
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinoleic acid metabolism activation in macrophages promotes the clearing of intracellular Staphylococcus aureus†
Bingpeng
Yan
 a,
Kingchun
Fung
a,
Sen
Ye
b,
Pok-Man
Lai
a,
Yuan Xin
Wei
a,
Kong-Hung
Sze
a,
Dan
Yang
bc,
Peng
Gao‡
*a and
Richard Yi-Tsun
Kao‡
*a
a,
Kingchun
Fung
a,
Sen
Ye
b,
Pok-Man
Lai
a,
Yuan Xin
Wei
a,
Kong-Hung
Sze
a,
Dan
Yang
bc,
Peng
Gao‡
*a and
Richard Yi-Tsun
Kao‡
*a
aDepartment of Microbiology, School of Clinical Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 21 Sassoon Road, Pokfulam, Hong Kong, China. E-mail: gaop0305@gmail.com; rytkao@hku.hk
bMorningside Laboratory for Chemical Biology and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China
cLaboratory of Chemical Biology and Molecular Medicine, School of Life Sciences, Westlake University, Hangzhou, Zhejiang, P. R. China
First published on 11th October 2022
Abstract
Multidrug-resistant bacterial pathogens pose an increasing threat to human health. Certain bacteria, such as Staphylococcus aureus, are able to survive within professional phagocytes to escape the bactericidal effects of antibiotics and evade killing by immune cells, potentially leading to chronic or persistent infections. By investigating the macrophage response to S. aureus infection, we may devise a strategy to prime the innate immune system to eliminate the infected bacteria. Here we applied untargeted tandem mass spectrometry to characterize the lipidome alteration in S. aureus infected J774A.1 macrophage cells at multiple time points. Linoleic acid (LA) metabolism and sphingolipid metabolism pathways were found to be two major perturbed pathways upon S. aureus infection. The subsequent validation has shown that sphingolipid metabolism suppression impaired macrophage phagocytosis and enhanced intracellular bacteria survival. Meanwhile LA metabolism activation significantly reduced intracellular S. aureus survival without affecting the phagocytic capacity of the macrophage. Furthermore, exogenous LA treatment also exhibited significant bacterial load reduction in multiple organs in a mouse bacteremia model. Two mechanisms are proposed to be involved in this progress: exogenous LA supplement increases downstream metabolites that partially contribute to LA's capacity of intracellular bacteria-killing and LA induces intracellular reactive oxygen species (ROS) generation through an electron transport chain pathway in multiple immune cell lines, which further increases the capacity of killing intracellular bacteria. Collectively, our findings not only have characterized specific lipid pathways associated with the function of macrophages but also demonstrated that exogenous LA addition may activate lipid modulator-mediated innate immunity as a potential therapy for bacterial infections.
Introduction
Staphylococcus aureus (S. aureus), as a major pathogen, could infect all of the organs through circulation around the body and lead to a lethal outcome. In the past few decades, the increasing prevalence of bacterial resistance to antibiotics is a rapidly growing concern, posing a challenge in eliminating resistant infections.1 Two years after the introduction of methicillin to fight against penicillinase-producing S. aureus, resistant S. aureus strains have ultimately appeared and become methicillin-resistant S. aureus (MRSA).1,2 This anti-resistant pathogen is commonly found in healthcare centers, especially hospitals where persons with wounds are likely at a higher risk of infection. Meanwhile, the drawbacks of conventional antibiotic such as unfavorable effects on the body, disruption on the microbiome and low efficacy in a single drug treatment have eventually led to a defeat at combatting the infections.3,4 Thus, the severity of resistance to single and even multiple drugs has driven a need for novel strategies to combat this pathogen.Enhancing host immunity is an ideal strategy with fewer side effects than antibiotics.5 Immune cells such as macrophages are generally the first defenders to encounter invading pathogens in the body. The cells engulf pathogens and other cell debris followed by degrading the cargo in phagosomes via phagocytosis.6 Increasing evidence indicates that S. aureus is capable of surviving within its hosts' cells through immune-evasive strategies to evade detection by professional phagocytes.7 A previous study reported that S. aureus strain Newman is able to escape the phagosome and persists within human monocyte-derived macrophages (hMDM), which resulted in host cell lysis on day 5 after infection.8 In addition, S. aureus could replicate in the phagosome and cytoplasm of its host cells. It induces cell apoptosis and even can induce antiapoptotic programs in phagocytes, and finally cause severe pneumonia. Therefore, the intracellular S. aureus has been accounted for a kind of pathogen resistant to the immune system via escaping detection by professional phagocytes.7 Lipids are a class of critical mediators in macrophages that respond to pathogenic bacteria.9 It is necessary to investigate the regulation of the specific lipid and how they promote the innate immunity upon macrophage responses to S. aureus infection.
In the last several years, several metabolomics and lipidomics studies on multiple pathogens including virus and bacteria have been performed.10–13 A previous metabolomics study demonstrated that exogenous L-valine promoted phagocytosis to kill multidrug resistant bacterial pathogens including Klebsiella pneumonia and Escherichia coli.5 Meanwhile some important lipid mediators were reported to involve in the inflammation progress and can further lead to either chronicity or self-resolving inflammation.14,15 Macrophage activation is intimately linked to metabolic reprogramming and lipid metabolism plays a critical role in the activation of both M1 and M2 macrophages.16,17 A recent report showed that human macrophage phenotypes (M1 and M2) each generate distinct lipid mediator profiles during S. aureus infection. S. aureus stimulated proinflammatory lipids (e.g., leukotriene B4 and prostaglandin E2) in M1 macrophages but stimulated M2 macrophages to produce pro-resolving lipids including resolvin D and maresin-1. Interestingly, M2 had higher bacterial killing capacity than the M1 macrophage.9 Although increasing evidence has been demonstrated that lipids are highly involved in S. aureus-infected macrophages, the function or effects of the lipid in the interaction between the macrophage and S. aureus infection still remain unclear.
In this study, we hypothesize that S. aureus infection will activate the macrophage, which will further regulate lipid metabolism to perform necessary cell functions. So a LC-MS based lipidomics study was applied to investigate the lipidome alteration upon S. aureus-infected macrophages at various infection time points. Furthermore, we performed pathway analysis and characterize perturbed lipid metabolism pathways (e.g., linoleic acid metabolism and sphingolipid metabolism pathways) that were associated with macrophage's function to eliminate intracellular S. aureus. Finally, we validated that upregulated linoleic acid (LA) and LA metabolism pathway activation could induce multiple types of reactive oxygen species (ROS), which can effectively clear intracellular S. aureus. Therefore, targeting lipid metabolism in macrophages may improve the outcome of S. aureus infected disease and the specific lipid metabolism was previously not known to be important.
Results
Characterization of the significantly changed lipidome upon an S. aureus infected macrophage
To characterize the dynamic host lipidome under S. aureus infection, we presented untargeted lipidomics analysis comprising serially collected cell samples at 1, 12, 36, and 48 hours, respectively (Fig. 1A). The coefficients of variation (CVs) of spiked standards and quality control (QC) samples were both lower than 20% (Table S1†). After proceeding to statistical analysis and structural confirmation, a total of 158 significantly altered lipids were finally identified during all collected time points (Table S2†). These 158 lipids belonged to 18 lipid classes, including ceramides (Cer), cardiolipin (CL), diacylglycerol (DG), ether-linked lysophosphatidylcholine (ether-LPC), ether-linked lyso-phosphatidylethanolamine (ether-LPE), ether-linked phosphatidylcholine (ether-PC), ether-linked phosphatidyl-ethanolamine (ether-PE), free fatty acid (FFA), lysophosphatidylcholine (LPC), lysophosphatidyl-ethanolamine (LPE), lysophosphatidylglycerol (LPG), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidylinositol (PI), phosphatidylserine (PS), sphingomyelin (SM) and sphingoid base (SPB). The dynamic lipid distribution revealed that most of the significantly changed lipids were observed in the later stage of infection such as 36 hours (71 lipids) and 48 hours (75 lipids), while only 3 and 9 lipids were significantly different in the 1 and 12 hours post-infection (hpi), respectively (Fig. 1B, and Table S2†). Collectively, the classes and number of significantly changed lipids increased gradually as the infectious time prolonged. The current results suggest that the interplay between intracellular bacteria and host cells would be more severe and strengthened as the infectious time passed.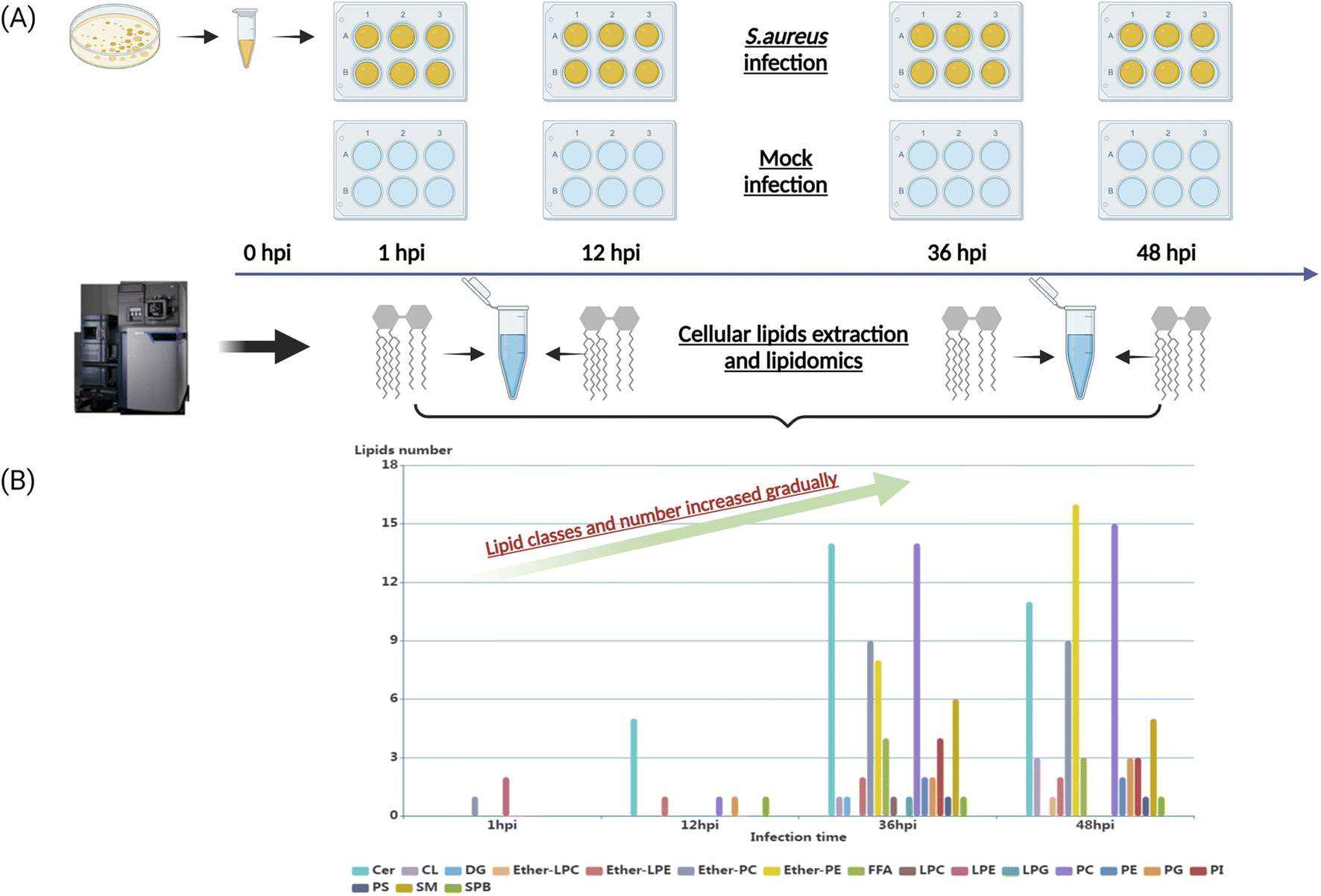 | ||
| Fig. 1 Time series lipidomics of S. aureus infected macrophage cells. (A) The schematics of LC-MS-based lipidomics of S. aureus infected macrophage cells at multiple time points. (B) The multi-histograms showing the distribution of significantly changed lipid classes and numbers at different time points. | ||
Perturbed lipid constitution and trend at the later stage of S. aureus infection
Because the later stage of S. aureus infection provided most of the perturbed lipids, here we only focus on those altered lipids at 36 and 48 hpi respectively. We firstly characterized perturbed lipid constitution, and similar results were observed at 36 and 48 hpi. Total 71 lipids belonging to 16 classes were perturbed at 36 hpi, and among them, PC, Cer, ether-PC and ether-PE were the four most perturbed lipid classes (Fig. 2A). Similarly, total 75 lipids belonging to 14 classes were perturbed at 48 hpi, and ether-PE, PC, Cer, and ether-PC were also the four most perturbed lipid classes (Fig. 2B).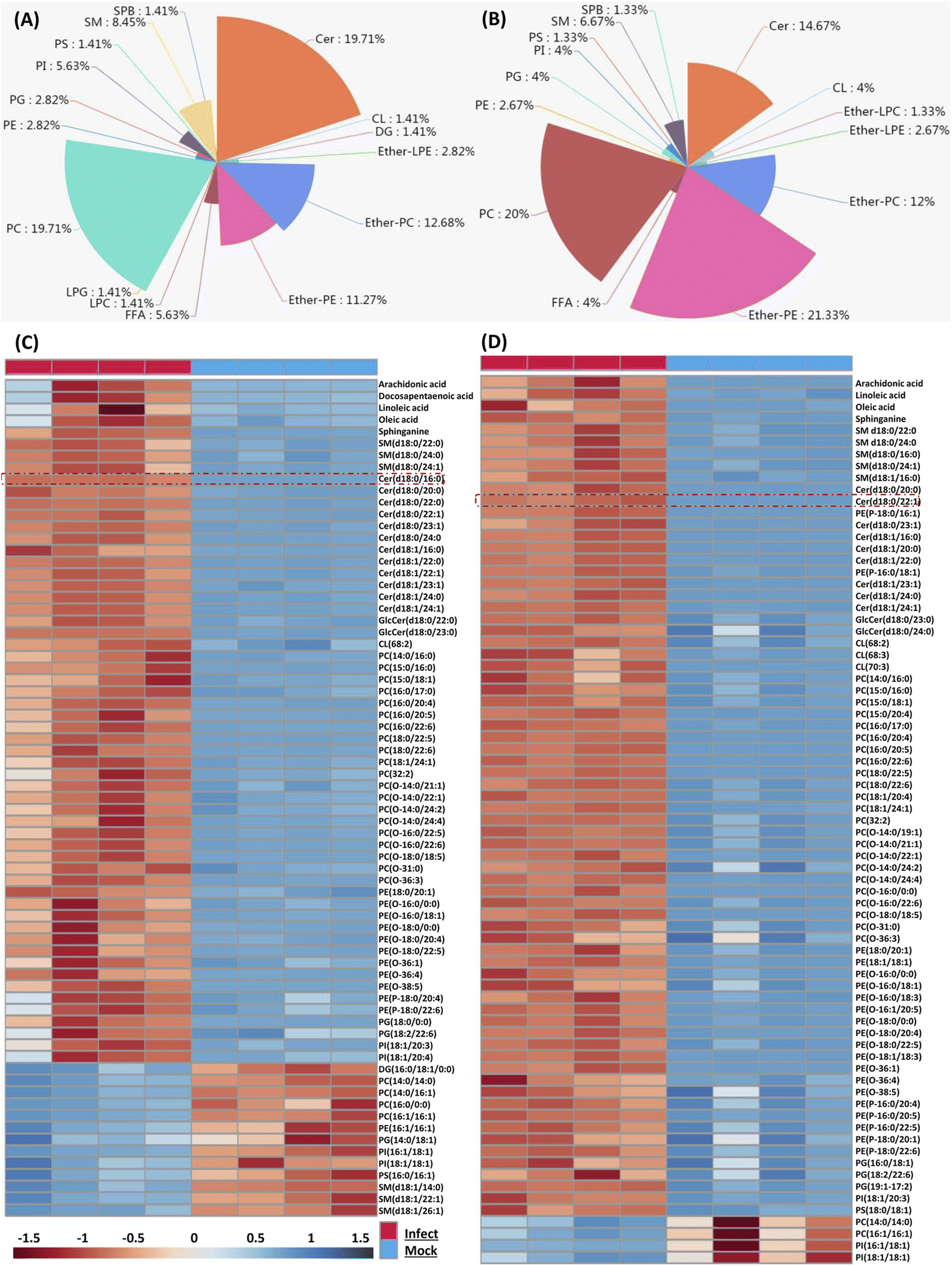 | ||
| Fig. 2 Composition and trend of significantly changed lipids upon S. aureus infected macrophage cells. (A and B) The pie charts showed significantly distinct lipid classes and corresponding lipid percentages were calculated at 36 hpi (A) and 48 hpi (B) respectively. (C and D) Hierarchical clustering analysis was performed based on all significantly changed lipids when we compared viral infections with mock infections at 36 hpi (C) and 48 hpi (D). Each bar represents an identified lipid colored by its average intensity on a normalized scale from blue (decreased level) to red (increased level). | ||
We next compared the trend and magnitude of these perturbed lipids at 36 and 48 hpi respectively (Fig. 2C and D). Interestingly, most of these perturbed lipids were consistently upregulated at both 36 and 48 hpi. Total 58 lipids (81.69%) were upregulated with a fold change in the range of 2.07 to 69.8 at 36 hpi, and the largest fold change of lipids was Cer(d18:0/16:0) (Fig. 2C). Astonishingly, almost all (94.66%) perturbed lipids were increased with a fold change in the range of 2.01 to 163.23 at 48 hpi, and the largest fold change of lipids was Cer(d18:0/22:1) (Fig. 2D). These increased lipid species mainly included FFA, sphingolipid (SM, Cer), PC, ether-PC, ether-PE and so on. Taken together, four lipid classes (PC, Cer, ether-PC, and ether-PE) were heavily perturbed and most of the perturbed lipids were significantly upregulated in the macrophage as time prolonged; especially sphingolipids such as ceramides were enriched in S. aureus infected macrophages.
Perturbed lipid pathway analyses and the role of sphingolipid metabolism in an S. aureus infected macrophage
The current pathway analysis revealed that linoleic acid metabolism, sphingolipid metabolism, glycerophospholipid metabolism and ether lipid metabolism were four mainly perturbed pathways after S. aureus infection (Fig. 3A). Intriguingly, the sphingolipid and linoleic acid pathways expressed higher pathway impact values than the others (Fig. 3A). To display these pathways with greater impact values, perturbed lipid pathway maps based on KEGG database pathway records were created (Fig. 3B). As shown in Fig. 3B, the sphingolipid metabolism pathway was significantly enhanced, as indicated by the upregulation of the main lipids in this route, such as sphinganine, dihydroceramide, ceramide, and sphingomyelin. Similarly, the linoleic acid metabolism pathway was also highly active, as the two most important regulatory lipids, linoleic acid (LA) and arachidonic acid (ADA), were both significantly elevated at 36 hpi and 48 hpi (Fig. 3B). In addition, we selected representative lipids from the two metabolic pathways to illustrate their abundance, trend, fold change and significance upon multiple infection time points (Fig. S1† and 3C). Similar to our lipid heatmaps (Fig. 2C and D), these representative lipids raised significantly at 36 phi and 48 hpi. Among these, Cer(d18:0/16:0) and arachidonic acid exhibited the greatest upregulation at 36 phi and 48 hpi, respectively, which also supported that our proposed sphingolipid and linoleic acid pathways were significant and warranted additional investigation.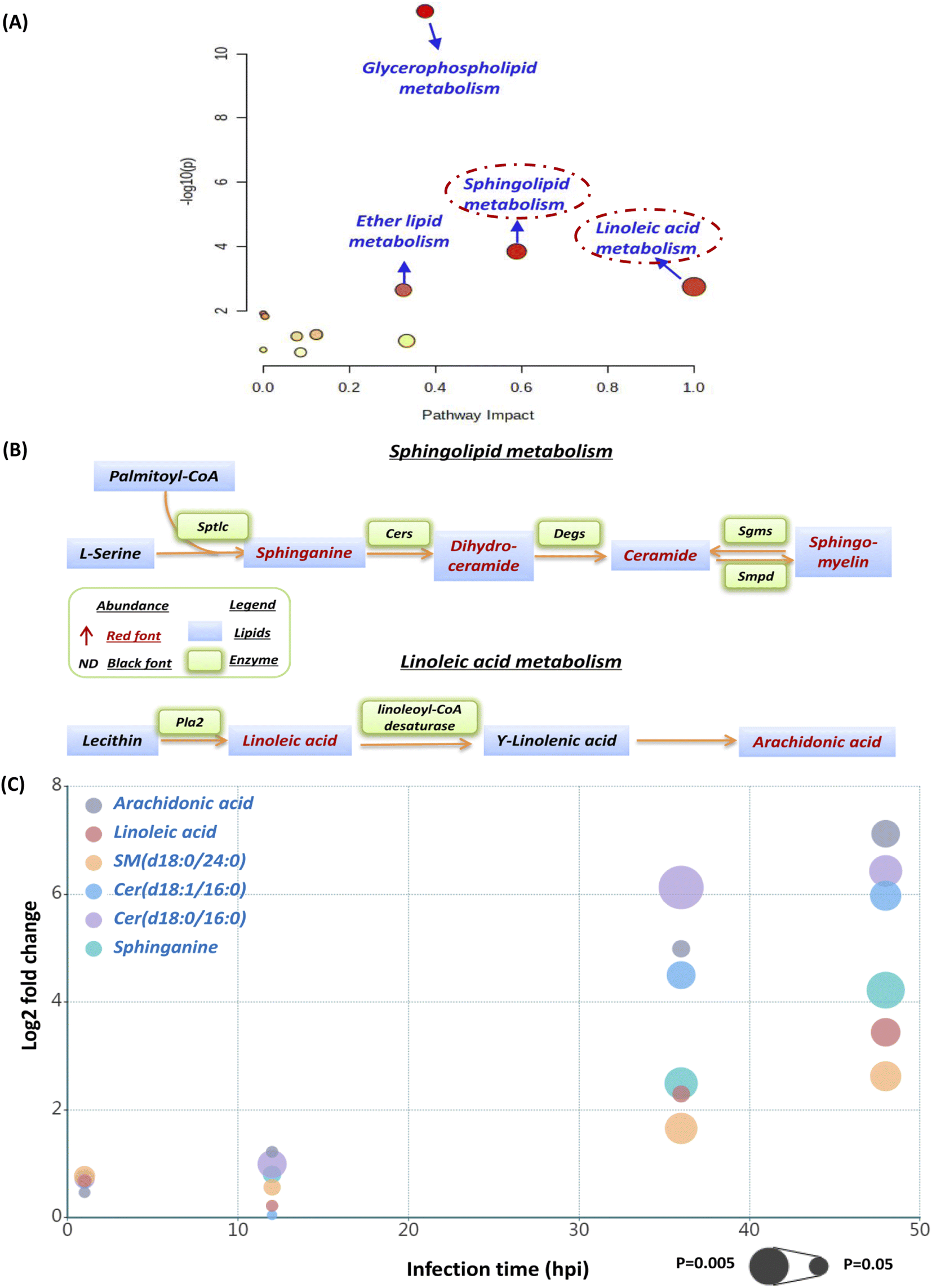 | ||
| Fig. 3 Perturbed lipid pathway analysis and targeted pathway maps. (A) Pathway analysis of S. aureus infected macrophages was carried out by using MetaboAnalyst. The Y-axis, “−log(p)”, represents the transformation of the original p-value calculated from the enrichment analysis. The X-axis, “Pathway Impact”, represents the value calculated from the pathway topology analysis. (B) The pathway maps were manually constructed based on the significantly changed lipids in the sphingolipid metabolism and linoleic acid metabolism with reference to the KEGG PATHWAY database. (C) The bubble plot showing that 6 representative lipids from sphingolipid and linoleic acid metabolic pathways were upregulated remarkably as the infection time prolongs. The x-axis and the y-axis in the bubble plot represent the infection time and log 2-fold change. The size of the bubbles represents the statistical significance of the change (FDR adjusted p-value from a multiple t-test with the bigger bubble size representing a smaller p-value). | ||
We firstly evaluated the impact of increased sphingolipids on intracellular S. aureus in macrophages. At 36 hpi and 48 hpi, dihydroceramide and ceramide had the largest fold change of all disturbed lipids in the sphingolipid pathway. Consequently, we used commercially available lipids such as Cer(d18:0/16:0) (dihydroceramide) and Cer(d18:1/16:0) (ceramide) to assess their effect on intracellular bacterial survival. However, both Cer(d18:0/16:0) and Cer(d18:1/16:0) didn't alter intracellular bacterial viability appreciably at a nontoxic concentration (Fig. S2†). We further selected sphinganine to investigate their role in the intracellular bacteria survival since it was consistently upregulated from 12 hpi to 48 hpi, but also no significant effect of sphinganine (spa) was observed (Fig. S3A†).
Moreover, we also investigate the effect of sphingolipid pathway inhibition on S. aureus infection. The serine-palmitoyl-transferase (SPT) and ceramide synthase (CERS) were involved in the initial steps of the sphingolipid pathway, which were validated to be blocked by myriocin and fumonisin B1, respectively.18,19 We pretreated the cells with the two inhibitors for 24 hours so that the pathway was effectively blocked prior to S. aureus infection. The internalization of S. aureus into macrophages was significantly reduced around 35% to 50%, when we suppressed SPT and CERS with various concentrations of myriocin and fumonisin B1, respectively (Fig. 4A). We then evaluated the two inhibitors' effect on intracellular bacteria for 24 hours. Interestingly, the inhibition of the sphingolipid pathway by using myriocin and fumonisin B1 boosted the intracellular bacteria survival in macrophages in a dose-dependent manner (Fig. 4B). Finally, we introduced a sphingolipid inhibitor into cells for 1 hour during the S. aureus infection to investigate whether the inhibitor affected bacterial adherence and phagocytosis. However, fumonisin B1 didn't significantly alter the amount of adherent and invaded bacteria during the infection, suggesting that the decrease of internalized bacteria was due to sphingolipid depletion (Fig. S3B†). Collectively, the current results indicated that these upregulated sphingolipids (sphinganine, ceramides, sphingomyelin, etc.) released from macrophages might be the outcome after S. aureus infection since they didn't affect intracellular bacteria survival. However, suppressed sphingolipid biosynthesis in the metabolism pathway not only impaired the phagocytosis of macrophages but also prevented intracellular killing of S. aureus in macrophages.
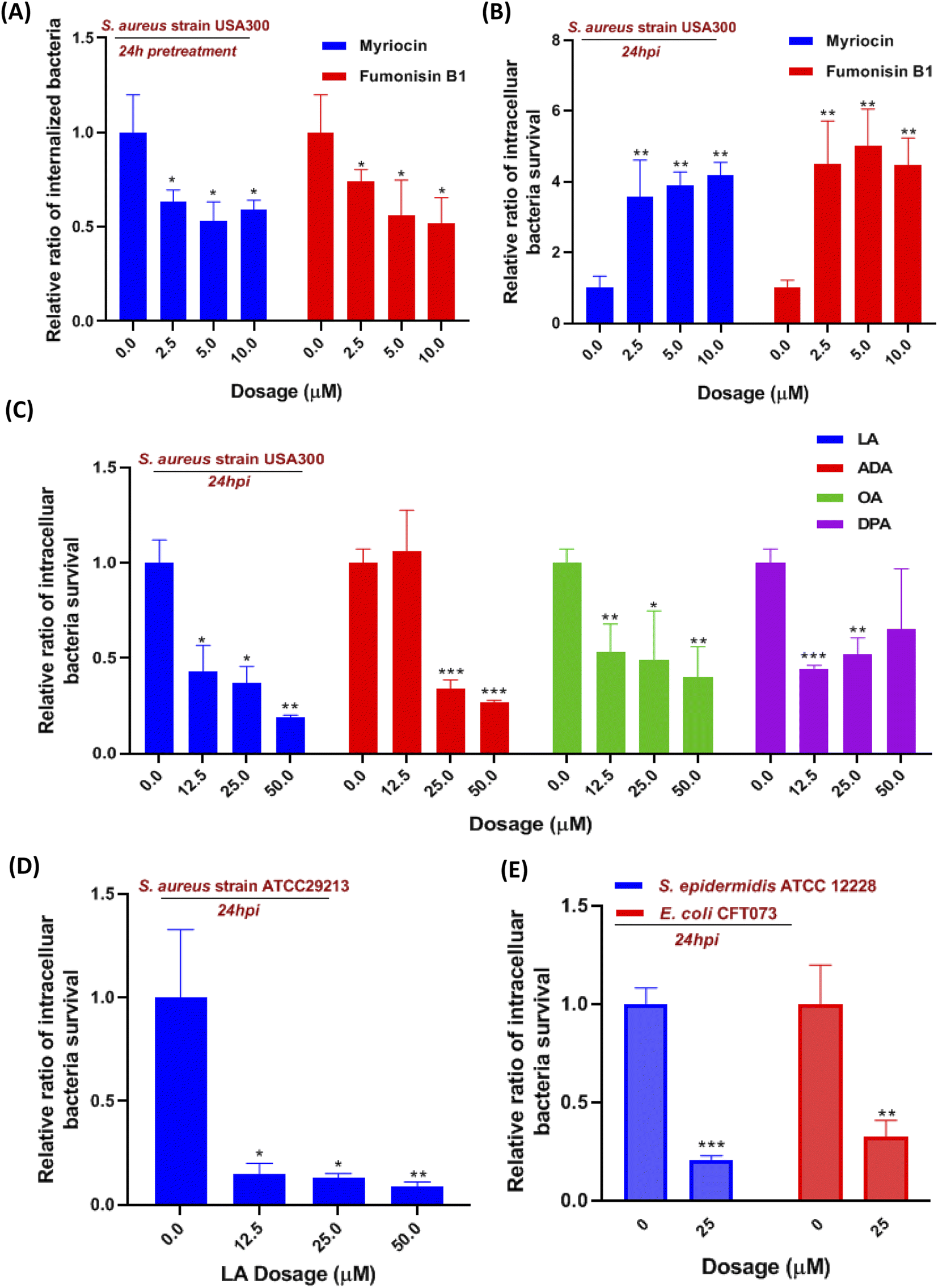 | ||
| Fig. 4 The effect of sphingolipid and linoleic acid metabolism pathways on S. aureus infected macrophages. (A and B) The relative ratios of internalized bacteria (A) or intracellular survival of S. aureus (B) determined for myriocin and fumonisin B1. For internalized bacteria assay, cells were treated with myriocin and fumonisin B1 for 24 hours before and during the infection. For intracellular survival, S. aureus (USA 300 strain) was determined after myriocin and fumonisin B1 treatments for 24 hours. (C) The relative ratios of intracellular survival of S. aureus (USA 300 strain) determined after treatment with four fatty acids for 24 hours. (D) The relative ratios of intracellular survival of S. aureus (ATCC29213 strain) determined after linoleic acid treatment for 24 hours. (E) The relative ratios of intracellular survival of Staphylococcus epidermidis (ATCC12228 strain) and Escherichia coli (CFT073 strain) determined after linoleic acid treatment for 24 hours. Data are given as mean ± SD of three independent experiments. One-way ANOVA when compared with the DMSO control group. *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
Linoleic acid metabolism pathway activation contributed to S. aureus clearance in macrophages
Although linoleic acid metabolism exhibited the highest pathway impact value, many studies also reported that LA as a precursor contributed to an inflammatory state and was involved in multiple inflammation processes.20–22 So we hypotheses that LA may be relevant to innate immunity in the S. aureus-infected macrophage. Linoleic acid (LA) and arachidonic acid (ADA) are the two critical lipids in the linoleic acid metabolism pathway (Fig. 3C), while oleic acid (OA) and docosapentaenoic acid (DPA) were not mapped in the LA pathway but both were upregulated at 36 hpi. Therefore, four fatty acids were incubated with S. aureus infected macrophages to evaluate their effects on intracellular bacteria survival. Utilizing the colony-forming unit (CFU) counting assay, we found a significant reduction of intracellular bacteria survival in all fatty acid treated cells, exhibiting generally higher dependence on LA and AA than the other two fatty acids (Fig. 4C). The result showed that LA metabolism pathway activation in macrophages could significantly reduce the intracellular bacterial load (S. aureus strains USA 300), which was further validated by using another S. aureus strain ATCC29213 (Fig. 4D). To investigate if LA-mediated bacterial killing could be observed in other bacterial strains, we also chose two representative bacterial strains e.g. S. epidermidis (Gram-positive) and Escherichia coli (Gram-negative). Infections with either S. epidermidis or E. coli demonstrated a substantial decrease in intracellular bacteria survival following treatment with 25 μM LA (Fig. 4E).Next, we also evaluated the impact of LA addition during infection on bacterial adherence and macrophage phagocytosis, but neither was affected (Fig. S4A†). Then we further investigated if LA could affect bacteria directly, and the minimum inhibitory concentration of LA in Dulbecco's Modified Eagle Medium (DMEM) containing bacteria was applied to evaluate the LA's effect. Surprisingly, bacteria in the medium didn't reduce even though the LA concentration increased to 500 μM (Fig. S4B†). We also performed the bacteria growth kinetics assay with or without LA addition. Interestingly, the bacteria growth results showed that two S. aureus strains (USA 300 and ATCC29213) co-incubated with LA exhibited a dose-dependent response and better growth curve than S. aureus alone (Fig. S5A and B†). The current results suggest that LA was beneficial for S. aureus growth without cell involvement, indicating that LA may potentiate intracellular bacterial clearance via an indirect way, such as downstream metabolites or regulating ROS production of macrophages.
Investigation of linoleic acid metabolism pathway's effect on S. aureus infected macrophages
Next, we explored if the capacity of intracellular bacteria killing was dominantly contributed by LA itself or the LA metabolism pathway. The targeted LC-MS method and chemical inhibitors were applied to monitor metabolites in the LA metabolic pathway upon exogenous LA addition to mock or S. aureus infected cells respectively (Fig. 5A). The targeted approach results indicated that all downstream metabolites of LA significantly increased in either mock or S. aureus infected cells (Fig. 5A and Table S3†). The current result also proved that the LA metabolism pathway was indeed enhanced upon LA supplement. Among them, AA showed the maximum increase (23.67/25.82 fold in infected/mock cells respectively), while 9(10)-EpOME exhibited the minimum upregulation (2.05/1.79 fold) after exogenous LA supplement. The result demonstrated that the LA metabolism and LA prefer to metabolize to AA, which might be a preferred metabolism pathway of LA. Because AA treatment also exhibited significantly reduced intracellular bacteria (Fig. 4C), we speculated that LA's capacity for intracellular bacteria-killing was partially contributed by AA. Then we further investigated another two downstream pathways that may contribute to the LA's anti-intracellular bacteria effect. Firstly, we couldn't propose a confirmed conclusion that EpOME metabolites contributed to LA's effect since SKF-525A alone (a cytochrome P450 inhibitor) treatment significantly increased intracellular bacteria (Fig. 5B). Even through the intracellular bacteria killing effect of LA was nullified by SKF-525A (Fig. 5B), we still speculated that it was largely due to the background effect of SKF-525A. Secondly, the HODE pathway was also validated by lipoxygenase inhibition and different results from the EpOME pathway were observed. LA remarkably reduced intracellular bacteria and this effect was partially blocked by nordihydroguaiaretic acid (NDGA, a lipoxygenase inhibitor) at dose response (Fig. 5C). Corroboratively, 5 μM NDGA treatment also decreased intracellular bacteria rather than increasing them (Fig. 5C). Thus, we speculated that the HODE pathway could also provide partial contribution to the LA's effect. Taken together, the current results demonstrated that two downstream pathways (AA and HODE pathways) were proved to partially contribute to LA's capacity of intracellular bacteria killing, which could be the combined effects from LA itself and the LA metabolism pathway.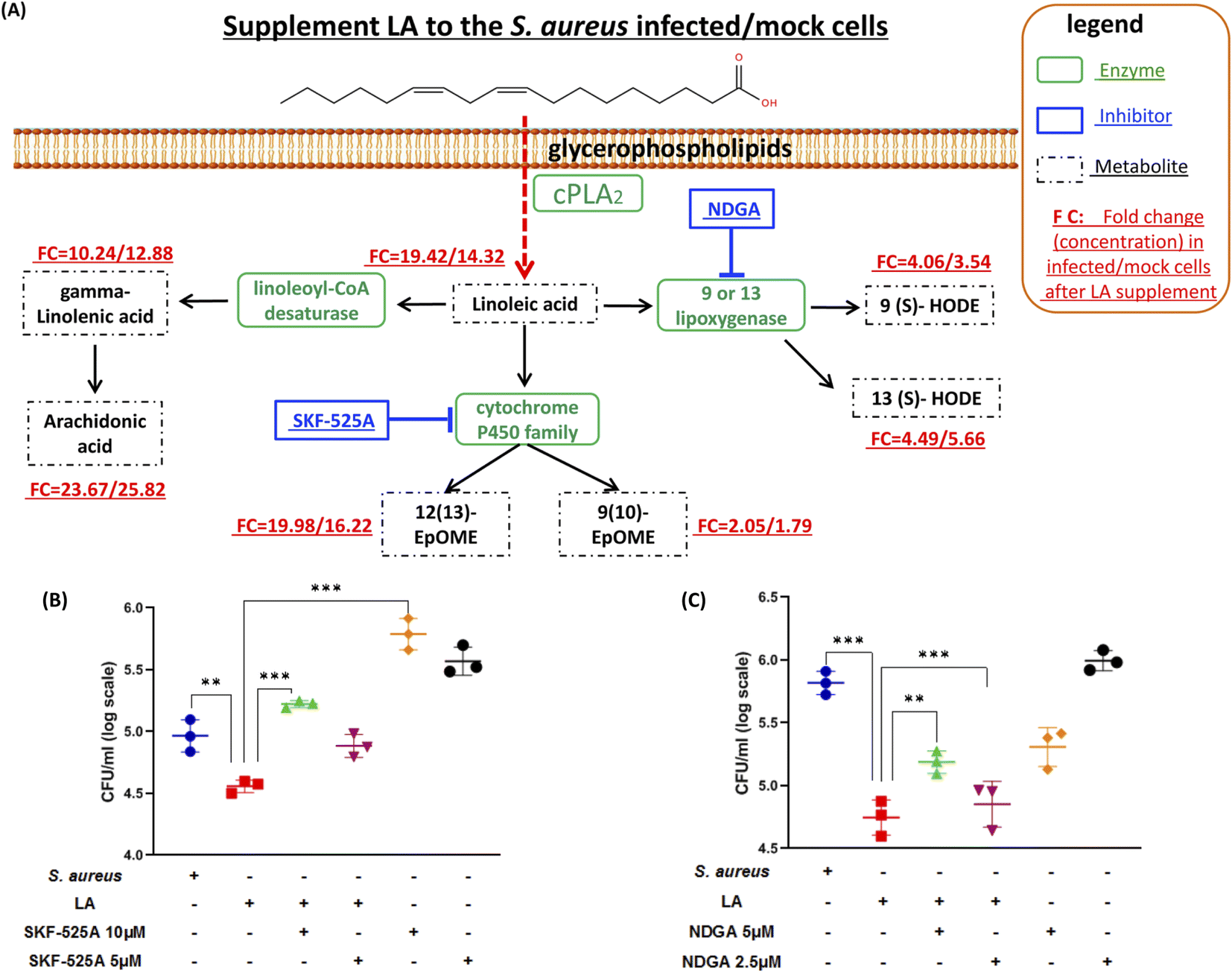 | ||
| Fig. 5 Targeted analysis of linoleic acid metabolism and investigation of the pathway's effect on an S. aureus/mock infected macrophage. (A) LC-MS/MS based target approach was applied to quantify these metabolites mapped in the linoleic acid metabolism with or without a LA supplement. The fold changes of selected metabolites were calculated from the ratio of absolute quantified concentration for specific metabolites in LA-treated and non-treated cells. (B) Intracellular bacterial loads were determined by counting CFUs after DMSO, LA, and different concentrations of SKF-525A treatments. (C) Intracellular bacterial loads determined by counting CFUs after DMSO, LA, and different concentrations of NDGA treatments. Data are shown as mean ± SD of three independent experiments. Unpaired t-test was used to analyze the significance of the observed differences. For all statistical analysis above, *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
Linoleic acid induced ROS production in macrophages
Reactive oxygen species (ROS) production in phagocytes is a major defense mechanism against pathogens. Thus, we firstly detected the intracellular superoxide by using a specific fluorescent probe HKSOX-1r since superoxide is the precursor to most other ROS.23 The superoxide result indicated that increasing concentrations of LA caused a dose-dependent enrichment of superoxide in the cell (Fig. 6A). Due to the short life of superoxide, it was induced by LA within 0.5 h and antimycin A (AMA) as a positive control exhibited a sustainable ROS effect at indicated time points (Fig. 6B). Moreover, the superoxide-generated pathway was also investigated by scavenged assay. Diphenylene iodonium (DPI) is a potent though nonselective NADPH oxidase (NOX) inhibitor that blocks the production of superoxide.24 Surprisingly, the LA induced ROS effect wasn't scavenged by DPI (Fig. 6C). Next, we further investigated the downstream ROS of superoxide, and 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), a general chemical probe, was used as an indicator for multiple ROS detection in cells. The H2DCFDA probe results indicated that LA remarkably induced ROS generation even more than phorbol myristate acetate (PMA), an activator of NADPH-oxidase,25 and the effect was sustained for 24 hours (Fig. 6D). Intriguingly, the ROS effects induced by LA at multiple time points were scavenged dramatically by antimycin A (Fig. 6E), which was an inhibitor of cellular respiration and binds to the Qi site of complex III.26 Taken together, the current ROS detection results illustrated that LA indeed increased intracellular superoxide, which quickly converted to other ROS and maintained the ROS effect for 24 hours. In addition, the pathway of ROS production induced by LA may relate to cellular respiration and the electron transport chain instead of the NADPH oxidase pathway.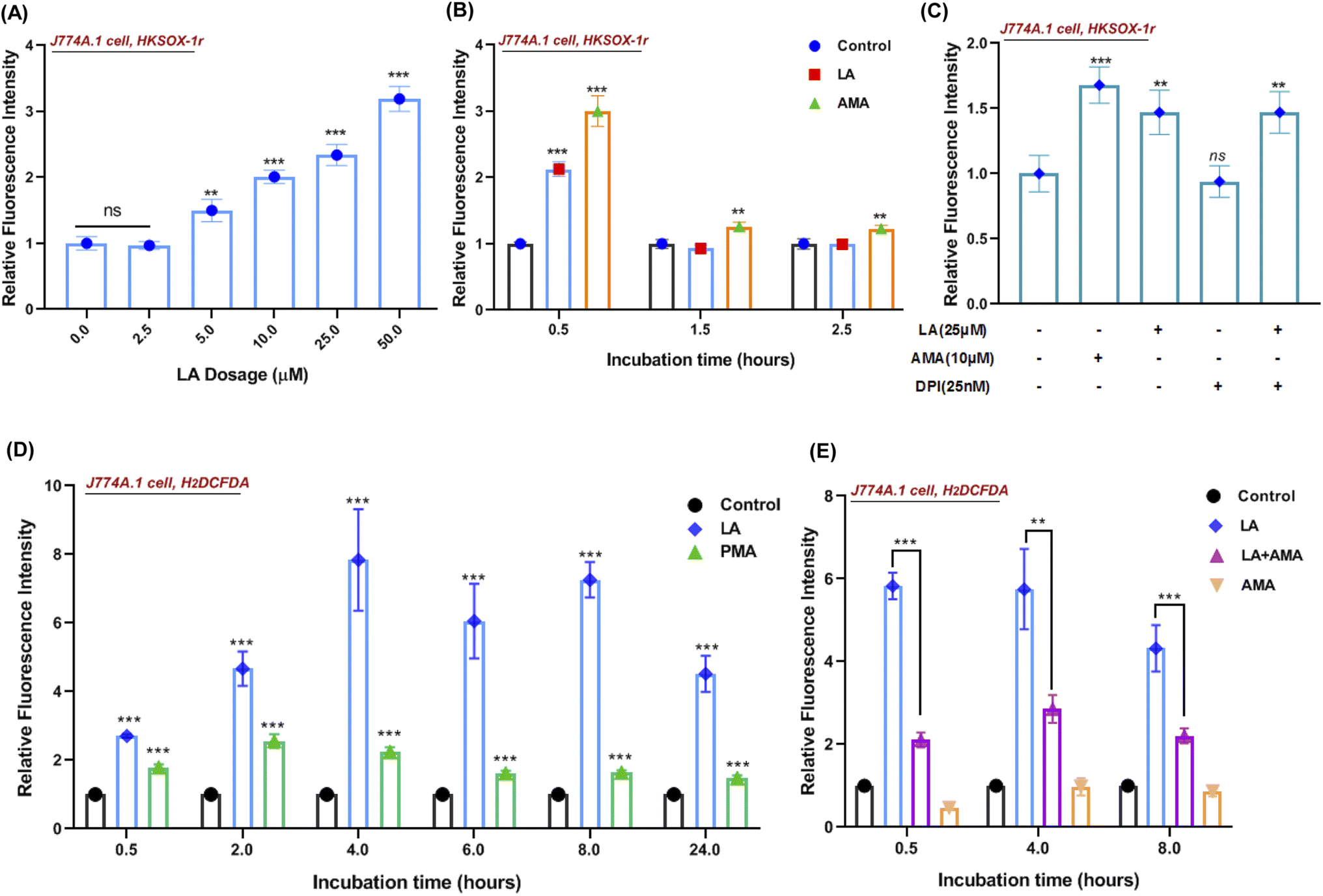 | ||
| Fig. 6 Linoleic acid induced ROS production in a macrophage. (A) Quantified relative fluorescence intensities of superoxide by (HKSOX-1r probe) upon a dose gradient of LA (2.5 μM to 50 μM) stimulation for 30 min incubation in J774A.1 cells. (B) Quantified relative fluorescence intensities of superoxide (HKSOX-1r probe) upon LA (25 μM) and AMA (5 μM) stimulation at indicated incubation times in J774A.1 cells. AMA, antimycin A. (C) Quantified relative fluorescence intensities of superoxide (HKSOX-1r probe) upon LA (25 μM) stimulation in the presence/absence of a NADPH oxidase (NOX) inhibitor (DPI, 25 nM) for 30 min incubation in J774A.1 cells. AMA (10 μM) was used as a positive control for reference. (D) Quantified relative fluorescence intensities of ROS (H2DCFDA probe) upon LA (25 μM) and PMA (100 nM) stimulation at different incubation times in J774A.1 cells. PMA (100 nM) was used as a positive control for reference. (E) Quantified relative fluorescence intensities of ROS (H2DCFDA probe) upon LA (25 μM), AMA (10 μM), and LA (25 μM) combined with AMA (10 μM) treatments at indicated incubation times in J774A.1 cells. Data are shown as mean ± SD of four independent experiments. Unpaired t-test was used to analyze the significance of the observed differences. For all statistical analysis above, *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
In addition, we also investigated whether linoleic acid supplementation may also stimulate ROS production in S. aureus. The results indicated that LA induced ROS production (H2DCFDA probe) only at 0.5 h in S. aureus and had no impact on the superoxide production (HKSOX-1r probe) at different time points (Fig. S6†). Furthermore, the results of bacteria growth and minimum inhibitory concentration assays (Fig. S4B and S5†) also revealed that LA didn't significantly affect bacterial survival at various concentrations. Collectively, these results demonstrated that the ROS production in macrophages, rather than the ROS production in S. aureus, played a dominant role in S. aureus killing.
LA induced ROS effect in multiple cell lines and improved S. aureus clearance in a mouse model
To visualize the ROS production induced by LA, we tested the performance of superoxide and general ROS with J774A.1 live cell imaging by using suitable fluorescent probes. As expected, unstimulated phagocytes loaded with HKSOX-1r, HKSOX-1m (specific for mitochondria targeting) and H2DCFDA probes showed a barely detectable background fluorescence signal (Fig. 7A–C). However, upon LA stimulation, the fluorescence signals were drastically enhanced in treated J774A.1 cells, indicating a superoxide burst in the cytosol and mitochondria (Fig. 7A and B) as well as general ROS production (Fig. 7C). The visualized ROS in living cells also support the evidence that LA induced ROS production was highly correlated with cellular respiration and the electron transport chain in mitochondria. Further positive control imaging experiments with AMA and PMA also exhibited significantly increased fluorescence signals in living J774A.1 cells (Fig. 7A–C).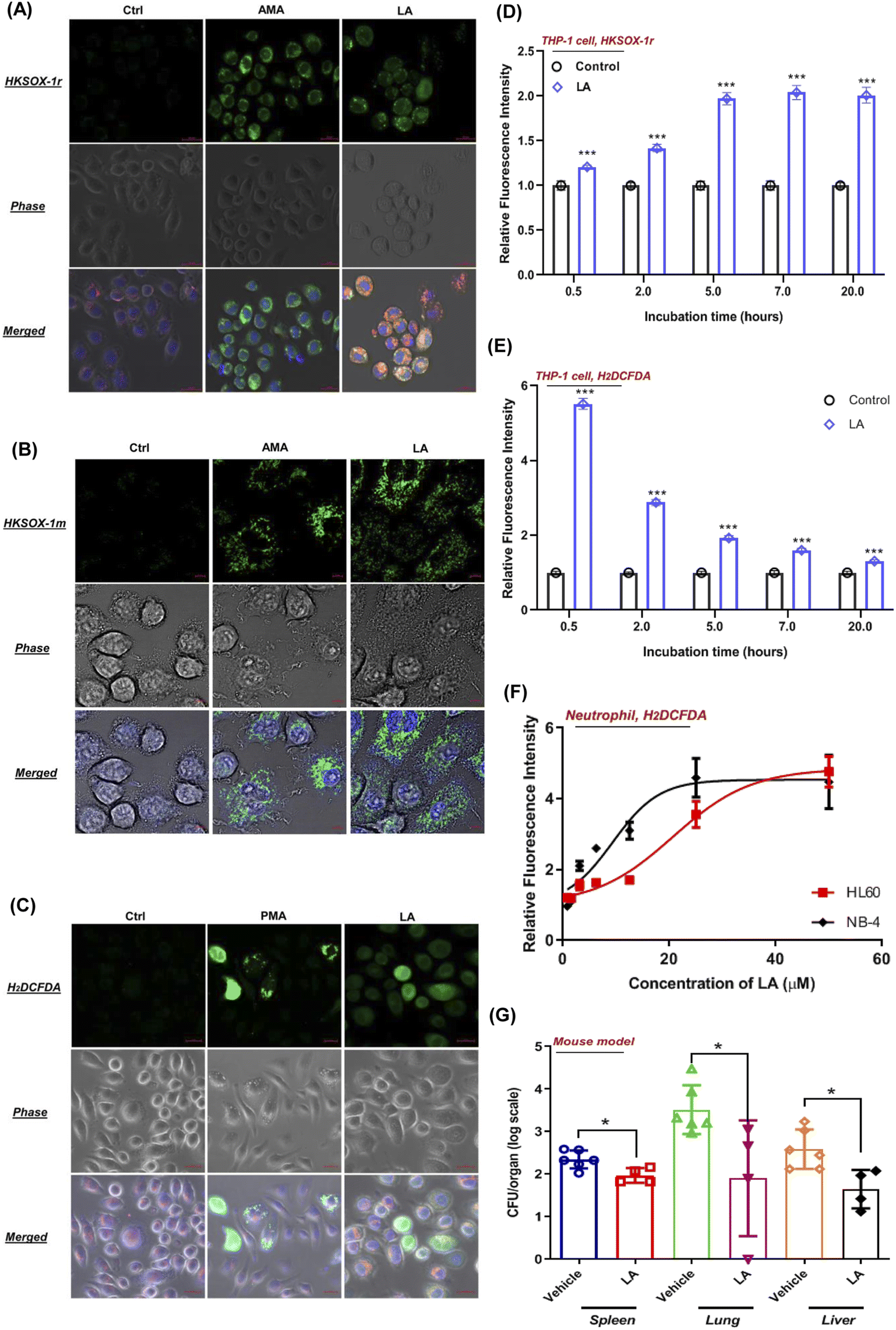 | ||
| Fig. 7 LA induced ROS production in living phagocytes and neutrophils. (A) Representative images of a J774A.1 cell stimulated with AMA (5 μM) or LA (25 μM), and coincubated with HKSOX-1r (2 μM) for 30 min before confocal imaging. (B) Representative images of a J774A.1 cell stimulated with AMA (5 μM) or LA (25 μM), and coincubated with HKSOX-1m (10 μM) for 30 min before confocal imaging. (C) Representative images of a J774A.1 cell stimulated with PMA (100 nM) or LA (25 μM) and coincubated with H2DCFDA (6 μM) for 30 min before confocal imaging. Cells were costained with mitochondrial dye MitoTracker Red (50 nM), nuclear DNA dye Hoechst 33342 (150 ng mL−1), and probes for 30 min before confocal imaging. The compound was kept incubated with cells during the image capture. Merged: all fluorescence images merged. Scale bar = 20 μm. (D and E) Fluorescence intensities of superoxide (D) and ROS (H2DCFDA probe) (E) upon LA (25 μM) induction at indicated incubation times in THP-1 cells. (F) Fluorescence intensities of ROS (H2DCFDA probe) upon LA induction at different concentrations in HL-60 and NB-4 differentiated neutrophils. (G) Mice organs such as lungs, livers and spleens were harvested, and bacterial load was analyzed by viable count respectively. Data represent mean ± SD. One-way ANOVA was used when compared with the vehicle group. For all statistical analysis above, *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
In addition, we also investigated LA induced ROS generation in other immune cells such as human THP-1 macrophages and neutrophil-like cells differentiated from HL-60 and NB-4. Surprisingly, LA induced significantly enriched superoxide from 0.5 hours to 20 hours in THP-1 cells, which was longer than superoxide production of J774A.1 cells (Fig. 7D). Like J774A.1 cells, LA also significantly induced sustainable H2DCFDA detected ROS enrichment in the THP-1 cells (Fig. 7E). Moreover, we also investigated the effect of LA's ROS induction on neutrophils by using a H2DCFDA probe. As shown in Fig. 7F, LA exhibited dose-dependent ROS generation effects in two kinds of human neutrophil-like cells (HL60 and NB-4). The current results confirmed that LA indeed induced intracellular ROS production including superoxide and other ROSs in multiple human cell lines, which suggest that ROS effects induced by LA could have potential applicable value for clinical S. aureus infection control. To further validate that LA induced ROS could reduce the bacteria load in vivo, we employed a mouse model to evaluate the in vivo effects of supplementing LA on S. aureus infection. In the mouse bacteremia model, after 7 doses of LA treatments, the vital organs, lungs, livers and spleen were harvested, and bacterial loads were analyzed by viable count on day 4. In LA-treated mice, significant decreases of the bacterial load in the lung (1.5 log, p = 0.03), liver (1 − log, p = 0.013) and spleen (0.5 log, p = 0.019) were shown when compared with the vehicle group (Fig. 7G). Collectively, our data demonstrated the in vitro and in vivo effects of LA and the corresponding metabolism pathway to ameliorate S. aureus infection.
Discussion
Lipid metabolism reprogramming upon S. aureus infected macrophages
In this study, we collected S. aureus infected cell samples at multiple time points to dynamically monitor intracellular lipid profile change. It is worth noting that 50% lipids are upregulated sphingolipids of all 12 significantly changed lipids at 1 hour post infection (hpi) and 12 hpi (Table S2†). The results revealed that the sphingolipid metabolism reprogramming was dominant at the early stage of S. aureus infected macrophages. Sphingolipids included multiple types such as sphingomyelins, ceramides, glycosphingolipids, etc., and exhibited that wide biological functions covered nearly all cell biological processes.27 Previous reports showed that sphingolipids were enriched in the plasma membrane and involved in various viruses' entry.28,29 Likewise, accumulating studies showed that the disruption of the sphingolipid biosynthesis pathway blocked the phagocytosis by phagocytes for bacteria such as Candida albicans and S. aureus.30,31 Acid sphingomyelinase plays a critical role in sphingolipid metabolism and is responsible for hydrolyzing sphingomyelin to ceramide and phosphorylcholine. Acid sphingomyelinase activation in macrophages facilitated the fusion of phagosomes with lysosomes and further killed intracellular bacteria more efficiently.32 Our study demonstrated that these dysregulated sphingolipids (e.g., sphinganine and ceramides) didn't affect the progression of bacterial infection. Consequently, we speculated that several different processes are responsible for the outcome. Firstly, these sphingolipids may be the physiological consequence of stimulating macrophages rather than signaling lipids (e.g., LA) that are able to eliminate intracellular bacteria. Secondly, ceramide-rich membrane regions promote structural changes in the plasma membrane such as membrane curvature and vesicle formation, fusion, and trafficking.33 Therefore, these upregulated ceramides in our study might be the result of structural alteration caused by bacterial infection. Finally, ceramides may potentially trigger a pro-apoptotic cascade in cells upon stimulation.34 Thus, we hypothesized that it might be the mechanism that macrophages kill intracellular bacteria by releasing ceramides to trigger cell apoptosis,35,36 which will be validated in future. Moreover, the current study proved that the inhibition of the sphingolipid pathway not only impaired the phagocytosis of macrophages but also weakened the capacity of S. aureus clearance. Therefore, the intact functional sphingolipid pathway in macrophages was critical for bacterial elimination and could maintain multiple functions such as phagocytosis and digest bacteria.Besides sphingolipids, we also found that the PCs (glycerophospholipid pathway), ether-PCs, ether-PEs (ether lipid pathway), LA and ADA (linoleic acid pathway) were increased at 36 hpi and 48 hpi. From the subsequent pathway analysis result, we could observe that linoleic acid and sphingolipid pathways exhibited higher pathway impact values than glycerophospholipid and ether lipid metabolism pathways (Fig. 3A). The pathway impact value was calculated by pathway topological analysis, which is based on the centrality measures of a metabolite in a given metabolic network. For example, the metabolite's position in the specific pathway is one of the criteria used to estimate the metabolite's importance or role in a metabolic network.37 LA is a precursor to (ADA) in the linoleic acid pathway, whereas ADA is the precursor to some prostaglandins, leukotrienes, and thromboxane in the arachidonic acid pathway.38–40 So the LA's position was very important as it could regulate both linoleic acid and arachidonic acid pathways (Fig. 3C). However, PCs, ether-PCs, and ether-Pes are all mapped in relative downstream positions in glycerophospholipid and ether lipid metabolism pathways. For lipid function, PCs are a major component of biological membranes and thought to be transported between membranes within the cell by phosphatidylcholine transfer protein (PCTP).41 Meanwhile ether lipids (e.g., ether-PCs and ether-PEs) are making up lipid raft microdomains in the membrane and affect membrane fluidity.42 In terms of lipid pathway's impact, regulatory role and function, we prefer to choose the LA metabolism pathway for further investigation.
Linoleic acid (LA) metabolism was the pathway with the highest impact value in our current study (Fig. 3A and C). LA is a polyunsaturated omega-6 fatty acid and is an essential fatty acid for humans, who must obtain it through their diet.43 Like all fatty acids, LA can be used either as a source of energy or esterified to form other lipids for storage such as phospholipids, triacylglycerols, and cholesterol esters. In addition, when external infection or stimulation is received by cells, LA is released from membrane phospholipids and can be enzymatically oxidized to a variety of derivatives involved in cell signaling.39 Our study indicated that LA and ADA significantly are upregulated in macrophages at 36 hpi and 48 hpi (Fig. 3C and Table S2†), which could be associated with the infectious state. Previous studies reported that mouse infections can be accompanied by an increase of the omega-6 fatty acid level in the plasma, spleen, and liver,44 contributing to an inflammatory state. Correspondingly, omega-6 fatty acids down-regulated the inflammation process, decreasing IL-6, TNF-α, and IL-1β levels, and CD86 expression.44 In addition, the downstream metabolites of LA such as gamma-linolenic acid (GLA) and ADA also attenuated the inflammatory state by M. tuberculosis infection or LPS stimulation in macrophages.22,45 Fewer studies reported LA's effect on the function of bacterially infected macrophages, and our current study provided the evidence that LA treatment activated all downstream metabolites and exhibited the in vitro and in vivo effects to ameliorate S. aureus infection.
Reactive oxygen species (ROS) have been known for many years as fundamental for macrophages to kill invasive microorganisms through the oxidative burst mediated by NADPH oxidase.46 However, accumulating studies demonstrated that mitochondrial ROS play essential roles in several innate immune functions.47 Likewise, our current study also showed that ROS production induced by LA could be related to the electron transport chain in mitochondria instead of the NADPH oxidase pathway. More importantly, the HKSOX-1m probe exhibited the co-localized capacity to prove that ROS production induced by LA occurred in the mitochondria. Other chemical probes such as HKSOX-1r are specific for superoxide located in cytosolic, and H2DCFDA is for general intracellular ROS. H2DCFDA could be used as an indicator for multiple ROS in cells such as hydrogen peroxide (H2O2), hydroxyl radical (HO˙), and peroxynitrite anion (OONO−).48 In addition, we also examined HOCl49 and H2O2 production by using specific probes, but no significant changes were observed after LA treatment (data not shown). Therefore, we could confirm that LA induced the superoxide and other types of ROS in multiple immune cell lines. A previous study revealed that LA could significantly reduce the expression of superoxide dismutase (SOD) and catalase (Cat) in THP-1 cells,50 while SOD and Cat are the two important enzymes that catalyze the dismutation of superoxide and hydrogen peroxide. This could explain why LA induced the sustainable superoxide and other ROS in THP-1 cells in our study.
Experimental
Chemical materials
HPLC-grade methanol, acetonitrile, chloroform and isopropanol were purchased from Merck (Darmstadt, Germany). HPLC grade water was prepared using a Milli-Q water purification system (Millipore, MA, USA). Analytical grade formic acid, ammonium formate, and commercial standards used for biomarker identification were purchased from Sigma Aldrich (MO, USA). Internal standards (IS) including arachidonic acid-d8 (AA-d8), platelet-activating factor C-16-d4 (PAF C-16-d4) 15(s)-HETE-d8 and leukotriene-B4-d4 were purchased from Cayman Chemical (Ann Arbor, MI, USA).Lipids for testing intracellular bacteria survival including linoleic acid (LA), arachidonic acid (ADA), docosapentaenoic acid (DPA), oleic acid (OA), sphinganine (SPA), Cer(d18:0/16:0) and Cer(d18:1/16:0) were purchased from Cayman Chemical (Ann Arbor, MI, USA). Chemical inhibitors such as myriocin (serine-palmitoyl-transferase inhibitor), fumonisin B1 (sphingosine N-acyltransferase inhibitor), nordihydroguaiaretic acid (NDGA, lipoxygenase inhibitor), and proadifen hydrochloride (SFK-525A, cytochrome P450 inhibitor) were also purchased from Cayman Chemical. The fluorescein probe H2DCFDA was purchased from Thermo Fisher (catalog number: D399). HKSOX-1r and HKSOX-1m probes for the detection of endogenous superoxide and ROS inducers or inhibitors including phorbol 12-myristate 13-acetate (PMA), dibenziodolium chloride (DPI) and antimycin A (AMA) were kindly donated by professor Yang Dan.
Bacterial strain, cells and culture conditions
The bacterial strains used in the current study were Staphylococcus aureus USA 300-FPR3757 (ATCC® BAA1556TM), Staphylococcus aureus subsp. aureus rosenbach (atcc® 29213) S. epidermidis (clinical isolate) and Escherichia coli (ATCC700928). The BHI broth and BHI agar plates were used for S. aureus. The bacterial strains were cultured from frozen stocks in BHI medium in a shaker bath at 37 °C. Bacterial cells from overnight cultures were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 into 5 mL of BHI medium for subculture. The cultures were harvested by centrifugation and the cell density was adjusted at an absorbance of 1.0 at 600 nm with a spectrophotometer (Hitachi U-2800). Then bacterial suspension was diluted to an appropriate density with DMEM medium before inoculation.
:100 into 5 mL of BHI medium for subculture. The cultures were harvested by centrifugation and the cell density was adjusted at an absorbance of 1.0 at 600 nm with a spectrophotometer (Hitachi U-2800). Then bacterial suspension was diluted to an appropriate density with DMEM medium before inoculation.
The used cell line included J774A.1 macrophage (ATCC® TIB-67™) and THP-1 (TIB-202™) and human promyelocytic leukemia HL-60 (ATCC CCL-240) and NB-4. The J774A.1 cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and incubated in 5% CO2 at 37 °C. J774A.1 cells were seeded onto 6 well plates for bacteria infected cell lipidomics study and 24 well plates for intracellular bacteria survival assay. J774A.1 cells, THP-1 cells, HL60 and NB-4 cells were seeded onto 96 well plates for ROS detection assay.
Lipid extraction for untargeted and target lipidomics
Confluent J774A.1 cells were mock infected or infected with S. aureus at a MOI of 10 and incubated in DMEM medium for indicated time points. After 1 hpi, the culture media were removed from the 6-well plate and the cells were rinsed with PBS, followed by incubation of the macrophages with 50 μg mL−1 gentamicin for 1 h at 37 °C. After another 1 hpi, low concentrated gentamicin (10 μg mL−1) was used to replace the medium and maintained for other testing time points.For untargeted lipidomics, cells were collected at 1, 12, 36 and 48 hpi, respectively. The lipid extraction was performed according to a previously described protocol with some modifications.12,13 Briefly, 1 mL of ice-cold PBS buffer containing butylated hydroxytoluene (BHT) was added to dissociate cells and 50 μL of cell suspension were used for DNA extraction for normalization.51 The cell suspension was sonicated on ice for 4 minutes at an amplitude of 30% in interval mode (work 10 seconds and cool down 20 seconds), followed by 0.22 μm filtering for removing intracellular bacteria. Four milliliters of chloroform/methanol (v/v 2:1, containing IS) were added to the cell suspension, followed by vortexing and centrifugation at 4500 rpm for 10 min at 4 °C. The bottom phase was transferred to glass vials and evaporated to dryness in a cold trap concentrator (Labconco Centrivap, MO, USA) for storage at −80 °C. The dried samples were reconstituted in 25 μL chloroform–methanol (1:1, v/v) and diluted to 10 times the original volume of cell lysate in 225 μL isopropanol–acetonitrile–water (2:1:1, v/v/v) for LC-MS analysis.52
For targeted analysis, cells were cultured and infected the same as described in the untargeted lipidomics experiments. At 24 hpi, the cells were gently washed twice with cold 60% methanol and 0.85% ammonium bicarbonate in water (quenching buffer, stored at −20 °C). Then 1 mL of the solution of ethanol: PBS (v/v, 1:3, pH 7.2) containing BHT and 100 μM AA-d8 was added to dissociate cells, vortexed and mixed well. After sonication, the cell lysates were applied to solid-phase extraction columns (SPE, Waters Oasis MAX 3 cc Vac Cartridge) for target lipid enrichment. Briefly, the SPE cartridges were washed with 3 mL of acetonitrile (ACN) and 25% ACN, respectively. Lipid extracts were then loaded onto cartridges and further washed with 3 mL of 25% ACN and ACN. Finally, the SPE cartridges were eluted with 1.3 mL ACN containing 0.1% formic acid into tubes containing 200 μL of 10% glycerol in methanol as a trap solution. Elute analytes were evaporated until only glycerol remains for storage at −80 °C until target analysis. 60 μL methanol/acetonitrile (v/v, 1:1) was added to the samples and mixed thoroughly for LC-MS analysis.53
Lipid treatment of S. aureus infection in vitro
For intracellular bacteria survival assay, the J774A.1 cells were infected with S. aureus at a MOI of 1. After killing extracellular bacteria with a high concentration of 50 μg mL−1 of gentamicin, the lipids or inhibitors and 10 μg mL−1 of gentamicin were co-incubated with the cell. At 24 hpi, the cells were extensively washed, lysed in ice-cold 0.025% Triton X-100 (ultrapure water) to release intracellular bacteria, pelleted by centrifugation at 8000 rpm, and resuspended in PBS. Aliquots were plated on Luria broth agar plates, and the CFUs were counted after growth overnight at 37 °C. For internalized bacteria assay, the J774A.1 cells were pretreated with lipids or inhibitors 24 hours before and during S. aureus infection. The internalized bacterial number was counted in the same way as intracellular bacteria survival.For adherence and phagocytosis assay, we added the lipids or inhibitors during the S. aureus infection for 1 hour. Then we removed the medium and washed the cells 3 times with 1 mL PBS. The cells were lysed in ice-cold 0.025% Triton X-100 (ultrapure water) to release intracellular bacteria for CFU counting and the same operation as the assay of intracellular bacteria survival.
Linoleic acid treatment of S. aureus infection in vivo
As previously described,54 we kept the 6 to 8 weeks-old BALB/c female mice in a biosafety level 2 animal facility. The mice were housed in microisolator cages, and they received food and water ad libitum. A standard operating procedure was followed for the ethically approved protocols (CULATR 5439-20). The experiments were conducted in a biosafety level 2 animal facility.S. aureus strain USA300 was grown to the early exponential phase and then washed and resuspended in PBS to a concentration of 5 × 106 CFU/100 microliters.
The 6 to 8 weeks-old female Balb/c mice were infected with S. aureus (1 × 107 per mouse) intravenously (i.v.) through the tail vein and randomized into two groups (6 mice for the vehicle and 4 mice for the LA treatment group). Half hour post infection, the mice received intraperitoneal injections of the compound LA (2.78 mg per kg per dose) or vehicle (PBS with 5% DMSO and 2% Tween-80) as a control injection. For every 12 hour period, the mice received the treatment of LA or the vehicle. The animals were euthanized from each group on day 4. Lungs, livers and spleens were collected and homogenized for bacterial counts.
Untargeted and targeted lipidomics analyses
Untargeted lipidomics analysis was using an Acquity UPLC system coupled to a Synapt G2-Si HDMS mass spectrometer system (Waters Corp., MA, USA). Chromatography was performed on a Waters ACQUITY CSH C18 column (100 × 2.1 mm; 1.7 μm) coupled to an ACQUITY CSH C18 VanGuard pre-column (5 × 2.1 mm; 1.7 μm) (Waters; Milford, MA, USA). The mobile phase and gradient are described in Table S4.†55 The column and autosampler temperature were maintained at 55 °C and 4 °C, respectively. The injection volume was 7 μL. The mass spectral data were acquired in both positive and negative modes. The capillary voltage was maintained at 2.0 kV (for negative) and 2.5 kV (for positive). Mass spectra were acquired over the m/z range of 100 to 1500. The SYNAPT G2-Si HDMS system was calibrated using sodium formate clusters and operated in sensitivity mode. MS/MS acquisition was performed with the same parameters as in MS acquisition. Collision energy was applied in the range from 30 to 45 eV for fragmentation to allow putative identification and structural elucidation of significant lipids.56,57For target lipidomics, an ultra-performance liquid chromatograph coupled to a triple quadrupole linear ion trap mass spectrometer (UPLC-Q-TRAP 5500) analytical platform (Applied Biosystems Instrument Corporation, Foster city, CA) was applied in the current study. The instrument was operated in negative multiple reaction monitoring (MRM) mode. Individual analyte standards were infused into the mass spectrometer and MRM transitions and source parameters were optimized for each analyte. Chromatography was performed on a Waters ACQUITY BEH C18 column (100 × 2.1 mm; 1.7 μm) coupled to an ACQUITY BEH C18 VanGuard pre-column (5 × 2.1 mm; 1.7 μm) (Waters; Milford, MA, USA). The column and autosampler temperature were maintained at 40 °C and 4 °C, respectively. The injection volume was 5 μL. The mobile phase, gradient and MRM transitions are described in Tables S5 and S6.†
Data processing, statistical data analysis and lipid identification
All lipidomics study data were processed to a useable data matrix by using MS-DIAL software for further statistical analysis.58 MetaboAnalyst 4.0 (https://www.metaboanalyst.ca) and SIMCA-P V12.0 (Umetrics, Umeå, Sweden) were used for univariate and multivariate analysis, respectively. For univariate analysis, the statistical significance of features was determined between the mock and infected groups using the Student's t-test and fold change. A false discovery rate (FDR) adjusted p-value < 0.05 and fold change >2 or <0.5 were used as the criteria for selecting significant features. For multivariate analysis, the features were subjected to Pareto scaling firstly and then partial least squares discriminant analysis (PLS-DA) was performed as a supervised method to find important variables with discriminative power. The PLS-DA model was evaluated with the relevant R2 and Q2 and the Variable Importance in Projection (VIP) >0.5 was used for significant feature selection.59MS/MS fragmentation was performed on the significant features with high abundances. The significant feature identification was carried out by searching accurate MS and MS/MS fragmentation pattern data in the MS-DIAL internal lipid database,60 MassBank of North America (MoNA, https://mona.fiehnlab.ucdavis.edu/), METLIN database (https://metlin.scripps.edu/) and LIPID MAPS (https://www.lipidmaps.org/). For the confirmation of lipid identity using an authentic chemical standard, the MS/MS fragmentation pattern of the chemical standard was compared with that of the candidate lipid under the same LC-MS conditions to reveal any matching.12,54 MetaboAnalyst and KEGG databases were used to perform significant lipid pathway analyses.
Reactive oxygen species (ROS) detection
The J774A.1 cells were plated in 96-well microplates (SKU: 33396; SPL Life Sciences), at a density of 4 × 104 cells per mL (100 μL per well in quadruplicate) one day before assay. The medium used was DMEM (Dulbecco's modified Eagle medium; high glucose) supplemented with 10% heat-inactivated FBS (fetal bovine serum).For H2DCFDA probe detection, lipids were dissolved in 10% FBS DMEM first. Then the medium containing lipids were loaded after the cells were washed briefly with PBS for indicated time points. At the end of lipid incubation, the lipid treatment medium was removed and washed with HBSS twice, followed by reloading HBSS containing lipids and the probe for another 30 min incubation.
For the neutrophil-like cell line, HL-60 and NB-4 were cultured in RPMI-1640 with 20% FBS. After being differentiated with 1.3% DMSO or all-trans retinoic acid (5 μM) for 6 days, live cells were separated with histopaque-1077 by centrifugation. After washing with HBSS, at a density of 1 × 105 cells per mL, the cells were added to each well of the black plate. Different concentrations of LA treatment for 30 min and then the same amount of probe for macrophage cells were added to each well for another 30 min.
For HKSOX-1r/1m probe detection, the measurements were performed according to a previously described protocol.12,54 All reagents and probe solutions were freshly dissolved in a HBSS mixture (HBSS supplemented with 0.6 mM L-arginine and 0.01% chloramphenicol). After lipid and probe incubation, the cells were briefly washed with PBS, and re-loaded with a probe-free HBSS mixture (100 μL per well) before fluorescence reading. Fluorescence measurement was performed on a DTX880 multimode detector (Beckman Coulter) with the following settings: excitation at 485 nm and emission at 535 nm, with an integration time of 400 ms. Data are presented with subtracting background fluorescence, as mean ± SD for fluorometric measurement with replicates (n = 4) in at least three independent experiments.
Cell imaging
For superoxide and general ROS induction, LA, positive control (AMA and PMA), and probe solutions were freshly dissolved in a HBSS mixture (HBSS supplemented with 0.6 mM L-arginine and 0.01% chloramphenicol) and co-incubated with a ROS fluorescent probe (HKSOX-1r, 1m or H2DCFDA) until imaging. The cells were typically incubated with probes for 30 min at 37 °C with 5% CO2. During imaging, the dish was mounted onto a live cell imaging module (Axiovision). Single-photosection images were acquired on a Zeiss LSM 880 confocal microscope by using the following settings: for HKSOX-1r, execution wavelength = 514 nm and execution wavelength = 527–559 nm (band-pass); for H2DCFDA, execution wavelength = 488 nm and execution wavelength = 520–560 nm (band-pass).Conclusions
In the present study, we examined the mechanisms by which macrophages destroy intracellular bacteria and investigated the critical lipid mediators involved, including LA, which was firstly found to be critical for bacterial clearance. Additional research into developing novel therapeutic strategies targeting the LA pathway may be an interesting direction to pursue. Additionally, our work demonstrates the utility of lipidomic analysis in elucidating new metabolic mechanisms underlying host–microbe interaction.Data availability
We will deposit our LC-MS raw data in the MetaboLights and the currently created study ID is MTBLS6150.Author contributions
Conceptualization: B. Y. and P. G. Methodology: B. Y. and P. G. Investigation: B. Y., K. F., P. G., P.-M. L., and Y. X. W. Visualization: S. Y. Funding acquisition: R. Y.-T. K., and P. G. Supervision: K.-H. S., D. Y., and R. Y.-T. K. Writing – original draft: B. Y. and K. F. Writing – review & editing: P. G., K.-H. S., and R. Y.-T. K.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by HMRF Commissioned Study Project Grant CID-HKU1-14 and RGC of the Hong Kong SAR Projects GRF No. 17104420, RIF R7070-18 and AoE/P-705/16 to RYK. The support from UGC-RMGS to RYK is also acknowledged. The authors also acknowledge the assistance of the University of Hong Kong Li Ka Shing Faculty of Medicine Faculty Core Facility.Notes and references
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS PubMed.
- D. G. Atashbeyk, B. Khameneh, M. Tafaghodi and B. S. Fazly Bazzaz, Pharm. Biol., 2014, 52, 1423–1428 CrossRef CAS PubMed.
- H. Li, X. Zhou, Y. Huang, B. Liao, L. Cheng and B. Ren, Front. Microbiol., 2020, 11, 622534 CrossRef PubMed.
- J. J. Wille and A. Kydonieus, Skin Pharmacol. Appl. Skin Physiol., 2003, 16, 176–187 CrossRef CAS PubMed.
- X. H. Chen, S. R. Liu, B. Peng, D. Li, Z. X. Cheng, J. X. Zhu, S. Zhang, Y. M. Peng, H. Li, T. T. Zhang and X. X. Peng, Front. Immunol., 2017, 8, 207 Search PubMed.
- S. P. Bernier and M. G. Surette, Front. Microbiol., 2013, 4, 20 Search PubMed.
- M. Fraunholz and B. Sinha, Front. Cell. Infect. Microbiol., 2012, 2, 43 Search PubMed.
- M. Kubica, K. Guzik, J. Koziel, M. Zarebski, W. Richter, B. Gajkowska, A. Golda, A. Maciag-Gudowska, K. Brix, L. Shaw, T. Foster and J. Potempa, PLoS One, 2008, 3, e1409 CrossRef PubMed.
- O. Werz, J. Gerstmeier, S. Libreros, X. De la Rosa, M. Werner, P. C. Norris, N. Chiang and C. N. Serhan, Nat. Commun., 2018, 9, 12 CrossRef PubMed.
- W. Hewelt-Belka, J. Nakonieczna, M. Belka, T. Baczek, J. Namiesnik and A. Kot-Wasik, J. Chromatogr. A, 2014, 1362, 62–74 CrossRef CAS PubMed.
- W. Hewelt-Belka, J. Nakonieczna, M. Belka, T. Baczek, J. Namiesnik and A. Kot-Wasik, J. Proteome Res., 2016, 15, 914–922 CrossRef CAS PubMed.
- B. Yan, H. Chu, D. Yang, K. H. Sze, P. M. Lai, S. Yuan, H. Shuai, Y. Wang, R. Y. Kao, J. F. Chan and K. Y. Yuen, Viruses, 2019, 11, 73 CrossRef CAS PubMed.
- B. Yan, Z. Zou, H. Chu, G. Chan, J. O. Tsang, P. M. Lai, S. Yuan, C. C. Yip, F. Yin, R. Y. Kao, K. H. Sze, S. K. Lau, J. F. Chan and K. Y. Yuen, Int. J. Mol. Sci., 2019, 20, 5952 CrossRef CAS PubMed.
- J. Z. Haeggstrom and C. D. Funk, Chem. Rev., 2011, 111, 5866–5898 CrossRef PubMed.
- C. N. Serhan, Nature, 2014, 510, 92–101 CrossRef CAS PubMed.
- A. Batista-Gonzalez, R. Vidal, A. Criollo and L. J. Carreno, Front. Immunol., 2019, 10, 2993 CrossRef CAS PubMed.
- J. Van den Bossche, L. A. O'Neill and D. Menon, Trends Immunol., 2017, 38, 395–406 CrossRef CAS PubMed.
- Y. Miyake, Y. Kozutsumi, S. Nakamura, T. Fujita and T. Kawasaki, Biochem. Biophys. Res. Commun., 1995, 211, 396–403 CrossRef CAS PubMed.
- S. K. Wang, S. Liu, L. G. Yang, R. F. Shi and G. J. Sun, Mol. Med. Rep., 2013, 7, 1970–1976 CrossRef CAS PubMed.
- E. Demetz, A. Schroll, K. Auer, C. Heim, J. R. Patsch, P. Eller, M. Theurl, I. Theurl, M. Theurl, M. Seifert, D. Lener, U. Stanzl, D. Haschka, M. Asshoff, S. Dichtl, M. Nairz, E. Huber, M. Stadlinger, A. R. Moschen, X. Li, P. Pallweber, H. Scharnagl, T. Stojakovic, W. Marz, M. E. Kleber, K. Garlaschelli, P. Uboldi, A. L. Catapano, F. Stellaard, M. Rudling, K. Kuba, Y. Imai, M. Arita, J. D. Schuetz, P. P. Pramstaller, U. J. F. Tietge, M. Trauner, G. D. Norata, T. Claudel, A. A. Hicks, G. Weiss and I. Tancevski, Cell Metab., 2014, 20, 787–798 CrossRef CAS PubMed.
- J. K. Innes and P. C. Calder, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2018, 132, 41–48 CrossRef CAS PubMed.
- L. Jordao, A. Lengeling, Y. Bordat, F. Boudou, B. Gicquel, O. Neyrolles, P. D. Becker, C. A. Guzman, G. Griffiths and E. Anes, Microbes Infect., 2008, 10, 1379–1386 CrossRef CAS PubMed.
- J. J. Hu, N. K. Wong, S. Ye, X. Chen, M. Y. Lu, A. Q. Zhao, Y. Guo, A. C. Ma, A. Y. Leung, J. Shen and D. Yang, J. Am. Chem. Soc., 2015, 137, 6837–6843 CrossRef CAS PubMed.
- Y. L. a. M. A. Trush, Biochem Bioph Res Co, 1998, 253, 295–299 CrossRef PubMed.
- A. Karlsson, J. B. Nixon and L. C. McPhail, J. Leukocyte Biol., 2000, 67, 396–404 CrossRef CAS PubMed.
- H. Kim, L. Esser, M. B. Hossain, D. Xia, C. A. Yu, J. Riso, D. van der Helm and J. Deisenhofer, J. Am. Chem. Soc., 1999, 121, 4902–4903 CrossRef CAS.
- Y. A. Hannun and L. M. Obeid, Nat. Rev. Mol. Cell Biol., 2018, 19, 175–191 CrossRef CAS PubMed.
- H. Wang, P. Yang, K. Liu, F. Guo, Y. Zhang, G. Zhang and C. Jiang, Cell Res., 2008, 18, 290–301 CrossRef CAS PubMed.
- E. Avota, J. Bodem, J. Chithelen, P. Mandasari, N. Beyersdorf and J. Schneider-Schaulies, Front. Physiol., 2021, 12, 715527 CrossRef PubMed.
- F. G. Tafesse, A. Rashidfarrokhi, F. I. Schmidt, E. Freinkman, S. Dougan, M. Dougan, A. Esteban, T. Maruyama, K. Strijbis and H. L. Ploegh, PLoS Pathog., 2015, 11, e1005188 CrossRef PubMed.
- C. Li, A. Wang, Y. Wu, E. Gulbins, H. Grassme and Z. Zhao, Cell. Physiol. Biochem., 2019, 52, 280–301 CrossRef CAS PubMed.
- C. Li, Y. Wu, A. Riehle, V. Orian-Rousseau, Y. Zhang, E. Gulbins and H. Grassme, Antioxid. Redox Signaling, 2018, 28, 916–934 CrossRef CAS PubMed.
- A. Burgert, J. Schlegel, J. Becam, S. Doose, E. Bieberich, A. Schubert-Unkmeir and M. Sauer, Angew. Chem., Int. Ed. Engl., 2017, 56, 6131–6135 CrossRef CAS PubMed.
- Y. A. Hannun and L. M. Obeid, Nat. Rev. Mol. Cell Biol., 2008, 9, 139–150 CrossRef CAS PubMed.
- S. M. Behar and V. Briken, Curr. Opin. Immunol., 2019, 60, 103–110 CrossRef CAS PubMed.
- X. H. Lai, Y. Xu, X. M. Chen and Y. Ren, Macrophage, 2015, 2, e779 Search PubMed.
- J. Xia and D. S. Wishart, Bioinformatics, 2010, 26, 2342–2344 CrossRef CAS PubMed.
- T. Terano, J. A. Salmon and S. Moncada, Prostaglandins, 1984, 27, 217–232 CrossRef CAS.
- J. Whelan and K. Fritsche, Adv. Nutr., 2013, 4, 311–312 CrossRef PubMed.
- P. Wlodawer and B. Samuelsson, J. Biol. Chem., 1973, 248, 5673–5678 CrossRef CAS PubMed.
- K. W. Wirtz, Annu. Rev. Biochem., 1991, 60, 73–99 CrossRef CAS PubMed.
- J. M. Dean and I. J. Lodhi, Protein Cell, 2018, 9, 196–206 CrossRef CAS PubMed.
- A. P. Simopoulos, Exp. Biol. Med., 2008, 233, 674–688 CrossRef CAS PubMed.
- M. R. Davanso, A. R. Crisma, G. Murata, P. Newsholme and R. Curi, Immunometabolism, 2020, 2.e200008 Search PubMed.
- C. S. Chang, H. L. Sun, C. K. Lii, H. W. Chen, P. Y. Chen and K. L. Liu, Inflammation, 2010, 33, 46–57 CrossRef CAS PubMed.
- M. Canton, R. Sanchez-Rodriguez, I. Spera, F. C. Venegas, M. Favia, A. Viola and A. Castegna, Front. Immunol., 2021, 12, 734229 CrossRef CAS PubMed.
- H. Sies and D. P. Jones, Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef CAS PubMed.
- M. Afzal, S. Matsugo, M. Sasai, B. Xu, K. Aoyama and T. Takeuchi, Biochem Bioph Res Co, 2003, 304, 619–624 CrossRef CAS.
- J. J. Hu, N. K. Wong, M. Y. Lu, X. Chen, S. Ye, A. Q. Zhao, P. Gao, R. Yi-Tsun Kao, J. Shen and D. Yang, Chem. Sci., 2016, 7, 2094–2099 RSC.
- M. Rybicka, E. Stachowska, I. Gutowska, M. Parczewski, M. Baskiewicz, B. Machalinski, A. Boron-Kaczmarska and D. Chlubek, J. Agric. Food Chem., 2011, 59, 4095–4103 CrossRef CAS PubMed.
- Measurement of DNA concentration as a normalization strategy for metabolomic data from adherent cell lines ed. L. P. Silva, P. L. Lorenzi, P. Purwaha, V. Yong, D. H. Hawke and J. N. Weinstein, 2013 Search PubMed.
- C. W. Damen, G. Isaac, J. Langridge, T. Hankemeier and R. J. Vreeken, J. Lipid Res., 2014, 55, 1772–1783 CrossRef CAS PubMed.
- J. Yang, K. Schmelzer, K. Georgi and B. D. Hammock, Anal. Chem., 2009, 81, 8085–8093 CrossRef CAS PubMed.
- P. Gao, P. L. Ho, B. P. Yan, K. H. Sze, J. Davies and R. Y. T. Kao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8003–8008 CrossRef CAS PubMed.
- T. Cajka and O. Fiehn, Metabolomics, 2016, 12, 34 CrossRef.
- B. Yan, Y. Deng, J. Hou, Q. Bi, M. Yang, B. Jiang, X. Liu, W. Wu and D. Guo, Mol. BioSyst., 2015, 11, 486–496 RSC.
- Z. Yang, J. J. Hou, P. Qi, M. Yang, B. P. Yan, Q. R. Bi, R. H. Feng, W. Z. Yang, W. Y. Wu and D. A. Guo, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1026, 193–203 CrossRef CAS PubMed.
- H. Tsugawa, T. Cajka, T. Kind, Y. Ma, B. Higgins, K. Ikeda, M. Kanazawa, J. VanderGheynst, O. Fiehn and M. Arita, Nat. Methods, 2015, 12, 523–526 CrossRef CAS PubMed.
- B. Galindo-Prieto, L. Eriksson and J. Trygg, J. Chemom., 2014, 28, 623–632 CrossRef CAS.
- T. Kind, K. H. Liu, D. Y. Lee, B. DeFelice, J. K. Meissen and O. Fiehn, Nat. Methods, 2013, 10, 755–758 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04307f |
| ‡ These authors are the corresponding authors. |
| This journal is © The Royal Society of Chemistry 2022 |