
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Syntheses, homeomorphic and configurational isomerizations, and structures of macrocyclic aliphatic dibridgehead diphosphines; molecules that turn themselves inside out†
Yun
Zhu
 ,
Michael
Stollenz‡
,
Samuel R.
Zarcone
,
Sugam
Kharel
,
Hemant
Joshi§
,
Nattamai
Bhuvanesh
,
Joseph H.
Reibenspies
and
John A.
Gladysz
*
,
Michael
Stollenz‡
,
Samuel R.
Zarcone
,
Sugam
Kharel
,
Hemant
Joshi§
,
Nattamai
Bhuvanesh
,
Joseph H.
Reibenspies
and
John A.
Gladysz
*
Department of Chemistry, Texas A&M University, PO Box 30012, College Station, Texas 77842-3012, USA. E-mail: gladysz@mail.chem.tamu.edu
First published on 20th October 2022
Abstract
The diphosphine complexes cis- or trans-![[upper bond 1 start]](https://www.rsc.org/images/entities/char_e010.gif) PtCl2(P((CH2)n)3P
PtCl2(P((CH2)n)3P![[upper bond 1 end]](https://www.rsc.org/images/entities/char_e011.gif) ) (n = b/12, c/14, d/16, e/18) are demetalated by MC
) (n = b/12, c/14, d/16, e/18) are demetalated by MC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) X nucleophiles to give the title compounds (P((CH2)n)3)P (3b–e, 91–71%). These “empty cages” react with PdCl2 or PtCl2 sources to afford trans-MCl2(P((CH2)n)3P). Low temperature 31P NMR spectra of 3b and c show two rapidly equilibrating species (3b, 86
X nucleophiles to give the title compounds (P((CH2)n)3)P (3b–e, 91–71%). These “empty cages” react with PdCl2 or PtCl2 sources to afford trans-MCl2(P((CH2)n)3P). Low temperature 31P NMR spectra of 3b and c show two rapidly equilibrating species (3b, 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14; 3c, 97:3), assigned based upon computational data to in,in (major) and out,out isomers. These interconvert by homeomorphic isomerizations, akin to turning articles of clothing inside out (3b/c: ΔH‡ 7.3/8.2 kcal mol−1, ΔS‡ −19.4/−11.8 eu, minor to major). At 150 °C, 3b, c, e epimerize to (60–51):(40–49) mixtures of (in,in/out,out):in,out isomers, which are separated via the bis(borane) adducts 3b, c, e·2BH3. The configurational stabilities of in,out-3b, c, e preclude phosphorus inversion in the interconversion of in,in and out,out isomers. Low temperature 31P NMR spectra of in,out-3b, c reveal degenerate in,out/out,in homeomorphic isomerizations (ΔG‡Tc 12.1, 8.5 kcal mol−1). When (in,in/out,out)-3b, c, e are crystallized, out,out isomers are obtained, despite the preference for in,in isomers in solution. The lattice structures are analyzed, and the D3 symmetry of out,out-3c enables a particularly favorable packing motif. Similarly, (in,in/out,out)-3c, e·2BH3 crystallize in out,out conformations, the former with a cycloalkane solvent guest inside.
:14; 3c, 97:3), assigned based upon computational data to in,in (major) and out,out isomers. These interconvert by homeomorphic isomerizations, akin to turning articles of clothing inside out (3b/c: ΔH‡ 7.3/8.2 kcal mol−1, ΔS‡ −19.4/−11.8 eu, minor to major). At 150 °C, 3b, c, e epimerize to (60–51):(40–49) mixtures of (in,in/out,out):in,out isomers, which are separated via the bis(borane) adducts 3b, c, e·2BH3. The configurational stabilities of in,out-3b, c, e preclude phosphorus inversion in the interconversion of in,in and out,out isomers. Low temperature 31P NMR spectra of in,out-3b, c reveal degenerate in,out/out,in homeomorphic isomerizations (ΔG‡Tc 12.1, 8.5 kcal mol−1). When (in,in/out,out)-3b, c, e are crystallized, out,out isomers are obtained, despite the preference for in,in isomers in solution. The lattice structures are analyzed, and the D3 symmetry of out,out-3c enables a particularly favorable packing motif. Similarly, (in,in/out,out)-3c, e·2BH3 crystallize in out,out conformations, the former with a cycloalkane solvent guest inside.
Introduction
Bicyclic compounds with two bridgehead heteroatoms are quite common for nitrogen but less familiar for other elements. The many dinitrogen examples include DABCO (1,4-diazobicyclo[2.2.2]octane), cryptands,1 and the macrocyclic diprotonated diamines I (Scheme 1).2 The last group represents touchstones for many of the phenomena detailed below.2 In contrast, the corresponding aliphatic dibridgehead diphosphines, or Brønsted or Lewis acid adducts thereof, are much less explored. Prior to the work herein, compounds of the formula P((CH2)n)3P were unknown for n > 4.3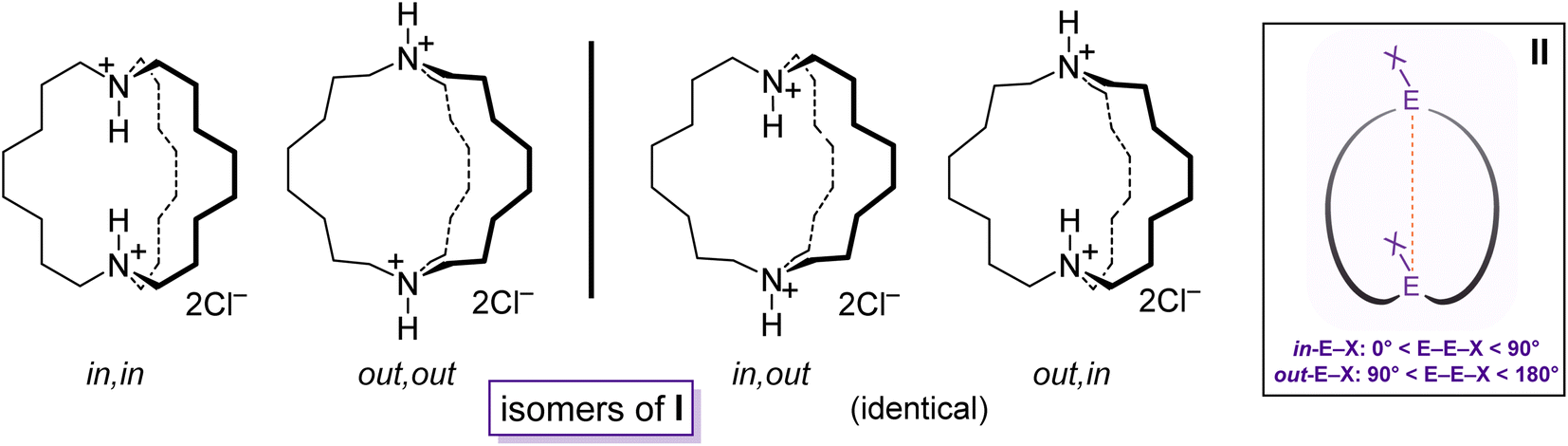 | ||
| Scheme 1 Dibridgehead diammonium salts that illustrate the limiting types of in/out isomers (I), and geometric criteria for in/out bridgeheads EX (II). | ||
Macrocyclic versions of such molecules, as well as analogs with carbon bridgeheads, can exist as in/out isomers4 differing in the relative orientations of the bridgehead substituents. As shown for I (Scheme 1), four limits apply: in,in, out,out, in,out, and out,in. When the northern and southern hemispheres are identical, the last two are degenerate. For structures that lack a C3 axis, in/out geometries can be assigned from the angles defined by the two bridgehead atoms and their substituents, as shown in II. For some time, in/out isomerism has been a curiosity, but the phenomenon has now been coupled to function, for example in the selective transport of metal dichloride fragments.5
The diprotonated dibridgehead diamines I represent the historical “ground zero” for the study of homeomorphic isomerization.4 This terminology, imported from the field of topology,6 denotes a dynamic process that is tantamount to turning a molecule inside out. Essentially, one of the tethers connecting the bridgehead atoms is “pulled through” the macrocycle defined by the other two, like reaching inside a rubber glove and pulling the inner surface outside. As depicted in Scheme 2 as well as a video (ESI†), this interconverts in,in and out,out isomers, and in,out and out,in isomers – in both cases, an apparent inversion of configuration at each bridgehead atom.7
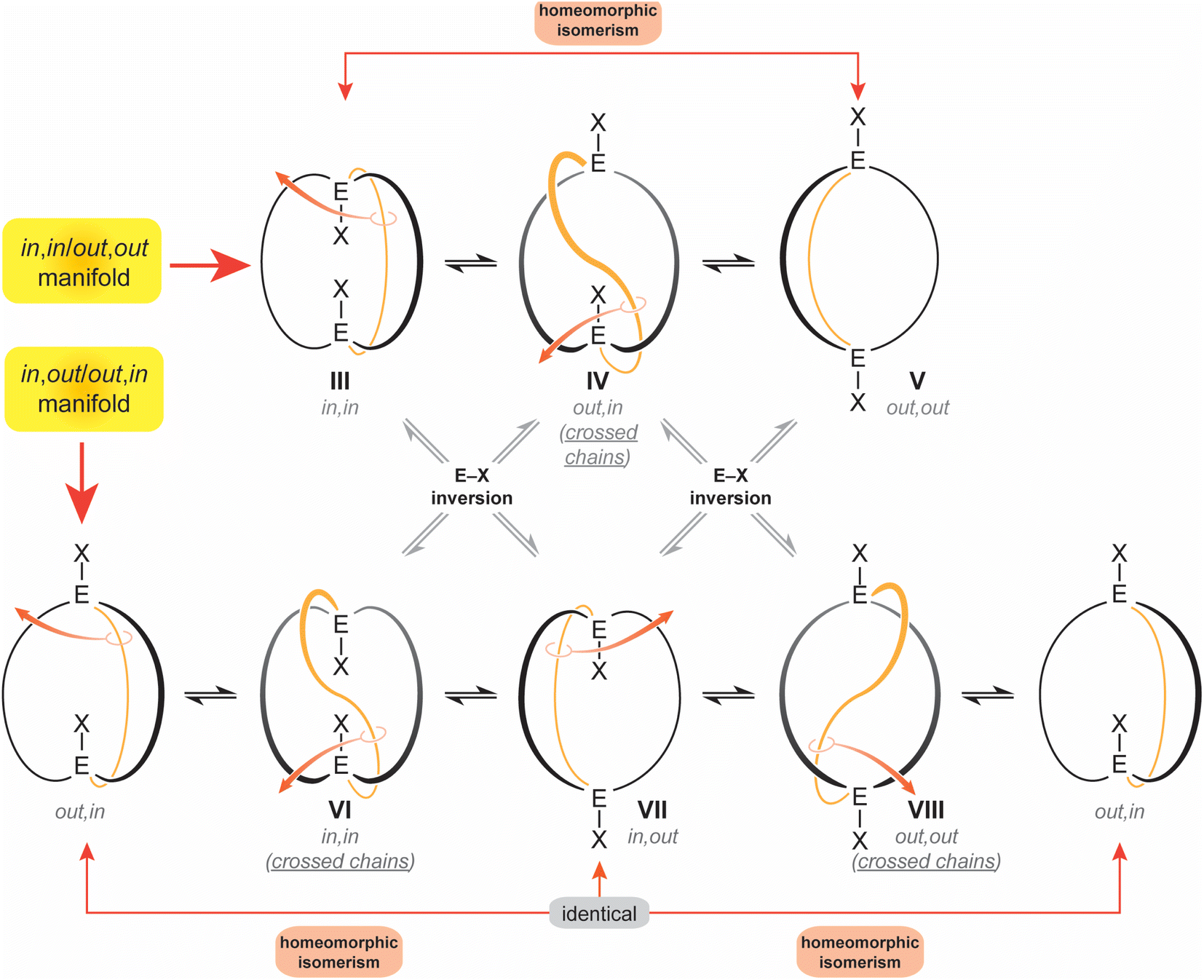 | ||
| Scheme 2 Conventional in,in (III), out,out (V), and in,out (VIII) isomers of a bicyclo[z.z.z] system with EX bridgeheads, and “crossed chain” variants IV, VII and VIII. | ||
Interestingly, data for the in,in, out,out and in,out equilibria of the diprotonated dibridgehead diamines I (Scheme 1) implicated an alternative mechanism: namely, proton dissociation, pyramidal inversion of the resulting trivalent nitrogen atom, and nitrogen reprotonation.2 Indeed, when the bridgeheads are trivalent group 15 heteroatoms (E:), simple pyramidal inversion can effect in/out isomerization, as represented by the “×” pathways connecting the upper and lower manifolds in Scheme 2. However, in the absence of unprecedented cooperative phenomena, only one inversion at a time would be expected, and given well established trends in inversion barriers,8,9 this would be rapid at room temperature only in the case of nitrogen.
For the sake of completeness, three species with “crossed chains” (IV, VI, VIII) are also depicted in Scheme 2. The conformational details of these isomerizations remain beyond the scope of this study, but such architectures receive support in dynamic simulation computations, which locate abundant numbers of local minima.10 As exemplified in the Discussion section, isomers of the type VIII have been isolated for P–X and Si–X systems with sterically demanding X groups.11–13 In all of these contexts, the “inside out” nature of homeomorphic isomerization means that functionality might be directed in a convergent manner towards an interior domain in one conformation, and externally in the other.
Apart from this work, only five molecular systems have been definitively shown to undergo homeomorphic isomerization,4,14,15 as defined by (1) the ability to observe both in,in and out,out forms and establish a direct path between them, or (2) the ability to observe inequivalent bridgehead atoms of an in,out isomer (the E–X in VII are not related by a symmetry operation), and their exchange at higher temperatures. In other cases, there can be a strong inference of homeomorphic isomerization from NMR properties, but with data for both limits remaining elusive.12,16,17 Particularly noteworthy are earlier efforts by Habicher and Bauer that encompass several types of bicyclic dibridgehead diphosphorus compounds with p-phenylene linkers in the tethers.16,17 They and Alder4 were pioneers in articulating some of the concepts expressed above.
Previous synthetic studies from the authors’ laboratory lay the groundwork for the effort detailed herein. As shown in Scheme 3 (top), platinum dichloride complexes with two trans or cis alkene containing phosphines of the formula P((CH2)mCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)3 undergo three fold intramolecular ring closing metathesis when treated with Grubbs’ catalyst.18,19 Subsequent hydrogenations give what are termed “gyroscope-like” (trans) or “parachute-like” (cis) complexes in modest yields. The trans isomers are adducts of in,in dibridgehead diphosphine ligands, and the cis isomers adducts of out,out ligands. This underscores the appreciable conformational flexibility of the ligands.
CH2)3 undergo three fold intramolecular ring closing metathesis when treated with Grubbs’ catalyst.18,19 Subsequent hydrogenations give what are termed “gyroscope-like” (trans) or “parachute-like” (cis) complexes in modest yields. The trans isomers are adducts of in,in dibridgehead diphosphine ligands, and the cis isomers adducts of out,out ligands. This underscores the appreciable conformational flexibility of the ligands.
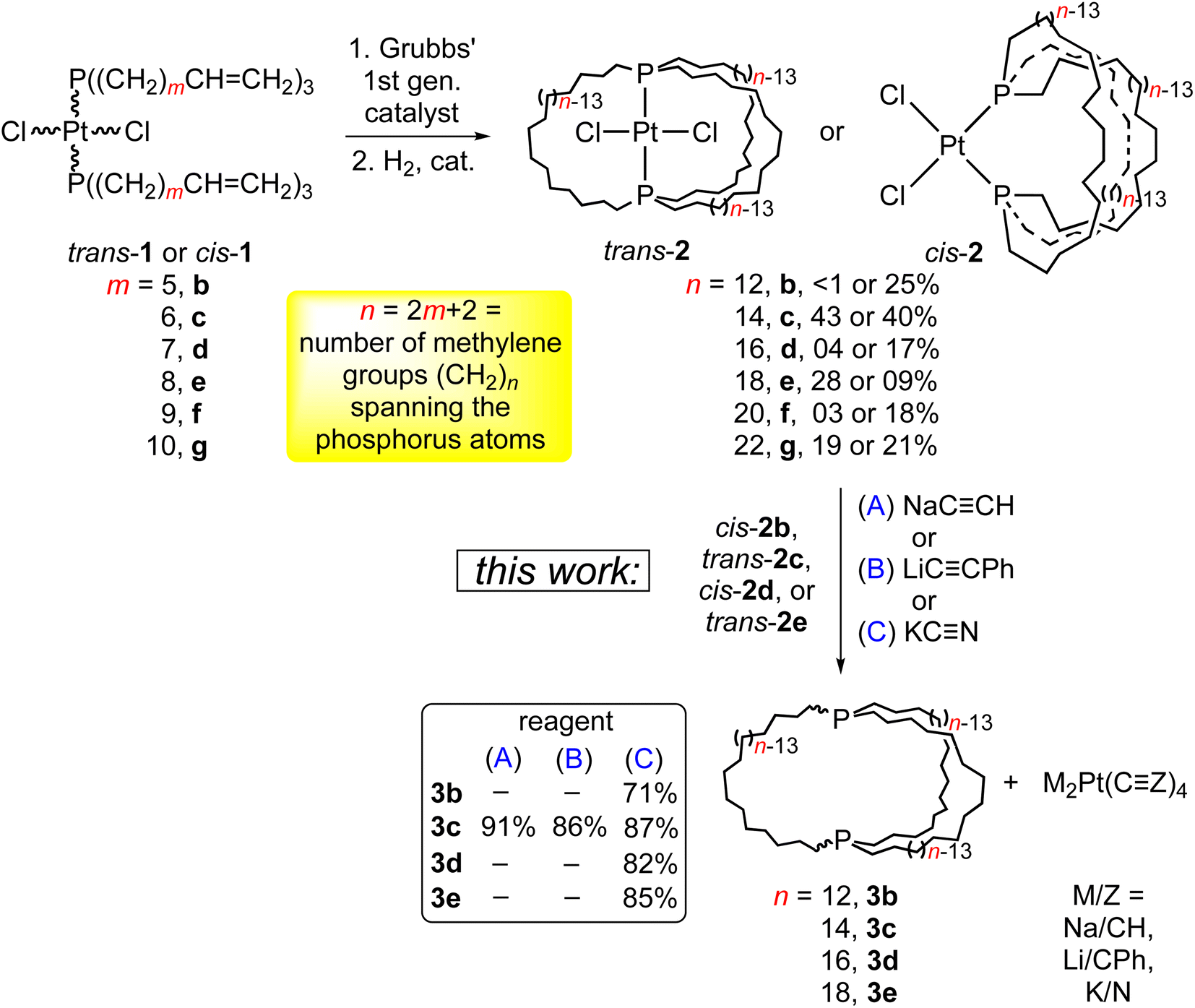 | ||
| Scheme 3 Syntheses of the title compounds (3b–e) from platinum complexes. | ||
In this paper, we report (1) the facile liberation of four aliphatic dibridgehead diphosphine ligands P((CH2)n)3P (3) from 2, (2) detailed variable temperature NMR analyses of 3, which in some cases allow the observation of distinct in,in and out,out isomers, (3) thermal epimerisations that equilibrate the in,in/out,out mixtures and in,out isomers, (4) the formation and deprotection of the corresponding bis(borane) adducts, (5) six crystals structures, and in-depth analyses thereof. Companion computational investigations that support certain assignments also deserve emphasis.10,20 Small portions of these data have been communicated, CCDC 838916 (out,out-3c·BH3·(C5H9CH3)) and CCDC 838917 (out,out-3c·2BH3·(C6H11CH3)).5,21
Results
Syntheses of title compounds 3
As shown in Scheme 3 (bottom), the platinum dichloride complex trans-2c was treated with excesses of NaCCH, LiCCPh, or KCN. Workups gave the dibridgehead diphosphine 3c (n = 14) in 86–91% yields. In the reaction with KCN, the concurrent formation of K2Pt(CN)4 was verified by 13C{1H} NMR and X-ray crystallography. In that with LiCCPh, the salt Li2Pt(CCPh)4·4THF could be isolated in 35% yield. A sample was independently synthesized from the reaction of LiCCPh and PtCl2(THT)2 (THT = tetrahydrothiophene). Similar reactions of cis-2b, d and trans-2e with KCN gave the diphosphines 3b, d, e in 71–85% yields. These feature 26- to 38-membered macrocycles.
Compounds 3b–e were isolated as low melting white solids that were moderately air sensitive, especially in solution.22 They were characterized by NMR spectroscopy (1H, 13C{1H}, 31P{1H}) and other techniques as described below. Data are summarized in the Experimental section. The three P![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H2H2H213C{1H} NMR signals showed comparable nJCP values (10–12 Hz), but more detailed assignments could not be made with certainty. Several cis-PtCl2(diphosphine) species similarly react with excess KCN to give K2Pt(CN)4.23
H2H2H213C{1H} NMR signals showed comparable nJCP values (10–12 Hz), but more detailed assignments could not be made with certainty. Several cis-PtCl2(diphosphine) species similarly react with excess KCN to give K2Pt(CN)4.23
Probes of stereochemistry and dynamic behavior
One initial question concerns the distribution of in/out stereoisomers of 3b–e produced in Scheme 3. The diphosphine ligands in trans-2c, e and cis-2b, d have in,in and out,out orientations, respectively, and the corresponding free ligands can interconvert by homeomorphic isomerizations as summarized in Scheme 2. As shown in Scheme 4, the diphosphines 3b, c, e reacted with PtCl2 sources to generate the gyroscope-like platinum complexes trans-2b, c, e. Comparable reactions of 3c and PdCl2 sources afforded trans-4c, which had been previously synthesized by a route analogous to trans-2c.18a Importantly, trans-2b is a new compound, unavailable in significant quantities by the direct route in Scheme 3.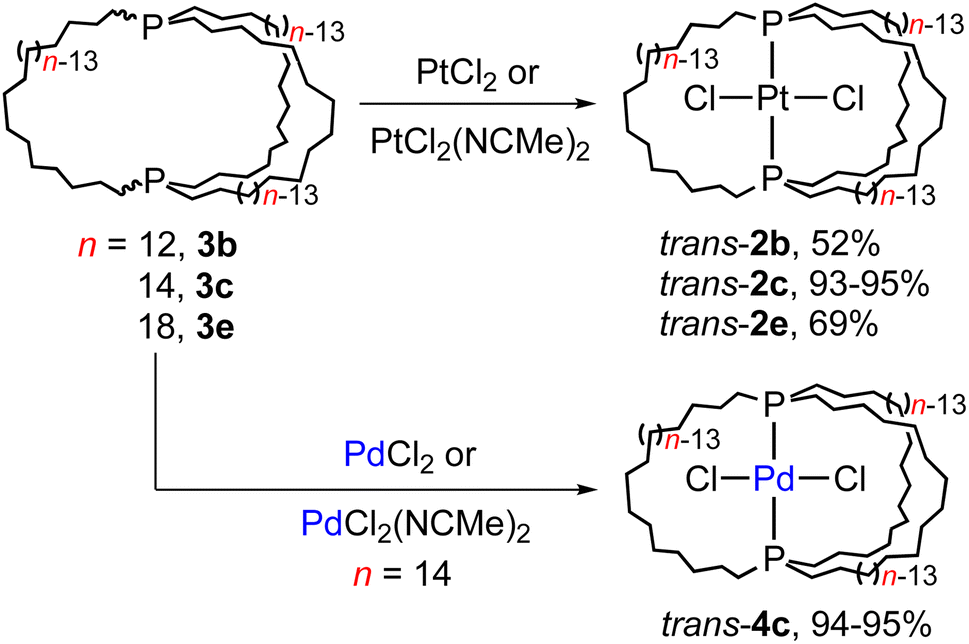 | ||
| Scheme 4 Reactions of title compounds with MCl2 sources. | ||
Trialkylphosphines normally exhibit appreciable pyramidal inversion barriers (ΔG‡403 K = 32–35 kcal mol−1),8 with temperatures of ≥140 °C typically required to effect racemization or epimerization. Hence, the efficient reconstitution of trans-2c, e under mild conditions in Scheme 4 seemingly excludes the formation of in,out-3b–e in Scheme 3. In separate studies, trans-2c–e have been shown to be much more stable than cis-2c–e (equilibration at ca. 160 °C, with ΔG for 2c 3–12 kcal mol−1 depending upon the medium and temperature by DFT).19,24 Hence, the absence of cis products in Scheme 4 is not surprising.
In the course of characterizing the reactivity of 3b–e, low temperature 31P{1H} NMR spectra were acquired. When toluene or toluene-d8 solutions of 3c were cooled, a small upfield peak reproducibly appeared as depicted in Fig. 1a and S1 (ESI†). The line widths for the major signal varied strongly (w1/2 5.8 to 93.0 Hz, 300 K to 233 K, decreasing to 6.3 Hz at 193 K; Fig. S5, ESI†). A 31P EXSY experiment (Fig. S2†) confirmed that the species responsible for the two peaks are in equilibrium. The area ratio at 193 K (−80 °C), 97:3, corresponded to a ΔG193K value of 1.33 kcal mol−1.
The smaller macrocycle 3b also exhibited two signals in toluene-d8 at lower temperatures (Fig. 1b and S3†). At 213 K (−60 °C), integration indicated a 86:14 isomer ratio, for a ΔG213K value of 0.77 kcal mol−1, with the downfield signal again dominant. These coalesced at approximately 303 K (30 °C), and 13C{1H} NMR signals also sharpened above this temperature (Fig. S4†). Two 31P{1H} NMR signals were also observed in mesitylene (85:15, 213 K; Tc near 313 K) and THF (91:9, 213 K; Tc near 263 K).25 For both 3b and c, the spectra were simulated, as exemplified for 3b in Fig. 1b and S3.† Eyring plots of the rate constants derived from the line shapes26 (Fig. S7†) gave ΔH‡ and ΔS‡ values of 7.3 kcal mol−1 and −19.4 eu for 3b (minor to major; ΔG‡213 K = 11.4 kcal mol−1 or 12.1 kcal mol−1 major to minor), and 8.2 kcal mol−1 and −11.8 eu for 3c (minor to major; ΔG‡193 K = 10.4 kcal mol−1, or 11.5 major to minor).26
Accordingly, the two 31P{1H} NMR signals are attributed to in,in and out,out isomers of 3b, c that rapidly interconvert by homeomorphic isomerization, and these descriptors are henceforth coupled to their alphanumeric designations. Perhaps counterintuitively, the major species have been assigned (for all macrocycle sizes) as in,in-3b–e. A strong rationale is provided in accompanying computational papers (which also show the 31P{1H} NMR signals of in P: bridgeheads to be 3.5–8.0 ppm downfield of out P: bridgeheads).10b,20,27 As elaborated below, dispersion forces are thought to play a key role. Analogous trends were found in earlier computational studies of the hydrocarbon bicyclo[6.6.6]eicosane and related species.29 However, the in,in thermodynamic preference is not reflected in crystal structures (vide infra), and of course cannot extend to smaller ring systems (e.g., DABCO).
BH3 adducts and thermal phosphorus epimerization
In principle, in,in-3b–e and out,out-3b–e could interconvert by sequential pyramidal inversions at each phosphorus atom. Although it was viewed as highly unlikely that such processes played any roles in the preceding phenomena, authentic samples of several of the potential intermediates, in,out-3b–e, were sought. If they were to be stable with respect to the in,in and out,out isomers at room temperature, their intermediacies could be definitively excluded.30As a prelude, (in,in/out,out)-3b, c, e were treated with moderate excesses of Me2S·BH3. As shown in Scheme 5, workups gave the diphosphine diboranes (in,in/out,out)-3b, c, e·2BH3 as white or light yellow analytically pure air stable solids or oils in 70–91% yields. These materials, unlike the diphosphines, can be purified chromatographically. They were characterized analogously and exhibited, like other phosphine boranes, broad boron-coupled 31P NMR and BH31H NMR signals.31 Their isomer distributions are also of interest, but this topic is deferred to future papers.
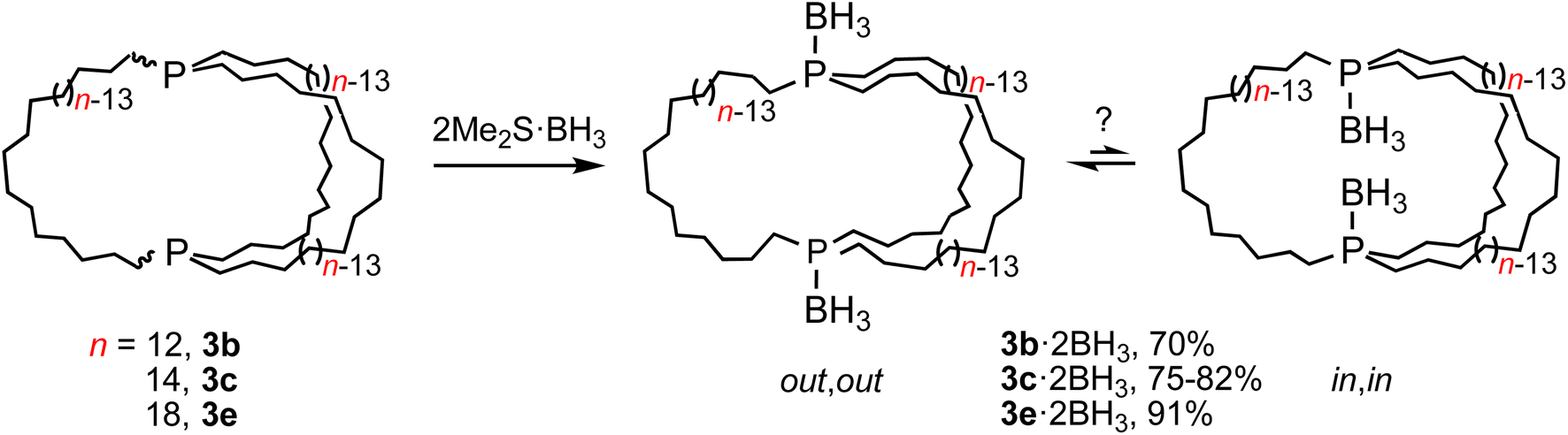 | ||
| Scheme 5 Conversion of title compounds to dibridgehead diphosphine diboranes. | ||
As depicted in Scheme 6, mesitylene solutions of (in,in/out,out)-3b, c, e were kept at 150 °C and monitored by 31P{1H} NMR. As exemplified in Fig. S8,† a new signal gradually appeared in each case. These were assigned to the epimerization products in,out-3b, c, e. After 30–60 h, 60:40 (3b) to 51:49 (3c, e) equilibrium mixtures were obtained ((in,in/out,out):in,out). The rate of epimerization of 3c (k1 = 1.47 × 10−5 s−1) gave a ΔG‡423 K value of 34.4 kcal mol−1 (ΔG423 K = 0.03 kcal mol−1), in good agreement with pyramidal inversion barriers of trialkyl monophosphines.8
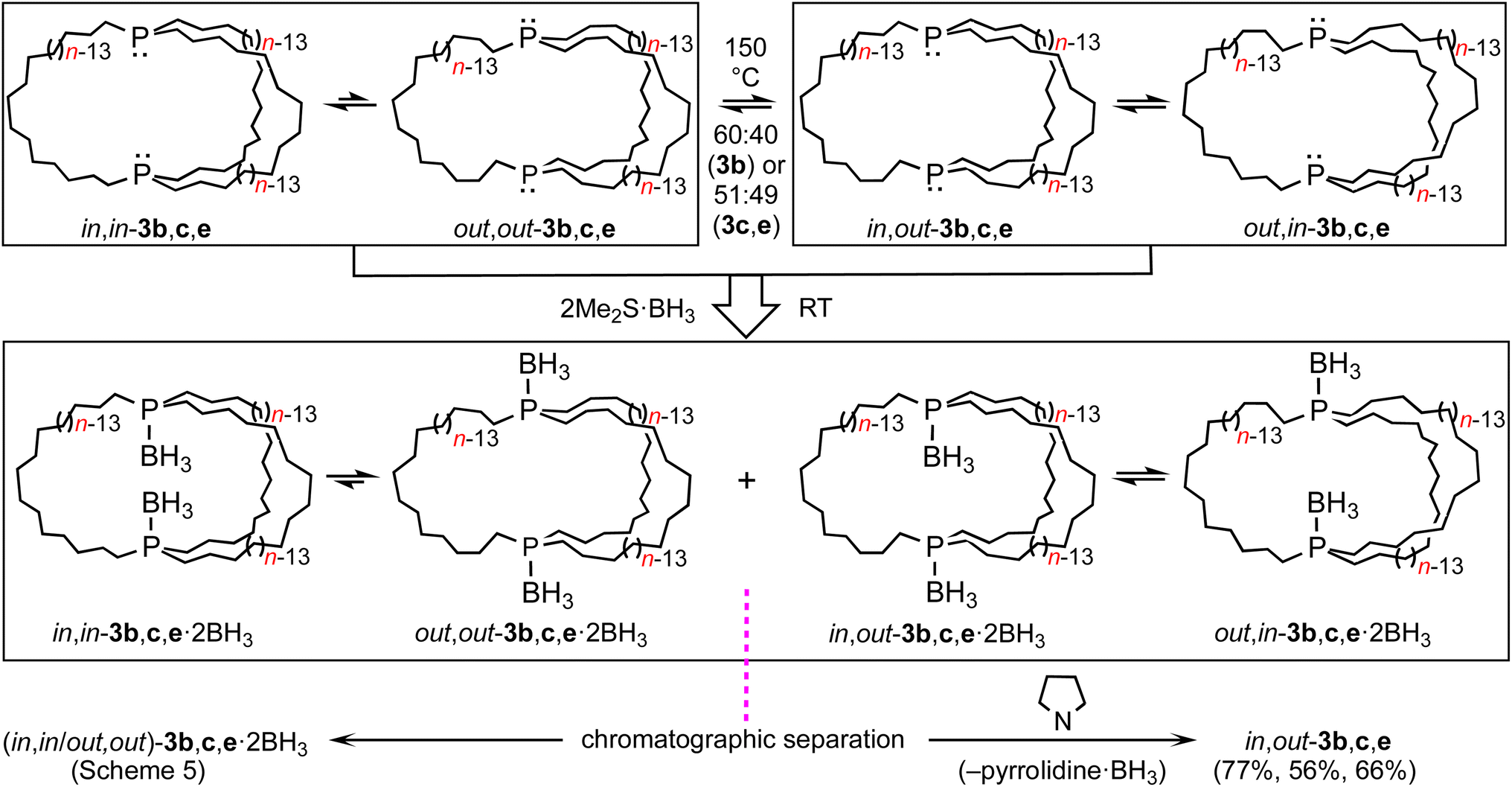 | ||
| Scheme 6 Thermal epimerization of (in,in/out,out)-3 to in,out-3 and separations via BH3 adducts. | ||
Practical direct separations of (in,in/out,out)-3b, c, e and in,out-3b, c, e could not be devised. Thus, as shown in Scheme 6, the samples were treated with excess Me2S·BH3. Silica gel chromatography gave the bis(borane) adducts (in,in/out,out)-3b, c, e·2BH3 (also prepared in Scheme 5) and in,out-3b, c, e·2BH3 in 43–23% and 42–16% yields, respectively. The latter were characterized analogously to the former,32 and representative 13C{1H} NMR spectra are compared in Fig. S9.† The phosphine boranes in,out-3b, c, e·2BH3 were deprotected using a standard protocol, neat refluxing pyrrolidine. Silica gel workups gave analytically pure in,out-3b, c, e in 77–56% yields as moderately air sensitive colorless oils.
Additional probes of equilibria
Importantly, in,out-3b, c, e exhibited a single 31P{1H} NMR signal at ≥290 K, although by symmetry two would have been expected. This implies rapid homeomorphic isomerization (Scheme 6, upper right, or Scheme 2, lower manifold) on the NMR time scale. Accordingly, CH2Cl2 solutions were cooled, and 31P{1H} NMR spectra were recorded. In the cases of in,out-3b, c, two signals of nearly equal intensities decoalesced as shown in Fig. 2 (Tc = 290 K and 200 K). The data yielded ΔG‡Tc values of 12.1 and 8.5 kcal mol−1, respectively. Thus, the activation energies increase as the macrocycles become smaller and degrees of freedom diminish.The chemical shifts of the decoalesced signals, and the Δppm values, were similar to those in Fig. 1. Thus, the downfield signals are provisionally assigned to in bridgeheads, and the upfield signals to out bridgeheads. The 31P{1H} NMR spectra of the diphosphine diboranes (in,in/out,out)-3c·2BH3 and in,out-3c·2BH3, as well as 1H and 13C{1H} NMR spectra of the latter, were recorded over a similar temperature range in CD2Cl2 (Fig. S10–S13†). Although the chemical shifts and peak widths varied, there were no well-defined decoalescence phenomena.
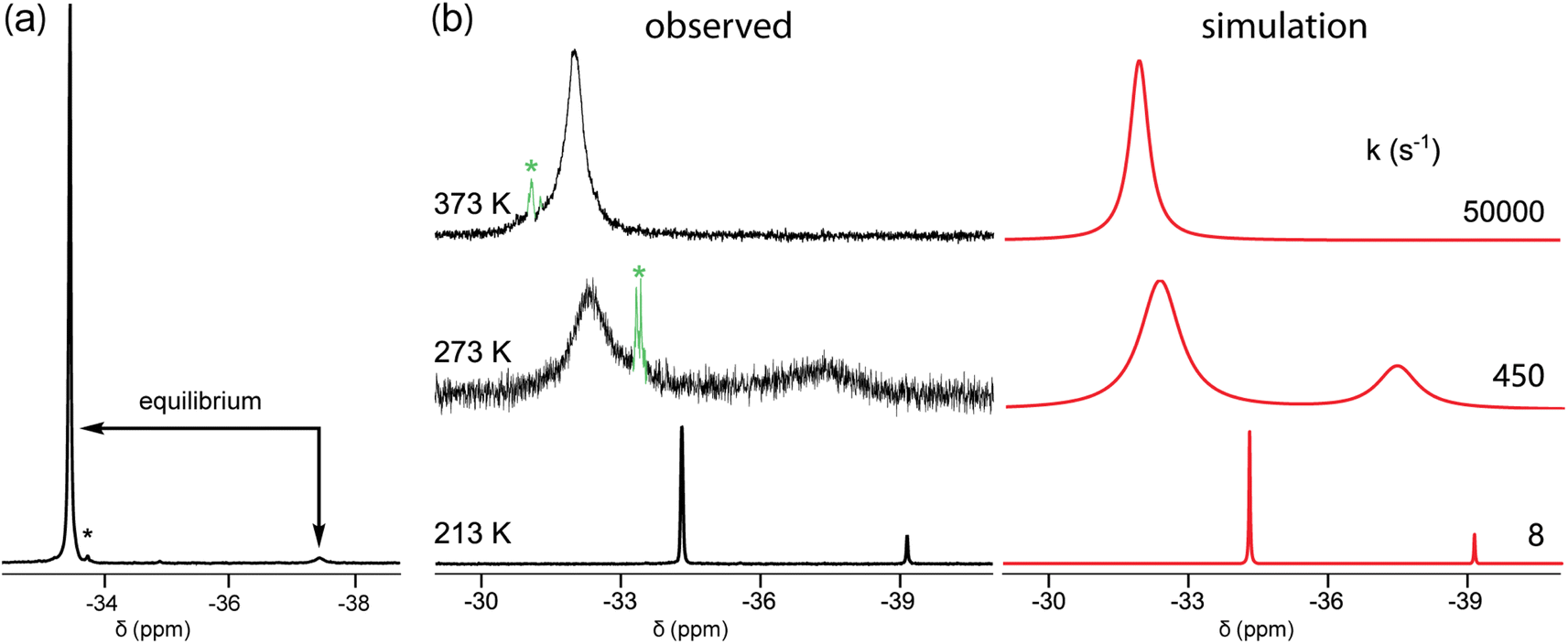 | ||
| Fig. 1 31P{1H} NMR spectra in toluene-d8: (a) 3c at 193 K; (b) 3b at 213 K, 273 K, and 373 K, together with simulated spectra. The label * indicates an impurity. | ||
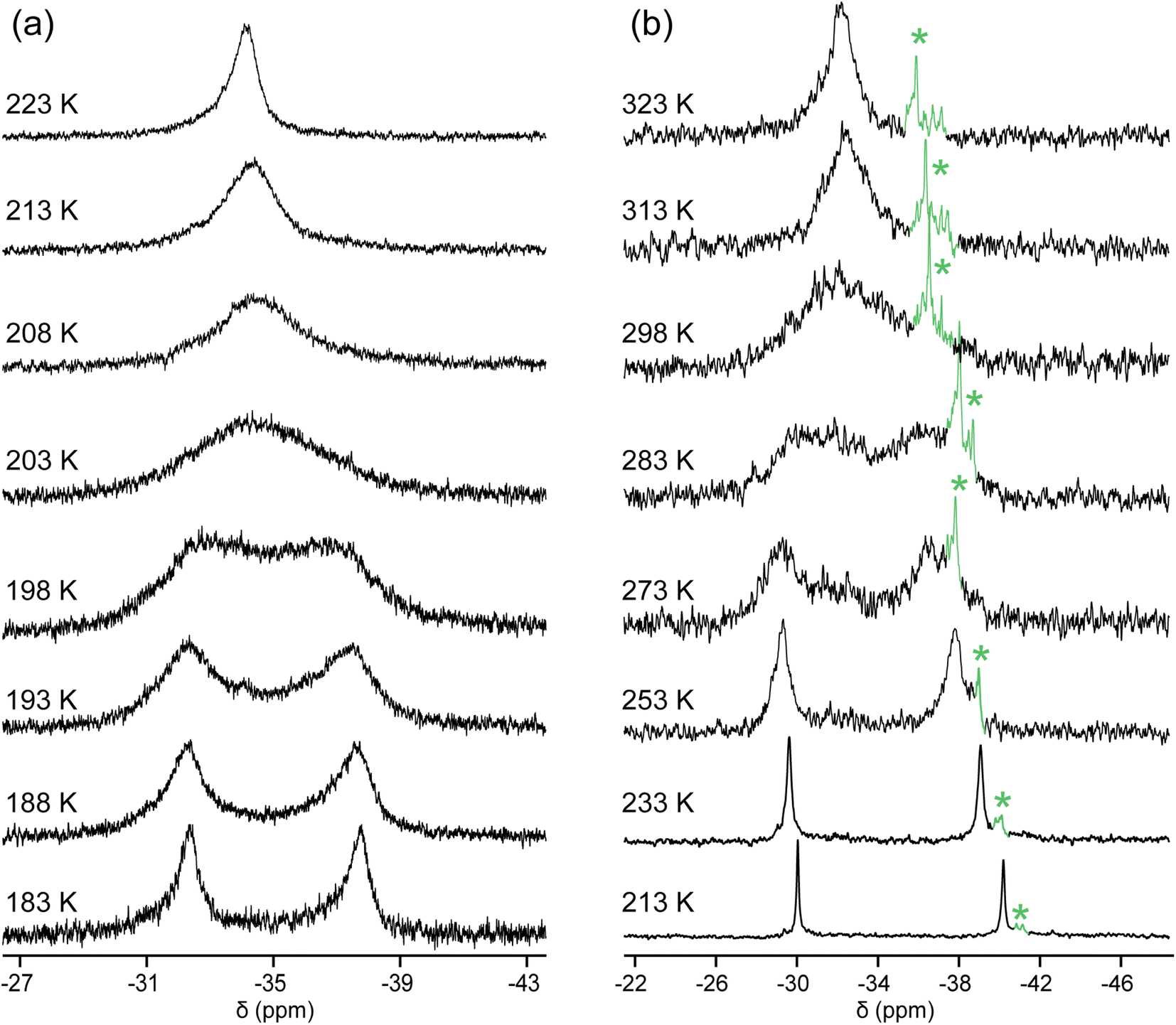 | ||
| Fig. 2 Variable temperature 31P{1H} NMR spectra (202 MHz) of (a) in,out-3c in CH2Cl2; (b) in,out-3b in CDCl3. The label * indicates an impurity. | ||
Crystal structures
Efforts were made to determine the crystal structures of as many of the preceding compounds as possible. Six were ultimately obtained as summarized in the ESI.† Key metrical parameters are presented in Table 1, and many additional distances and angles are tabulated in the ESI.† Interestingly, all compounds exhibited out,out geometries. Consider first the diphosphines out,out-3b, c, e, the thermal ellipsoid plots of which are compared in Fig. 3.| out,out-3b | out,out-3c | out,out-3e | out,out-3c·2BH3·(C5H9CH3) | out,out-3c·2BH3·(C6H11CH3) | out,out-3e·2BH3 (1)a | out,out-3e·2BH3 (2)a | out,out-3e·2BH3 (3)a | |
|---|---|---|---|---|---|---|---|---|
| a The multiple columns refer to the three independent molecules in the unit cell. b The corresponding value in the platinum complex trans-2c is 4.61 Å. c Sum of the three C–P–C bond angles (360° = planar or sp2 limit, 328.4° = tetrahedral or sp3 limit; 270° = unhybridized px/py/pz limit. | ||||||||
| P–Pb | 10.8271(8) | 12.948(3) | 17.978(1) | 13.212(4) | 13.220(4) | 19.407(3) | 19.800(3) | 19.540(3) |
| P–C | 1.8535(15) | 1.8361(15) | 1.848(2) | 1.840(11) | 1.823(8) | 1.825(8) | 1.827(8) | 1.805(8) |
| 1.8544(15) | 1.87(3) | 1.770(11) | 1.827(8) | 1.817(8) | 1.802(8) | |||
| 1.8526(15) | 1.832(2) | 1.812(8) | 1.929(12) | 1.801(8) | 1.814(8) | 1.814(9) | ||
| 1.8541(16) | 1.708(13) | 1.837(10) | 1.825(8) | 1.822(9) | 1.819(8) | |||
| 1.8530(15) | 1.850(2) | 1.92(3) | 1.738(12) | 1.819(8) | 1.779(10) | 1.825(9) | ||
| 1.8543(15) | 1.852(10) | 1.825(11) | 1.831(8) | 1.817(9) | 1.830(8) | |||
| P–B | — | — | — | 1.906(12) | 1.864(12) | 1.903(10) | 1.903(10) | 1.907(10) |
| 1.900(12) | 1.881(12) | 1.903(9) | 1.920(10) | 1.910(10) | ||||
| C–P–C | 98.67(7) | 98.64(6) | 102.06(10) | 105.9(4) | 105.7(5) | 107.2(4) | 105.8(4) | 106.9(4) |
| 98.68(7) | 102.0(16) | 102.6(5) | 103.9(4) | 104.4(4) | 101.7(4) | |||
| 98.02(7) | 100.30(11) | 106(2) | 107.0(5) | 106.6(4) | 105.7(4) | 110.0(4) | ||
| 97.65(7) | 101.7(5) | 108.5(6) | 103.7(4) | 105.8(5) | 107.1(4) | |||
| 99.21(7) | 100.32(10) | 114.4(10) | 105.0(5) | 109.3(4) | 107.1(4) | 108.2(4) | ||
| 98.11(7) | 97.1(11) | 102.3(5) | 107.8(4) | 111.3(5) | 103.3(4) | |||
| C–P–B | — | — | — | 117.1(6) | 115.9(5) | 111.5(4) | 112.2(4) | 110.7(4) |
| 109.2(15) | 120.4(6) | 112.7(5) | 112.8(4) | 116.0(4) | ||||
| 114.8(5) | 103.7(5) | 114.4(4) | 115.1(4) | 111.1(4) | ||||
| 118.8(7) | 112.8(6) | 112.8(4) | 106.4(5) | 112.8(5) | ||||
| 109.7(10) | 116.5(6) | 113.2(4) | 113.9(5) | 109.4(5) | ||||
| 112.9(6) | 110.7(6) | 109.6(5) | 112.2(5) | 114.7(4) | ||||
| P–P-lone pair or P–P–B | 172.9 | 180.0 | 108.5 | 174.2(4) | 176.1(4) | 149.2(3) | 153.0(3) | 149.2(3) |
| 173.5 | 157.6(4) | 158.4(4) | 133.1(3) | 129.2(4) | 133.3(4) | |||
| Phosphorus pyramidalizationc | 295.4 | 295.9 | 302.7 | 313.9 | 315.3 | 317.7 | 315.9 | 318.6 |
| 295.0 | 313.2 | 315.8 | 320.8 | 324.2 | 319.2 | |||
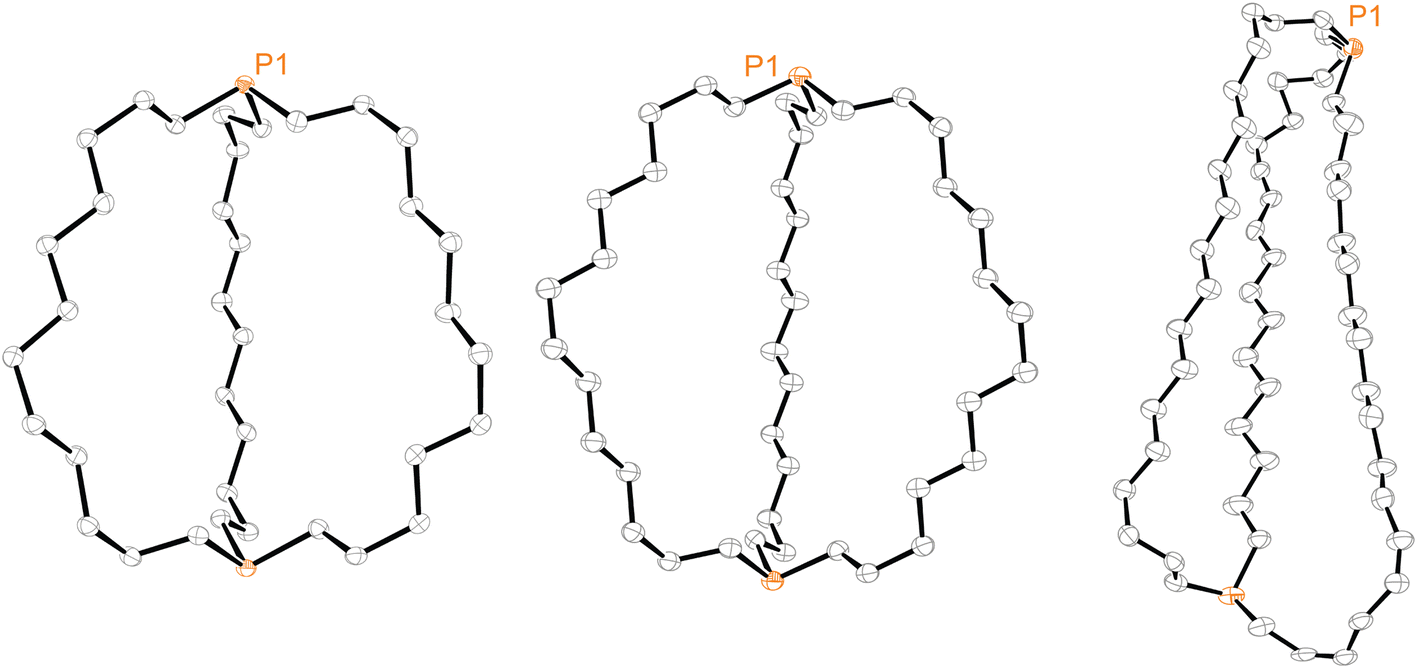 | ||
| Fig. 3 Thermal ellipsoid plots (50% probability level) for out,out-3b (left), out,out-3c (middle), and out,out-3e (right, dominant conformation). | ||
Crystalline out,out-3c exhibits unusual molecular symmetry (D3), such that the positions of all 44 non-hydrogen atoms can be defined from the atomic coordinates of only eight (P(CH2)7). As depicted in panel (a) of Fig. 4, a C3 axis passes through the two phosphorus atoms, and three C2 axes lie in a perpendicular plane (one of which runs perpendicular to the plane of the paper in panel (b)). However, the conformation remains chiral, with no internal mirror planes, although as required by the achiral space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c, both enantiomers are present in the unit cell. Panel (c) highlights the cage like nature and attendant interior space, which in the lattice is partially occupied by neighboring molecules (vide infra).
c, both enantiomers are present in the unit cell. Panel (c) highlights the cage like nature and attendant interior space, which in the lattice is partially occupied by neighboring molecules (vide infra).
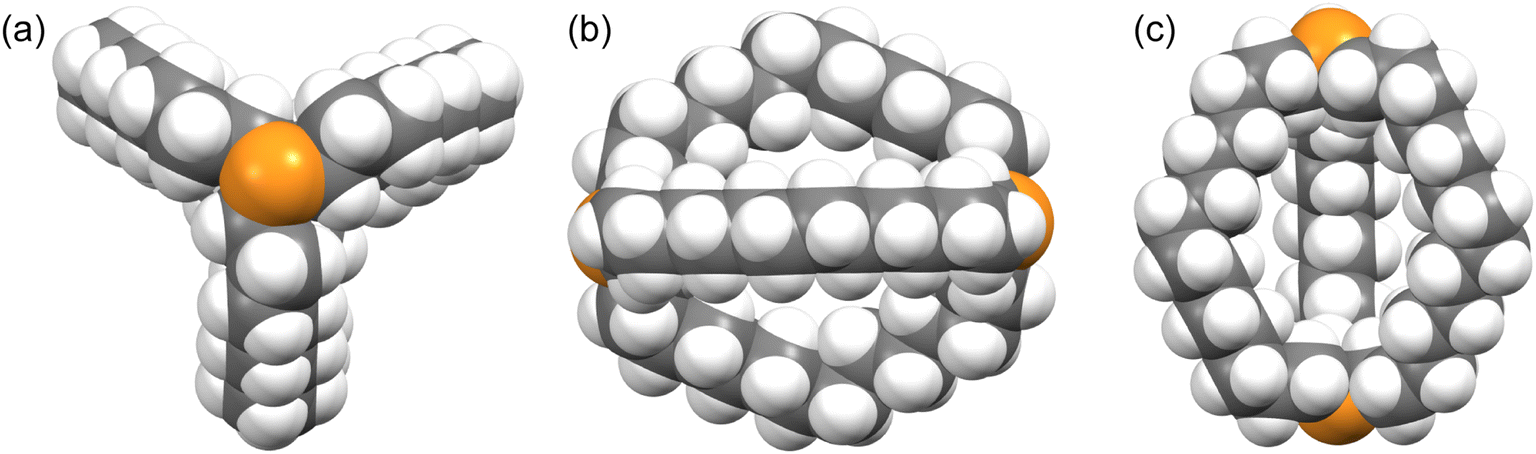 | ||
| Fig. 4 Space filling representations of out,out-3c from three orthogonal perspectives. | ||
In the case of out,out-3b, there is a “near miss” with respect to D3 symmetry. Space filling views comparable to those in Fig. 4 are provided in Fig. S14,† and visually most deviations are slight. With out,out-3e (Fig. 3), a C2 axis passes through the midpoint of one P(CH2)18P chain and exchanges the phosphorus atoms and the other two chains. As detailed in the Experimental section, there is disorder in two chains but this is easily modeled and only the dominant conformation (77%) is treated. As may be facilitated by the larger ring sizes in out,out-3e, the chains partially collapse in on each other, stopping just short of van der Waals contacts and retaining a smidgen of interior space (Fig. S15†). Furthermore, the two phosphorus atoms no longer occupy geometric apices.
Features associated with the phosphorus–phosphorus vectors of out,out-3b, c, e are of interest. As summarized in Table 1, their lengths increase from 10.8271(8) to 12.948(3) to 17.978(1) Å. This constitutes an immense expansion of the phosphorus–phosphorus distances in the crystalline platinum complexes 2c, g (4.611–4.620 Å), emphasizing the structural flexibility of the diphosphines. The phosphorus–phosphorus-lone pair angles in out,out-3c are both 180°, whereas in out,out-3b they decrease slightly to 173.5°–172.9°. However, in out,out-3e the angles are both 108.5°, approaching the 90° cutoff for out,out and in,in isomers diagrammed in II (Scheme 1).
Consider next the diphosphine diboranes out,out-3c, e·2BH3. The former could be crystallized as both methylcyclopentane33 and methylcyclohexane monosolvates, out,out-3c·2BH3·(C5H9CH3) and out,out-3c·2BH3·(C6H11CH3). These exhibited very similar unit cell parameters (Table S1†) and molecular structures, with the solvent molecules occupying the interior of the diphosphine cages. The former (the better structure) is depicted in Fig. 5, but both are quite similar. Solvent is analogously incorporated into the diphosphine cages of crystalline digold complexes of the type out,out-3c·2AuX.11,34 The phosphorus–phosphorus vectors in out,out-3c·2BH3·(C5H9CH3) and out,out-3c·2BH3·(C6H11CH3) are 2.1% longer than that in the parent diphosphine out,out-3c (13.212(4)–13.220(4) Å vs. 12.948(3) Å), with one P–P–B angle close to 180° (174.2–176.1°) and the other somewhat smaller (157.6(4)–158.4(4)°).
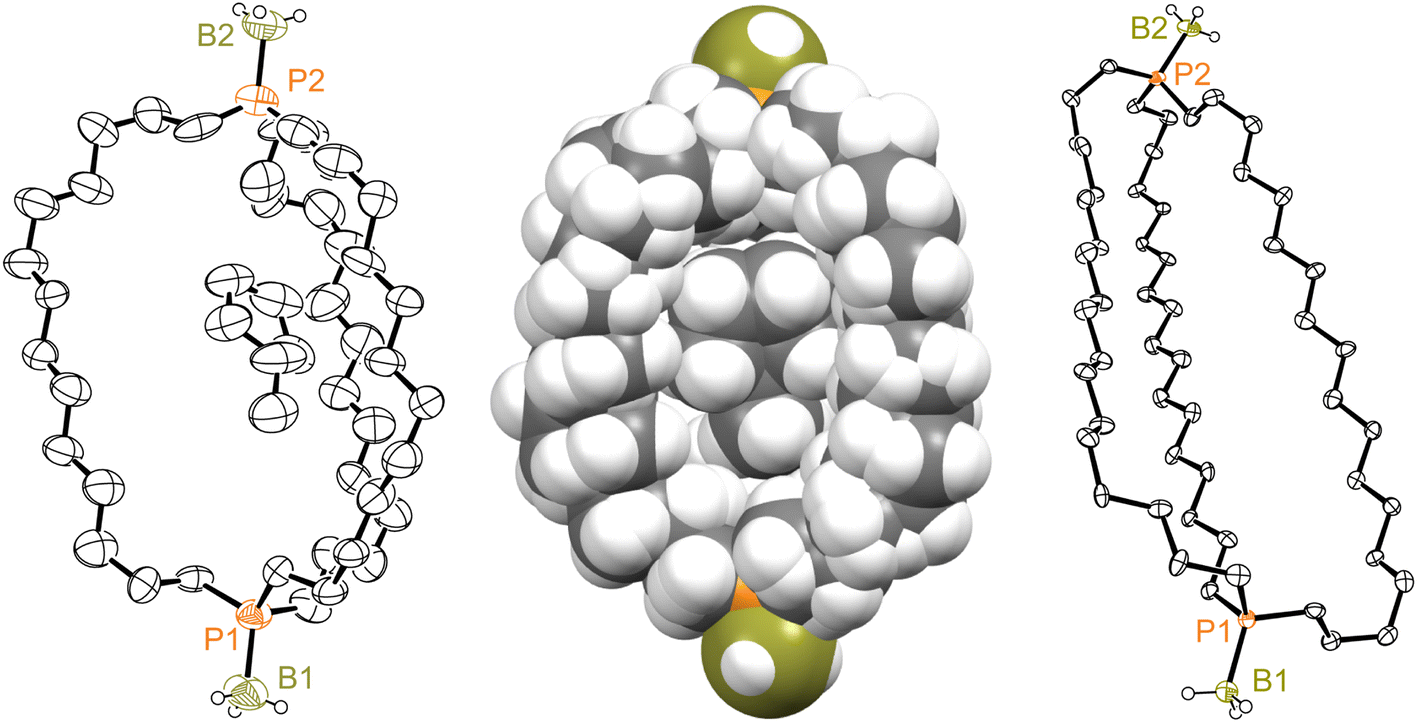 | ||
| Fig. 5 Thermal ellipsoid plots (50% probability level) for out,out-3c·2BH3·(C5H9CH3) (left, dominant conformation) and out,out-3e·2BH3 (right, one of three independent molecules in the unit cell), and a space filling representation of out,out-3c·2BH3·(C5H9CH3) (middle). | ||
Although out,out-3e·2BH3 crystallized without solvent incorporation, three independent molecules were present in the unit cell. One has been arbitrarily selected for Fig. 5. As with the congener out,out-3e, the cages in all three molecules have collapsed inward, with the P(CH2)18P chains close to but not quite in van der Waals contact. As expected, the phosphorus–phosphorus vectors (19.407(3)–19.800(3) Å) are much longer than those of the out,out-3c·2BH3 monosolvates, and for conformational reasons longer than that in out,out-3e (17.978(1) Å). The P–P–B angles (153.0(3)–129.2(4)°; avg 141.2°) are smaller than the P–P–(B or lone pair) angles of all the compounds except out,out-3e (108.5°).
Crystal lattices
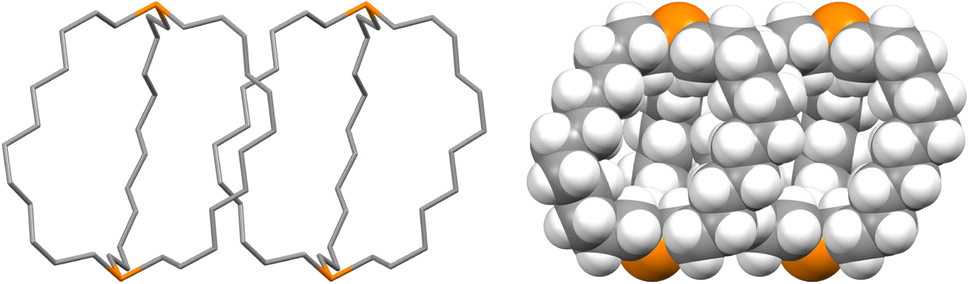
The unexpected uniformity with which the preceding compounds crystallized as out,out isomers prompted close inspections of the crystal lattices for potential “packing forces”. The crystal systems exhibited by out,out-3b, c, e (triclinic, rhombohedral, monoclinic; Z = 2, 6, 4) differed. However, the unit cells always featured one axis that was very much longer than the others (Table S1†), as particularly pronounced for out,out-3c (c = 89.58(2) Å vs. 2 × 9.1903(19) Å). When viewed along this axis, the phosphorus–phosphorus vectors are aligned, as highlighted in orange in panel (a) of Fig. 6. Each stack is surrounded by six others, all equidistant and in a hexagon motif.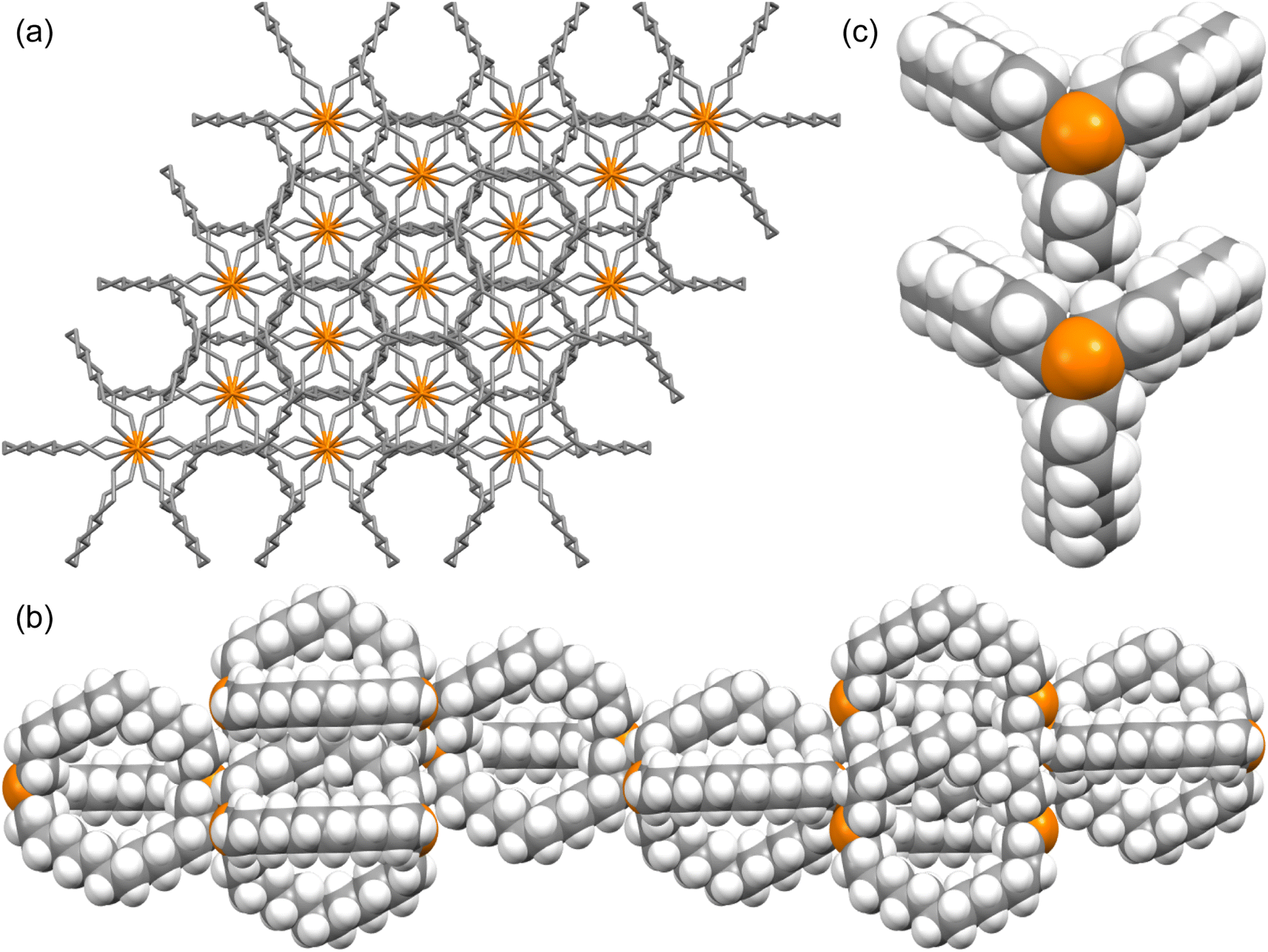 | ||
| Fig. 6 Views of the crystal lattice of out,out-3c with the long c axis perpendicular to the plane of the paper (a and c) or in the plane of the paper (b). | ||
There are six (CH2)14 segments radiating with 60° spacings from each phosphorus–phosphorus stack, the PCH2 segments of which generate apparent rhomboids, followed by “tails” representing the remaining CH2 groups. Three are associated with one layer of molecules aligned along the c axis, and the other three with a layer that is three layers above or below (Fig. 6, panel (b)). Within each layer, the (CH2)14 bridge of one molecule intercalates between two (CH2)14 bridges of a neighboring molecule (panel (c)). Between adjacent layers, the molecules are offset, the phosphorus atoms of one abutting the 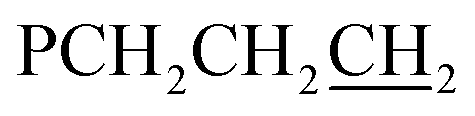 groups of the other. However, the closest intermolecular distances fall just short of van der Waals contacts.
groups of the other. However, the closest intermolecular distances fall just short of van der Waals contacts.
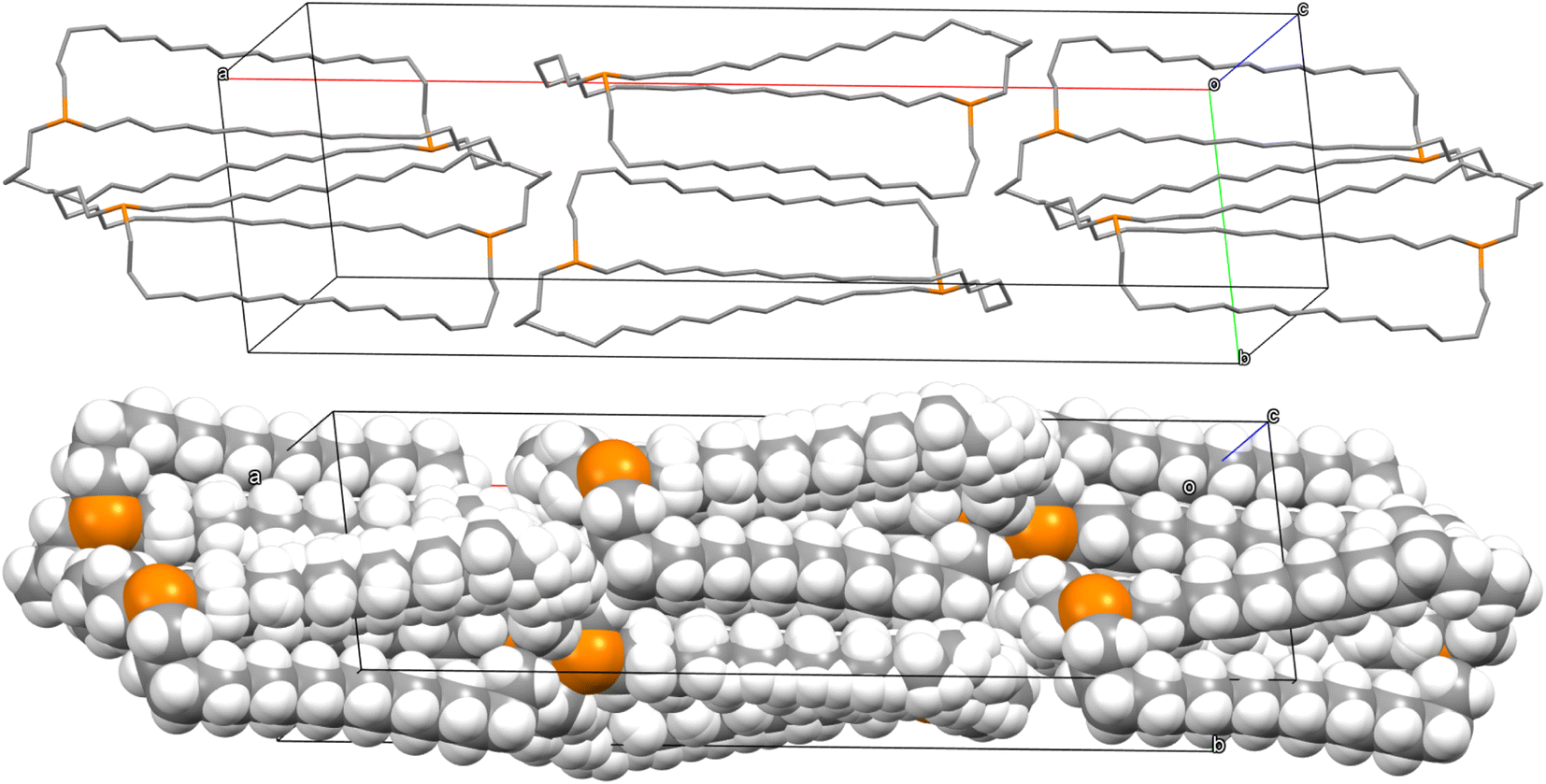
Although out,out-3b does not give as symmetrical a lattice as out,out-3c, the (CH2)12 chains similarly nest within the interstices generated by two (CH2)12 chains of a neighboring molecule (Fig. 7). The two bis(borane) adducts out,out-3c·2BH3·(C5H9CH3) and out,out-3c·2BH3·(C6H11CH3) pack similarly. Here, the interior solvent molecules require expression as out,out isomers, so crystal packing forces cannot be playing a direct role.
 | ||
| Fig. 7 Two adjacent molecules in the crystal lattice of out,out-3b. | ||
The long dimensions of the P(CH2)18P systems out,out-3e and out,out-3e·2BH3 are also roughly aligned in the respective crystal lattices. However, as illustrated in Fig. 8, there is no intercalation as in Fig. 6. Nonetheless, the intermolecular spacings between (CH2)18 chains are comparable to the intramolecular spacings, which as noted above are slightly greater than van der Waals contacts. Interestingly, out,out-3e and out,out-3e·2BH3 exhibit the highest densities in each series (e.g., ρ 1.022 (out,out-3e) vs. 0.991–0.990 (out,out-3b, c) Mg m−3).
 | ||
| Fig. 8 Adjacent molecules in the crystal lattice of out,out-3e (dominant conformations only). | ||
Discussion
Syntheses of dibridgehead diphosphines
As shown in Scheme 3, this study has established the synthetic availability of an extensive family of aliphatic macrocyclic dibridgehead diphosphines P((CH2)n)3P (3) that can exist as in,in, out,out, and in,out isomers. There currently seems to be no obstacle to extending this chemistry to n ≥ 20, or ≥42 membered macrocycles. As depicted in Scheme 7 (top), isomers of 3c (n = 14) have also been accessed via three-fold intermolecular olefin metatheses of the metal-free phosphine borane H3B·P((CH2)6CHCH2)3.32 However, the metathesis steps proceed in much lower yields than the platinum templated pathway, and the overall yields are miniscule.
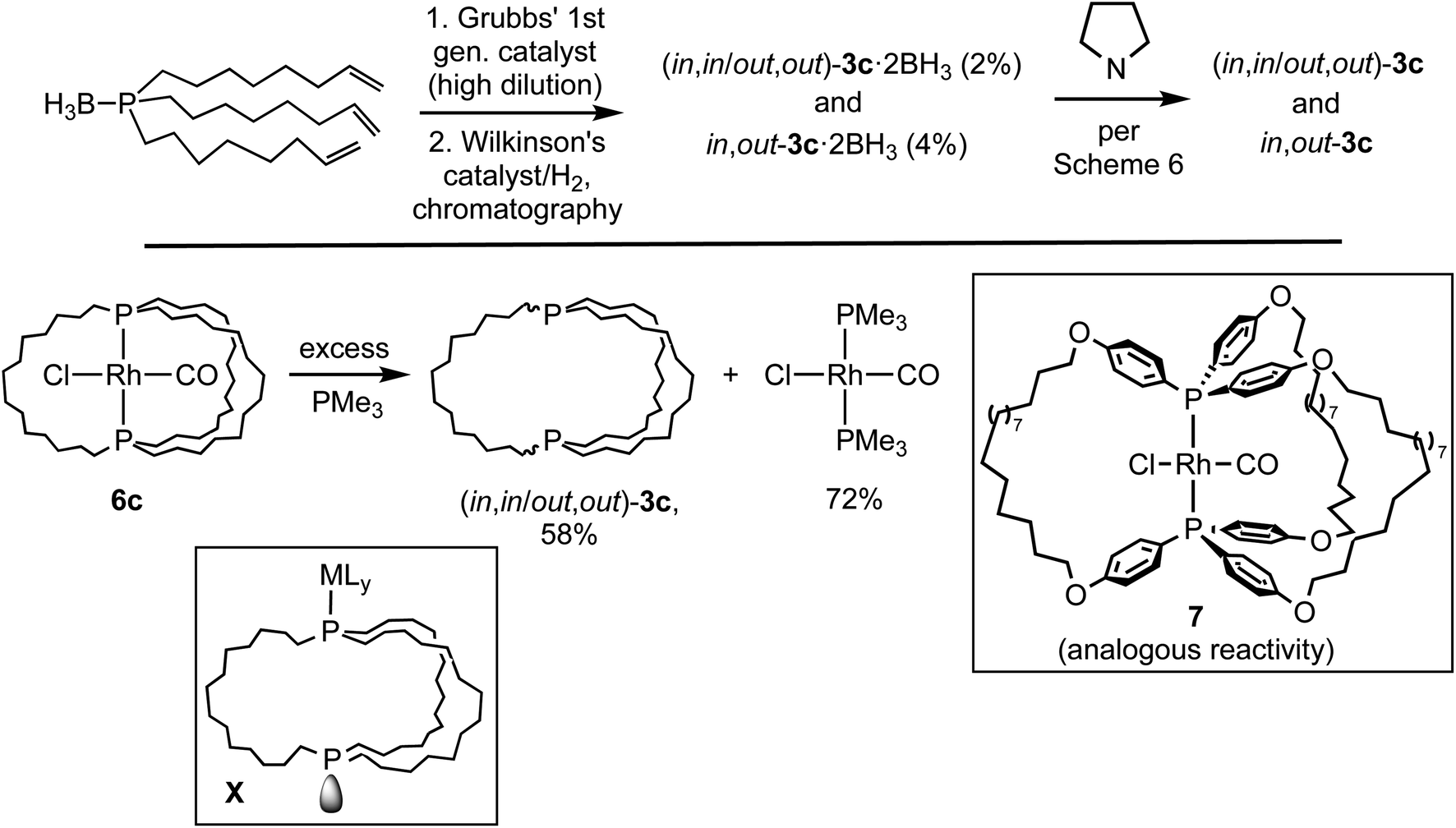 | ||
| Scheme 7 Additional routes to the title molecules and related species. | ||
As exemplified by 6c in Scheme 7 (bottom), demetallation of gyroscope-like rhodium(I) complexes can also be effected,35 but in efforts to date yields have not been superior to the routes in Scheme 3. Precursors such as 7 afford related dibridgehead di(triaryl)phosphines.35b However, there have recently been promising developments regarding alternative routes that involve inexpensive metals.28 These have included the iron-based syntheses of the dibridgehead diarsines As((CH2)n)3As (n = 10, 12, 14).36
The mechanisms of these demetalations, which are receiving ongoing attention, are beyond the scope of this study. However, since metal fragments can generally be reintroduced (Scheme 4), intermediates such as X (Scheme 7) derived from metal–phosphorus bond cleavage and homeomorphic isomerization have been suggested. The platinum byproducts M2Pt(CX)4 generated in Scheme 3 would be derived from the displacement of all the metal–ligand bonds in X by –CX nucleophiles. Accordingly, excess KCN has been shown to convert various cis-PtCl2(diphosphine) adducts to K2Pt(CN)4.23
The most closely related dibridgehead diphosphorus macrocycles in the literature are depicted in Scheme 8.17b The dibridgehead di(triaryl)phosphine dioxides 8·2O were constructed via Williamson ether syntheses that afforded both (in,in/out,out) and in,out isomers. Subsequent reductions gave 8, which were characterized in situ due to their air sensitivity. It has not yet proved possible to quantify equilibrium ratios or obtain crystal structures for any of these species. Nonetheless, the in/out isomers could be assigned based upon rate trends in Scheme 8 and derivatization reactions. Recently, several additional types of novel compounds exhibiting in/out isomerism have been reported.15,37
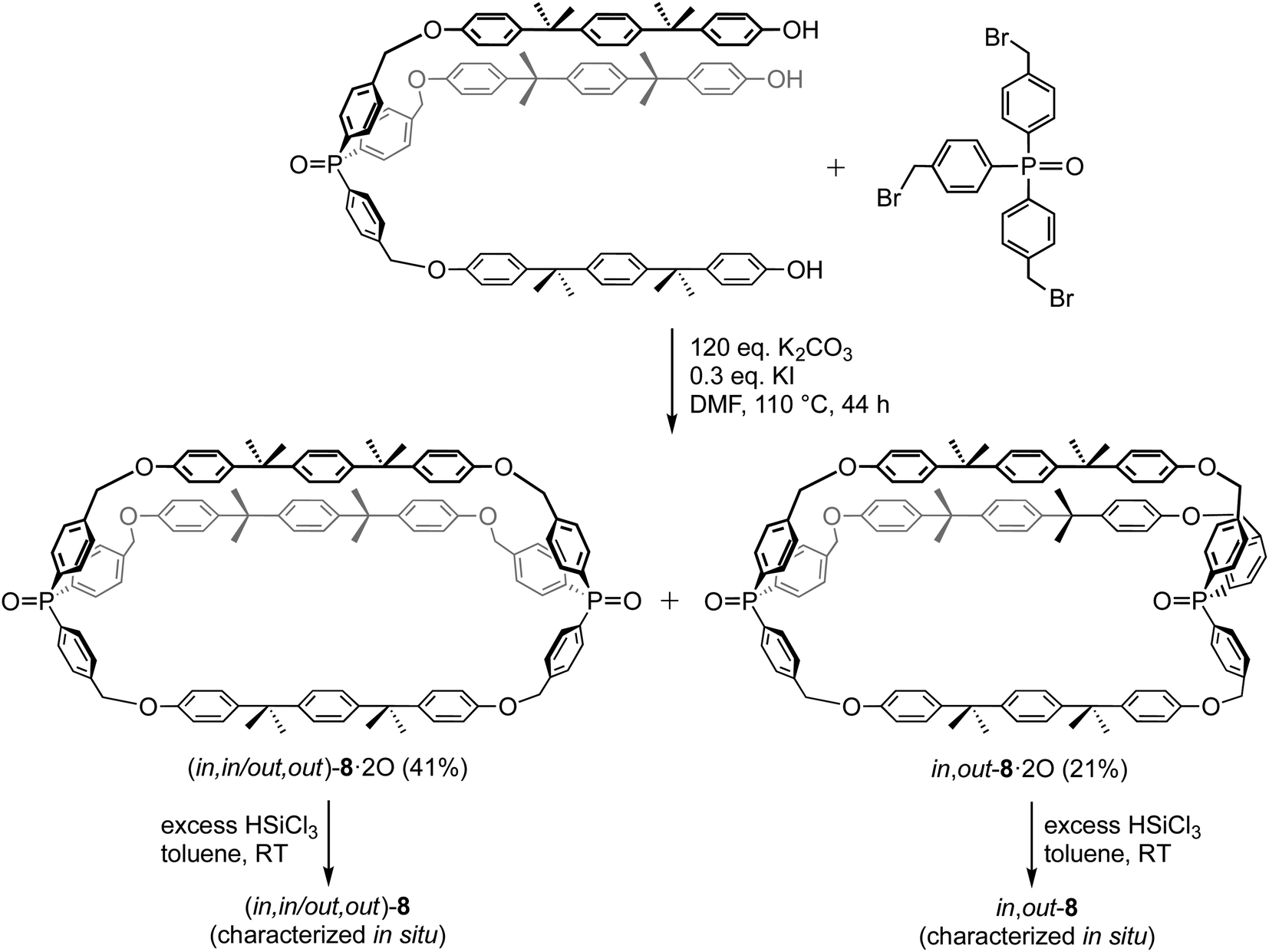 | ||
| Scheme 8 Macrocyclic dibridgehead diphosphorus compounds reported by Bauer.17b | ||
Isomer assignments
In certain cases, unequivocal isomer assignments are possible for the preceding compounds. For example, the thermal epimerisation of 3b, c, e in Scheme 4 give species that can exhibit two 31P{1H} NMR signals of equal areas at low temperatures. These can only be in,out isomers that undergo rapid homeomorphic isomerization. The other isomers must therefore represent the in,in/out,out manifold as mapped in Scheme 2. Similarly, there are two cases in which the in,in/out,out isomers exhibit two 31P{1H} NMR signals (unequal areas) at low temperature. One of these must represent an in,in isomer, and the other out,out. Our rationale for assigning the dominant isomer as in,in, which is perhaps counterintuitive, is as follows.First, the earliest computational probes of such equilibria, conducted with the hydrocarbons HC((CH2)n)3CH, pointed to an increasing and eventually dominant proportion of in,in isomers as n is increased.29 Second, DFT calculations with (P(CH2)n)3P (n = 8–20) have always given parallel results.10,21 As reported separately, DFT has also been used to compute the 31P NMR chemical shifts (n = 10–20),10b,20 and two relationships emerge: (1) the chemical shifts of the in,in isomers are downfield of the out,out isomers, and (2) the chemical shifts of the in bridgeheads of the in,out isomers are downfield of the out bridgeheads (Δppm similar to experiment in both cases).27 Inverting the assignments made above would contradict these findings.
Molecular dynamics simulations have also been carried out.10 These indicate an abundance of conformers that are relatively closely spaced in energy for all limiting isomers. Several factors point to increased attractive intramolecular dispersion (van der Waals) forces in the in,in as opposed to out,out isomers. For example, a reduced surface area should translate into a reduced void space in these cage-like structures. The latter should in turn increase the dispersive (attractive) van der Waals forces between methylene linkers. Accordingly, the Connolly contact surfaces38 generated using various probe radii (e.g., 4.0 Å, comparable to a small solvent molecule) are on the average lower for the ensemble of low-energy conformations for the in,in isomers. We note in passing that additional computational studies have addressed other aspects of in/out isomerism.12,15,37
Further analyses of equilibria and dynamic properties
The in,in and out,out equilibrium ratios for 3b, c, 86:14 and 97:3 (213 K and 193 K, toluene-d8), correspond to small ΔG values (0.77, 1.33 kcal mol−1) at the temperatures of measurement. Importantly, the proportion of out,out isomer increases in the smaller macrocycle,27 as would be expected as a limiting bicyclo[2.2.2]octane-based structure such as DABCO is approached. The equilibrium ratios of (in,in/out,out)-3b, c, eversus in,out-3b, c, e, as established at 150 °C (Scheme 6), are not very dependent upon the macrocycle size. The slightly greater bias of 3b towards the (in,in/out,out)-isomers (60:40 vs. 51:49) is consistent with a small decrease in the relative stabilities of in,out isomers with decreasing ring sizes.
The ΔG‡ values for the homeomorphic isomerization of out,out-3 to in,in-3 increase as the ring sizes decrease (10.4 kcal mol−1 for 3c, 193 K vs. 11.4 kcal mol−1 for 3b, 213 K). Importantly, the ΔS‡ values become more negative as the ring sizes decrease (−11.8 eu, 3c; −19.4 eu, 3b). This is consistent with a more pronounced loss of degrees of freedom in turning the smaller macrocycle inside out. However, the ΔH‡ values decrease (8.2 kcal mol−1, 3c; 7.3 kcal mol−1, 3b), hinting at a possible isokinetic relationship.39 Interestingly, the ΔG‡ value for the degenerate homeomorphic isomerization of in,out-3c (8.5 kcal mol−1, 200 K) is less than those for the non-degenerate isomerizations of in,in-3c and out,out-3c (10.4 or 11.5 kcal mol−1, 193 K, depending upon direction). The trend for in,out-3b compared to in,in- and out,out-3b is analogous but less pronounced (290 K: 12.1 vs. 13.1 or 13.8 kcal mol−1).
The epimerisations of (in,in/out,out)-3b, c, e (Scheme 6) require similar temperatures and time scales. As noted above, the ΔG‡ value for (in,in/out,out)-3c (34.4 kcal mol−1, 150 °C or 423 K) is typical for inversions of acyclic trialkylphosphines. This suggests a common bridgehead inversion mechanism, as opposed to unconventional pathways involving phosphorus–phosphorus interactions that could show a dependency upon macrocycle size. Reactions of dibridgehead diphosphines with P(CH2)nP linkages with n ≤ 4 can afford species with phosphorus–phosphorus bonds, for which inversions at phosphorus have been documented.40
Of course, all the same questions can be posed with regard to the diphosphine diboranes 3·2BH3. These are beyond the scope of this paper, but it is clear that equilibria involving in,in and out,out isomers, or degenerate in,out species, remain rapid on NMR time scales. Despite the greater steric demand of a bridgehead BH3 substituent as opposed to a lone pair, we believe that the diphosphines in this study provide ample clearance for in,in and in,out isomers. However, when the BH3 groups of in,out-3c·2BH3 are replaced by bulky gold(I) Lewis acids AuAr as in 9 (Scheme 9), steric interactions greatly increase. Accordingly, the structure “flips” into an out,out form with crossed chains (see VIII, Scheme 2), both in solution and the solid state.11
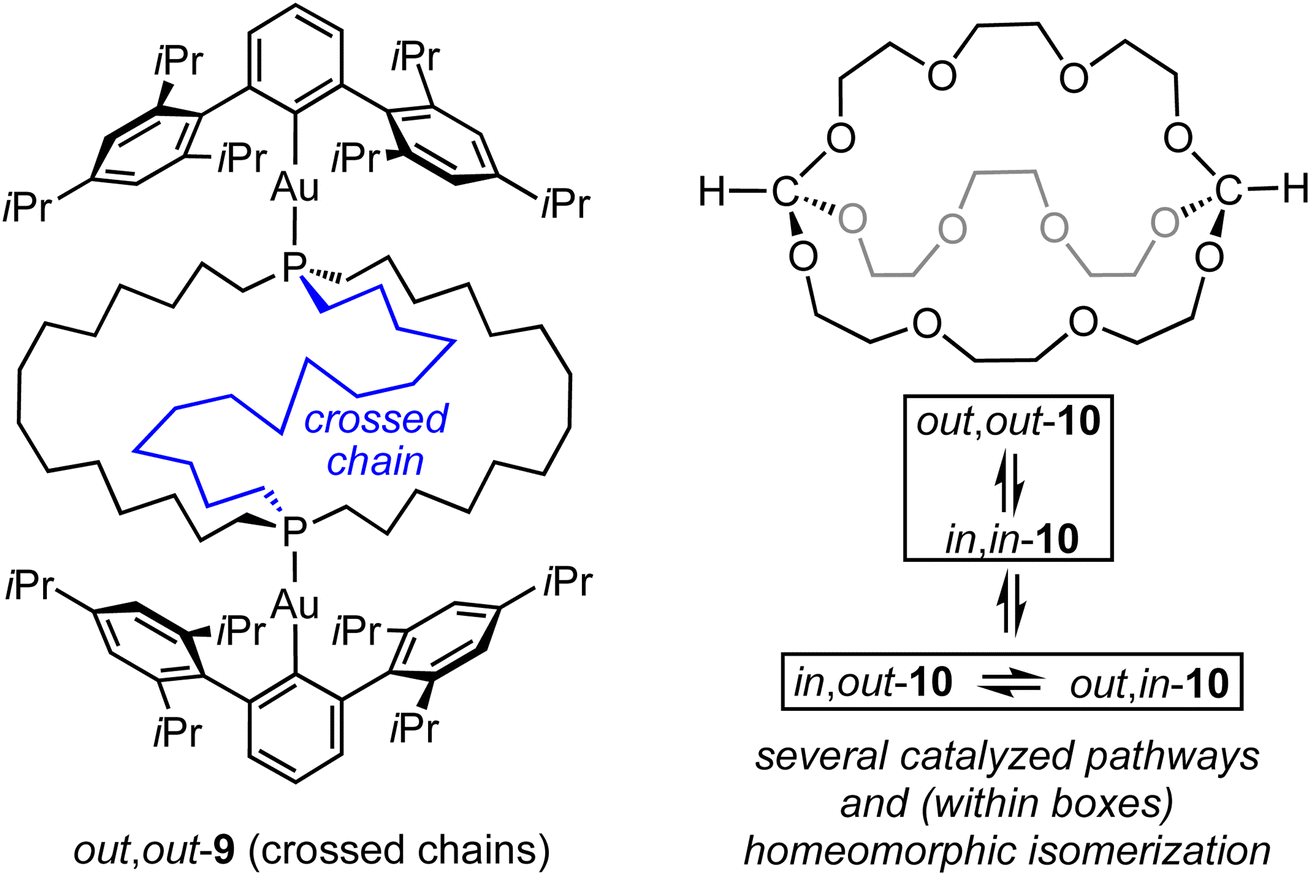 | ||
| Scheme 9 Additional types of compounds exhibiting in/out isomerism. | ||
Finally, it should be kept in mind that a wide variety of mechanisms may be operative in in/out isomerizations. For example, dibridgehead diorthoesters with (OCH2CH2)n bridges have recently been prepared, as exemplified by 10 in Scheme 9.15 All three in/out orientations of the HC(OR)3 units can be observed. These can equilibrate by both homeomorphic isomerizations and catalyzed pathways involving cleavage of the carbon–oxygen bonds.
Crystal structures
As shown in Fig. 3, the dibridgehead diphosphines 3b, c, e all crystallize as out,out isomers, despite the conclusions in the preceding sections that in,in isomers dominate in solution. There are many cases where a compound crystallizes as the less stable of two possible isomers, which is commonly attributed to packing forces. However, this is rarely observed for an entire series of compounds. In the case of out,out-3c, Fig. 6 convincingly documents a highly symmetric molecular structure (D3) that affords an exceptional lattice with optimal intermolecular contacts as analyzed above. The dibridgehead diarsine As((CH2)14)3As crystallizes analogously.41Crystalline out,out-3b as well as the arsenic homolog As((CH2)12)3As36 are just a few CH2–CH2 rotations removed from D3 symmetry, and share several packing features with out,out-3c (e.g., Fig. 7). As shown in Fig. 8, out,out-3e crystallizes in a much different motif, but still with a visually impressive degree of van der Waals contacts or near-contacts. In any event, these apparently trump the greater intramolecular dispersion forces posited for individual in,in isomers. Nonetheless, this dichotomy remains an obvious focus for further study and interpretation.
The bridgehead phosphorus atoms in out,out-3b, c, e are much more pyramidalized than in out,out-3c, e·2BH3, as evidenced by the sums of the three carbon–phosphorus–carbon bond angles (295.0–302.7° vs. 313.2–324.3°; Table 1). The solvent occupied cages in the two structures of out,out-3c·2BH3 raise the issue of whether any of the equilibria or other phenomena described above might be affected by encapsulated solvent. We view the solvate molecules as simple consequences of crystal growth, as our gyroscope-like complexes (e.g., trans-2g in Scheme 3)18b occasionally but by no means routinely crystallize with a solvent molecule within a macrocycle.34 Also, attempts to detect toluene-dn adducts of in,in-3c or out,out-3c at low temperatures in CD2Cl2 and other solvents have been unsuccessful.
Conclusion
This study has brought heretofore unavailable definition to equilibria and dynamic and configurational processes involving the most fundamental type of large-ring aliphatic bicyclic compounds with bridgehead heteroatoms, XE((CH2)n)3EX. The new dibridgehead diphosphines 3b–e are highly flexible with extensive manifolds of conformations and coordination modes. They represent very promising building blocks for both monometallic and polymetallic or polymeric systems – research directions that will be facilitated as improved syntheses are developed.28 Monometallic adducts of in,in isomers have been termed gyroscope-like and are attractive springboards for molecular gyroscopes.42 The dynamic properties of 3c, e have already been put to use in metal transport protocols,5 and all of the preceding directions of inquiry are actively being extended to diarsine36,41 and phosphine oxide analogs (EX = As, PO).13 These and related themes will be the subject of future reports from this laboratory.Experimental section
(in,in/out,out)-P((CH2)12)3P ((in,in/out,out)-3b)
A Schlenk flask was charged with cis-PtCl2(P((CH2)12)3P (cis-2b;19 0.1010 g, 0.1213 mmol), KCN (0.097 g, 1.489 mmol), THF (15 mL) and degassed water (0.5 mL). The mixture was stirred (24 h). The solvent was removed from the filtrate by oil pump vacuum. The residue was filtered through a pad of silica (1.5 × 1 cm). The filter cake was washed with CH2Cl2 (2 × 10 mL). The solvent was removed from the filtrate by oil pump vacuum to give (in,in/out,out)-3b (0.0488 g, 0.0861 mmol, 71%) as a white solid, mp 52–55 °C. Anal. calcd for C36H72P2 (566.90): C, 76.27; H, 12.80; found: C, 76.49; H, 12.82.
NMR (CDCl3, δ/ppm): 1H (500 MHz) 1.46–1.35, 1.35–1.25 (2 br m, 72H, C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2); 13C{1H} (126 MHz) 30.9 (d, JCP = 9.5 Hz, H2), 28.5 (s, H2), 28.4 (s, H2), 28.1 (s, H2), 26.3–25.9 (br s, H2), 25.0–24.7 (br s, H2); 31P{1H} (202 MHz) −29.7 to −32.1 (br s).
2); 13C{1H} (126 MHz) 30.9 (d, JCP = 9.5 Hz, H2), 28.5 (s, H2), 28.4 (s, H2), 28.1 (s, H2), 26.3–25.9 (br s, H2), 25.0–24.7 (br s, H2); 31P{1H} (202 MHz) −29.7 to −32.1 (br s).
NMR (toluene-d8, δ/ppm, 373 K): 1H (500 MHz) 1.45–1.35, 1.35–1.23 (2 br m, 72H, C2); 13C{1H} (126 MHz) 30.8 (d, JCP = 10.1 Hz, H2), 28.71 (s, H2), 28.70 (s, H2), 28.67 (s, H2), 28.3 (d, JCP = 14.6 Hz, H2), 26.0 (d, JCP = 13.4 Hz, H2); 31P{1H} (202 MHz) −31.9 (br s).
(in,in/out,out)-P((CH2)14)3P ((in,in/out,out)-3c)
A Schlenk flask was charged with trans-PtCl2(P((CH2)14)3P (trans-2c;18a 0.4356 g, 0.475 mmol), KCN (0.4674 g, 7.177 mmol), THF (20 mL) and degassed water (0.5 mL). The mixture was stirred (24 h) and filtered to remove a yellow precipitate. The solvent was removed from the filtrate by oil pump vacuum to give in,in/out,out-3c (0.2692 g, 0.411 mmol, 87%) as a white solid, mp 68 °C. Anal. calcd for C42H84P2 (651.06): C, 77.48; H, 13.00; found: C, 77.67; H, 13.08.
NMR (CDCl3, δ/ppm): 1H (500 MHz) 1.37–1.32, 1.31–1.23 (2 br m, 84H, C2); 13C{1H} (126 MHz) 31.2 (d, JCP = 10.3 Hz, H2), 29.2 (s, H2), 29.14 (s, H2), 29.08 (s, H2), 28.7 (s, H2), 26.3 (d, JCP = 12.2 Hz, H2), 25.2 (d, JCP = 11.0 Hz, H2); 31P{1H} (202 MHz) −30.1 (s).
A 13C{1H} NMR spectrum of the yellow precipitate showed signals (D2O, δ/ppm) for K2Pt(CN)4 (126.5; lit43 126.5) and KCN (166.8). The precipitate was dissolved in water (5 mL) and the solution allowed to slowly concentrate. After 7 d, thin colorless plates of K2Pt(CN)4 were obtained, as verified by X-ray crystallography.44
(in,in/out,out)-P((CH2)16)3P ((in,in/out,out)-3d)
A Schlenk flask was charged with cis-PtCl2(P((CH2)16)3P (cis-2d;19 0.2453 g, 0.245 mmol), KCN (0.2393 g, 3.675 mmol), THF (15 mL), and degassed water (0.5 mL). The mixture was stirred. After 24 h, the mixture was filtered. The filter cake was washed with THF (2 × 5 mL). The solvent was removed from the filtrate by oil pump vacuum, and CH2Cl2 (25 mL) added to the solid residue. The sample was filtered through a pad of silica (1.5 × 1 cm). The filter cake was washed with CH2Cl2 (2 × 10 mL). The solvent was removed from the filtrate by oil pump vacuum to give (in,in/out,out)-3d (0.1476 g, 0.201 mmol, 82%) as a white solid, mp 56–59 °C.
NMR (CDCl3, δ/ppm) 1H (500 MHz) 1.46–1.33, 1.33–1.23 (2 br m, 96H, C2); 13C{1H} (126 MHz) 31.2 (d, JCP = 10.2 Hz, H2), 29.2 (s, 2 × H2), 29.1 (s, H2), 29.0 (s, H2), 28.8 (s, H2), 26.6 (d, JCP = 12.0 Hz, H2), 25.4 (d, JCP = 11.6 Hz, H2); 31P{1H} (202 MHz) −30.7 (s).
(in,in/out,out)-P((CH2)18)3P ((in,in/out,out)-3e)
A Schlenk flask was charged with trans-PtCl2(P((CH2)18)3P (trans-2e;18 0.2529 g, 0.233 mmol), KCN (0.2319 g, 3.561 mmol), THF (15 mL), and degassed water (0.5 mL) with stirring. After 24 h, the mixture was filtered. The filter cake was washed with THF (2 × 5 mL). The solvent was removed from the filtrate by oil pump vacuum, and CH2Cl2 (25 mL) added to the solid residue. The sample was filtered through a pad of silica (1.5 × 1 cm). The filter cake was washed with CH2Cl2 (2 × 10 mL). The solvent was removed from the filtrate by oil pump vacuum to give (in,in/out,out)-3e (0.1622 g, 0.198 mmol, 85%) as a white solid, mp 54–57 °C. Anal. calcd for C54H108P2 (819.38): C, 79.15; H, 13.29; found: C, 79.16; H, 13.48.
NMR (C6D6, δ/ppm): 1H (500 MHz) 1.60–1.52 (br m, 12H, C2), 1.49–1.40 (br m, 24H, C2), 1.40–1.29 (br m, 72H, C2); 13C{1H} (126 MHz) 32.1 (d, JCP = 10.5 Hz, H2), 30.34 (s, H2), 30.32 (s, H2), 30.3 (s, H2), 30.2 (s, H2), 30.1 (s, H2), 30.0 (s, H2), 28.4 (d, JCP = 13.6 Hz, H2), 26.8 (d, JCP = 13.0 Hz, H2); 31P{1H} (202 MHz) −32.8 (s). IR (cm−1, powder film): 2916 (s), 2847 (s), 1466 (s), 1442 (s), 725 (s).
(in,in/out,out)-3b·2BH3
A Schlenk flask was charged with (in,in/out,out)-3b (0.0705 g, 0.124 mmol) and THF (10 mL). Then Me2S·BH3 (2.0 M in THF; 0.13 mL, 0.26 mmol) was added with stirring. After 1 d, the solvent was removed by oil pump vacuum. The residue was chromatographed on a silica column (4 × 30 cm) using hexanes/CH2Cl2 (1:2 v/v). The solvents were removed from the product fractions by oil pump vacuum to give (in,in/out,out)-3b·2BH3 (0.0521 g, 0.0876 mmol, 70%) as a light yellow oil. Anal. calcd for C36H78B2P2 (594.57): C, 72.72; H, 13.22; found: C, 72.72; H, 13.01.
NMR (CDCl3, δ/ppm): 1H (500 MHz) 1.63–1.53 (m, 12H, C2), 1.53–1.44 (m, 12H, C2), 1.41–1.37 (m, 12H, C2), 1.37–1.23 (m, 36H, C2), 0.48 and 0.27 (br apparent d, 6H, B3); 13C {1H} (126 MHz) 30.3 (d, JCP = 11.9 Hz, H2), 28.9 (s, H2), 28.5 (s, H2), 28.1 (s, H2), 22.4 (d, JCP = 34.0 Hz, H2), 21.9 (d, JCP = 2.9 Hz, H2); 31P{1H} (202 MHz): 15.0–13.7 (br s).31b
(in,in/out,out)-3c·2BH3
A J. Young NMR tube was charged with in,in/out,out-3c (0.0271 g, 0.0416 mmol), CDCl3 (0.6 mL), and Me2S·BH3 (0.20 mL, 2.0 M in THF, 0.40 mmol). After 1 d, the solvent was removed in vacuo. The residue was passed through silica gel (1 × 15 cm) using hexanes/CH2Cl2 (2:1 v/v). The solvent was removed in vacuo to give in,in/out,out-3c·2BH3 (0.0211 g, 0.0311 mmol, 75%) as a white solid, mp 112 °C. DSC (Ti/Te/Tp/Tc/Tf): 95.8/110.6/112.7/114.2/117.5 °C (endotherm). TGA: onset of mass loss, 282 °C. Anal. calcd for C42H90B2P2 (678.73): C, 74.32; H, 13.37; found C, 74.01; H, 13.08.
NMR (CDCl3, δ/ppm): 1H (500 MHz) 1.60–1.49 (m, 12H, C2), 1.50–1.41 (m, 12H, C2), 1.41–1.32 (m, 12H, C2), 1.32–1.20 (m, 48H, C2), 0.38 and 0.26 (br apparent d, 6H, B3); 13C{1H} (126 MHz) 30.7 (d, JCP = 12.1 Hz, H2), 29.31 (s, H2), 29.25 (s, H2), 29.0 (s, H2), 28.5 (s, H2), 22.5 (d, JCP = 34.1 Hz, H2), 22.2 (d, JCP = 2.6 Hz, H2); 31P{1H} (202 MHz) 16.2–14.4 (br s).31b IR (cm−1, powder film): 2922 (s), 2853 (s), 2366 (m), 1467 (w), 1061 (m), 718 (m). MS (EI): 678 (M+, 1%), 665 ([M − BH3]+, 38%), 651 ([M − 2BH3]+, 100%).
(in,in/out,out)-3e·2BH3
A Schlenk flask was charged with (in,in/out,out)-3e (0.2147 g, 0.262 mmol) and THF (15 mL). Then Me2S·BH3 (2.0 M in THF; 0.65 mL, 1.3 mmol) was added with stirring. After 1 d, the solvent was removed by oil pump vacuum. The residue was chromatographed on a silica column (4 × 46 cm) using hexanes/CH2Cl2 (1:0 to 0:1 v/v). The solvents were removed from the product fractions by oil pump vacuum to give (in,in/out,out)-3e·2BH3 (0.2027 g, 0.239 mmol, 91%) as a white powder, mp 67–69 °C. Anal. calcd for C54H114P2B2 (847.07): C, 76.57; H, 13.57; found: C, 76.28; H, 13.62.
NMR (CDCl3, δ/ppm): 1H (500 MHz): 1.60–1.52 (m, 12H, C2), 1.52–1.42 (m, 12H, C2), 1.42–1.34 (m, 12H, C2), 1.34–1.21 (m, 72H, C2), 0.45 and 0.28 (br apparent d, 6H, B3); 13C{1H} (126 MHz): 31.0 (d, JCP = 12.0 Hz, H2), 29.60 (s, H2), 29.58 (s, H2), 29.50 (s, H2), 29.45 (s, H2), 29.2 (s, H2), 28.8 (s, H2), 22.8 (d, JCP = 34.2 Hz, H2), 22.4 (d, JCP = 2.7 Hz, H2); 31P{1H} 14.9–13.6 (br s).31b IR (cm−1, powder film): 2915 (s), 2846 (m), 2361 (m), 1468 (m), 1063 (m), 716 (m).
Epimerization of 3b; in,out-3b·2BH3 and (in,in/out,out)-3b·2BH3
A Schlenk flask was charged with (in,in/out,out)-3b (0.0488 g, 0.0861 mmol) and mesitylene (10 mL). The solution was stirred at 150 °C. After 60 h, a 31P{1H} NMR spectrum showed a 60:40 (in,in/out,out)-3b/in,out-3b mixture. The solvent was removed by oil pump vacuum, the residue dissolved in THF (15 mL), and Me2S·BH3 (2.0 M in THF; 0.30 mL, 0.60 mmol) added with stirring. After 24 h, the solvent was removed by oil pump vacuum. The residue was chromatographed on a silica column (1 × 26 cm) using hexanes/CH2Cl2 (1:1 v/v). The solvents were removed from the product fractions by oil pump vacuum. First in,out-3b·2BH3 eluted (colorless oil, 0.0131 g, 0.0220 mmol, 26%). Anal. calcd for C36H72P2 (594.57): C, 72.72; H, 13.22; found: C, 73.02; H, 13.13.
NMR (CDCl3, δ/ppm): 1H (500 MHz) 1.64–1.54 (m, 12H, C2), 1.54–1.45 (m, 12H, C2), 1.45–1.37 (m, 12H, C2), 1.37–1.24 (m, 36H, C2), 0.51 and 0.29 (br apparent d, 6H, B3); 13C{1H} (126 MHz) 30.4 (d, JCP = 11.2 Hz, H2), 27.7 (s, H2), 27.6 (s, H2), 27.5 (s, H2), 23.4 (d, JCP = 34.3 Hz, H2), 22.2 (s, H2); 31P{1H} (202 MHz): 15.6–14.1 (br s).31b
Next (in,in/out,out)-3b·2BH3 eluted (colorless oil, 0.0194 g, 0.326 mmol, 38%). The NMR data agreed with that from the independent synthesis above.
in,out-P((CH2)12)3P (in,out-3b)
A Schlenk flask was charged with in,out-3b·2BH3 (0.0070 g, 0.0117 mmol) and pyrrolidine (2 mL). The mixture was refluxed (60 °C) for 3 d. The solvent was removed by oil pump vacuum. Toluene (5 mL) was added, and the suspension passed through a pad of silica gel using a pipette (0.7 × 3 cm). The filter cake was washed with toluene (10 mL). The solvent was removed from the filtrate by oil pump vacuum to give in,out-3b (0.0052 g, 0.009 mmol, 77%) as a colorless oil.NMR (CDCl3, δ/ppm): 1H (500 MHz) 1.49–1.36, 1.36–1.23 (2 br m, 72H, C2); 13C{1H} (126 MHz) 30.9 (d, JCP = 9.7 Hz, H2), 28.7 (s, 2 × H2), 28.6 (s, H2), 27.0–26.5 (br s, H2), 25.5 (d, JCP = 10.6 Hz, H2); 31P{1H} (202 MHz) −29.8 to −36.9 (br s) and (from Fig. 2b) −30.1/−40.4 (s/s, 213 K), −29.4/−38.0 (br s/br s, 253 K), −29.2/−36.5 (br s/br s, 273 K), −32.2 (br s, 298 K), −32.3 (br s, 323 K).
Epimerization of 3c; in,out-3c·2BH3 and (in,in/out,out)-3c·2BH3
A Schlenk flask was charged with (in,in/out,out)-3c (0.252 g, 0.387 mmol) and mesitylene (10 mL). The solution was stirred at 150 °C. After 40 h, a 31P{1H} NMR spectrum showed a 51:49 (in,in/out,out)-3c/in,out-3c mixture. The solvent was removed by oil pump vacuum, the residue dissolved in THF (15 mL), and Me2S·BH3 (2.0 M in THF; 1.3 mL, 2.6 mmol) added with stirring. After 2 d, the solvent was removed by oil pump vacuum. The residue was chromatographed on a silica column (4 × 46 cm) using hexanes/CH2Cl2 (1:0 to 0:1 v/v). The solvents were removed from the product fractions by oil pump vacuum. First eluted in,out-3c·2BH3 (colorless oil, 0.110 g, 0.162 mmol, 42%). Anal. calcd for C42H90B2P2 (678.73): C, 74.32; H, 13.37; found: C, 73.86; H, 13.49.
NMR (CDCl3, δ/ppm): 1H (500 MHz) 1.56–1.51 (m, 12H, C2), 1.49–1.42 (m, 12H, C2), 1.39–1.33 (m, 12H, C2), 1.31–1.21 (m, 48H, C2), 0.45 and 0.27 (br apparent d, 6H, B3); 13C {1H} (126 MHz) 30.5 (d, JCP = 11.3 Hz, H2), 28.35 (s, H2), 28.28 (s, H2), 28.2 (s, H2), 28.1 (s, H2), 23.0 (d, JCP = 34.3 Hz, H2), 22.2 (d, JCP = 1.9 Hz, H2); 31P{1H} (202 MHz): 15.8–15.4 (br m).31b
Next eluted (in,in/out,out)-3c·2BH3 (colorless oil, 0.114 g, 0.168 mmol, 43%), which solidified to a white powder. The NMR data agreed with that from the independent synthesis above.
in,out-P((CH2)14)3P (in,out-3c)
A Schlenk flask was charged with in,out-3c·2BH3 (0.048 g, 0.071 mmol) and pyrrolidine (3 mL). The mixture was refluxed (60 °C) for 11 d. The solvent was removed by oil pump vacuum. Toluene (5 mL) was added, and the suspension passed through a pad of silica gel on a Schlenk frit (1.5 × 1 cm). The filter cake was washed with toluene (3 × 4 mL). The solvent was removed from the filtrate by oil pump vacuum to give in,out-3c (0.026 g, 0.040 mmol, 56%) as a colorless oil. Anal. calcd for C42H84P2 (651.06): C, 77.48; H, 13.00; found: C, 77.66; H, 13.09.NMR (CDCl3, δ/ppm): 1H (500 MHz) 1.45–1.35, 1.32–1.23 (2 br m, 84H, C2); 13C{1H} (126 MHz) 30.9 (d, JCP = 10.3 Hz, H2), 29.0 (s, 2 × H2), 28.94 (s, H2), 28.86 (s, H2), 27.0 (d, JCP = 11.5 Hz, H2), 25.6 (d, JCP = 11.8 Hz, H2); 31P{1H} (202 MHz) −31.3 (s).
NMR (CDCl2F,45δ/ppm, 263 K): 1H (500 MHz) 1.46–1.25 (br m, 84H, C2); 13C{1H} (126 MHz) 31.0 (d, JCP = 10.3 Hz, H2), 29.0 (s, 2 × H2), 28.98 (s, H2), 28.90 (s, H2), 26.8 (d, JCP = 10.9 Hz, H2), 25.5 (d, JCP = 11.5 Hz, H2); 31P{1H} (202 MHz) −32.5 (s).
31P{1H} NMR (CH2Cl2, δ/ppm, 202 MHz selected data from Fig. 2a) −32.3/−37.7 (br s/br s, 183 K), −32.4/−37.4 (br s/br s, 193 K), −34.5 (br s, 208 K), −34.1 (br s, 223 K).
Epimerization of 3e; in,out-3e·2BH3 and (in,in/out,out)-3e·2BH3
A Schlenk flask was charged with (in,in/out,out)-3e (0.1934 g, 0.2360 mmol) and mesitylene (10 mL). The solution was stirred at 150 °C. After 30 h, a 31P{1H} NMR spectrum showed a 51:49 (in,in/out,out)-3e/in,out-3e mixture. The solvent was removed by oil pump vacuum, the residue dissolved in THF (15 mL), and Me2S·BH3 (2.0 M in THF; 0.24 mL, 0.48 mmol) added with stirring. After 24 h, the solvent was removed by oil pump vacuum. The residue was chromatographed on a silica column (4 × 46 cm) using hexanes/CH2Cl2 (1:3 v/v). The solvents were removed from the product fractions by oil pump vacuum. First eluted in,out-3e·2BH3 (colorless oil, 0.0310 g, 0.0366 mmol, 16%). Anal. calcd for C54H114B2P2 (847.07): C, 76.57; H, 13.57; found: C, 76.53; H, 13.45.
NMR (CDCl3, δ/ppm): 1H (500 MHz) 1.59–1.52 (m, 12H, C2), 1.52–1.44 (m, 12H, C2), 1.41–1.34 (m, 12H, C2), 1.34–1.22 (m, 72H, C2), 0.49 and 0.29 (br apparent d, 6H, B3); 13C{1H} (126 MHz) 31.0 (d, JCP = 11.9 Hz, H2), 29.4 (s, H2), 29.31 (s, H2), 29.28 (s, H2), 29.27 (s, H2), 29.0 (s, H2), 28.8 (s, H2), 23.1 (d, JCP = 34.5 Hz, H2), 22.5 (d, JCP = 2.4 Hz, H2); 31P{1H} (202 MHz): 15.1–13.9 (br s).31b
Next eluted (in,in/out,out)-3e·2BH3 (colorless oil, 0.0464 g, 0.0548 mmol, 23%), which solidified (6–8 h) to a white powder. The NMR data agreed with that from the independent synthesis above.
in,out-P((CH2)18)3P (in,out-3e)
A Schlenk flask was charged with in,out-3e·2BH3 (0.031 g, 0.0366 mmol) and pyrrolidine (2 mL). The mixture was refluxed (60 °C) for 3 d. The solvent was removed by oil pump vacuum. Toluene (5 mL) was added, and the suspension passed through a pad of silica gel on a Schlenk frit (1.5 × 2 cm). The filter cake was washed with toluene (20 mL). The solvent was removed from the filtrate by oil pump vacuum to give in,out-3e (0.0197 g, 0.024 mmol, 66%) as a colorless oil. Anal. calcd for C54H108P2 (819.38): C, 79.15; H, 13.28; found: C, 79.02; H, 13.29.NMR (CDCl3, δ/ppm): 1H (500 MHz) 1.46–1.33, 1.32–1.22 (2 br m, 108H, C2); 13C{1H} (126 MHz) 31.2 (d, JCP = 10.5 Hz, H2), 29.43 (s, H2), 29,42 (s, H2), 29.39 (s, H2), 29.38 (s, H2), 29.3 (s, H2), 29.1 (s, H2), 27.2 (d, JCP = 11.9 Hz, H2), 25.8 (d, JCP = 12.0 Hz, H2); 31P{1H} (202 MHz) −31.5 (s).
Conversion of (in,in/out,out)-3b to trans-2b
A J. Young NMR tube was charged with (in,in/out,out)-3b (0.0110 g, 0.0194 mmol), PtCl2 (0.0054 g, 0.020 mmol), and C6D6 (0.6 mL) in a glove box. The mixture was kept at 55 °C for 24 h and chromatographed (SiO2 column, 0.7 × 3 cm, 5:1 v/v hexanes/CH2Cl2). The solvent was removed from the product fractions by rotary evaporation to give trans-2b (0.0084 g, 0.0101 mmol, 52%) as a yellow powder, mp 160–161 °C. Anal. calcd for C36H72P2Cl2Pt (832.89): C, 51.91; H, 8.71; found: C, 53.25; H, 8.98.46
NMR (C6D6, δ/ppm): 1H (500 MHz) 1.92–1.82, (br m, 12H, PCH2C2), 1.72–1.62 (br m, 12H, PC2), 1.55–1.41 (br m, 48H, remaining C2); 13C{1H} (126 MHz) 30.2 (virtual t, JCP = 6.9 Hz, PCH2CH2H2), 27.9 (s, H2), 27.8 (s, H2), 27.0 (s, H2), 25.4 (virtual t, JCP = 16.3 Hz, PH2), 24.3 (s, P CH2H2); 31P{1H} (202 MHz) 9.3 (s, JPPt (satellite) = 2442 Hz).
Conversion of (in,in/out,out)-3c to trans-2c
(A) A round bottom flask was charged with (in,in/out,out)-3c (0.0635 g, 0.097 mmol), PtCl2 (0.0306 g, 0.115 mmol), and CH2Cl2 (7 mL) in a glove box. The mixture was stirred for 24 h and chromatographed (SiO2 column, 1 × 5 cm, 4:1 v/v hexanes/CH2Cl2). The solvent was removed from the product fractions by rotary evaporation to give trans-2c (0.0825 g, 0.090 mmol, 93%) as a yellow powder.47 (B) A round bottom flask was charged with in,in/out,out-3c (0.0629 g, 0.096 mmol), PtCl2(NCCH3)2 (0.0400 g, 0.115 mmol), and THF (7 mL) in a glove box. The mixture was stirred for 6 h and worked up as in A to give trans-2c (0.0835 g, 0.091 mmol, 95%) as a yellow powder.47
Conversion of (in,in/out,out)-3e to trans-2e
A round bottom flask was charged with (in,in/out,out)-3e (0.0788 g, 0.096 mmol), PtCl2 (0.0260 g, 0.098 mmol), and CH2Cl2 (10 mL) in a glove box. The mixture was stirred for 24 h and chromatographed (SiO2 column, 1 × 10 cm, 5:1 v/v hexanes/CH2Cl2). The solvent was removed from the product fractions by rotary evaporation to give trans-2e (0.0719 g, 0.0662 mmol, 69%) as a yellow powder.47
Conversion of (in,in/out,out)-3c to trans-![[upper bond 1 start]](https://www.rsc.org/images/entities/h3_char_e010.gif) PtCl2(P((CH2)14)3P
PtCl2(P((CH2)14)3P![[upper bond 1 end]](https://www.rsc.org/images/entities/h3_char_e011.gif) (trans-4c)
(trans-4c)
(A) A round bottom flask was charged with in,in/out,out-3c (0.0668 g, 0.102 mmol), PdCl2 (0.0216 g, 0.122 mmol), and CH2Cl2 (7 mL) in a glove box. The mixture was stirred for 24 h and chromatographed (SiO2 column, 1 × 5 cm, 4:1 v/v hexanes/CH2Cl2). The solvent was removed from the product fractions by rotary evaporation to give trans-4c (0.0795 g, 0.096 mmol, 94%) as a yellow powder.45 (B) A round bottom flask was charged with in,in/out,out-3c (0.0740 g, 0.113 mmol), PdCl2(NCCH3)2 (0.0311 g, 0.120 mmol), and THF (7 mL) in a glove box. The mixture was stirred for 6 h and worked up as in A to give trans-4c (0.0886 g, 0.107 mmol, 95%) as a yellow powder.47
Data availability
Additional experimental data are deposited in the ESI† associated with this publication and any further data are available upon request.Author contributions
All authors except J.A.G. contributed to the experimental work. All authors participated in data analysis. Overall supervision and funding acquisition was carried out by J.A.G. The manuscript was written by J.A.G., Y.Z., and M.S. with contributions by all authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the US National Science Foundation (CHE-1153085, CHE-1566601, CHE-1900549) for support, Dr Agnieszka J. Nawara-Hultzsch for preliminary observations, Dr Michał Barbasiewicz for assistance with the characterization of (in,in/out,out)-3c·2BH3, and Dr Andreas Ehnbom for helpful discussions and drafting key graphics. This paper is dedicated to Prof. Dr Wolf-Dieter Habicher (Technische Universität Dresden)16 on the occasion of his 83rd birthday (31 December 2022).Notes and references
- L. Fabbizzi, Cryptands and Cryptates, World Scientific, Hackensack, New Jersey, 2018. ISBN 9781786343697 Search PubMed.
- C. H. Park and H. E. Simmons, Macrobicyclic Amines. II. out-out ⇌ in-in Prototropy in l,(k + 2)-Diazabicyclo[k.l.m] alkaneammonium Ions, J. Am. Chem. Soc., 1968, 90, 2429–2431 CrossRef CAS . See also the immediately preceding and following communications in this issue..
- R. W. Alder, C. P. Butts, A. G. Orpen, D. Read and J. M. Oliva, Superbasic bridgehead diphosphines: the effects of strain and intrabridgehead P⋯P bonding on phosphine basicity, J. Chem. Soc., Perkin Trans. 2, 2001, 282–287 RSC , and references therein..
- R. W. Alder and S. P. East, In/Out Isomerism, Chem. Rev., 1996, 96, 2097–2011 CrossRef CAS PubMed.
- S. Kharel, H. Joshi, S. Bierschenk, M. Stollenz, D. Taher, N. Bhuvanesh and J. A. Gladysz, Homeomorphic Isomerization as a Design Element in Container Molecules; Binding, Displacement, and Selective Transport of MCl2 Species (M = Pt, Pd, Ni), J. Am. Chem. Soc., 2017, 139, 2172–2175 CrossRef CAS PubMed.
- Merriam-Webster definition of homeomorphism: a function that is a one-to-one mapping between sets such that both the function and its inverse are continuous and that in topology exists for geometric figures which can be transformed one into the other by an elastic deformation..
- However, when one assigns an arbitrary 1/2/3 Cahn-Ingold-Prelog priority to each bridge prior to a homeomorphic isomerization, an R configured bridgehead remains R and an S remans S, as easily verified with molecular models. Hence, a chiral substrate would not racemize, apropos to a reaction coordinate with no bond breaking or pyramidal inversion..
- R. D. Baechler and K. Mislow, The Effect of Structure on the Rate of Pyramidal Inversion of Acyclic Phosphines, J. Am. Chem. Soc., 1970, 92, 3090–3093 CrossRef CAS.
- (a) R. D. Baechler, J. D. Andose, J. Stackhouse and K. Mislow, Linear Free Energy Correlations of Barriers to Pyramidal Inversion, J. Am. Chem. Soc., 1972, 94, 8060–8065 CrossRef CAS; (b) J. D. Andose, A. Rauk and K. Mislow, Semiempirical Calculation of Barriers to Pyramidal Inversion. Extension to the Third Row of the Periodic Table, J. Am. Chem. Soc., 1974, 96, 6904–6907 CrossRef CAS.
- (a) A. Ehnbom, J. E. Kuszynski, M. B. Hall and J. A. Gladysz, Manuscript in preparation; (b) A. Ehnbom, Doctoral dissertation, Texas A&M University, 2021, Chapter 5.
- M. Stollenz, D. Taher, N. Bhuvanesh, J. H. Reibenspies, Z. Baranová and J. A. Gladysz, Steric control of the in/out sense of bridgehead substituents in macrobicyclic compounds: isolation of new “crossed chain” variants of in/out isomers, Chem. Commun., 2015, 51, 16053–16056 RSC.
- Y. Ikeda, Y. Inagaki and W. Setaka, Simultaneous synthesis and characterization of in/out-isomers of disilabicyclo[14.14.14]alkanes, Chem. Commun., 2021, 57, 7838–7841 RSC.
- See also S. Kharel, T. Jia, N. Bhuvanesh, J. H. Reibenspies, J. Blümel and J. A. Gladysz, A Nontemplated Route to Macrocyclic Dibridgehead Diphosphorus Compounds: Crystallographic Characterization of a “Crossed Chain” Variant of in/out Stereoisomers, Chem. – Asian J., 2018, 13, 2632–2640 CrossRef CAS PubMed.
- (a) A. H. Haines and P. Karntiang, Synthesis of Some out,in- and out,out-Macrobicyclic Polyethers derived from Glycerol. Out,in-in,out Isomerism, J. Chem. Soc., Perkin Trans. 1, 1979, 2577–2587 RSC; (b) R. S. Wareham, J. D. Kilburn, D. L. Turner, N. H. Rees and D. S. Holmes, Homeomorphic Isomerism in a Peptidic Macrobicycle, Angew. Chem., Int. Ed. Engl., 1995, 34, 2660–2662 CrossRef CAS , Übergangsmetallkomplexe als Wirt und als Gast: Koordination eines Platin-Ammin-Komplexes in zweiter Sphäre an einen Platin-haltigen Azakronenether, Angew. Chem., 1995, 107, 2902–2904; (c) M. Saunders and N. Krause, The Use of Stochastic Search in Looking for Homeomorphic Isomerism: Synthesis and Properties of Bicyclo[6.5.1]tetradecane, J. Am. Chem. Soc., 1990, 112, 1791–1795 CrossRef CAS; (d) R. W. Alder, E. Heilbronner, E. Honegger, A. B. McEwan, R. E. Moss, E. Olefirowicz, P. A. Petillo, R. B. Sessions, G. R. Weisman, J. M. White and Z.-Z. Yang, The out,out to out,in Transition for 1,(n+2)-Diazabicyclo[n.3.1]alkanes, J. Am. Chem. Soc., 1993, 115, 6580–6591 CrossRef CAS.
- H. Löw, E. Mena-Osteritz, K. M. Mullen, C. M. Jäger and M. von Delius, Self-Assembly, Adaptive Response, and in,out-Stereoisomerism of Large Orthoformate Cryptands, ChemPlusChem, 2020, 85, 1008–1012 CrossRef PubMed.
- I. Bauer and W. D. Habicher, out-Isomerism of Phosphorus Bridgehead Cage Compounds. A Review, Collect. Czech. Chem. Commun., 2004, 69, 1195–1230 CrossRef CAS.
- (a) F. Däbritz, A. Jäger and I. Bauer, Synthesis, Derivatisation and Structural Characterisation of a New Macrobicyclic Phosphane Oxide Cryptand, Eur. J. Org. Chem., 2008, 5571–5576 CrossRef; (b) F. Däbritz, G. Theumer, M. Gruner and I. Bauer, New conformational flexible phosphane and phosphane oxide macrobicycles, Tetrahedron, 2009, 65, 2995–3002 CrossRef.
- (a) A. J. Nawara-Hultzsch, M. Stollenz, M. Barbasiewicz, S. Szafert, T. Lis, F. Hampel, N. Bhuvanesh and J. A. Gladysz, Gyroscope-Like Molecules Consisting of PdX2/PtX2 Rotators within Three-Spoke Dibridgehead Diphosphine Stators: Syntheses, Substitution Reactions, Structures, and Dynamic Properties, Chem.– Eur. J., 2014, 20, 4617–4637 CrossRef CAS PubMed; (b) H. Joshi, S. Kharel, N. Bhuvanesh and J. A. Gladysz, Syntheses, Structures, and Thermal Properties of Gyroscope Like Complexes Consisting of PtCl2 Rotators Encased in Macrocyclic Dibridgehead Diphosphines P((CH2)n)3P with Extended Methylene Chains (n = 20/22/30), and Isomers Thereof, Organometallics, 2018, 37, 2991–3000 CrossRef.
- (a) H. Joshi, S. Kharel, A. Ehnbom, K. Skopek, G. D. Hess, T. Fiedler, F. Hampel, N. Bhuvanesh and J. A. Gladysz, Three Fold Intramolecular Ring Closing Alkene Metatheses of Square Planar Complexes with cis Phosphorus Donor Ligands P(X(CH2)mCH=CH2)3 (X/m = –/5-10, O/3-5); Syntheses, Structures, and Thermal Properties of Macrocyclic Dibridgehead Diphosphorus Complexes, J. Am. Chem. Soc., 2018, 140, 8463–8478 CrossRef CAS PubMed; (b) A workup improvement has raised the yield of cis-2b in Scheme 3 from 6% to 20-30%, and a revised procedure is supplied in the ESI.†.
- A. Ehnbom, M. B. Hall and J. A. Gladysz, manuscript in preparation.
- M. Stollenz, M. Barbasiewicz, A. J. Nawara-Hultzsch, T. Fiedler, R. M. Laddusaw, N. Bhuvanesh and J. A. Gladysz, Dibridgehead Diphosphines that Turn Themselves Inside Out, Angew. Chem., Int. Ed., 2011, 50, 6647–6651 CrossRef CAS PubMed; M. Stollenz, M. Barbasiewicz, A. J. Nawara-Hultzsch, T. Fiedler, R. M. Laddusaw, N. Bhuvanesh and J. A. Gladysz, Dreifach-verbrückte Diphosphine mit von innen nach außen invertierender Konfiguration, Angew. Chem. 2011, 123, 6777–6781 Search PubMed.
- As will be detailed in future papers, solutions stirred under air slowly give diphosphine monoxides and the diphosphine dioxides..
- G. Petöcz, L. Jánosi, W. Weissensteiner, Z. Csók, Z. Berente and L. Kollár, Synthesis and NMR investigation of Pt(CN)2(diphosphine) and [Pt(CN)(triphosphine)]Cl complexes, Inorg. Chim. Acta, 2000, 303, 300–305 CrossRef.
- Y. Zhu, A. Ehnbom, T. Fiedler, Y. Shu, N. Bhuvanesh, M. B. Hall and J. A. Gladysz, Platinum(II) Alkyl Complexes of Chelating Dibridgehead Diphosphines P((CH2)n)3P (n = 14, 18, 22); Facile cis/trans Isomerizations Interconverting Gyroscope and Parachute like Adducts, Dalton Trans., 2021, 50, 12457–12477 RSC.
- Line widths were carefully examined in many series of variable temperature NMR experiments in order to ensure that coalescence phenomena were not being overlooked (e.g., (in,in/out,out)-3c in CH2Cl2 and CDCl2F; (in,in/out,out)-3e·in toluene, CH2Cl2, and CDCl3). Representative data are provided in Fig. S5 and S6.†.
- The error limits on the rate constants so derived are commonly viewed as ca. 5%, leading to a corresponding uncertainty in the ln(k/T) vs. 1/T plots in Fig. S7:† P. M. Morse, M. D. Spencer, S. R. Wilson and G. S. Girolami, A Static Agostic α-CH⋯M Interaction Observable by NMR Spectroscopy: Synthesis of the Chromium(II) Alkyl [Cr2(CH2SiMe3)6]2− and Its Conversion to the Unusual “Windowpane” Bis(metallacycle) Complex [Cr(κ2C,C'-CH2SiMe2CH2)2]2−, Organometallics, 1994, 13, 1646–1655 CrossRef CAS.
- While the work was under review, a new synthetic route to 3 was developed that furthermore allows access to the smaller macrocycle P((CH2)10)3P (3a).28 This species again shows two 31P{1H} NMR signals (ca. −32 and −39 ppm, mesitylene), but now the upfield peak (assigned to the out,out isomers of 3b, 3c) dominates. This is in accord with the expected crossover to more stable out,out isomers as the ring size decreases, strengthening all proposed assignments..
- S. R. Zarcone and J. A. Gladysz, manuscript in preparation.
- M. Saunders, Stochastic Search for the Conformations of Bicyclic Hydrocarbons, J. Comput. Chem., 1989, 10, 203–208 CrossRef CAS.
- A second mechanistic test is in theory possible. When one assigns an arbitrary 1/2/3 Cahn-Ingold-Prelog priority to each bridge prior to a double pyramidal inversion, an R configured bridgehead inverts to S and an S becomes R (racemization). As noted above, a homeomorphic isomerization of such an educt would proceed with retention.7.
- (a) J. M. Brunel, B. Faure and M. Maffei, Phosphane-boranes: synthesis, characterization, and synthetic applications, Coord. Chem. Rev., 1998, 178–180, 665–698 CrossRef CAS , see section 5.1.2.; (b) While the overall shapes of the P·BH331P{1H} and 1H NMR signals of 3b, c, e·2BH3 (e.g., Fig. S10 and S11†) are in accord with those in the literature, most couplings are not resolved..
- Independent but much longer and lower yielding syntheses of these compounds are briefly described in the discussion section: see T. Fiedler, M. Barbasiewicz, M. Stollenz and J. A. Gladysz, Non-Metal-Templated Approaches to bis(Borane) Derivatives of Macrocyclic Dibridgehead Diphosphines via Alkene Metathesis, Beilstein J. Org. Chem., 2018, 14, 2354–2365 CrossRef CAS PubMed.
- The methylcyclopentane was unexpected as the crystals were grown from a hexanes solution. However, methylcyclopentane is often a significant component of technical grade hexanes. Of the several methylcyclopentane solvates in the Cambridge Structural Database, at least one was also obtained from hexanes: M. Vale, M. Pink and A. Rajci, Synthesis, Structure, and Conformation of Aza[1n]metacyclophanes, J. Org. Chem., 2008, 73, 27–35 CrossRef CAS PubMed , see pages S4, S12 therein..
- M. Stollenz, N. Bhuvanesh, J. H. Reibenspies and J. A. Gladysz, Syntheses and Structures of Digold Complexes of Macrobicyclic Dibridgehead Diphosphines that can turn themselves Inside Out, Organometallics, 2011, 30, 6510–6513 CrossRef CAS.
- (a) A. E. Estrada, Y. Wang, N. Bhuvanesh, F. Hampel and J. A. Gladysz, Syntheses, Structures, Reactivities, and Dynamic Properties of Gyroscope Like Complexes Consisting of Rh(CO)(X) or Rh(CO)2(I) Rotators and Cage Like trans Aliphatic Dibridgehead Diphosphine Stators, Organometallics, 2022, 41, 733–749 CrossRef CAS; (b) A. L. Estrada, Y. Wang, G. Hess, F. Hampel and J. A. Gladysz, Square Planar and Octahedral Gyroscope-Like Metal Complexes Consisting of Dipolar Rotators Encased in Dibridgehead Di(triaryl)phosphine Stators: Syntheses, Structures, Dynamic Properties, and Reactivity, Inorg. Chem., 2022, 61 DOI:10.1021/acs.inorgchem.2c02855.
- S. R. Zarcone, P. J. Verardi, N. Bhuvanesh and J. A. Gladysz, A Surprise Landing on the Terra Incognita of Macrocyclic Dibridgehead Diorganoarsines: Syntheses, Structures, and Reactivities, Chem. Commun., 2022, 58, 8694–8697 RSC.
- (a) T. L. Walker, I. S. Taschner, S. M. Chandra, M. J. Taschner, J. T. Engle, B. R. Schrage, C. J. Ziegler, X. Gao and S. E. Wheeler, Lone-Pair-Induced Topicity Observed in Macrobicyclic Tetra-thia Lactams and Cryptands: Synthesis, Spectral Identification, and Computational Assessment, J. Org. Chem., 2018, 83, 10025–10036 CrossRef CAS PubMed; (b) I. S. Taschner, T. L. Walker, S. M. Chandra, B. R. Schrage, C. J. Ziegler, X. Gao and S. E. Wheeler, Topomeric aza/thia cryptands: synthesis and theoretical aspects of in/out isomerism using n-alkyl bridging, Org. Chem. Front., 2020, 7, 1164–1176 RSC.
- M. L. Connolly, Solvent-accessible surfaces of proteins and nucleic-acids, Science, 1983, 221, 709–713 CrossRef CAS PubMed.
- L. Liu and Q.-X. Guo, Isokinetic Relationship, Isoequilibrium Relationship, and Enthalpy-Entropy Compensation, Chem. Rev., 2001, 101, 673–696 CrossRef CAS PubMed.
- (a) R. W. Alder and D. Read, Remarkable In/out Inversions at Bridgehead Phosphorus Atoms, Angew. Chem., Int. Ed., 2000, 39, 2879–2882 ( Angew. Chem. , 2000 , 112 , 3001–3004 ) CrossRef CAS; (b) See also R. W. Alder, C. P. Butts, A. G. Orpen and D. Read, Bridgehead phosphorus chemistry: in–out inversion, intrabridgehead P⋯P bonding, and reactivity, J. Chem. Soc., Perkin Trans. 2, 2001, 288–295 RSC.
- S. R. Zarcone, N. Bhuvanesh and J. A. Gladysz, Unpublished crystal structures.
- A. Ehnbom and J. A. Gladysz, Gyroscopes and the Chemical Literature, 2002-2020: Approaches to a Nascent Family of Molecular Devices, Chem. Rev., 2021, 121, 3701–3750 CrossRef CAS PubMed.
- C. Brown, B. T. Heaton and J. Sabounchei, Oxidative addition reactions of [Pt(CN)4]2−; A 13C and 195Pt NMR study, J. Organomet. Chem., 1977, 142, 413–421 CrossRef CAS.
- C. Mühle, J. Nuss and M. Z. Jansen, Crystal Structure of Tetracyanoplatinate(IV) dihydrate, Pt(CN)4·2H2O, Z. Kristallogr. – New Cryst. Struct., 2009, 224, 9–10 Search PubMed.
- J. S. Siegel and F. A. L. Anet, Dichlorofluoromethane-d: a versatile solvent for VT-NMR experiments, J. Org. Chem., 1988, 53, 2629 CrossRef CAS.
- Although this sample did not give a satisfactory microanalysis, the best available data are given. It was judged to be >96% pure by NMR..
- The 1H and 31P{1H} NMR spectra (CDCl3) were identical with those reported earlier.18a.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional preparative, crystallographic, NMR, and rate data, and a video representing the interconversion of III, IV, and VI in Scheme 2. The EX (or P:) bridgeheads are depicted as triangles, initially with the blue sides “in” and the white sides “out”. Threading one (CH2)n chain through the macrocycle defined by the other two reverses these relationships (without pyramidal inversion or bond-breaking). This is termed “homeomorphic isomerization”. CCDC 2190116 (out,out-3b), 2190438 (out,out-3c), 2190439 (out,out-3e), and 2190440 (out,out-3e·2BH3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04724a |
| ‡ Current address: Department of Chemistry and Biochemistry, Kennesaw State University 370 Paulding Avenue NW, MD #1203, Kennesaw, GA 30144, USA. |
| § Current address: Department of Chemistry, School of Chemical Sciences and Pharmacy, Central University of Rajasthan, Ajmer, Rajasthan 305817 India. |
| This journal is © The Royal Society of Chemistry 2022 |