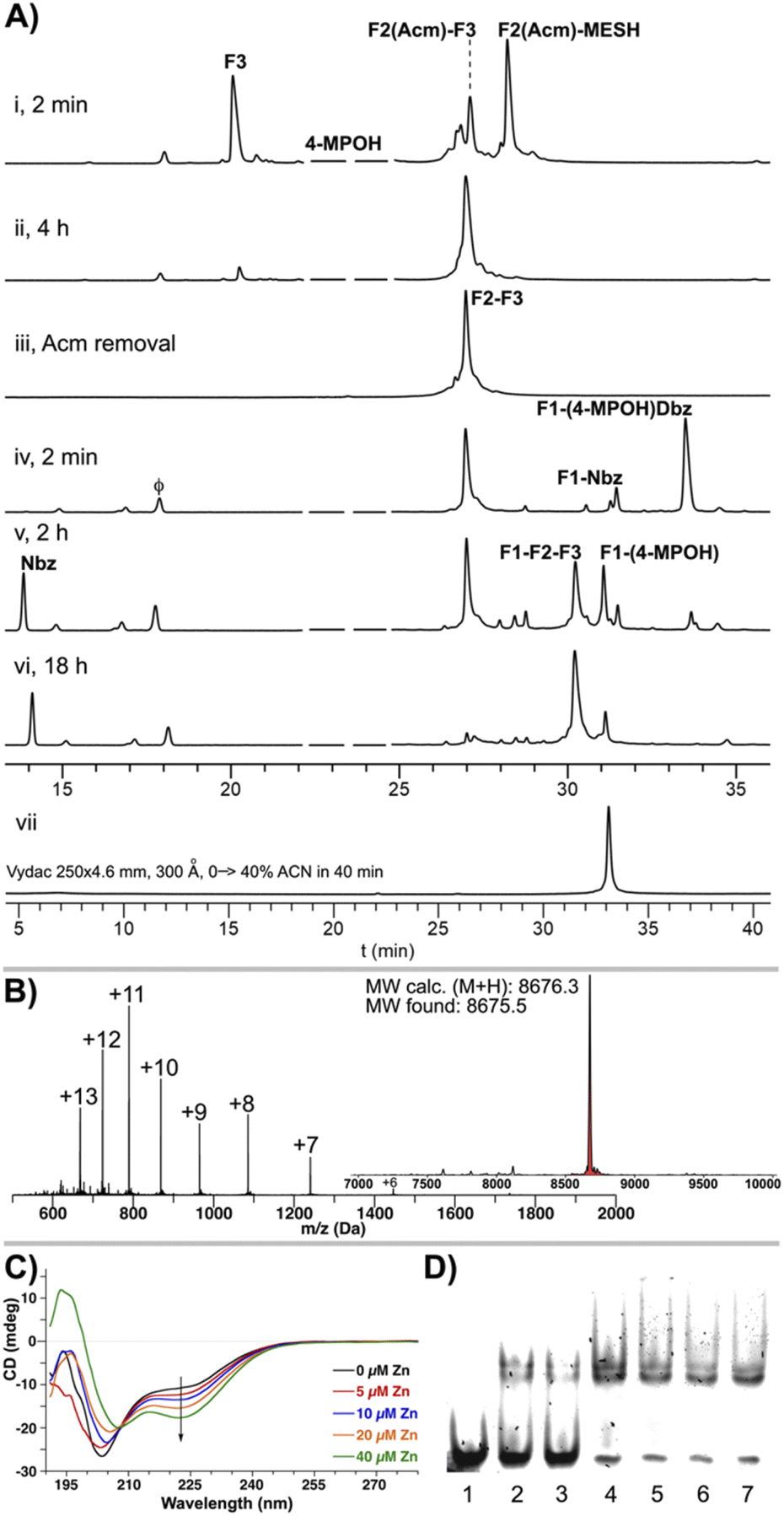 | ||
| Fig. 6 (A) HPLC traces at 220 nm of the CPS of the GR DBD: (i) F2(Acm)–MESH + F3 at 2 min and (ii) at 4 h, (iii) Acm removal from F2(Acm)–F3, (iv) F1-(pCN-Phoc)Dbz + F2–F3 at 2 min, (v) 2 h and (vi) 6 h, (vii) analytical HPLC trace at 220 nm of a purified sample of F1–F2–F3. ACN = acetonitrile. ϕ = p-cyanophenol. (B) ESIMS of F1–F2–F3 (Lys419–Lys495) and the deconvoluted mass spectrum. (C) Circular dichroism spectra of the GR DBD (35 μM) in the presence of increasing concentrations of ZnCl2: 0, 5, 10, 20, and 40 μM. (D) Electrophoretic mobility shift assay of the GR DBD and GRE. [GR DBD] lanes 1–7 = 0, 0.6, 0.8, 1.0, 1.4, 1.8, and 2.3 μM. | ||
Following the condensation of F2(Acm) and F3, the Acm group was removed under standard conditions (50 mM AgOTf in H2O:AcOH, Fig. 6A(iii), 71%). Next, F1–(pCN-Phoc)Dbz was ligated to F2–F3 to achieve the synthesis of the linear sequence (46%, Fig. 6A(iv–vi)). A closer inspection of the final ligation after 2 h of reaction (Fig. 6A(v)) revealed all the intermediates resulting from the mechanism depicted above (Fig. 2D), including also the possible cyclic thiolactones formed by the intramolecular attack of the internal Cys of F1 to the C-terminal Thr. The ESIMS corroborated the expected linear F1–F2–F3 GR Zn finger domain (Lys419–Lys495, Fig. 6B).
Zn finger domains adopt a particular folding upon incubation with Zn2+, represented by the prototypical TFIIIA from Xenopus.55,56 The metal in the Zn finger of TFIIIA is tetracoordinated by two Cys and two His residues. In the GR DBD, the metal also adopts a tetrahedral sphere, but instead it is coordinated by four Cys residues that fold the resulting structure in a different conformation. This configuration displays a particular circular dichroism signature that mainly reflects the contribution of the predominant α-helical motifs. When the synthetic GR DBD linear peptide (35 μM) was incubated in the presence of increasing concentrations of ZnCl2 (0–40 μM), an increase in the band at 222 nm was observed, while at 208 nm, it decreased, indicative of the formation of an α-helix induced by the nucleation of the Zn atom (Fig. 6C). Electrophoretic mobility shift assays in non-denaturing polyacrylamide–bisacrylamide gels showed a complex between the GR DBD and the GRE cognate sequence (Fig. 6D). In addition, MALDI-TOF mass spectrometry showed the Zn finger folded structure by the detection of the [M + Zn]+ and [M + 2Zn]+ ions (Fig. S64†).
Conclusions
In summary, here we have presented the Dbz, a simplified version of the o-aminoanilide moieties for CPS. Coupled with the PAL linker, it leverages the monoacylation selectivity imparted by the MeDbz–COOH and delivers o-aminoanilide peptides that can be cleaved directly from the resin to give the peptide–Dbz or first carbamoylated and then cleaved to afford the peptide–(aryloxycarbonyl)Dbz. The resulting activated peptide–Nbz and peptide–Bt are more stable than the corresponding peptide–Nbz/MeNbz–CONH–G and peptide–Bt–CONH–G but also labile enough to undergo thiolysis. In particular, the peptide–Bt is more functional and provides fast (<5 min) and scalable (mg quantities) peptide–COSR products. Very remarkably, the superior acylating properties of the peptide–Bt over the peptide–Nbz also permit the selenoesterification in the presence of benzeneselenol at a similar rate to that of the thioesterification. Selenoesters have found a wide range of applications in the recent years including faster kinetics at sterically demanding residues and diselenide-selenoester ligations. We foresee that the Dbz approach will find wide application within all the selenoester-based technologies for peptide ligations.Data availability
All experimental data and detailed experimental procedures are available in the ESI,† and from the corresponding author upon request.Author contributions
I. S.-C.: conceptualization, investigation, formal analysis, methodology, visualization and writing. J. M.-G.: investigation and formal analysis. P. K.: investigation and formal analysis. J. B. B.-C.: conceptualization, investigation, methodology, visualization, funding acquisition, supervision and writing. All authors have read, commented and given approval to the final version of the manuscript.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work has received funding from the grants RTI2018-096323-I00, PDI2021-128902OB-I00 (Spanish Ministerio de Ciencia e Innovación) and LCF/PR/HR20/52400006 (‘la Caixa’ Foundation). I. S.-C. thanks the AGAUR (Generalitat of Catalunya) for a predoctoral fellowship (2021 FI-B 00142), and P. K. thanks the Erasmus+ programme (European Commission) for a mobility fellowship. Rosaria Ragozzino (Erasmus+, University of Naples) is acknowledged for the preliminary electrophoretic experiments.Notes and references
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776 CrossRef CAS.
- P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923 CrossRef CAS PubMed.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705 CrossRef CAS PubMed.
- L. E. Canne, S. J. Bark and S. B. H. Kent, J. Am. Chem. Soc., 1996, 118, 5891 CrossRef CAS.
- J. Offer, C. N. C. Boddy and P. E. Dawson, J. Am. Chem. Soc., 2002, 124, 4642 CrossRef CAS PubMed.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046 RSC , and references cited therein..
- O. Fuchs, S. Trunschke, H. Hanebrink, M. Reimann and O. Seitz, Angew. Chem., Int. Ed., 2021, 60, 19483 CrossRef CAS PubMed.
- T. Durek and P. F. Alewood, Angew. Chem., Int. Ed., 2011, 50, 12042 CrossRef CAS PubMed.
- N. J. Mitchell, L. R. Malins, X. Liu, R. E. Thompson, B. Chan, L. Radom and R. J. Payne, J. Am. Chem. Soc., 2015, 137, 14011 CrossRef CAS.
- Y. Shin, K. A. Winans, B. J. Backes, S. B. H. Kent, J. A. Ellman and C. R. Bertozzi, J. Am. Chem. Soc., 1999, 121, 11684 CrossRef CAS.
- R. Ingenito, E. Bianchi, D. Fattori and A. Pessi, J. Am. Chem. Soc., 1999, 121, 11369 CrossRef CAS.
- A. P. Tofteng, K. K. Sørensen, K. W. Conde-Frieboes, T. Hoeg-Jensen and K. J. Jensen, Angew. Chem., Int. Ed., 2009, 48, 7411 CrossRef CAS.
- H. E. Elashal, Y. E. Sim and M. Raj, Chem. Sci., 2017, 8, 117 RSC.
- G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645 CrossRef CAS.
- N. Ollivier, J. Dheur, R. Mhidia, A. Blannpain and O. Melnyk, Org. Lett., 2010, 12, 5238 CrossRef CAS PubMed.
- P. Botti, M. Villain, S. Manganiello and H. Gaertner, Org. Lett., 2004, 26, 4681 Search PubMed.
- J. D. Warren, J. S. Miller, S. J. Keding and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6576 CrossRef CAS PubMed.
- K. Sato, A. Shigenaga, K. Tsuji, S. Tsuda, Y. Sumikawa, K. Sakamoto, A. Otaka, K. Sato, A. Shigenaga, K. Tsuji, S. Tsuda, Y. Sumikawa, K. Sakamoto and A. Otaka, ChemBioChem, 2011, 12, 1840 CrossRef CAS PubMed.
- J.-S. Zheng, H.-N. Chang, F.-L. Wang and L. Liu, J. Am. Chem. Soc., 2011, 133, 11080 CrossRef CAS PubMed.
- J.-X. Wang, G.-M. Fang, Y. He, D.-L. Qu, M. Yu, Z.-Y. Hong and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 2194 CrossRef CAS.
- J. Weidmann, E. Dimitrijević, J. D. Hoheisel and P. E. Dawson, Org. Lett., 2016, 18, 164 CrossRef CAS PubMed.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851 CrossRef CAS PubMed.
- J. B. Blanco-Canosa, B. Nardone, F. Albericio and P. E. Dawson, J. Am. Chem. Soc., 2015, 137, 7197 CrossRef CAS.
- J. Palà-Pujadas, F. Albericio and J. B. Blanco-Canosa, Angew. Chem., Int. Ed., 2018, 57, 16120 CrossRef PubMed.
- D. T. Flood, J. J. Hintzen, M. J. Bird, P. A. Cistrone, J. Ason, S. Chen and P. E. Dawson, Angew. Chem., Int. Ed., 2018, 57, 11634 CrossRef CAS PubMed.
- S. K. Mahto, C. J. Howard, J. C. Shimko and J. J. Ottesen, ChemBioChem, 2011, 12, 2488 CrossRef CAS PubMed.
- J. Mannuthodikayil, S. Singh, A. Biswas, A. Kar, W. Tabassum, P. Vydyam, M. K. Bhattacharyya and K. Mandal, Org. Lett., 2019, 21, 9040 CrossRef CAS PubMed.
- K. S. A. Kumar, L. Spasser, L. A. Erlich, S. N. Bavikar and A. Brik, Angew. Chem., Int. Ed., 2010, 48, 9126 CrossRef.
- S. Gunasekera, T. L. Aboye, W. A. Madian, H. R. El-Seedi and U. Göransson, Int. J. Pept. Res. Ther., 2013, 19, 43 CrossRef CAS PubMed.
- B. Fauvet, S. M. Butterfield, J. Fuks, A. Brik and H. A. Lashuel, Chem. Commun., 2013, 49, 9254 RSC.
- L. Dery, P. S. Reddy, R. Mousa, O. Ktorza, A. Talhami and N. Metanis, Chem. Sci., 2017, 8, 1922 RSC.
- Z. Zhao, R. Mousa and N. Metanis, Chem.–Eur. J., 2022, 28, e202200279 CAS.
- A. Baral, A. Asokan, V. Bauer, B. Kieffer and V. Torbeev, Tetrahedron, 2019, 75, 703 CrossRef CAS.
- A. R. Katritzky, A. A. Shestopalov and K. Suzuki, Synthesis, 2004, 2004, 1806 CrossRef.
- D. Bang, B. L. Pentelute and S. B. H. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985 CrossRef CAS PubMed.
- C. A. Arbour, T. D. Kondasinghe, H. Y. Saraha, T. L. Vorlicek and J. L. Stockdill, Chem. Sci., 2018, 9, 350 RSC.
- C. A. Arbour, R. E. Stamatin and J. L. Stockdill, J. Org. Chem., 2018, 83, 1797 CrossRef CAS PubMed.
- C. A. Arbour, L. G. Mendoza and J. L. Stockdill, Org. Biomol. Chem., 2020, 18, 7253 RSC.
- A. Selvaraj, H.-T. Chen, A. Y.-T. Huang and C.-L. Kao, Chem. Sci., 2018, 9, 345 RSC.
- T. Ohara, M. Kaneda, T. Saito, N. Fujii, H. Ohno and S. Oishi, Bioorg. Med. Chem. Lett., 2018, 28, 1283 CrossRef CAS PubMed.
- G. A. Acosta, L. Murray, M. Royo, B. G. de la Torre and F. Albericio, Front. Chem., 2020, 8, 298 CrossRef CAS PubMed.
- J. Weidmann, M. Schnölzer, P. E. Dawson and J. D. Hoheisel, Cell Chem. Biol., 2019, 26, 645 CrossRef CAS.
- F. J. Ferrer-Gago and L. Q. Koh, Pept. Sci., 2021, 113, e24194 CAS.
- F. Albericio and G. Barany, Int. J. Pept. Protein Res., 1987, 30, 206 CrossRef CAS PubMed.
- R. Kaplánek and V. Krchnák, Tetrahedron Lett., 2013, 54, 2600 CrossRef.
- P. W. R. Harris and M. A. Brimble, Biopolymers, 2013, 100, 356 CrossRef CAS.
- The Nbz was first described by Pascal as a carboxyl-protecting and activating group in peptides: R. Pascal, D. Chauvey and R. Sola, Tetrahedron Lett., 1994, 35, 6291 CrossRef CAS.
- Some peptideXaa1–Bt (Xaa1 = V, P) derivatives could be isolated by HPLC purification, although they slowly hydrolyze in the form of lyophilized powders.
- Y. Li, J. Liu, Q. Zhou, J. Zhao and P. Wang, Chin. J. Chem., 2021, 39, 1861 CrossRef CAS.
- S. S. Kulkarni, E. E. Watson, J. W. C. Maxwell, G. Niederacher, J. Johansen-Leete, S. Huhmann, S. Mukherjee, A. R. Norman, J. Kriegesmann, C. F. W. Becker and R. J. Payne, Angew. Chem., Int. Ed., 2022, 61, e202200163 CrossRef CAS PubMed.
- E. R. Weikum, M. T. Knuesel, E. A. Ortlund and K. R. Yamamoto, Nat. Rev. Mol. Cell Biol., 2017, 18, 159 CrossRef CAS PubMed.
- V. L. Chandler, B. A. Maler and K. R. Yamamoto, Cell, 1983, 33, 489 CrossRef CAS PubMed.
- B. F. Luisi, W. X. Xu, Z. Otwinowski, L. P. Freedman, K. R. Yamamoto and P. B. Sigler, Nature, 1991, 352, 497 CrossRef CAS PubMed.
- S. H. Meijsing, M. A. Pufall, A. Y. So, D. L. Bates, L. Chen and K. R. Yamamoto, Science, 2009, 324, 407 CrossRef CAS PubMed.
- J. Miller, A. D. McLachlan and A. Klug, EMBO J., 1985, 4, 1609 CrossRef CAS PubMed.
- R. S. Brown, C. Sander and P. Argos, FEBS Lett., 1985, 186, 274 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc04158h |
| ‡ Present address: School of Chemistry, University College Dublin (UCD), Belfield, Dublin 4, D04 N2E2, Ireland. |
| This journal is © The Royal Society of Chemistry 2022 |