
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating the frontier orbitals of L(X)Ga-substituted diphosphenes [L(X)GaP]2 (X = Cl, Br) and their facile oxidation to radical cations†
Mahendra K.
Sharma
 a,
Sonia
Chabbra
b,
Christoph
Wölper
a,
Hanns M.
Weinert
a,
Edward J.
Reijerse
b,
Alexander
Schnegg
b and
Stephan
Schulz
*ac
a,
Sonia
Chabbra
b,
Christoph
Wölper
a,
Hanns M.
Weinert
a,
Edward J.
Reijerse
b,
Alexander
Schnegg
b and
Stephan
Schulz
*ac
aInstitute of Inorganic Chemistry, University of Duisburg-Essen, Universitätsstraße 5-7, D-45141, Essen, Germany. E-mail: stephan.schulz@uni-due.de; Web: https://www.uni-due.de/ak_schulz/index_en.php
bEPR Research Group, Max Planck Institute for Chemical Energy Conversion, Stiftstrasse 34-36, Mülheim an der Ruhr, D-45470, Germany
cCenter for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Carl-Benz-Straße 199, 47057 Duisburg, Germany
First published on 12th October 2022
Abstract
Modulating the electronic structures of main group element compounds is crucial to control their chemical reactivity. Herein we report on the synthesis, frontier orbital modulation, and one-electron oxidation of two L(X)Ga-substituted diphosphenes [L(X)GaP]2 (X = Cl 2a, Br 2b; L = HC[C(Me)N(Ar)]2, Ar = 2,6-i-Pr2C6H3). Photolysis of L(Cl)GaPCO 1 gave [L(Cl)GaP]22a, which reacted with Me3SiBr with halide exchange to [L(Br)GaP]22b. Reactions with MeNHC (MeNHC = 1,3,4,5-tetramethylimidazol-2-ylidene) gave the corresponding carbene-coordinated complexes L(X)GaPP(MeNHC)Ga(X)L (X = Cl 3a, Br 3b). DFT calculations revealed that the carbene coordination modulates the frontier orbitals (i.e. HOMO/LUMO) of diphosphenes 2a and 2b, thereby affecting the reactivity of 3a and 3b. In marked contrast to diphosphenes 2a and 2b, the cyclic voltammograms (CVs) of the carbene-coordinated complexes each show one reversible redox event at E1/2 = −0.65 V (3a) and −0.36 V (3b), indicating their one-electron oxidation to the corresponding radical cations as was confirmed by reactions of 3a and 3b with the [FeCp2][B(C6F5)4], yielding the radical cations [L(X)GaPP(MeNHC)Ga(X)L]B(C6F5)4 (X = Cl 4a, Br 4b). The unpaired spin in 4a (79%) and 4b (80%) is mainly located at the carbene-uncoordinated phosphorus atoms as was revealed by DFT calculations and furthermore experimentally proven in reactions with nBu3SnH, yielding the diphosphane cations [L(X)GaPHP(MeNHC)Ga(X)L]B(C6F5)4 (X = Cl 5a, Br 5b). Compounds 2–5 were fully characterized by NMR and IR spectroscopy as well as by single crystal X-ray diffraction (sc-XRD), and compounds 4a and 4b were further studied by EPR spectroscopy, while their bonding nature was investigated by DFT calculations.
Introduction
Stable radicals of main-group elements have attracted significant interest due to their interesting electronic nature, bonding situation, reactivity, and physical properties, which render them promising reagents in synthetic chemistry and materials science.1 Therefore, the isolation of stable main-group element radicals is of high interest and many stable main-group element-based radicals have been reported in recent years,1,2 including kinetically-stabilized neutral,3 anionic,4 and cationic5 phosphorous-centered radicals. The use of sterically demanding ligands and/or singlet carbenes such as N-heterocyclic carbenes (NHCs) and cyclic (alkyl)(amino)-carbenes (cAACs) as well as π-delocalization of the unpaired electron spin density have been proven to stabilize these radicals. Phosphorus-centred radical cations Trip2(R)P˙+I (R = iPr3C6H2, Me3C6H2) and tetraaryldiphosphane radical cation (Trip2PPTrip2)˙+II (Trip = iPr3C6H2) were isolated by one-electron oxidation of the corresponding (di)phosphanes (Fig. 1),5a,b whereas base-stabilized radical cations III–VI were isolated using strong σ-donor NHCs, cAACs and N-heterocyclic vinylidenes (NHV, IX), respectively.5c,e,5l,10a In addition, four-membered π-delocalized cyclic radical cations VII and VIII as well as several metal-coordinated radical compounds (X, XI) were reported.5k,m,n,11a,12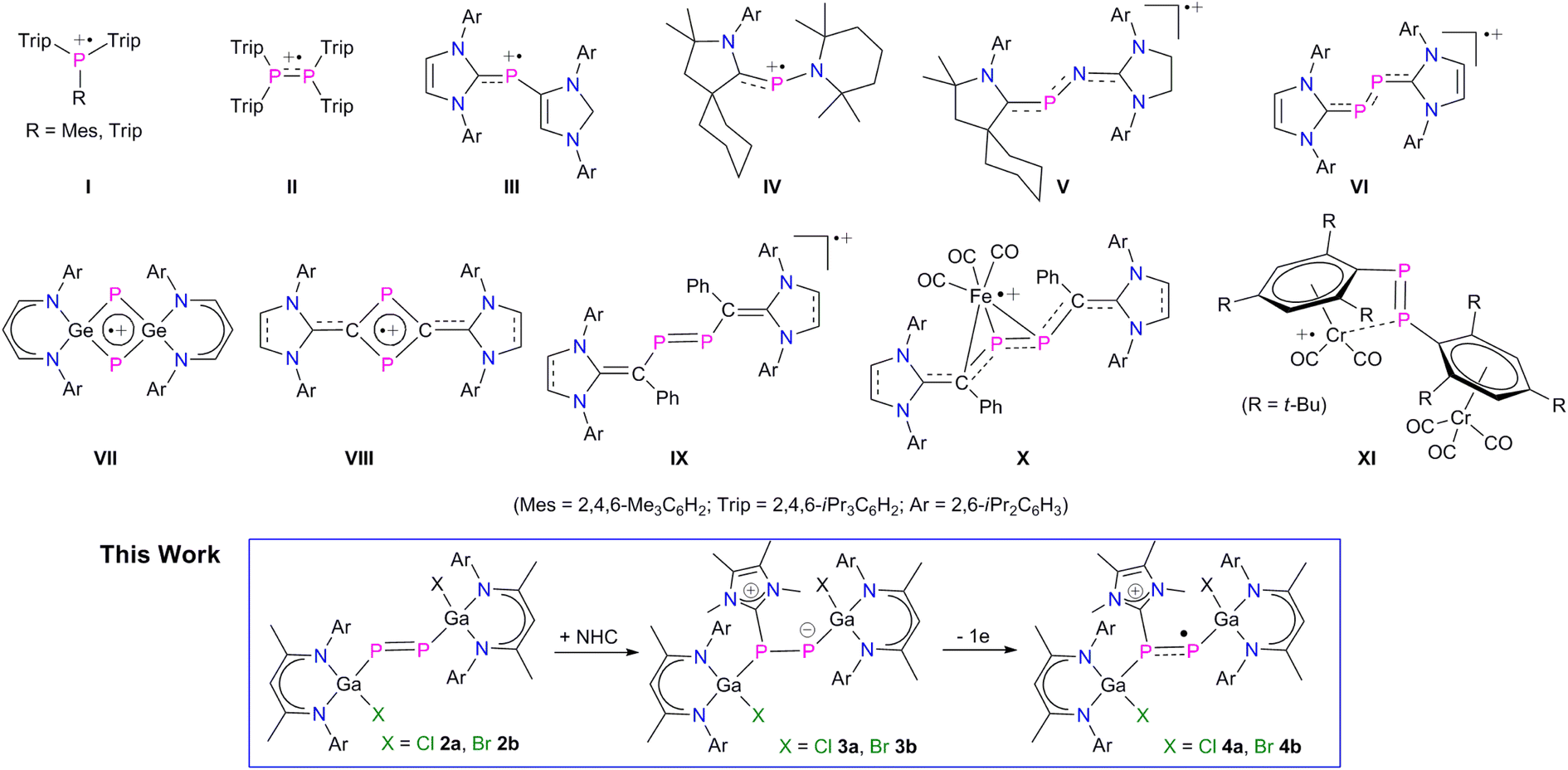 | ||
| Fig. 1 Selected examples of phosphorus radical cations I–VIII, diphosphene radical cation IX, diphosphene stabilized metal-centered radical cations X–XI, and L(X)Ga-substituted diphosphenes radical cations (this work). | ||
Diphosphenes (RPPR) containing a P–P double bond are analogues to alkenes and have received substantial fundamental interest in molecular main-group chemistry due to their small HOMO–LUMO energy gaps compared to alkenes, resulting in an increased reactivity and potential applicability in synthetic chemistry.6 In 1981, Yoshifuji et al. isolated the first stable diphosphene, [Mes*P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PMes*] (Mes* = 2,4,6-t-Bu3-C6H2),7 followed by subsequent reports on other kinetically stabilized diphosphenes.8 In general, the LUMO of diphosphenes typically consists of the low-lying π* orbital of the PP double bond, whereas the HOMO comprises the electron lone pair orbital of the phosphorus atoms.6–8 Therefore, one-electron reduction of diphosphenes to the corresponding radical anions is feasible.9 In marked contrast, the one-electron oxidation to the corresponding radical cations remains a challenging task.6–8 To the best of our knowledge, the divinyldiphosphene radical cations [{(NHC)C(Ph)}P]2˙+ (IX) reported by Ghadwal et al. represent the only genuine example (Fig. 1),10 whereas radical cations X and XI are rather metal-centered radical cations according to quantum chemical calculations (Fig. 1).11,12
PMes*] (Mes* = 2,4,6-t-Bu3-C6H2),7 followed by subsequent reports on other kinetically stabilized diphosphenes.8 In general, the LUMO of diphosphenes typically consists of the low-lying π* orbital of the PP double bond, whereas the HOMO comprises the electron lone pair orbital of the phosphorus atoms.6–8 Therefore, one-electron reduction of diphosphenes to the corresponding radical anions is feasible.9 In marked contrast, the one-electron oxidation to the corresponding radical cations remains a challenging task.6–8 To the best of our knowledge, the divinyldiphosphene radical cations [{(NHC)C(Ph)}P]2˙+ (IX) reported by Ghadwal et al. represent the only genuine example (Fig. 1),10 whereas radical cations X and XI are rather metal-centered radical cations according to quantum chemical calculations (Fig. 1).11,12
The scarcity of diphosphene radical cations as well as our general interest in the reactivity of L(X)Ga-substituted, electron-rich dipnictenes13 prompted us to synthesize L(X)Ga-substituted diphosphenes and investigate their redox properties. We herein report on the synthesis of two L(X)Ga-substituted diphosphenes [L(X)GaP]2 (X = Cl 2a, Br 2b) and their reactions with an N-heterocyclic carbene. Carbene-coordination in 3a and 3b resulted in a modulation of their frontier orbitals with respect to those of the uncoordinated diphosphenes, allowing one-electron oxidation reactions to the corresponding diphosphene radical cations [L(X)GaPP(MeNHC)Ga(X)L]B(C6F5)4 (X = Cl 4a, Br 4b, Fig. 1). Radical cations 4a and 4b further react with nBu3SnH to diphosphane cations 5a and 5b, respectively.
Results and discussion
Phosphaketenes (RPCO) contain weak P–CO bonds and readily undergo decarbonylation upon photolysis, thermolysis and in reactions with Lewis bases.14 We recently prepared the gallaphosphaketene L(Cl)GaPCO 1 and reported on its decarbonylation reaction with LGa to an unprecedented gallaphosphene L(Cl)GaPGaL and its remarkable activity in small molecule activation.15 We here extended our studies to the photolysis of 1. UV treatment of a colorless toluene solution of 1 occurred with decarbonylation at ambient temperature and yielded the desired L(Cl)Ga-substituted diphosphene [L(Cl)GaP]22a in 65% isolated yield, which reacted with an excess of Me3SiBr with halide exchange and quantitative formation of [L(Br)GaP]22b (Scheme 1).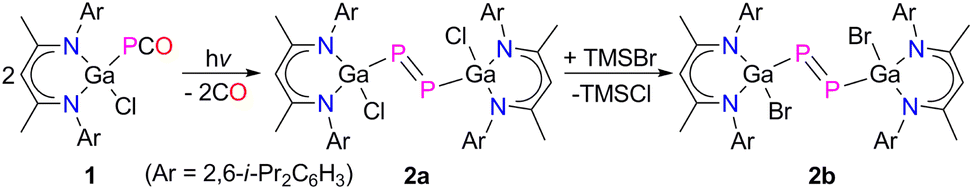 | ||
| Scheme 1 Synthesis of diphosphenes 2a and 2b. | ||
Diphosphenes 2a and 2b are green crystalline solids, which are stable in solution and solid states under inert gas atmosphere and poorly soluble in common organic solvents such as THF, toluene, fluorobenzene, benzene, and n-hexane. Their 1H and 13C{1H} NMR spectra each exhibit one set of resonances for the β-diketiminate ligand, indicating a symmetric nature of the molecules in solution. The 31P{1H} NMR spectrum of 2a (+761.6 ppm) and 2b (+766.8 ppm) each displays a sharp singlet for the phosphorus atoms, which are consistent with the previously reported metal-substituted diphosphenes.16 The solid-state molecular structures of 2a and 2b (Fig. 2) revealed trans-bent geometries along the P–P double bonds. Both compounds reside at a crystallographic center of inversion and crystallize in the monoclinic space group P21/n.17 The phosphorus atoms are twofold- and the gallium atoms fourfold-coordinated. The P1–P1′ bond lengths of 2a (2.0381(5) Å) and 2b (2.0282(8) Å) are in the typical range of diphosphenes (2.02–2.08 Å)6–8 and agree well with sum of the calculated P–P double-bond radii (2.04 Å).18a The Ga–X bond lengths of 2a (X = Cl, 2.2207(3) Å) and 2b (X = Br, 2.3680(2) Å) are comparable to those of LGaX2 [X = Cl 2.218(1), 2.228(1) Å; Br 2.286(1), 2.330(1) Å].19
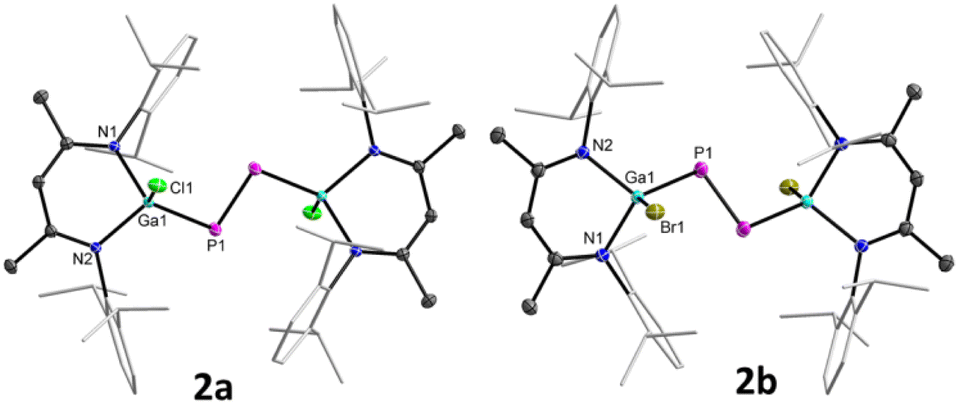 | ||
| Fig. 2 Molecular structures of 2a and 2b. Ellipsoids set at 50% probability; hydrogen atoms and alternate positions of the disordered parts (in 2a) are omitted for clarity. Symmetry operations used to generate equivalent atoms: −x, −y, −z + 1 (2a), and −x + 1, −y, −z (2b). | ||
To gain further insights into the electronic structures of compounds 2a and 2b, we performed DFT calculations at the B3LYP-D3BJ/def2-TZVP level of theory.20 The DFT optimized geometries of compounds 2 are in good agreement with their solid-state molecular structures (Tables 1 and S4†). The calculations revealed that the HOMO of 2a (−5.22 eV) and 2b (−5.26 eV) each corresponds to the Ga–P σ-bonds with a small contribution from the non-bonding electron lone pair of the P atoms, while the HOMO−3 (−6.23 eV 2a, −6.28 eV 2b) involves the π type orbital of the P–P double bond (Fig. 4, and S47 and S48†) and the LUMOs (−2.37 eV 2a, −2.44 eV 2b) are mainly the π* orbital of the P–P double bond. Due to our general interest in the redox properties of L(X)Ga-substituted dipnictenes,21 we measured the cyclic voltammograms (CVs) of 2a and 2b. Both compounds exhibited two irreversible reduction events at Epc = −2.30, −2.78 V (2a) and −2.07, −2.58 V (2b) vs. Fc/Fc+, respectively, which can be assigned to the corresponding radical anions and dianions (Fig. S29 and S30†). Unfortunately, all attempts to reduce 2a and 2b failed and yielded only LGa as the main product.
| P1–P2/P1′ | Ga1–P1 | Ga2–P2/P1′ | P1–C59 | |
|---|---|---|---|---|
| 2a | 2.0381(5) [2.040] | 2.3131(3) [2.332] | 2.3131(3) [2.334] | — |
| 3a | 2.1871(8)/2.1835(8) [2.158] | 2.3328(7)/2.3433(6) [2.325] | 2.2747(6)/2.2695(7) [2.280] | 1.862(2)/1.864(2) [1.852] |
| 4a | 2.142(8) [2.130] | 2.3479(13) [2.358] | 2.3456(10) [2.354] | 1.823(3) [1.816] |
| 5a | 2.2227(14) | 2.3655(16) | 2.3466(16) | 1.838(4) |
| 2b | 2.0282(8) [2.040] | 2.3120(4) [2.333] | 2.3120(4) [2.333] | — |
| 3b | 2.1623(5) [2.158] | 2.3205(4) [2.327] | 2.2664(4) [2.280] | 1.8570(13) [1.852] |
| 4b | 2.1336(7) [2.133] | 2.3591(8) [2.359] | 2.3522(7) [2.357] | 1.8262(19) [1.817] |
| 5b | 2.223(3) | 2.367(3) | 2.339(3) | 1.829(8) |
Since NHCs are known as strong σ-donor ligands, which have been reported to enhance the reactivity of diphosphenes,22 we reacted compounds 2a and 2b with MeNHC (MeNHC = 1,3,4,5-tetramethylimidazol-2-ylidene). The reaction proceeded at ambient temperature with an immediate color change from green to red and formation of the corresponding MeNHC-coordinated diphosphenes L(X)GaPP(MeNHC)Ga(X)L (X = Cl 3a, Br 3b) in high (>92%) yields (Scheme 2). Compounds 3a and 3b are soluble in common organic solvents and stable under argon atmosphere at ambient temperature, but rapidly decompose upon exposure to air. The 1H and 13C{1H} NMR spectra of compounds 3a and 3b exhibit two distinct sets of resonances for the β-diketiminate ligand as was observed for L(X)Ga-substituted dipnictenes,13 dipnictanes,23 gallapnictenes,15a,24 and other complexes.25 The 31P{1H} NMR spectra of 3a (−85.8 ppm, 1JPP = 353.0 Hz, and −258.5 ppm, 1JPP = 353.0 Hz) and 3b (−77.7 ppm, 1JPP = 358.9 Hz, and −247.5 ppm, 1JPP = 358.9 Hz) each display two doublets for the electronically nonequivalent phosphorus atoms, which are upfield shifted with respect to diphosphenes 2a (+761.6 ppm) and 2b (+766.8 ppm).
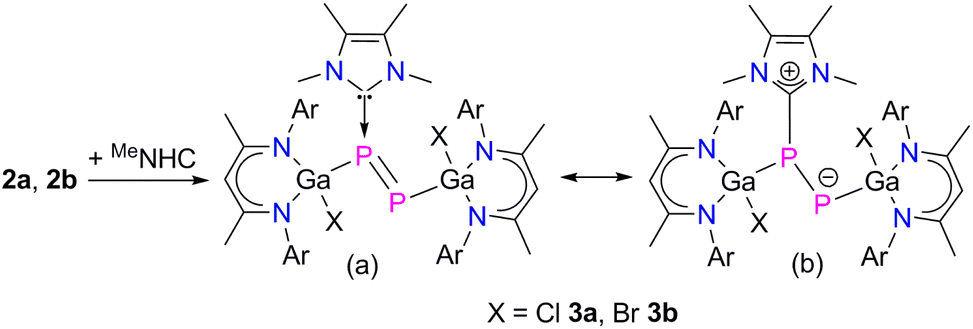 | ||
| Scheme 2 Synthesis of MeNHC-diphosphenes 3 (a), and likely resonance structures (b). | ||
The molecular structures of complexes 3a and 3b in the solid state confirmed the carbene coordination to only one phosphorus atom and revealed a similar trans-bent geometry along the P–P double bond as was observed for 2a and 2b (Fig. 3).
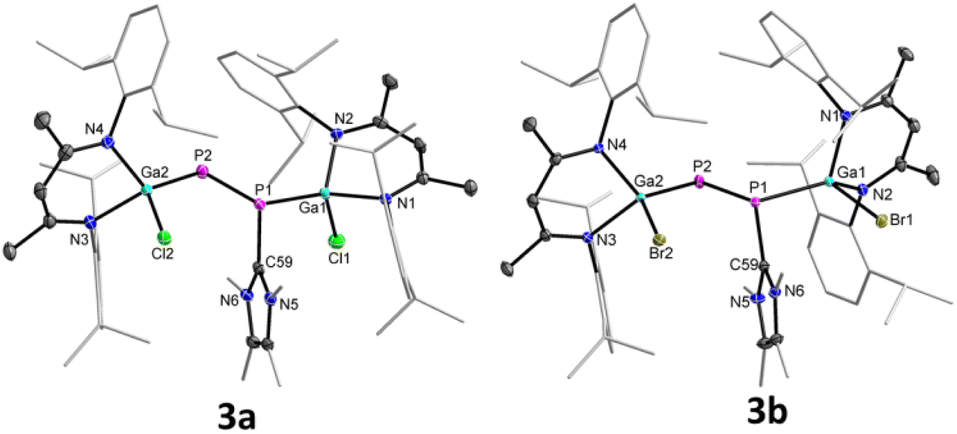 | ||
| Fig. 3 Molecular structures of 3a and 3b. Ellipsoids set at 50% probability; hydrogen atoms, alternate positions of the disordered parts (in 3b) are omitted for clarity. | ||
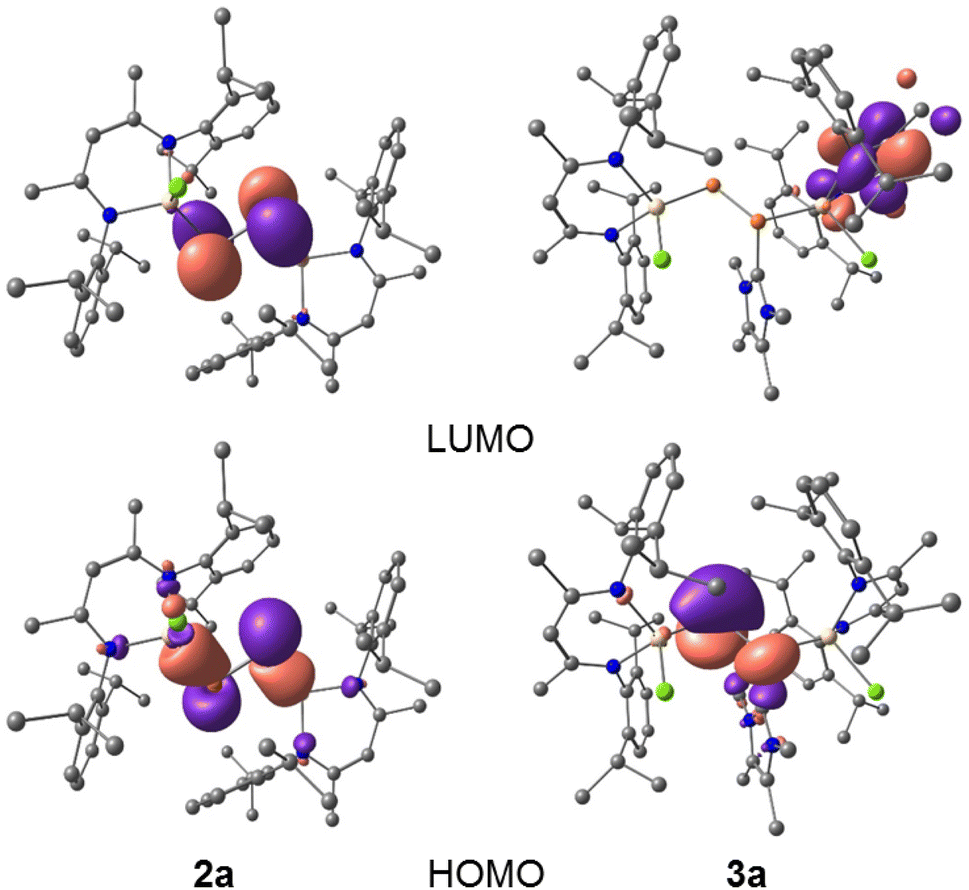 | ||
| Fig. 4 HOMO and LUMO of compounds 2a and 3a calculated at B3LYP-D3BJ/def2-TZVP level of theory (isovalue 0.0432 a.u.). Hydrogen atoms are omitted for clarity. | ||
Compound 3a crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and 3b in the monoclinic space group P21/n.17 The P1–P2 bond lengths in 3a (2.1871(8)/2.1835(8) Å) and 3b (2.1623(5) Å) are significantly elongated compared to those of diphosphenes 2a (2.0381(5) Å) and 2b (2.0282(8) Å), but still shorter than the calculated P–P single bond length (2.22 Å).18b The Ga1–P1 bonds in 3a (2.3328(7)/2.3433(6) Å) and 3b (2.3205(4) Å) are slightly elongated and the Ga2–P2 bonds slightly shortened (3a 2.2747(6)/2.2695(7) Å, 3b 2.2664(4) Å) compared to those in diphosphenes 2a and 2b (Table 1), respectively; but they are comparable to the sum of the calculated Ga–P single-bond radii18b and previously reported Ga–P single bond lengths.15
and 3b in the monoclinic space group P21/n.17 The P1–P2 bond lengths in 3a (2.1871(8)/2.1835(8) Å) and 3b (2.1623(5) Å) are significantly elongated compared to those of diphosphenes 2a (2.0381(5) Å) and 2b (2.0282(8) Å), but still shorter than the calculated P–P single bond length (2.22 Å).18b The Ga1–P1 bonds in 3a (2.3328(7)/2.3433(6) Å) and 3b (2.3205(4) Å) are slightly elongated and the Ga2–P2 bonds slightly shortened (3a 2.2747(6)/2.2695(7) Å, 3b 2.2664(4) Å) compared to those in diphosphenes 2a and 2b (Table 1), respectively; but they are comparable to the sum of the calculated Ga–P single-bond radii18b and previously reported Ga–P single bond lengths.15
DFT calculations at the B3LYP-D3BJ/def2-TZVP level of theory20 revealed that the HOMOs of complexes 3a and 3b are mainly composed of p-type orbitals of the two-fold-coordinated phosphorus atoms (P2), while the LUMOs are predominantly located on the ligand backbone (Fig. 4, S49 and S50†).
The electron donation from MeNHC to the π*-orbitals of 2a and 2b leads to charge separation between the P atoms, hence, the P2 atoms show higher negative NPA charges (3a −0.84e, 3b −0.83e) than the P1 atoms (3a −0.09e, 3b −0.09e). The P–P bond in 3a and 3b reflects single bond character with less π-bonding contribution than in the diphosphenes 2a and 2b, as was confirmed by quantum chemical calculations. Calculated MBOs (Mayer bond orders) for the P1–P2 bond of 2a (1.881) and 2b (1.883) are significantly larger than for 3a (1.208) and 3b (1.219), respectively, hence the electronic situation in 3a and 3b is best illustrated by the zwitterionic structure depicted in Scheme 2. In addition, the MeNHC coordination significantly reduces the HOMO–LUMO energy gap of diphosphenes 2a (−7.60 eV) and 2b (−7.70 eV) to −5.25 eV (3a) and −5.32 eV (3b), respectively. The HOMOs of 3a (−3.91 eV) and 3b (−3.95 eV) are high-lying and therefore expected to allow the oxidation of 3a and 3b to the corresponding radical cations.
We therefore measured CVs of 3a and 3b to get deeper insights into their redox properties. The CVs of 3a (Fig. S31†) and 3b (Fig. S32†) each show one reversible redox event at E1/2 = −0.65 V and −0.36 V vs. Fc/Fc+, respectively, which are fully consistent with their one-electron oxidation to the corresponding radical cations. It is worth mentioning here that the MeNHC coordination not only changed the frontier orbitals of 2a and 2b in compounds 3a and 3b, respectively, but also significantly altered their redox properties (see above). Indeed, reactions of 3a and 3b with [FeCp2][B(C6F5)4] gave the radical cations [L(X)GaPP(MeNHC)Ga(X)L][B(C6F5)4] (X = Cl 4a, Br 4b) as yellow crystalline solids in good yields (Scheme 3). Compounds 4a and 4b are soluble in common organic solvents (toluene, fluorobenzene, THF) and stable under an inert gas atmosphere at ambient temperature, but rapidly decompose when exposed to air and moisture.
 | ||
| Scheme 3 One-electron oxidation of compounds 3 to radical cations 4 and reactions of 4 with nBu3SnH. | ||
The molecular structures of 4a and 4b were determined by sc-XRD (Fig. 5). Suitable single crystals were obtained upon storage of saturated toluene solutions of 4a and 4b at ambient temperature. Compound 4a crystallizes in the triclinic space group P and 4b in the monoclinic space group P21/c.17
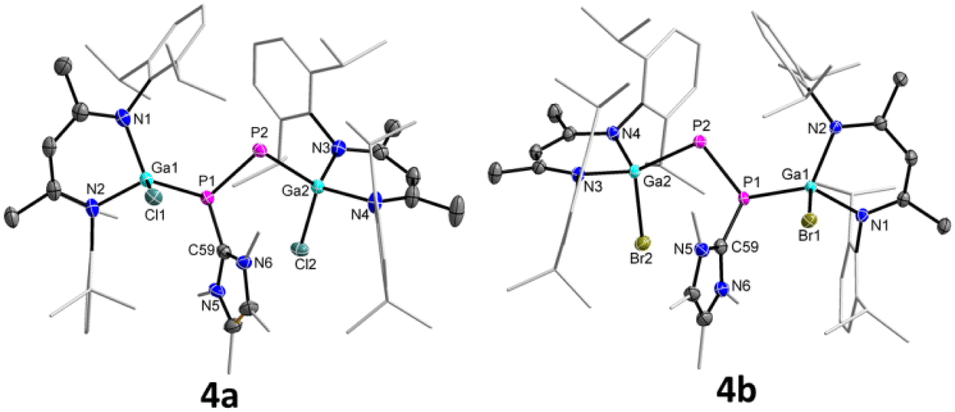 | ||
| Fig. 5 Molecular structures of radical cations 4a and 4b. Ellipsoids set at 50% probability; hydrogen atoms, alternate positions of the disordered parts, anion (BArF4), and solvent molecule (toluene in 4b) are omitted for clarity. | ||
Upon one-electron oxidation, the P1–P2 bond lengths of the NHC-coordinated complexes 3a (2.1871(8) Å) and 3b (2.1623(5) Å) became slightly shorter in the radical cations 4a (2.1423(8) Å) and 4b (2.1336(7) Å), which is most likely caused by the slight delocalization of the unpaired electron within the P2 unit (see below). Despite the slightly increased π-bonding contribution in the radical cations 4a and 4b compared to the carbene-coordinated complexes 3a and 3b, the unpaired electrons in 4a and 4b are mostly located at the carbene-free P atoms. 4a and 4b are therefore best described as phosphorus-centered radicals, in which the delocalization of the unpaired electron is disturbed by the coordination of NHC. The Ga1–P1 bonds in 4a (2.3479(13) Å) and 4b (2.3591(8) Å) are virtually identical with those of 3a (2.3328(7) Å) and 3b (2.3205(4) Å), whereas the Ga2–P2 bonds in 4a (2.3456(10) Å) and 4b (2.3522(7) Å) are substantially elongated compared to those of 3a (2.2747(6) Å) and 3b (2.2664(4) Å), respectively. In compounds 3a and 3b, the P2 atoms are far more negatively charged (−0.84 3a, −0.84 3b) than those in 4a (−0.33) and 4b (−0.32), respectively, which rather contain only one unpaired electron (Table S6†). Therefore, the short Ga2–P2 bond lengths in 3a and 3b most likely result from electrostatic attractions between the negatively charged P and positively charged Ga atoms. The P1–C59 bonds in 4a (1.823(3) Å) and 4b (1.8262(19) Å) are rather contracted compared to those of 3a (1.862(2) Å) and 3b (1.8570(13) Å), in accordance with an increased electron density transfer from the carbene carbon atom to the coordinated P atom in 4a and 4b upon oxidation.
The DFT optimized geometries of compounds 2–4 are in good agreement with their solid-state molecular structures (Tables 1 and S4†).17,20 The calculated MBOs (Mayer bond orders) for the P1–P2 bond of 2a (1.8810), 2b (1.8830), 3a (1.2075), 3b (1.2193), 4a (1.2938), and 4b (1.2534) are fully consistent with the experimentally observed changes of the P–P bond lengths (Tables 1 and S4†).26 Similarly, the MBOs for the P1–C59 bond of 3a (1.0712), 3b (1.0670), 4a (1.0013), and 4b (1.0268) exhibit the expected trend (Tables 1 and S4†). Upon one-electron oxidation, the HOMOs of 3a and 3b become the corresponding SOMOs in the radicals 4a and 4b, while the LUMO remains the same. Furthermore, the topology of the SOMOs in 4a and 4b also remains identical to the HOMO of 3a and 3b.
To gain additional insight into the electronic structures of radicals 4a and 4b, Spin Hamiltonian (SH) parameters (g- and hyperfine (A)-tensors with main values [gx, gy,gz] and [Ax, Ay,Az], respectively) were obtained from quantum chemical calculations and compared to experimental values obtained from EPR spectroscopy. Fig. 6 depicts continuous wave (CW) X-band EPR spectra of 4a and 4b measured in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 solution of fluorobenzene and toluene at 290 K (A) and 80 K (B), respectively. The 290 K multi-line spectra centered at an effective g = 2.015 ± 0.002 is assigned to hyperfine splitting of the unpaired electron with two 31P nuclei (I = 1/2) and two 69/71Ga nuclei (I = 3/2). At room temperature, the splittings in the EPR spectra are dominated by the isotropic part of the A-tensors since their anisotropies are averaged by rotational motion of the molecules. In the frozen solution spectra obtained at 80 K, all components of the g- and A-tensors contribute to the EPR spectrum. Due to the large number of couplings, an overall less structured spectrum and an increase of the total linewidth by 60%, as compared to the 290 K spectrum, is observed.
:9 solution of fluorobenzene and toluene at 290 K (A) and 80 K (B), respectively. The 290 K multi-line spectra centered at an effective g = 2.015 ± 0.002 is assigned to hyperfine splitting of the unpaired electron with two 31P nuclei (I = 1/2) and two 69/71Ga nuclei (I = 3/2). At room temperature, the splittings in the EPR spectra are dominated by the isotropic part of the A-tensors since their anisotropies are averaged by rotational motion of the molecules. In the frozen solution spectra obtained at 80 K, all components of the g- and A-tensors contribute to the EPR spectrum. Due to the large number of couplings, an overall less structured spectrum and an increase of the total linewidth by 60%, as compared to the 290 K spectrum, is observed.
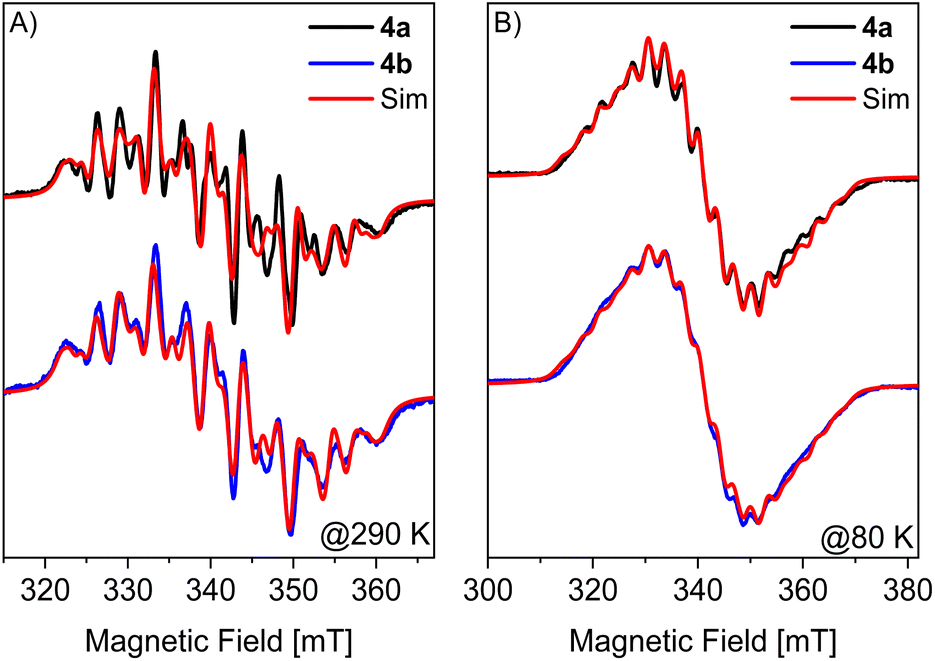 | ||
| Fig. 6 X-band CW EPR spectra of 4a (black), 4b (blue) and corresponding simulations in red at (A) 290 K and (B) 80 K. Simulation parameters for 290 K: giso = 2.015, GaAiso = 92.6 MHz and −59.7 MHz, PAiso = 298.7 MHz and 192.7 MHz. For 4a and 4b the same parameters were used except the Lorentzian full width at half maximum (FWHM) linewidth, which was adjusted to 2.25 mT (4a) and 2.5 mT (4b). Simulation parameters for 80 K: giso = 2.017, GaA = [72.2, 104.4, 72.7] MHz and [−98.1, −21.2, −95.3] MHz, PA = [255.9, 229.9, 352.0] MHz and [−114.4, −55.3, 595.0] MHz and Lorentzian FWHM linewidths of 2.06 mT (4a) and 2.8 mT (4b). | ||
EPR spectra at 290 K and 80 K were successfully simulated using isotropic g-values (giso) and isotropic (Aiso for 290 K) and anisotropic A-tensors (for 80 K) for the phosphorus and gallium atoms, respectively (Fig. 6, and Table S10†). Here, the A-tensor of the 69Ga (i.e., highest natural abundance of 60.11%) isotope is reported. The corresponding values of the 71Ga isotope (39.89% natural abundance) can be obtained by scaling the nuclear gyromagnetic ratios of the two isotopes A(71Ga) = A(69Ga)gn(71Ga)/gn(69Ga) = A(69Ga) × 1.2706. Despite an overall satisfying match between experiment and simulation the obtained Aiso for the different nuclei (and isotopes) was found to differ for liquid and frozen solution spectra, which likely originates from a strong correlation of the different A-tensor components in the fit.
The calculated Mulliken atomic spin-populations and the plots of SOMO (at B3LYP-D3BJ/def2-TZVP level of theory)20 suggest that the unpaired electron in 4a (79.7%) and 4b (80.4%) are mainly localized in the p-orbitals of the twofold-coordinated phosphorus atoms (P2) with smaller contributions at P1 (4a 12.2% and 4b 11.7%). The individual SOMO p-populations are given in Table S11.† The spin-population at the gallium and the carbene carbon atoms is very small. However, the total electron spin-densities are covering both phosphorus atoms (Fig. 7).
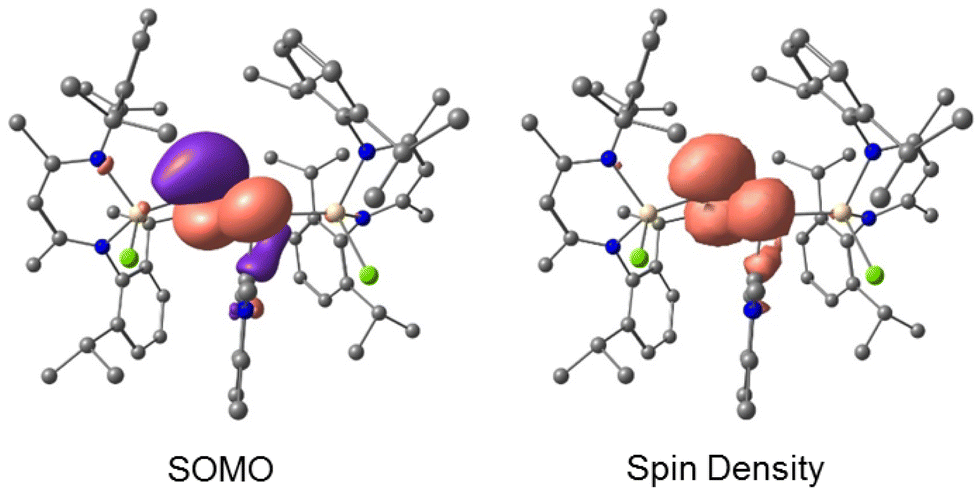 | ||
| Fig. 7 Calculated SOMO (isovalue 0.0432 a.u.) and spin-density distribution (isovalue 0.0025 a.u.) of radical cation 4a at the B3LYP-D3BJ/ZORA-def2-TZVPP level of theory. Hydrogen atoms are omitted for clarity. | ||
To further assign the A-tensors derived from EPR spectra of 4a and 4b to their two P and Ga atoms, experimental values were compared to theoretical values obtained by DFT calculations (Table S10†). This comparison allows for a tentative assignment of the most anisotropic 31P A-tensor to P2 (consistent with the large p-orbital contribution to the spin population), while P1 is assumed to exhibit a large and less anisotropic A-tensor. The assignment of the Ga1 HFI to the most isotropic tensor seems to be straightforward since the correspondence between the DFT predicted values and the experimental ones is good (Table S10†). However, the situation for Ga2 is somewhat ambiguous. The DFT calculations predict a tensor with axial anisotropy and smaller values as compared to Ga1. The experimental values are, however, relatively large but do show an axial anisotropy. Possibly, the isotropic component in the fits of the frozen solution spectra is overestimated.
To probe the chemical reactivity of radical cations 4a and 4b, we reacted them with a slight excess of nBu3SnH in fluorobenzene at ambient temperature. The solutions of 4a and 4b immediately turned colorless and diphosphanes 5a and 5b were isolated as colorless crystalline solids. The 1H (Fig. S19 and S24†) and 31P (Fig. S21, S22, S26 and S27†) NMR spectra of both 5a and 5b display the expected signals of the β-diketiminate ligand and the carbene unit. The 31P NMR spectra of 5a (−147.5 ppm) (dd, 1JPP = 85.3, 2JPH = 25.3 Hz) and −228.0 ppm (dd, 1JPP = 85.3, 1JPH = 180.9 Hz), and 5b (−142.4 ppm) (dd, 1JPP = 88.2, 2JPH = 24.7 Hz) and −225.3 ppm (dd, 1JPP = 88.2, 1JPH = 181.1 Hz) both exhibited doublets of doublets for the P–H units, while the same resonances in the 1H NMR spectra overlapped with the signals of the β-diketiminate ligand and therefore could not be assigned. The molecular structures of compounds 5a and 5b were confirmed by sc-XRD (Fig. S45 and S46†). Both compounds crystallized in the triclinic space group P.17 The P1–P2 bond lengths in 5a (2.223(1) Å), and 5b (2.223(3) Å) are significantly elongated compared to the radical cations 4a (2.142(8) Å) and 4b (2.134(7) Å), respectively, and are consistent with the calculated P–P single bond radii (2.22 Å)18b and previously reported P–P single bond lengths.27 More important, the isolation of compounds 5a and 5b clearly confirmed that the unpaired spin in radical cations 4a and 4b is mainly localized at the carbene-free phosphorus atom (P2).
Conclusions
L(X)Ga-substituted diphosphenes 2a and 2b reacted with MeNHC to the corresponding MeNHC-coordinated complexes 3a and 3b. In contrast to compounds 2a and 2b, the MeNHC-coordinated complexes 3a and 3b reacted in one-electron oxidation reactions to the corresponding radical cations 4a and 4b. Quantum chemical calculations and EPR spectroscopy revealed that the spin-population in 4a and 4b mainly resides at carbene-free phosphorus atoms (P2), which was further confirmed in reactions with nBu3SnH, yielding diphosphanes 5a and 5b, respectively. The study emphasizes on the reactivity enhancement of diphosphenes using a carbene to isolate the cationic P-centered radicals. As a variety of carbenes with different donor properties are available, the reactivity of dipnictenes can likely be tuned and various dipnictene radicals may be isolated in the future.Data availability
Crystallographic data for all compounds (2a/b–4a/b) has been deposited at the CCDC under 2184009–2184012, 2184019, 2184020, 2184142 and 2184320. Additional analytical datasets supporting this article have been uploaded as part of the ESI.†Author contributions
MKS performed the experiments and wrote the manuscript draft, HMW performed CV experiments and CW the sc-XRD studies, SS supervised the work and finalized the manuscript. EPR data was measured by SC and analyzed by SC, ER and AS. DFT calculations were performed and analysed by ER. SC, ER and AS jointly wrote the EPR and DFT sections.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the DFG (SCHU 1069/27-1), the University of Duisburg-Essen (S. S.) and the Max Planck Institute for Chemical Energy Conversion (SC, ER and AS) is gratefully acknowledged.References
- (a) R. G. Hicks, Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, John Wiley & Sons Ltd, Chichester, 2010 CrossRef; (b) P. P. Power, Chem. Rev., 2003, 103, 789–809 CrossRef CAS PubMed; (c) J. Konu and T. Chivers, Stable Radicals: Fundamental and Applied Aspects of Odd-Electron Compounds, R. G. Hicks, John Wiley & Sons, Ltd, 2010, pp. 381–406 Search PubMed; (d) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed; (e) G. W. Tan and X. P. Wang, Chin. J. Chem., 2018, 36, 573–586 CrossRef CAS; (f) V. Y. Lee, M. Nakamoto and A. Sekiguchi, Chem. Lett., 2008, 37, 128–133 CrossRef CAS.
- (a) S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Chem. Sci., 2019, 10, 4727–4741 RSC; (b) K. C. Mondal, S. Roy and H. W. Roesky, Chem. Soc. Rev., 2016, 45, 1080–1111 RSC; (c) Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS; (d) Y. Kim and E. Lee, Chem.–Eur. J., 2018, 24, 19110–19121 CrossRef CAS PubMed; (e) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; (f) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC; (g) F. Breher, Coord. Chem. Rev., 2007, 251, 1007–1043 CrossRef CAS; (h) H. Grützmacher and F. Breher, Angew. Chem., Int. Ed., 2002, 41, 4006–4011 CrossRef; (i) C. Helling and S. Schulz, Eur. J. Inorg. Chem., 2020, 34, 3209–3221 CrossRef.
- For neutral phosphorus radicals: (a) S. L. Hinchley, C. A. Morrison, D. W. H. Rankin, C. L. B. Macdonald, R. J. Wiacek, A. H. Cowley, M. F. Lappert, G. Gundersen, J. A. C. Clyburne and P. P. Power, Chem. Commun., 2000, 2045–2046 RSC; (b) A. Armstrong, T. Chivers, M. Parvez and R. T. Boeré, Angew. Chem., Int. Ed., 2004, 43, 502–505 CrossRef CAS PubMed; (c) S. Ito, M. Kikuchi, M. Yoshifuji, A. J. Arduengo III, T. A. Konovalova and L. D. Kispert, Angew. Chem., Int. Ed., 2006, 45, 4341–4345 CrossRef CAS PubMed; (d) P. Agarwal, N. A. Piro, K. Meyer, P. Müller and C. C. Cummins, Angew. Chem., Int. Ed., 2007, 46, 3111–3114 CrossRef CAS PubMed; (e) O. Back, B. Donnadieu, M. von Hopffgarten, S. Klein, R. Tonner, G. Frenking and G. Bertrand, Chem. Sci., 2011, 2, 858–861 RSC; (f) T. Beweries, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 8974–8978 CrossRef CAS PubMed; (g) A. Hinz, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 668–672 CrossRef CAS PubMed; (h) L. Gu, Y. Zheng, E. Haldón, R. Goddard, E. Bill, W. Thiel and M. Alcarazo, Angew. Chem., Int. Ed., 2017, 56, 8790–8794 CrossRef CAS PubMed; (i) X. Chen, A. Hinz, J. R. Harmer and Z. Li, Dalton Trans., 2019, 48, 2549–2553 RSC; (j) Z. Li, Y. Hou, Y. Li, A. Hinz, J. R. Harmer, C.-Y. Su, G. Bertrand and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 198–202 CrossRef CAS PubMed; (k) A. M. Tondreau, Z. Benkő, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC; (l) M. Scheer, C. Kuntz, M. Stubenhofer, M. Linseis, R. F. Winter and M. Sierka, Angew. Chem., Int. Ed., 2009, 48, 2600–2604 CrossRef CAS PubMed; (m) S. Ishida, F. Hirakawa and T. Iwamoto, J. Am. Chem. Soc., 2011, 133, 12968–12971 CrossRef CAS PubMed.
- For phosphorus radical anions: (a) T. Cantat, F. Biaso, A. Momin, L. Ricard, M. Geoffroy, N. Mézailles and P. Le Floch, Chem. Commun., 2008, 874–876 RSC; (b) C. Pi, Y. Wang, W. Zheng, L. Wan, H. Wu, L. Weng, L. Wu, Q. Li and P. v. R. Schleyer, Angew. Chem., Int. Ed., 2010, 49, 1842–1845 CrossRef CAS PubMed; (c) X. Pan, X. Wang, Y. Zhao, Y. Sui and X. Wang, J. Am. Chem. Soc., 2014, 136, 9834–9837 CrossRef CAS PubMed; (d) G. Tan, S. Li, S. Chen, Y. Sui, Y. Zhao and X. Wang, J. Am. Chem. Soc., 2016, 138, 6735–6738 CrossRef CAS PubMed.
- For phosphorus radical cations: (a) X. Pan, X. Chen, T. Li, Y. Li and X. Wang, J. Am. Chem. Soc., 2013, 135, 3414–3417 CrossRef CAS PubMed; (b) X. Pan, Y. Su, X. Chen, Y. Zhao, Y. Li, J. Zuo and X. Wang, J. Am. Chem. Soc., 2013, 135, 5561–5564 CrossRef CAS PubMed; (c) O. Back, M. A. Celik, G. Frenking, M. Melaimi, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 10262–10263 CrossRef CAS PubMed; (d) O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369–373 CrossRef CAS PubMed; (e) R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930–5933 CrossRef CAS PubMed; (f) Y. Su, X. Zheng, X. Wang, X. Zhang, Y. Sui and X. Wang, J. Am. Chem. Soc., 2014, 136, 6251–6254 CrossRef CAS PubMed; (g) A. Brückner, A. Hinz, J. B. Priebe, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 7426–7430 CrossRef PubMed; (h) X. Pan, X. Wang, Z. Zhang and X. Wang, Dalton Trans., 2015, 44, 15099–15102 RSC; (i) G. Ménard, J. A. Hatnean, H. J. Cowley, A. J. Lough, J. M. Rawson and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 6446–6449 CrossRef PubMed; (j) M. K. Sharma, S. Blomeyer, B. Neumann, G. Stammler, A. Hinz, M. van Gastel, T. Glodde and R. S. Ghadwal, Chem. Sci., 2020, 11, 1975–1984 RSC; (k) H. Cui, D. Xiao, L. Zhang, H. Ruan, Y. Fang, Y. Zhao, G. Tan, L. Zhao, G. Frenking, M. Driess and X. Wang, Chem. Commun., 2020, 56, 2167–2170 RSC; (l) K. Schwedtmann, S. Schulz, F. Hennersdorf, T. Strassner, E. Dmitrieva and J. J. Weigand, Angew. Chem., Int. Ed., 2015, 54, 11054–11058 CrossRef CAS PubMed; (m) Z. Li, X. Chen, D. M. Andrada, G. Frenking, Z. Benkő, Y. Li, J. R. Harmer, C. Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 5744–5749 CrossRef CAS PubMed; (n) D. Rottschäfer, B. Neumann, H. G. Stammler and R. S. Ghadwal, Chem.–Eur. J., 2017, 23, 9044–9047 CrossRef PubMed.
- (a) L. Weber, F. Ebeler and R. S. Ghadwal, Coord. Chem. Rev., 2022, 461, 214499 CrossRef CAS; (b) R. S. Ghadwal, Acc. Chem. Res., 2022, 55, 457 CrossRef CAS PubMed; (c) L. Weber, Chem. Rev., 1992, 92, 1839–1906 CrossRef CAS; (d) N. Djawed and O. Andreas, Eur. J. Inorg. Chem., 2016, 709–717 Search PubMed; (e) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed; (f) H.-L. Peng, J. L. Payton, J. D. Protasiewicz and M. C. Simpson, J. Phys. Chem. A, 2009, 113, 7054–7063 CrossRef CAS PubMed; (g) T. Sasamori and N. Tokitoh, Dalton Trans., 2008, 1395–1408 RSC; (h) T. Busch, W. W. Schoeller, E. Niecke, M. Nieger and H. Westermann, Inorg. Chem., 1989, 28, 4334–4340 CrossRef CAS; (i) L. P. Ho, A. Nasr, P. G. Jones, A. Altun, F. Neese, G. Bistoni and M. Tamm, Chem.–Eur. J., 2018, 24, 18922–18932 CrossRef CAS PubMed.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- (a) A. H. Cowley, J. E. Kilduff, T. H. Newman and M. Pakulski, J. Am. Chem. Soc., 1982, 104, 5820–5821 CrossRef CAS; (b) M. Yoshifuji, Eur. J. Inorg. Chem., 2016, 607–615 CrossRef CAS; (c) L. L. Liu, L. L. Cao, J. Zhou and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 273–277 CrossRef CAS PubMed; (d) N. Hayakawa, K. Sadamori, S. Tsujimoto, M. Hatanaka, T. Wakabayashi and T. Matsuo, Angew. Chem., Int. Ed., 2017, 56, 5765–5769 CrossRef CAS PubMed; (e) R. C. Smith, E. Urnezius, K.-C. Lam, A. L. Rheingold and J. D. Protasiewicz, Inorg. Chem., 2002, 41, 5296–5299 CrossRef CAS PubMed; (f) L. P. Ho, A. Nasr, P. G. Jones, A. Altun, F. Neese, G. Bistoni and M. Tamm, Chem.–Eur. J., 2018, 24, 18922–18932 CrossRef CAS PubMed; (g) E. Niecke, R. Rüger, M. Lysek, S. Pohl and W. Schoeller, Angew. Chem., Int. Ed. Engl., 1983, 22, 486–487 CrossRef; (h) S.-S. Asami, M. Okamoto, K. Suzuki and M. Yamashita, Angew. Chem., Int. Ed., 2016, 55, 12827–12831 CrossRef CAS PubMed.
- (a) M. Culcasi, G. Gronchi, J. Escudie, C. Couret, L. Pujol and P. Tordo, J. Am. Chem. Soc., 1986, 108, 3130–3132 CrossRef CAS; (b) T. Sasamori, E. Mieda, N. Nagahora, K. Sato, D. Shiomi, T. Takui, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 12582–12588 CrossRef CAS PubMed; (c) S.-s. Asami, S. Ishida, T. Iwamoto, K. Suzuki and M. Yamashita, Angew. Chem., Int. Ed., 2017, 56, 1658–1662 CrossRef CAS PubMed; (d) A. J. Bard, A. H. Cowley, J. E. Kilduff, J. K. Leland, N. C. Norman, M. Pakulski and G. A. Heath, J. Chem. Soc., Dalton Trans., 1987, 249–251 RSC; (e) S. Shah, S. C. Burdette, S. Swavey, F. L. Urbach and J. D. Protasiewicz, Organometallics, 1997, 16, 3395–3400 CrossRef CAS; (f) N. Nagahora, T. Sasamori, Y. Hosoi, Y. Furukawa and N. Tokitoh, J. Organomet. Chem., 2008, 693, 625–632 CrossRef CAS.
- (a) M. K. Sharma, D. Rottschäfer, S. Blomeyer, B. Neumann, H.-G. Stammler, M. van Gastel, A. Hinz and R. S. Ghadwal, Chem. Commun., 2019, 55, 10408–10411 RSC; (b) D. Rottschäfer, M. K. Sharma, B. Neumann, H. G. Stammler, D. M. Andrada and R. S. Ghadwal, Chem.–Eur. J., 2019, 25, 8127–8134 CrossRef PubMed.
- (a) W. Wang, C. Q. Xu, Y. Fang, Y. Zhao, J. Li and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 9419–9424 CrossRef CAS PubMed; (b) W. Wang, X. Wang, Z. Zhang, N. Yuan and X. Wang, Chem. Commun., 2015, 51, 8410–8413 RSC.
- M. K. Sharma, D. Rottschäfer, B. Neumann, H. G. Stammler, S. Danes, D. M. Andrada, M. van Gastel, A. Hinz and R. S. Ghadwal, Chem.–Eur. J., 2021, 27, 5803–5809 CrossRef CAS PubMed.
- (a) L. Tuscher, C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 10657–10661 CrossRef CAS PubMed; (b) L. Tuscher, C. Helling, C. Ganesamoorthy, J. Krüger, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem.–Eur. J., 2017, 23, 12297–12304 CrossRef CAS PubMed; (c) L. Tuscher, C. Helling, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem.–Eur. J., 2018, 24, 3241–3250 CrossRef CAS PubMed.
- J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS PubMed.
- (a) M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 6784–6790 CrossRef CAS PubMed; (b) M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 21784–21788 CrossRef CAS PubMed; (c) M. K. Sharma, C. Wölper and S. Schulz, Dalton Trans., 2022, 51, 1612–1616 RSC; (d) M. K. Sharma, P. Dhawan, C. Helling, C. Wölper and S. Schulz, Chem.–Eur. J., 2022, e202200444 CAS.
- (a) L. Liu, D. A. Ruiz, F. Dahcheh, G. Bertrand, R. Suter, A. M. Tondreauc and H. Grützmacher, Chem. Sci., 2016, 7, 2335–2341 RSC; (b) D. Wilson, W. Myers and J. M. Goicoechea, Dalton Trans., 2020, 49, 15249–15255 RSC.
- Full crystallographic data of all structurally characterized compounds described herein as well as central bond lengths and angles (Tables S1–S3 and Fig. S37–S44) are given in the Supporting Information.†.
- (a) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed; (b) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- M. Stender, B. E. Eichler, N. J. Hardman, P. P. Power, J. Prust, M. Noltemeyer and H. W. Roesky, Inorg. Chem., 2001, 40, 2794–2799 CrossRef CAS PubMed.
- The ORCA quantum chemistry package was used and details regarding the quantum chemical calculations are given in the ESI.† (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1–6 Search PubMed; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- H. Weinert, C. Wölper and S. Schulz, Organometallics, 2021, 40, 3486–3495 CrossRef CAS.
- (a) N. Hayakawa, K. Sadamori, S. Tsujimoto, M. Hatanaka, T. Wakabayashi and T. Matsuo, Angew. Chem., Int. Ed., 2017, 56, 5765–5769 CrossRef CAS PubMed; (b) D. Dhara, P. Kalita, S. Mondal, R. S. Narayanan, K. R. Mote, V. Huch, M. Zimmer, C. B. Yildiz, D. Scheschkewitz, V. Chandrasekhar and A. Jana, Chem. Sci., 2018, 9, 4235–4243 RSC.
- C. Helling, C. Wölper and S. Schulz, Eur. J. Inorg. Chem., 2020, 4225–4235 CrossRef CAS.
- (a) C. Helling, C. Wölper and S. Schulz, J. Am. Chem. Soc., 2018, 140, 5053–5056 CrossRef CAS PubMed; (b) J. Krüger, C. Ganesamoorthy, L. John, C. Wölper and S. Schulz, Chem.–Eur. J., 2018, 24, 9157–9164 CrossRef PubMed; (c) J. Schoening, L. John, C. Wölper and S. Schulz, Dalton Trans., 2019, 48, 17729–17734 RSC.
- (a) J. Krüger, C. Wölper, L. John, L. Song, P. R. Schreiner and S. Schulz, Eur. J. Inorg. Chem., 2019, 1669–1678 CrossRef; (b) L. Tuscher, C. Helling, C. Ganesamoorthy, J. Krüger, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem.–Eur. J., 2017, 23, 12297–12304 CrossRef CAS PubMed; (c) C. Ganesamoorthy, J. Krüger, C. Wölper, A. S. Nizovtsev and S. Schulz, Chem.–Eur. J., 2017, 23, 2461–2468 CrossRef CAS PubMed; (d) L. Tuscher, C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 10657–10661 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- (a) S. Burck, K. Götz, M. Kaupp, M. Nieger, J. Weber, J. S. a. d. Günne and D. Gudat, J. Am. Chem. Soc., 2009, 131, 10763–10774 CrossRef CAS PubMed; (b) V. Gandon, J. B. Bourg, F. S. Tham, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 155–159 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2184009–2184012, 2184019–2184020, 2184142 and 2184320. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04207j |
| This journal is © The Royal Society of Chemistry 2022 |