DOI:
10.1039/D2SC04637G
(Edge Article)
Chem. Sci., 2022,
13, 12078-12086
From mixed group 13 cations [M(AlCp*)3]+ (M = Ga/In/Tl) to an Al4+ cluster†
Received
19th August 2022
, Accepted 28th September 2022
First published on 30th September 2022
Abstract
AlCp*-complexes with transition metals have shown to be highly reactive and enable C–H or Si–H bond activation. Yet, complexes of AlCp* with low-valent main-group metals are scarce. Here, we report the syntheses of [M(AlCp*)3][Al(ORF)4] (RF = C(CF3)3) with M = Ga, In, Tl, which include the first covalent Al–In and Al–Tl bonds. For M = Ga, AlCp*-coordination induced the formation of the dication [Ga2(AlCp*)6]2+ in the solid state, which exhibits a solvent and temperature dependent monomer–dimer equilibrium in solution. By contrast, the In and Tl complexes are monomeric and prone to reduction to the metal by the electron-rich AlCp*-moieties. The QTAIM analysis suggests that the metal centres are already highly reduced in the complexes, while the positive charge is distributed onto the AlCp* units. Addition of Me3TACN (1,4,7-trimethyl-1,4,7-triazacyclononane) to the Ga- and Tl-complex salts resulted in an isomerization to the novel low-valent Al4+ cation [(Me3TACN)Al(AlCp*)3][Al(ORF)4]. Intermittently formed tetrahedral GaAl3+ clusters could be structurally characterized. From a detailed mechanistic study of this isomerization, the very high yield and clean preparation of [(Me3TACN)Al(AlCp*)3][Al(ORF)4] was devised from [M(Me3TACN)][Al(ORF)4] (M = Ga, Tl) and [(AlCp*)4].
Introduction
Metallo-ligands have found wide scientific interest due to their electronic flexibility and ligand-cooperativity.1 Here, silylenes have recently developed into a highly versatile and widely applied ligand class.2,3 In contrast, examples of ligands with aluminium – the second most abundant metal in the earth crust after silicon – as donor atom are scarce.3,4 Here, the discovery of [(AlCp*)4] as first molecular Al(I) compound represented a milestone for inorganic coordination and cluster chemistry.5 Although being tetrameric in the solid state, the room-temperature stable, air-sensitive [(AlCp*)4] can reversibly dissociate into its monomers in solution to undergo bond formations with various small molecules or transition metals.6 Hence, a large variety of AlCp*-coordination complexes with electron-rich, late transition metals have been reported (Cr,7 W,8 Fe,9,10 Ru,9,11 Co,12 Rh,13 Ni,14,15 Pd,16,17 Pt,17,18 Cu19). Due to the diverse reactivity of AlCp*, which is isolobal to CO, complexes with terminal AlCp*-units, bridging AlCp*-ligands in binuclear complexes as well as large clusters could be prepared. [M(AlCp*)x] formation was often challenged by competing C–H activation, most likely due to the highly electrophilic nature of the coordinated Al atom in AlCp*.8,9 Complexes of AlCp* with electron-poor metals are scarce and limited to the f-block metals (Eu,20 Yb,20 U21). Quantum-chemical analysis of the Al–M bonding in heterobimetallic complexes revealed a dominant electrostatic character for the bonding interactions, where the covalent part largely constitutes to a ligand-to-metal σ-donation from the lone-pair at AlCp* and diminished π-back-bonding.10,15,20,22
However, no bimetallic cluster with [MxAl4−x] core, mimicking the tetrahedral structure of [(AlCp*)4], is known to date. In addition, and despite the year-long research on the coordination chemistry of AlCp*, complexes to electron-rich, low-valent main-group metals have only been reported with heavy group 15 metals.23 Hence, we were interested in bonding interactions between AlCp* and its heavier homologues in low-oxidation states. Examples for complexes with Al–Ga or Al–In bonds are scarce. The first characterized Al–Ga bond was reported by Cowley in 2005 with the donor–acceptor complex [Cp*Ga–Al(C6F5)3] I.24 One year later, Schulz described the synthesis of the mixed group 13 donor–acceptor complexes of type [Cp*M1–M2(tBu)3] (I–III: M1/M2 = Ga/Al, Al/Ga, In/Al).25 Here, the Cp*In–Al(tBu)3 molecule features the only structurally characterized In–Al bond known in literature. Intriguingly, Schulz reported the synthesis of the group 13 Lewis acid–base adduct [(BDI)Ga→Al(C6F5)] (V, BDI = β-diketiminiate), which readily inserted in benzaldehyde.26 Recently, the observation of the first non-dative bonding interaction between gallium and aluminium was described by Okuda in the complex salt [(BDI)Ga(H)–Al(H)(tmeda)][B(3,5-Me2–C6H3)4] VI.27 With the high reactivity of low-valent transition metal–AlCp* complexes as well as the absence of covalent Al–In and Al–Tl bonds in literature, we were interested to study the interaction of the strongly electron-donating AlCp* moiety with the M+ cations of low-valent gallium, indium and thallium salts of the weakly coordinating anion [Al(ORF)4]− (ORF = C(CF3)3).28–31 Very recently, we isolated the first accessible low-valent aluminium complex salt [Al(AlCp*)3][Al(ORF)4] VII prepared via removal of LiCp* from (AlCp*)4 with Li+.32 Here, we report independent syntheses and characterizations of the novel salts [M(AlCp*)3][Al(ORF)4] (M = Ga (1), In (2), Tl (3)). Unexpectedly, but very useful, all the complex salts isomerize upon addition of 1,4,7-trimethyl-1,4,7-triazacyclononane (Me3TACN) via unprecedented mixed group 13 clusters to the novel Al4+ cluster [(Me3TACN)Al(AlCp*)3][Al(ORF)4] in high yields (Fig. 1).
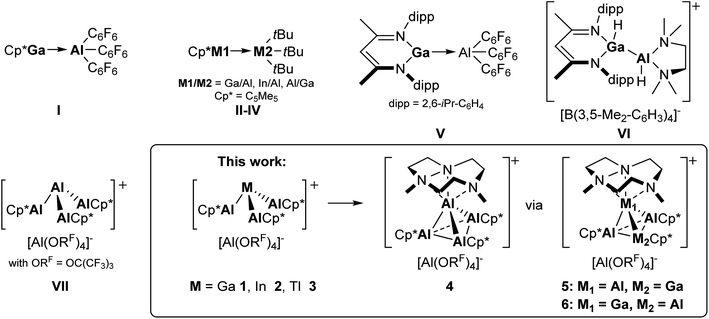 |
| | Fig. 1 Known compounds with Al–Ga or Al–In bonds as well as novel complex salts of type [M(AlCp*)3][Al(ORF)4] (ORF = C(CF3)3). | |
Results and discussion
Syntheses of [M(AlCp*3)][Al(ORF)4]
The complex salts 1–3 were synthesized starting from [(AlCp*)4] and the readily accessible fluorobenzene complexes of gallium and indium [M(PhF)2–3][Al(ORF)4] (M = Ga,31 In28,29) as well as Tl[Al(ORF)4] (Scheme 1).30
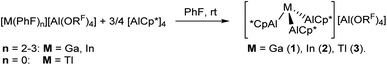 |
| | Scheme 1 Synthesis of the complex salts [M(AlCp*)3][Al(ORF)4]. | |
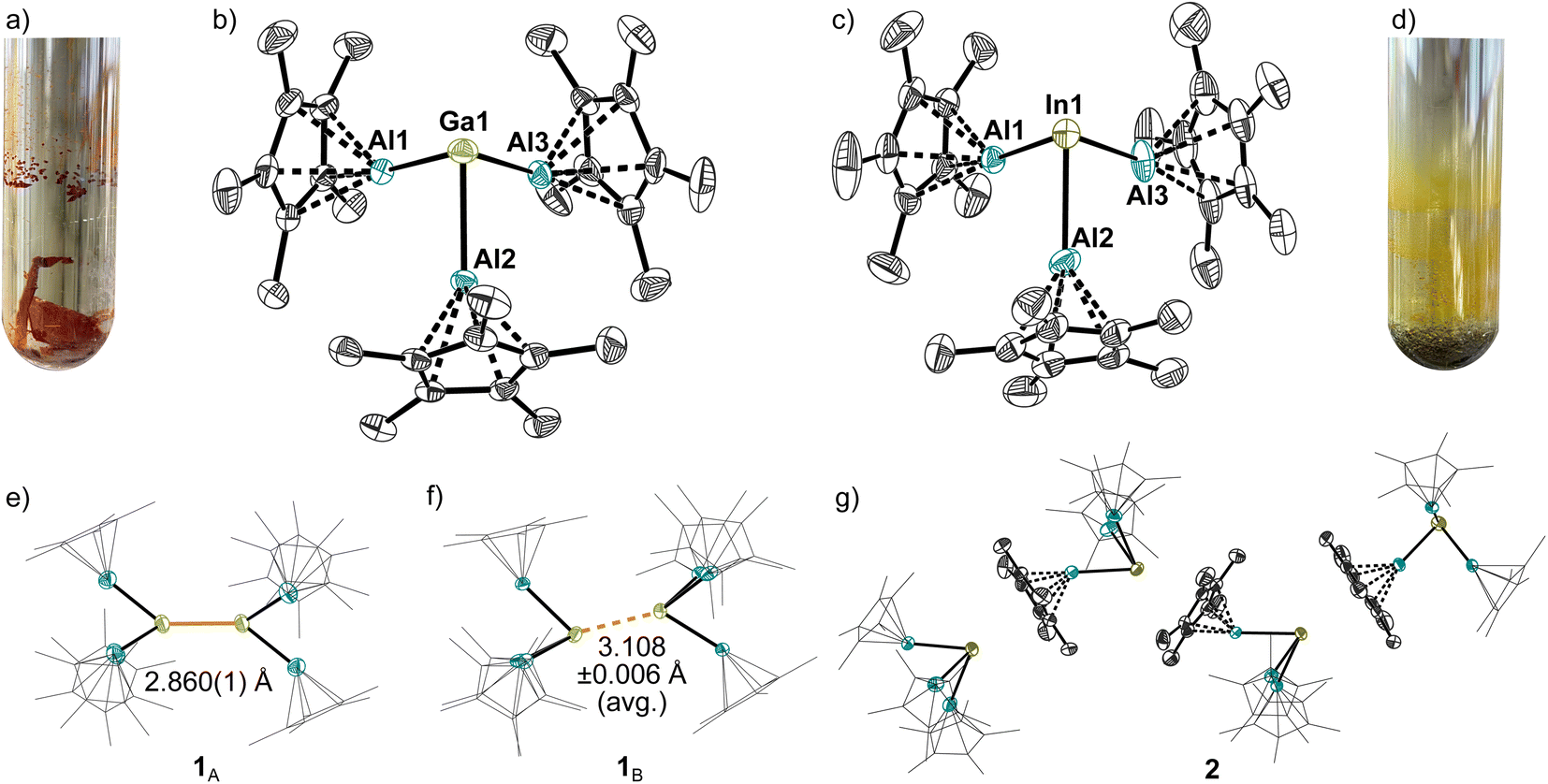
In a first experiment, [Ga(PhF)2–3][Al(ORF)4] was reacted with [(AlCp*)4] in 1,2-DFB (DFB = difluorobenzene) at room temperature to yield an orange solution from which orange crystals were isolated upon layering with n-heptane at −30 °C. Single crystal X-ray structure determination (sc-XRD) revealed the formation of the dicationic cluster [Ga2(AlCp*)6]([Al(ORF)4])2 (1A, vide infra). Yet, formation of a metallic precipitate was observed at room temperature already after a few minutes. This was prevented by conducting and keeping the reaction at −30 °C. Interestingly, switching to the less-polar, but more strongly-coordinating solvent fluorobenzene, a yellow solution was obtained from which no metal precipitation occurred at room temperature. Crystallization at room temperature by layering with n-pentane yielded orange crystals of [Ga2(AlCp*)6]([Al(ORF)4])2, however, now in a second modification with a slipped Ga+–Ga+ interaction (1B, Fig. 2a, vide infra).
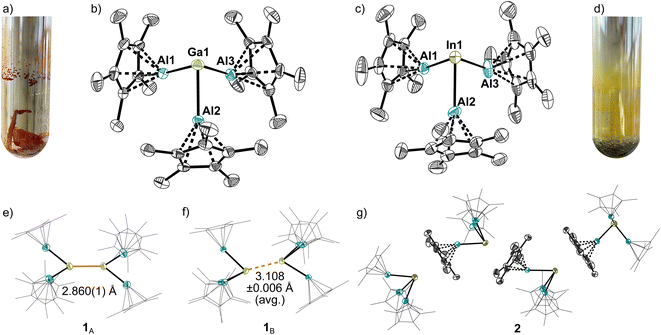 |
| | Fig. 2 (a) Orange crystals of [Ga2(AlCp*)6]([Al(ORF)4])21A obtained by crystallization of 1 from PhF/n-pentane at room temperature. (b) Molecular structure of a [Ga(AlCp*)3]+ unit in the scXRD structure of 1A. Hydrogen atoms and [Al(ORF)4]− anions omitted for clarity. Thermal displacement of the ellipsoids was set at 50% probability. (c) Molecular structure of a [In(AlCp*)3]+ unit in 2. Hydrogen atoms and [Al(ORF)4]− anions omitted for clarity. Thermal displacement of the ellipsoids was set at 50% probability. (d) Yellow crystals of [In(AlCp*)3][Al(ORF)4] 2 along with indium metal mirror. (e and f) Visualization of the two independent dicationic [Ga2(AlCp*)6]2+ units obtained at −30 °C from 1,2-DFB/n-heptane (1A) and at room temperature from PhF/n-pentane (1B). Hydrogen atoms and [Al(ORF)4]− anions omitted for clarity. Thermal displacement of the ellipsoids was set at 50% probability. (g) Visualization of the intermolecular In+–Cp* interaction in the molecular structure of 2. Hydrogen atoms and [Al(ORF)4]− anions omitted for clarity. Thermal displacement of the ellipsoids was set at 50% probability. | |
The reactions of [In(PhF)2–3][Al(ORF)4] and Tl[Al(ORF)4] with [(AlCp*)4] in fluorinated arenes yielded yellow solutions from which yellow crystals of monomeric [In(AlCp*)3][Al(ORF)4] 2 (Fig. 2d) and [Tl(AlCp*)3][Al(ORF)4] 3 separated. However, crystallization was always accompanied with decomposition and formation of some metallic indium and thallium. Hence, the respective complexes are best prepared in situ. Nevertheless, 3 could be isolated in high purity by precipitation via cannulation of the PhF-solutions onto n-heptane at room temperature.
Molecular structures
Related to the independently prepared complex salt [Al(AlCp*)3][Al(ORF)4],32 scXRD analyses on the obtained crystals yield a trigonal-pyramidal coordination of the formal cationic gallium or indium atom by three AlCp* groups (Fig. 2b and c). In the monomeric units [M(AlCp*)3]+, the average Al–Al distances are long (3.074 ± 0.009) Å in 1A, 3.09 ± 0.04 Å in 1B, 3.12 ± 0.06 Å in 2,33 which precludes a strong covalent bonding interaction. By contrast, the Ga1–Al bond lengths in both molecular structures are similarly short and average to 2.520 ± 0.006 Å in 1A and 2.532 ± 0.007 Å in 1B. Hence, the Ga–Al bond lengths are close to the bond length reported by Okuda for complex VI (2.5238(9) Å)27 as well as in the formal Lewis-adducts between AlIII(C6F5)3 and GaI-based Lewis bases GaI(BDI) (2.5482(4) Å)26 or GaICp* (2.515(11) Å).25 As expected, the average In–Al bond lengths are longer at 2.75 ± 0.01 Å. Still, the observed In–Al distances are significantly shorter than the previously reported dative InI→AlIII bond length of 2.843(2) Å in III.25 Interestingly, molecular structures of the indium and gallium complex salts reveal decisive differences in the intermolecular interaction between the [M(AlCp*)3]+ cations in the solid state. For 1, the dimerization to [Ga2(AlCp*)6]2+([Al(ORF)4])2 followed in the molecular structures 1A and 1B, with a shorter Ga–Ga distance of 2.860(1) Å in 1A (Fig. 2e). By contrast, 1B displays a “slipped” interaction between the [GaAl3]+ tetrahedra with a longer Ga–Ga distance of 3.108 ± 0.006 Å (avg., Fig. 2f). The Ga–Ga distances in 1 are significantly longer compared to literature known covalent “Ga+–Ga+” bonds as in [Ga5(dmap)5]5+ (2.495 Å (avg.), dmap = dimethylaminopyridine),34 [Ga4(tBuNC)8]4+ (2.495 Å (avg.))35 and [Ga4(dmpe)4]4+ (2.485 Å (avg.)).36 Notably, in the latter complexes the gallium cations are only coordinated by two strongly electron donating ligands that induce cluster formation via delocalization of the cationic charge onto the ligand. In contrast to 1, the [In(AlCp*)3]+ cations in 2 form coordination oligomers with the indium atom loosely coordinating to the Cp*-ligand of a neighbouring cation. Related coordination environments were reported for the solid-state structures of M(C5H5) (M = In, Tl) and TlCp*.37 The molecular structure of [Tl(AlCp*)3][Al(ORF)4] 3 shows similar interionic interactions as the indium cluster. Yet, extensive disorder of the cations and anions precludes a detailed discussion of the bond lengths.
Monomer–dimer equilibria
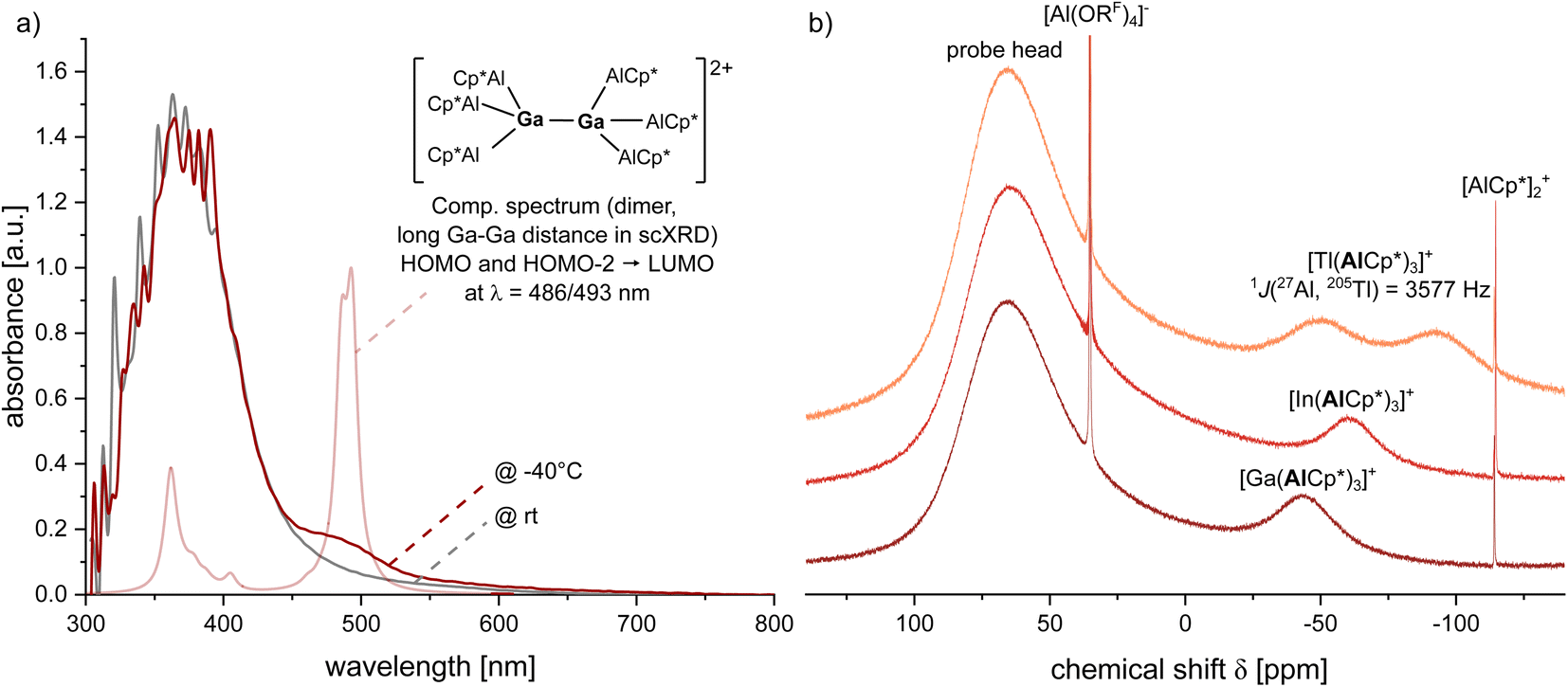
Interestingly, the dimerization of the gallium complex 1 in 1,2-DFB solution is an equilibrium reaction. Orange solutions matching the colour of the crystals prevail at higher concentrations and/or low temperatures. In the UV/VIS spectrum at −40 °C (Fig. 3) a broad UV/VIS band was observed at 476 nm, which fits to HOMO/HOMO−2 → LUMO excitations of the [Ga2(AlCp*)6]2+ dimer-dication computed by TD-DFT at 486 and 493 nm. No excitations in this spectral region were computed for the monomer. Warming the solution to room temperature is accompanied by a disappearance of the UV/VIS band at 476 nm and colour change to yellow, which suggests a break-up of the dimers into monomeric [Ga(AlCp*)3]+ cations. This monomer–dimer equilibrium is also dependent on the polarity of the solvent: in contrast to the equilibria noted in polar 1,2-DFB (εr (295 K) = 13.8), dimer formation is supressed in the less polar fluorobenzene (εr (295 K) = 5.7) by its Coulomb explosion into the monocations (see ESI, Fig. S64†). Hence, suppression of dimerization in fluorobenzene allows to handle solutions of 1 at room temperature. By contrast and as expected from the distinct differences of their solid-state structures, no colour changes that would indicate a dimerization were observed for solutions of the (monomeric) indium or thallium complex 2 and 3.
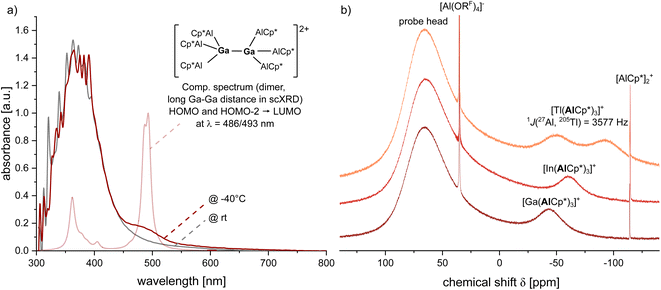 |
| | Fig. 3 (a) UV/VIS spectrum of a solution of 1 in 1,2-DFB (c = 19 mg mL−1) measured at rt and −40 °C as well as the computed spectrum for the dimer with the long Ga–Ga distance (TD-DFT with bp86-d3bj/def2-svp, 20 singlet excitations with Fermi exchange). (b) 27Al NMR spectra of PhF solutions of the complex salts 1, 2 and 3. [AlCp*2]+ represents a common decomposition product of AlCp*. | |
NMR-spectroscopy
NMR spectra of the crystals of 1 as well as of the precipitated powders of 2 and 3 in PhF indicate a high purity, since only one Cp*-resonance is detected and no NMR signals of the starting materials were observed. Moreover, the 27Al NMR spectra of the [M(AlCp*)3]+ complex show distinct resonances of the (AlCp*)3 units (Fig. 3): they shift to higher field following 1 (δ27Al = −43) > 2 (δ27Al = −60) > 3 (δ27Al = −70). These observed 27Al NMR shifts lie in between the shifts observed for the (AlCp*)3 units in [Al(AlCp*)3]+ (δ27Al = −40) and free [(AlCp*)4] (δ27Al = −79).32 Since both isotopes, 203Tl and 205Tl (29.5 and 70.5% natural abundance) possess a nuclear spin of ½, the 27Al NMR resonance of AlCp* in 3 is observed as a doublet with a coupling constant of 1J(27Al–205Tl) = 3577 Hz. As 3 represents the first reported compound with a Tl–Al bond, no other 1J(27Al–205Tl) are literature-known. In addition, only few coupling constants of Tl(I) are known due to the typically highly ionic nature of Tl(I) complexes making 3 rather the exception than the rule.38 The quantitative decomposition of solutions of 2 and 3 with formation of a metallic precipitate took a few days at RT. The spectra of NMR-scale reactions showed a clean conversion to [(AlCp*)4] and [AlCp*2][Al(ORF)4]. Hence, a disproportionation of [M(AlCp*)3][Al(ORF)4] into elemental indium or thallium and [AlIIICp*2]+ has occurred. Related observations were made for highly concentrated solutions of [Al(AlCp*)3][Al(ORF)4]. Moreover, characteristic Raman bands corresponding to symmetric and asymmetric stretching vibrations of the M–Al bonds in the MAl3+ units were detected (see ESI, Fig. S7, S16 and S24†). Here, the trends (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) sym: 515 cm−1 (1) > 496 cm−1 (2) > 495 cm−1 (3); asym: 471 cm−1 (1) > 460 cm−1 (2) > 451 cm−1 (3)) reflect the decreasing bond strength of the M–Al3 cluster bonds in the order Ga > In > Tl.
sym: 515 cm−1 (1) > 496 cm−1 (2) > 495 cm−1 (3); asym: 471 cm−1 (1) > 460 cm−1 (2) > 451 cm−1 (3)) reflect the decreasing bond strength of the M–Al3 cluster bonds in the order Ga > In > Tl.
Addition of Me3TACN
To investigate the effects of cation coordination on the group 13 intermetallic clusters 1–3, Me3TACN (1,4,7-trimethyl-1,4,7-triazacyclononane) was added to fluorobenzene solutions giving yellow solutions for the gallium and indium complexes, from which yellow crystals were obtained along a few colourless, unidentifiable crystals. The reaction with 3 yielded a black, metallic precipitate and yellow crystals. Surprisingly, in all reactions the yellow crystals were identified by scXRD measurements as the novel, cationic Al4+ cluster [(Me3TACN)Al(AlCp*)3][Al(ORF)4] 4 (Fig. 4). Unexpectedly, the molecular structure of 4 includes a symmetrically bound Al4+ cluster with Al–Al bond lengths between 2.736(1)–2.789(1) Å and very similar average AlCp*–AlCp* and Al+–AlCp* distances of 2.75 ± 0.03 and 2.765 ± 0.009 Å. Apparently, the neutral ligand Me3TACN mimics almost ideally the electronics of a Cp* ligand and matches in 4 the average Al–Al bond lengths of 2.758 Å found in (AlCp*)4. This contrasts with the other known32 coordinated Al4+ clusters that for cdp- and tmeda-ligands exhibit shorter Al+–AlCp* bonds of 2.662(1) Å (cdp = C(PPh3)2) and 2.695(1) Å (tmeda = Me2NC2H4NMe2) with slightly longer AlCp*–AlCp* separations of 2.773(1) and 2.782(2) Å. For the (dmap)3-coordinated Al4+ tetrahedron, the situation is inverted: d(Al+–AlCp*) = 2.802(1) Å, d(AlCp*–AlCp*) = 2.671(1) Å. Hence, the electronics of the Al4+-cluster bonding is highly flexible.
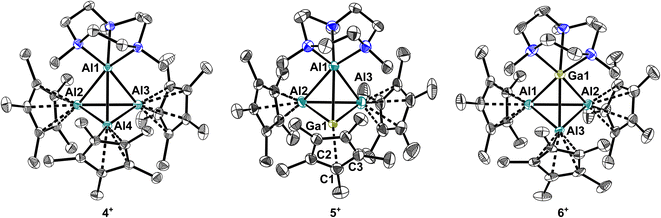 |
| | Fig. 4 Molecular structures of cationic clusters in [(Me3TACN)Al(AlCp*)3][Al(ORF)4] 4, [(Me3TACN)Al(AlCp*)2(GaCp*)][Al(ORF)4] 5 and [(Me3TACN)Ga(AlCp*)3][Al(ORF)4] 6. Hydrogen atoms and [Al(ORF)4]− anions were omitted for clarity. Thermal displacement of the ellipsoids was set at 50% probability. | |
While the proton resonances at the Cp* and Me3TACN residues in 4 are visible in the 1H NMR spectrum, the 27Al NMR spectrum only shows a broad signal attributed to the (AlCp*)3 units at δ = −77. Yet, in contrast to the highly labile (dmap)3 complex, 4 is stable in solution over weeks and hence will be an interesting starting compound to explore the potential of these clusters as source of a cationic low-valent aluminium.
Aiming for the isolation of a mixed GaAl3+ cluster, the reaction mixture was layered directly after addition of Me3TACN with pentane. Along with crystals of 4, some yellow crystals of the mixed cluster [(Me3TACN)Al(AlCp*)2(GaCp*)][Al(ORF)4] 5 were obtained (Fig. 4). Here, an isomerization occurred, yielding a formally cationic Al+ coordinated by Me3TACN along with a GaCp* unit in the tetrahedral base. In 5, the Cp* ring is bound very loosely, almost η1 to the gallium atom with a short Ga–C distance of d(Ga–C1) = 2.440(4) Å. Interestingly, the shortest M–M bonds in the tetrahedron are the Ga–AlCp* bonds of on avg. 2.726(2) Å, whereas all Al–Al and the Ga–AlTACN distances are longer: AlTACN–ACp* (d(Al1–Al2) = 2.800(2) Å, d(Al1–Al3) = 2.837(2) Å), AlCp*–AlCp* (2.826 (2) Å) and Ga–AlTACN (2.832 (2) Å). Performing the reaction and crystallization at low temperatures yielded yellow crystals of the cluster [(Me3TACN)Ga(AlCp*)3][Al(ORF)4] 6 before isomerization and with coordination of Me3TACN to the formally cationic gallium atom. Here, the Ga–Al distances ranging from 2.726(2) to 2.759(2) Å with an average value of 2.75 ± 0.01 Å reflect the symmetric bonding of the Ga(Me3TACN) unit to the three AlCp* moieties. Whereas the average AlCp*–AlCp* bond lengths of 2.79 ± 0.06 Å (range 2.765(4)–2.833(4) Å) are similar compared to the bond lengths observed in the Al4+ cluster 4, more significant differences are observed upon comparison of the average Ga–N bonds in 6 and Al–N bonds in 4 with average lengths of 2.33 ± 0.02 Å and 2.153 ± 0.009 Å.
In situ NMR study
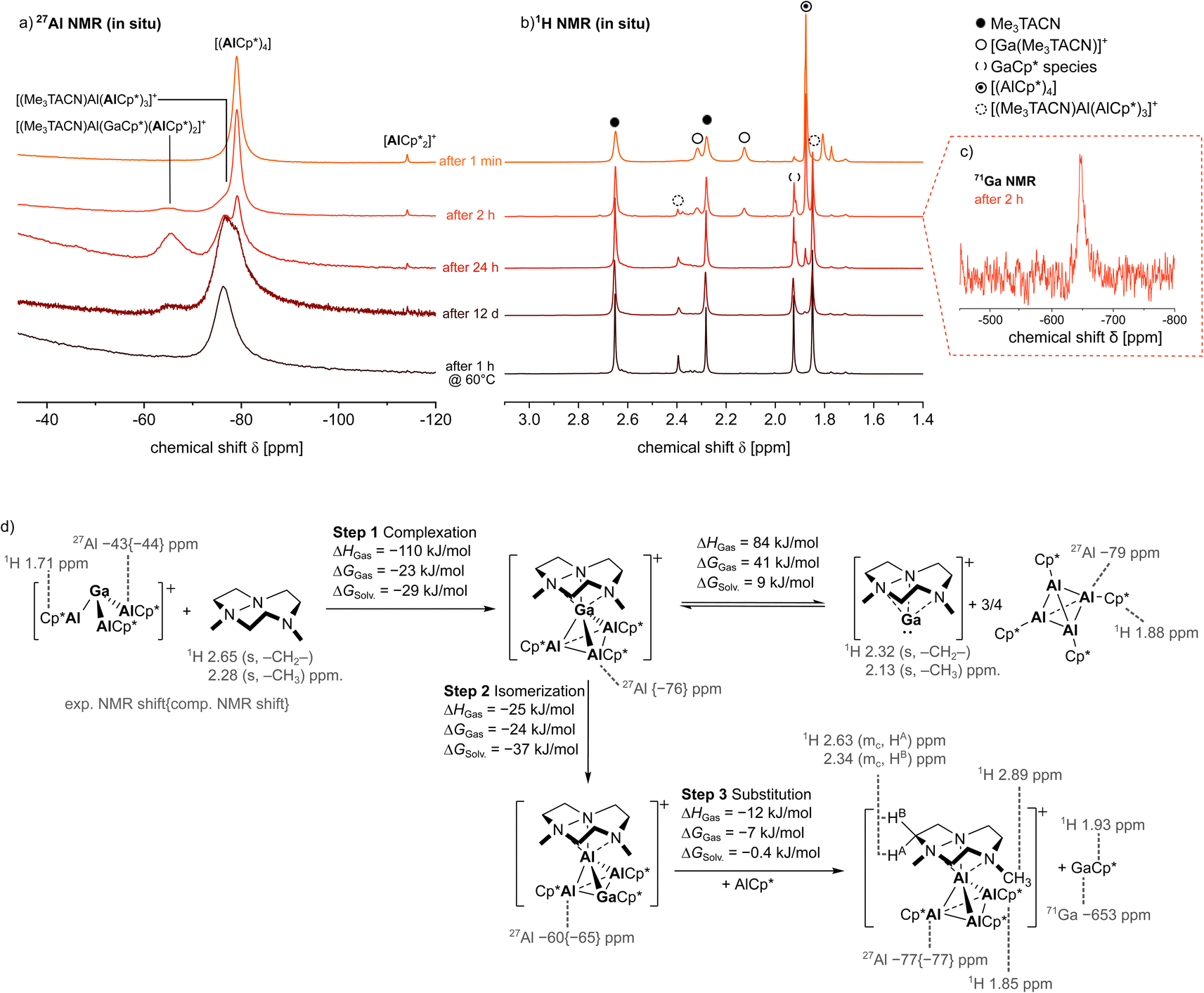
To investigate the mechanism of the formation of 4, the reaction of 1 with excess Me3TACN was studied by in situ NMR spectroscopy (Fig. 5). Here, already one minute after addition of Me3TACN to a solution of 1 in PhF, a quantitative consumption of the starting material 1 and formation of [(AlCp*)4] was observed in the 27Al NMR spectrum. Hence, the Me3TACN ligand displaces the three AlCp*-ligands at Ga+ to form the respective [Ga(Me3TACN)]+ complex. This is supported by the 1H NMR spectrum, where proton resonances fitting to the signals detected for individually prepared [Ga(Me3TACN)][Al(ORF)4] 7 can be observed (see ESI† for characterization). After 2 h, NMR resonances attributed to the product 4 were observed: a broad shoulder in the [(AlCp*)4] signal and the resonances of the Me3TACN- and Cp*-methyl-groups of [(Me3TACN)Al(AlCp*)3]+ were detected as singlets at δ(Me3TACN) = 2.40 and 1.85 (Cp*) in the 1H NMR spectrum. With appearance of the product peaks of 4, the signals assigned to [(AlCp*)4] and [Ga(Me3TACN)]+ reduce in intensity. Moreover, the 71Ga NMR spectra show the formation of monomeric GaCp*, which resonates as a characteristic singlet at δ = −647 (Lit.: δ71Ga = −653, δ1H = 1.93 in C7D8;39 [Ga(PhF)2–3][Al(ORF)4]: δ71Ga = −756 (ref. 31)). At this stage, three overlapping singlets appear at δ1H = 1.92, 1.93 and 1.94, which hint to the presence of several GaCp* species. Concomitantly, a novel broad singlet was observed at δ27Al = −65, potentially assigned to the mixed GaAl3+5 cluster (δ27Alcalc. = −60) structurally characterised independently. After 24 h, the 1H NMR spectrum was completely depleted from [Ga(Me3TACN)]+ and only a small peak of free [(AlCp*)4] was left, whereas the signals assigned to the products GaCp* (δ1H = 1.93) and the Al4+ cluster 4 represent the major resonances. In the 27Al NMR spectrum, signals of 4 and (potentially) 5 became significantly more intense. In the course of the next 11 days, a slow decrease of the signals attributed to [(AlCp*)4] and 5 was observed. Since no quantitative conversion could be achieved, the NMR tube was warmed to 60 °C for one hour. Subsequently, the 27Al NMR spectrum displays only the broad signal of the product 4 at δ = −77. In the 1H NMR spectrum, the respective proton resonances of 4 are observed next to proton resonances of remaining free ligand and GaCp* as side-product of the isomerization (δ = 1.93). Interestingly, orange crystals formed in the NMR tube upon storage, which were identified by scXRD analysis to be the Al4+ cluster 4, as well as traces of a metallic precipitate.
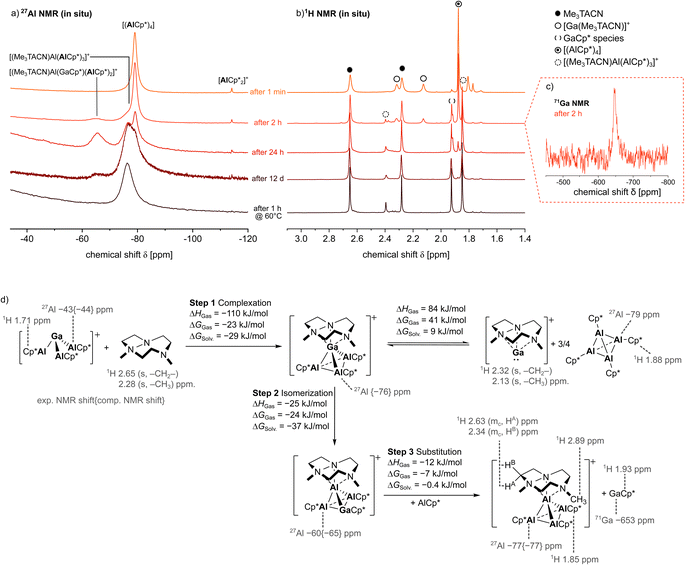 |
| | Fig. 5 (a)–(c) Mechanistic studies on the reaction of [Ga(AlCp*)3]+[Al(ORF)4]−1 with Me3TACN by 27Al NMR (a), 1H NMR (b) spectroscopy and 71Ga NMR (c) spectroscopy. (d) Postulated mechanism of the formation of 4 on the basis of NMR studies and scXRD structures and computed thermodynamics for the postulated reaction pathway (b3lyp-d3bj/def2-tzvpp//bp86-d3bj/def2-svp gas phase energies with Cosmo RS solvation energies). | |
Mechanism of the formation of 4
The results of these NMR studies together with the molecular structure of the mixed GaAl3+ cluster allow for the formulation of a mechanism underlying the cluster rearrangement reaction (Fig. 5). At first, Me3TACN reacts rapidly with [Ga(AlCp*)3]+ to form [(AlCp*)4] and [Ga(Me3TACN)]+. Subsequently, the [Ga(Me3TACN)]+ cation replaces an AlCp* unit from the cluster to form the elusive, structurally characterized [(Me3TACN)Ga(AlCp*)3]+6 (δ27Alcalc. = −76), which rapidly isomerizes to give the structurally characterized [(Me3TACN)Al(AlCp*)2(GaCp*)]+ in 5. From the latter a slow exchange of GaCp* for AlCp* occurs, finally yielding the Al4+ cluster 4 and GaCp* as products. To further elucidate the reaction, Me3TACN and the thallium cluster 3 were investigated by in situ NMR spectroscopy (see ESI, Section S2†). Similarly, the quick initial formation of [(AlCp*)4] and [Tl(Me3TACN)]+ was observed. Subsequently, [(AlCp*)4] was quantitatively transformed into 4 during only 10 hours. The reaction was accompanied by formation of a black precipitate, most likely elemental Tl. Unfortunately, no intermediates were observed in the 27Al NMR spectra, hinting to a greater lability of the mixed TlAl3+ clusters. The proposed overall mechanism delineated in Fig. 5 is supported by computational thermodynamics.
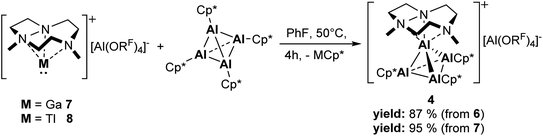
Optimized route to 4
Since we identified the initial formation of the Me3TACN complexes as the first step of the reaction mechanism, we tested the preparation of the cationic Al4+ cluster by reaction of the easily accessible complex salts [M(Me3TACN)][Al(ORF)4] (M = Ga (7), Tl (8); cf. ESI†) with [(AlCp*)4] at 50 °C. Thereby, complex salt 4 could be obtained in only 4 h and in high yields exceeding 87%, significantly improving on the low yield, two-step procedure towards Al4+ clusters reported previously.32 Hence, these exchange reactions represent a highly promising route towards isolation of novel cationic low-valent aluminium complexes.
DFT calculations on the bonding in the mixed clusters
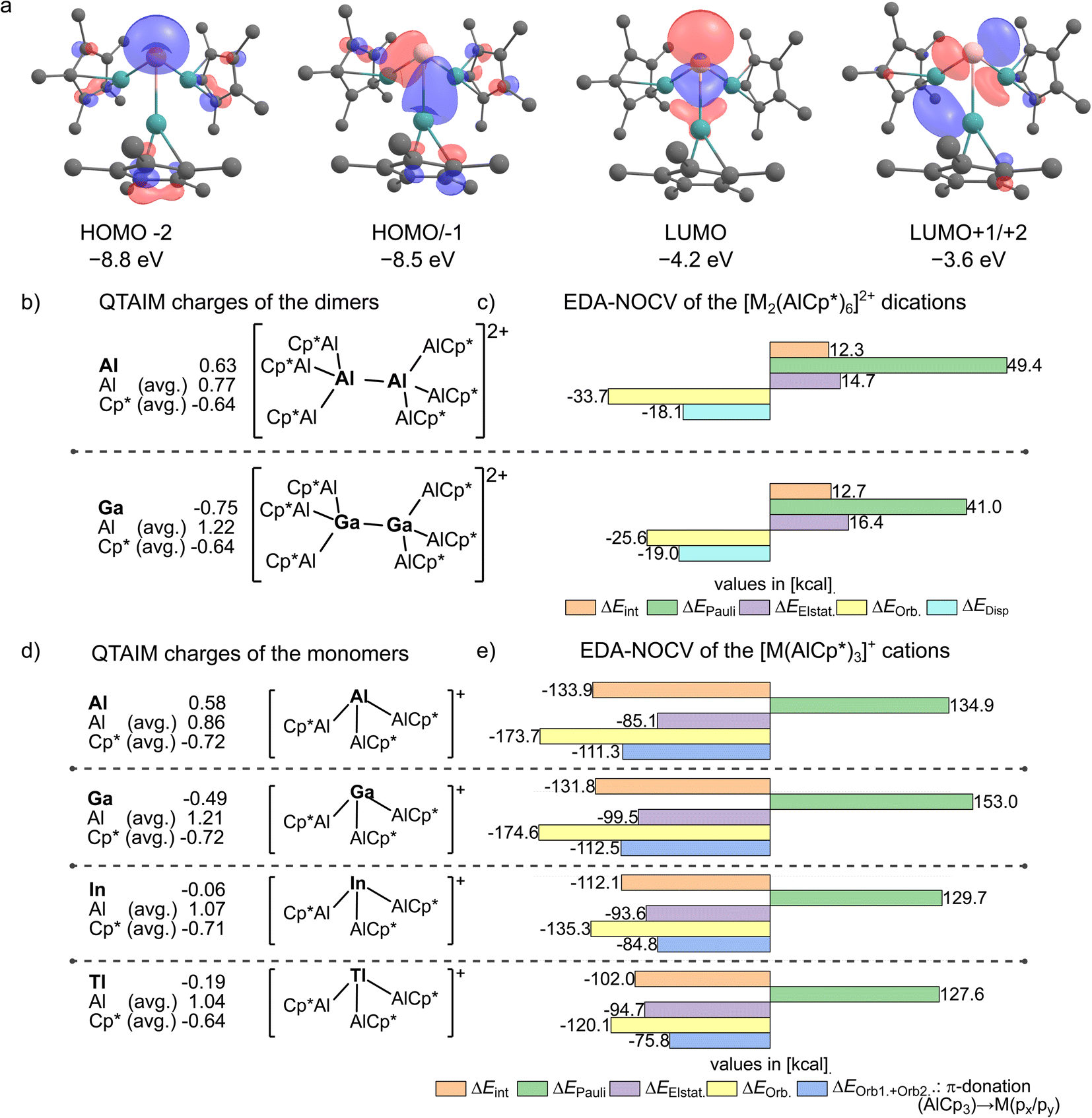
To investigate differences in bonding and reactivity of the isolated complex cations, a computational DFT analysis was performed. Here, similar frontier orbitals are computed for the [M(AlCp*)3]+ cations (e.g. for M = Ga in Fig. 6a). The lone-pair at M is represented by the HOMO−2 orbital. The two degenerate HOMO/−1 display the interaction of the AlCp*-lone pairs with the px- and py-orbitals at the unique metal atom M. The LUMO has a strong pz-character at the heavier group 13 metals. The calculated HOMO/−1/−2 energies decrease from Ga over In to Tl (HOMO−2/HOMO/−1: In at −8.9/−8.3 eV; Tl at −9.1/−8.2 eV). Due to the increase of the HOMO–LUMO gaps for the In and Tl compounds as well as the more diffuse orbitals, the formation of dimeric structures of type [M2(AlCp*)6]2+ is not feasible for In and Tl. For the observed dimer dication [Ga2(AlCp*)6]2+, an EDA-NOCV (energy decomposition analysis with natural orbitals for chemical valence) revealed a rather weak σ-bonding interaction with a significant contribution of dispersion forces on the total attractive interactions (ΔEOrb. = −25.6 kcal mol−1, ΔEDisp. = −18.97 kcal mol−1, Fig. 6c see ESI, Table S2†). Furthermore, the differences between the Ga and the In/Tl based cations are also reflected in the EDA-NOCV analyses (Fig. 6e, more detail see ESI, Section 4†).
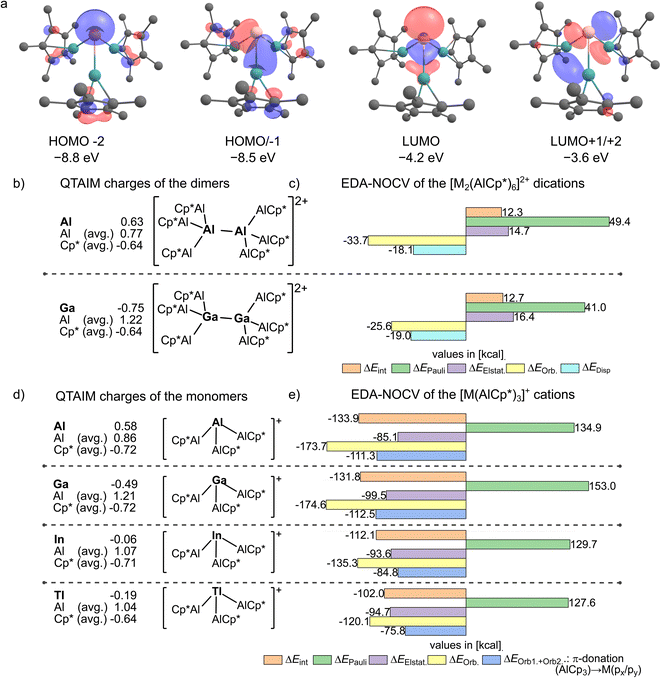 |
| | Fig. 6 (a) Kohn–Sham orbitals of [Ga(AlCp*)3]+ computed at pbe0-d3bj/def2-tzvpp//bp86-d3bj/def2-svp level of DFT (isovalue 0.05). (b) QTAIM charges computed for the dimeric [M2(AlCp*)6]2+ cations (scXRD-structures with shortest M+–M+ distances used). (c) EDA-NOCV results for the interaction of two [M(AlCp*)3]+ fragments (S) in [M2(AlCp*)6]2+ (scXRD-structures with shortest M+–M+ distances used) at bp86-d3bj/tz2p//bp86-d3bj/def2-svp. (d) QTAIM charges computed for the [M(AlCp*)3]+ cations (gas-phase optimized structures). (e) EDA-NOCV results for the interaction of M+ (S, (s2)(p0)) with (AlCp*)3 (S) computed at bp86-d3bj/tz2p//bp86-d3bj/def2-svp with ZORA. | |
Here, the total interaction energies between the formal M+ and (AlCp*)3 fragments are lower for 2+ and 3+ compared to 1+, which originates from a significant drop of the total orbital interaction energy. Nevertheless, even in the indium and thallium complexes, the orbital interaction energy is greater than the electrostatic contribution to the total interaction energy. This reveals the covalent nature of the M–Al bonds (M = Al, Ga, In, Tl), which contrasts the dominant electrostatic character of TM–AlCp* bonds.10,15,20 Moreover, the gallium complex 1+ shows an even greater total orbital interaction than the previously reported [Al(AlCp*)3]+ complex.32 This observation can be attributed to the similar covalent radius40 of aluminium (rcov. = 1.21) and gallium (rcov. = 1.22) and the even higher electronegativity41 of gallium (XP = 1.81) compared to aluminium (XP = 1.61). The EDA-NOCV results agree with the QTAIM analysis, where a negative QTAIM charge of qGa = −0.49 was calculated (Fig. 6b). With the dampened covalent interactions in 2+ and 3+, also greatly reduced negative QTAIM charges of −0.06 and −0.19 follow for M. Hence, a formal reduction of the metal atoms by the AlCp* units occurred. In contrast, the aluminium atoms possess positive QTAIM charges, in particular for the gallium complex. This combination of a reduced metal centre covalently bonded to a positively charged and hence potentially Lewis-acidic atom suggests a promising reactivity.
Conclusion and outlook
Complexes between low-valent Al and Ga/In/Tl have been largely unexplored. Here we report the synthesis and characterization of such elusive complex salts [M(AlCp*)3][Al(ORF)4] (M = Ga, In, Tl). Interestingly, the GaAl3+ complex dimerises in solution and in the solid state, but no dimerization is observed for the In and Tl complexes. Addition of Me3TACN to all the mixed group 13 cations resulted in an isomerization to the novel low-valent Al4+ cation [(Me3TACN)Al(AlCp*)3]+. Here, first mixed GaAl3+ clusters were structurally characterized, representing intermediates in the isomerization reaction. The novel Al4+ cluster could be isolated in exceptionally high yields by the optimized route starting from [M(Me3TACN)]+ and ¾ (AlCp*)4 delineated in Scheme 2. As already suggested by the decomposition of complexes in solution under formation of metallic Ga/In/Tl, negative QTAIM values indicate a formal reduction of the heavier group 13 metal cations in the complexes. With the adjacent positively charged aluminium atoms, a potentially bimetallic reactivity of the complexes will be studied in future research.
 |
| | Scheme 2 Optimized synthesis procedure for the cationic Al4+ cluster [(Me3TACN)Al(AlCp*)3][Al(ORF)4] 4. | |
Data availability
Reference numbers CCDC 2193060 (1A), 2193049 (1B), 2194540 (2), 2194543 (3), 2193851 (4), 2194404 (5) and 2194403 (6). All other data supporting the findings are contained in the main text or the ESI.†
Author contributions
PD planned and carried out all experiments and analysed the data (conceptualization/investigation). PD carried out the single-crystal X-ray diffraction measurements and conducted the computational investigations (formal analysis). PD and IK wrote the manuscript (visualization/writing – original draft).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Fonds of the Chemical Industry FCI for a fellowship for Philipp Dabringhaus, the German Research Foundation (DFG) for the funding of project KR2046/35-1 and the Albert-Ludwigs-University Freiburg for supporting the work. We thank Julie Willrett for help with the experiments. Furthermore, we thank Manuel Schmitt for help with the quantum-chemical calculations and Burkhard Butschke for help with structure refinement. We acknowledge Harald Scherer and Fadime Bitgül for measurement of NMR spectra. Furthermore, the authors acknowledge support by the state of Baden-Württemberg through bwHPC and the DFG through grant no. INST 40/575-1 FUGG (JUSTUS 2 cluster).
References
- J. F. Berry and C. C. Lu, Inorg. Chem., 2017, 56, 7577 CrossRef CAS PubMed.
-
(a)
S. Inoue, in Discovering the future of molecular sciences, ed. B. Pignataro, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, pp. 243–273 Search PubMed;
(b) Y.-P. Zhou and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 3715 CrossRef CAS PubMed;
(c) B. Blom, D. Gallego and M. Driess, Inorg. Chem. Front., 2014, 1, 134 RSC.
- J. Takaya, Chem. Sci., 2020, 12, 1964 RSC.
-
(a) R. Y. Kong and M. R. Crimmin, Dalton Trans., 2021, 50, 7810 RSC;
(b) M. Batuecas, N. Gorgas and M. R. Crimmin, Chem. Sci., 2021, 12, 1993 RSC.
- C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed., 1991, 30, 564 CrossRef.
- H. W. Roesky and S. S. Kumar, Chem. Commun., 2005, 4027 RSC.
- Q. Yu, A. Purath, A. Donchev and H. Schnöckel, J. Organomet. Chem., 1999, 584, 94 CrossRef CAS.
- M. Molon, C. Gemel and R. A. Fischer, J. Organomet. Chem., 2014, 751, 573 CrossRef CAS.
- T. Steinke, M. Cokoja, C. Gemel, A. Kempter, A. Krapp, G. Frenking, U. Zenneck and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 2943 CrossRef CAS PubMed.
- J. Weiss, D. Stetzkamp, B. Nuber, R. A. Fischer, C. Boehme and G. Frenking, Angew. Chem., Int. Ed., 1997, 36, 70 CrossRef CAS.
- T. Cadenbach, T. Bollermann, C. Gemel and R. A. Fischer, Dalton Trans., 2009, 322 RSC.
- C. Üffing, A. Ecker, R. Köppe and H. Schnöckel, Organometallics, 1998, 17, 2373 CrossRef.
- T. Cadenbach, C. Gemel, T. Bollermann and R. A. Fischer, Inorg. Chem., 2009, 48, 5021 CrossRef CAS PubMed.
-
(a) T. Steinke, C. Gemel, M. Cokoja, M. Winter and R. A. Fischer, Angew. Chem., Int. Ed., 2004, 43, 2299 CrossRef CAS PubMed;
(b) M. Molon, T. Bollermann, C. Gemel, J. Schaumann and R. A. Fischer, Dalton Trans., 2011, 40, 10769 RSC;
(c) C. Dohmeier, H. Krautscheid and H. Schnöckel, Angew. Chem., Int. Ed., 1995, 33, 2482 CrossRef.
- J. Hornung, J. Weßing, P. Jerabek, C. Gemel, A. Pöthig, G. Frenking and R. A. Fischer, Inorg. Chem., 2018, 57, 12657 CrossRef CAS PubMed.
- B. Buchin, T. Steinke, C. Gemel, T. Cadenbach and R. A. Fischer, Z. Anorg. Allg. Chem., 2005, 631, 2756 CrossRef CAS.
- T. Steinke, C. Gemel, M. Winter and R. A. Fischer, Chem.–Eur. J., 2005, 11, 1636 CrossRef CAS PubMed.
- D. Weiss, T. Steinke, M. Winter, R. A. Fischer, N. Fröhlich, J. Uddin and G. Frenking, Organometallics, 2000, 19, 4583 CrossRef CAS.
-
(a) C. Ganesamoorthy, J. Weßing, C. Kroll, R. W. Seidel, C. Gemel and R. A. Fischer, Angew.
Chem., Int. Ed., 2014, 53, 7943 CrossRef CAS PubMed;
(b) M. Schütz, C. Gemel, M. Muhr, C. Jandl, S. Kahlal, J.-Y. Saillard and R. A. Fischer, Chem. Sci., 2021, 12, 6588 RSC.
- M. T. Gamer, P. W. Roesky, S. N. Konchenko, P. Nava and R. Ahlrichs, Angew. Chem., Int. Ed., 2006, 45, 4447 CrossRef CAS PubMed.
- S. G. Minasian, J. L. Krinsky, V. A. Williams and J. Arnold, J. Am. Chem. Soc., 2008, 130, 10086 CrossRef CAS PubMed.
- G. Frenking, Coord. Chem. Rev., 2003, 238–239, 55 CrossRef CAS.
-
(a) C. Ganesamoorthy, J. Krüger, E. Glöckler, C. Helling, L. John, W. Frank, C. Wölper and S. Schulz, Inorg. Chem., 2018, 57, 9495 CrossRef CAS PubMed;
(b) C. K. F. von Hänisch, C. Üffing, M. A. Junker, A. Ecker, B. O. Kneisel and H. Schnöckel, Angew. Chem., Int. Ed., 1996, 35, 2875 CrossRef.
- J. D. Gorden, C. L. B. Macdonald and A. A. H. Cowley, Main Group Chem., 2005, 4, 33 CrossRef CAS.
- S. Schulz, A. Kuczkowski, D. Schuchmann, U. Flörke and M. Nieger, Organometallics, 2006, 25, 5487 CrossRef CAS.
- C. Ganesamoorthy, M. Matthias, D. Bläser, C. Wölper and S. Schulz, Dalton Trans., 2016, 45, 11437 RSC.
- L. J. Morris, A. Carpentier, L. Maron and J. Okuda, Chem. Commun., 2021, 57, 9454 RSC.
- A. Higelin, U. Sachs, S. Keller and I. Krossing, Chem.–Eur. J., 2012, 18, 10029 CrossRef CAS PubMed.
- S. Welsch, M. Bodensteiner, M. Dušek, M. Sierka and M. Scheer, Chem.–Eur. J., 2010, 16, 13041 CrossRef CAS.
- M. Gonsior, I. Krossing and N. Mitzel, Z. Anorg. Allg. Chem., 2002, 628, 1821 CrossRef CAS.
- J. M. Slattery, A. Higelin, T. Bayer and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 3228 CrossRef CAS.
- P. Dabringhaus, J. Willrett and I. Krossing, Nat. Chem., 2022, 14, 1151–1157 CrossRef CAS PubMed.
-
The ± sign was used to describe a standard deviation computed for average bond lengths
Search PubMed.
- K. Glootz, D. Himmel, D. Kratzert, B. Butschke, H. Scherer and I. Krossing, Angew. Chem., Int. Ed., 2019, 58, 14162 CrossRef CAS PubMed.
- K. Glootz, D. Kratzert, D. Himmel, A. Kastro, Z. Yassine, T. Findeisen and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 14203 CrossRef CAS PubMed.
- A. Barthélemy, H. Scherer and I. Krossing, Chem.–Eur. J., 2022, 28 DOI:10.1002/chem.202201369.
-
(a) O. T. Beachley, J. C. Pazik, T. E. Glassman, M. R. Churchill, J. C. Fettinger and R. Blom, Organometallics, 1988, 7, 1051 CrossRef CAS;
(b) H. Werner, H. Otto and H. J. Kraus, J. Organomet. Chem., 1986, 315, C57–C60 CrossRef CAS;
(c) F. Olbrich and U. Behrens, Z. Kristallogr. - New Cryst. Struct., 1997, 212, 47 CAS.
-
J. F. Hinton, K. R. Metz and R. W. Briggs, in Annual reports on NMR spectroscopy, ed. G. A. Webb, Academic Press, London, 1982, vol. 13, pp. 211–318 Search PubMed.
- D. Loos and H. Schnöckel, J. Organomet. Chem., 1993, 463, 37 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
-
L. Pauling, The nature of the chemical bond and the structure of molecules and crystals. An introduction to modern structural chemistry, Cornell Univ. Press, Ithaca, NY, 3rd edn, 1960 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Ingo
Krossing
and
Ingo
Krossing