
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure-guided design and characterization of a clickable, covalent PARP16 inhibitor†
Daniel S.
Bejan
 a,
Sunil
Sundalam
a,
Haihong
Jin
a,
Rory K.
Morgan
a,
Ilsa T.
Kirby
a,
Ivan R.
Siordia
a,
Barr
Tivon
b,
Nir
London
b and
Michael S.
Cohen
*a
a,
Sunil
Sundalam
a,
Haihong
Jin
a,
Rory K.
Morgan
a,
Ilsa T.
Kirby
a,
Ivan R.
Siordia
a,
Barr
Tivon
b,
Nir
London
b and
Michael S.
Cohen
*a
aDepartment of Chemical Physiology and Biochemistry, Oregon Health & Science University, Portland, OR 97239, USA. E-mail: cohenmic@ohsu.edu
bDepartment of Chemical and Structural Biology, The Weizmann Institute of Science, Rehovot, 7610001, Israel
First published on 16th November 2022
Abstract
PARP16—the sole ER-resident PARP family member—is gaining attention as a potential therapeutic target for cancer treatment. Nevertheless, the precise function of the catalytic activity of PARP16 is poorly understood. This is primarily due to the lack of inhibitors that are selective for PARP16 over other PARP family members. Herein, we describe a structure-guided strategy for generating a selective PARP16 inhibitor by incorporating two selectivity determinants into a phthalazinone pan-PARP inhibitor scaffold: (i) an acrylamide-based inhibitor (DB008) designed to covalently react with a non-conserved cysteine (Cys169, human numbering) in the NAD+ binding pocket of PARP16 and (ii) a dual-purpose ethynyl group designed to bind in a unique hydrophobic cavity adjacent to the NAD+ binding pocket as well as serve as a click handle. DB008 exhibits good selectivity for PARP16 versus other PARP family members. Copper-catalyzed azide–alkyne cycloaddition (CuAAC) confirmed that covalent labeling of PARP16 by DB008 in cells is dependent on Cys169. DB008 exhibits excellent proteome-wide selectivity at concentrations required to achieve saturable labeling of endogenous PARP16. In-cell competition labeling experiments using DB008 provided a facile strategy for evaluating putative PARP16 inhibitors. Lastly, we found that PARP16 is sequestered into a detergent-insoluble fraction under prolonged amino acid starvation, and surprisingly, treatment with PARP16 inhibitors prevented this effect. These results suggest that the catalytic activity of PARP16 regulates its solubility in response to nutrient stress.
Introduction
ADP-ribosylation is a critical post-translational modification carried out by a family of enzymes known as PARPs. Upon binding nicotinamide adenine dinucleotide (NAD+), PARPs cleave the nicotinamide group and transfer the resulting ADP-ribose (ADPr) to targets—primarily on proteins but also on nucleic acids.1 The PARP family is divided based on the ability to transfer ADPr in the form of polymers (referred to as poly-ADP-ribosylation or PARylation) or monomers (referred to as mono-ADP-ribosylation or MARylation). PARP1, PARP2, and the tankyrases—TNKS1 (PARP5a) and TNKS2 (PARP5b)—catalyze PARylation, while the remaining members (PARP 3, 4, 6–12, 14–16) catalyze MARylation, with the exception of PARP13 being inactive.2 The role of PARP1/2-mediated PARylation has been extensively studied, particularly in the context of DNA double strand break (DSB) repair in cancer cells, resulting in four clinically approved PARP1/2 inhibitors (i.e., olaparib, rucaparib, niraparib, talazoparib) that are particularly effective in DSB repair defective ovarian and breast cancers.3 In contrast, the physiological and pathophysiological roles for MARylation are far less understood, in large part due to the general lack of appropriately selective inhibitors of MARylating PARPs. However, there have been recent advances in inhibitor development focusing on a subset of MARylating PARPs (i.e., PARP7, 10, 11, 14, 15)4 as some of these PARPs are being implicated in diseases such as cancer and inflammation. For example, PARP11 has been shown to be induced in cytotoxic T lymphocytes to regulate the immunosuppressive tumor microenvironment in various mouse cancer models.5 Our group has developed ITK7,6 an inhibitor for PARP11. Additionally, inhibitors for PARP7 (RBN-2397)7 and PARP14 (RBN-3143) have recently entered clinical trials for the treatment of lung cancer (NCT05127590) and atopic dermatitis (NCT05215808), respectively.PARP16 is an example of another MARylating PARP that has been gaining attention as a novel therapeutic target. PARP16 contains a C-terminal transmembrane (TM) domain that localizes to the endoplasmic reticulum (ER) membrane, and a N-terminal catalytic domain facing the cytoplasm. Upon ER stress, PARP16 has been shown to MARylate two sensors of the unfolded protein response (UPR), PKR-like ER kinase (PERK) and inositol-requiring enzyme 1α (IRE1α), leading to a suppression of protein synthesis.8 The role of PARP16 in UPR has motivated additional studies to explore opportunities for therapeutic intervention, since the UPR pathway is implicated in a variety of diseases including diabetes, neurodegeneration, and cancer.9 Two recent studies have revealed PARP16-dependent vulnerabilities that can be exploited to treat cancer. In one study, Palve et al. used a chemoproteomics approach to show that the clinically approved PARP1/2 inhibitor, talazoparib (Tal), targets PARP16 in small cell lung cancer (SCLC).10 Consistent with the target engagement studies, Tal inhibited recombinant PARP16 with sub-micromolar potency. Knockdown of PARP16 alone reduced SCLC cell viability, which was exacerbated by selective inhibition of PARP1/2 by olaparib. These results suggest that PARP16 inhibition by Tal contributes to the increased efficacy of Tal versus olaparib in SCLC. In the second study, Challa et al. identify a surprising role for PARP16 in ovarian cancer cell survival.11 Knockdown or knockout of PARP16 in various ovarian cancer cell lines increased protein synthesis rates, forcing the cancer cell to enter a state of proteotoxic stress, ultimately leading to a loss of viability in vitro and in xenograft models in mice.11 One mechanism proposed for the effects of PARP16 on protein synthesis rates is via MARylation of ribosomes by PARP16, yet direct (i.e., in vitro MARylation of ribosomes by PARP16) evidence for this is lacking.
While these studies highlight PARP16 as a potential therapeutic target in certain cancers, whether or not targeting the catalytic activity of PARP16 to elicit the same effect as knockdown or knockout of PARP16 is not known. Moreover, our understanding of the role of PARP16-mediated MARylation in cancer cell proteostasis and survival, as well as the UPR, is inadequate to draw any firm conclusions about the physiological and pathophysiological function of PARP16 catalytic activity. To address these gaps in knowledge we need selective, membrane-permeable PARP16 inhibitors. To date, reported PARP16 inhibitors lack the selectivity and potency required to be a useful chemical probe of PARP16 catalytic activity in cells.12,13 Herein, we describe the structure-based design of a clickable, covalent PARP16 inhibitor (DB008) that exhibits exquisite selectivity for PARP16 in the covalent binding mode in cells.
Results and discussion
Structure-based design of a covalent PARP16 inhibitor
Developing selective inhibitors for PARPs is challenging due to the highly conserved NAD+-binding site shared between PARP family members. One solution to this challenge, for another family of enzymes, namely protein kinases, has been to target non-conserved cysteines within the highly conserved ATP-binding site.14 This has led to the generation of potent and selective covalent kinase inhibitors, several of which are FDA-approved drugs.15 Thus, we were inspired to search for the presence of a non-conserved cysteine in the NAD+ binding site of PARP16 that could potentially be covalently modified by an appropriately positioned electrophilic PARP inhibitor. Previous studies from our lab and others showed that the phthalazinone-based inhibitor olaparib, a nanomolar inhibitor of PARP1-4,6,16 is also moderately potent against PARP16 (IC50 = 2–3 μM).10,17 Crystal structures of PARP1-bound olaparib overlaid with PARP16 show that the cyclopropyl amide emanating from the piperazine substituent is in close proximity (3.7 Å) to Cys169 (human PARP16 number) (Fig. 1A). Cys169 resides in the donor-loop (D-loop), a conserved structural element in the ADP-ribose sub-pocket of the NAD+ binding site. Critically, a structure-based alignment of the entire PARP family reveals that a cysteine at this position of the D-loop is unique to PARP16 (Fig. 1B).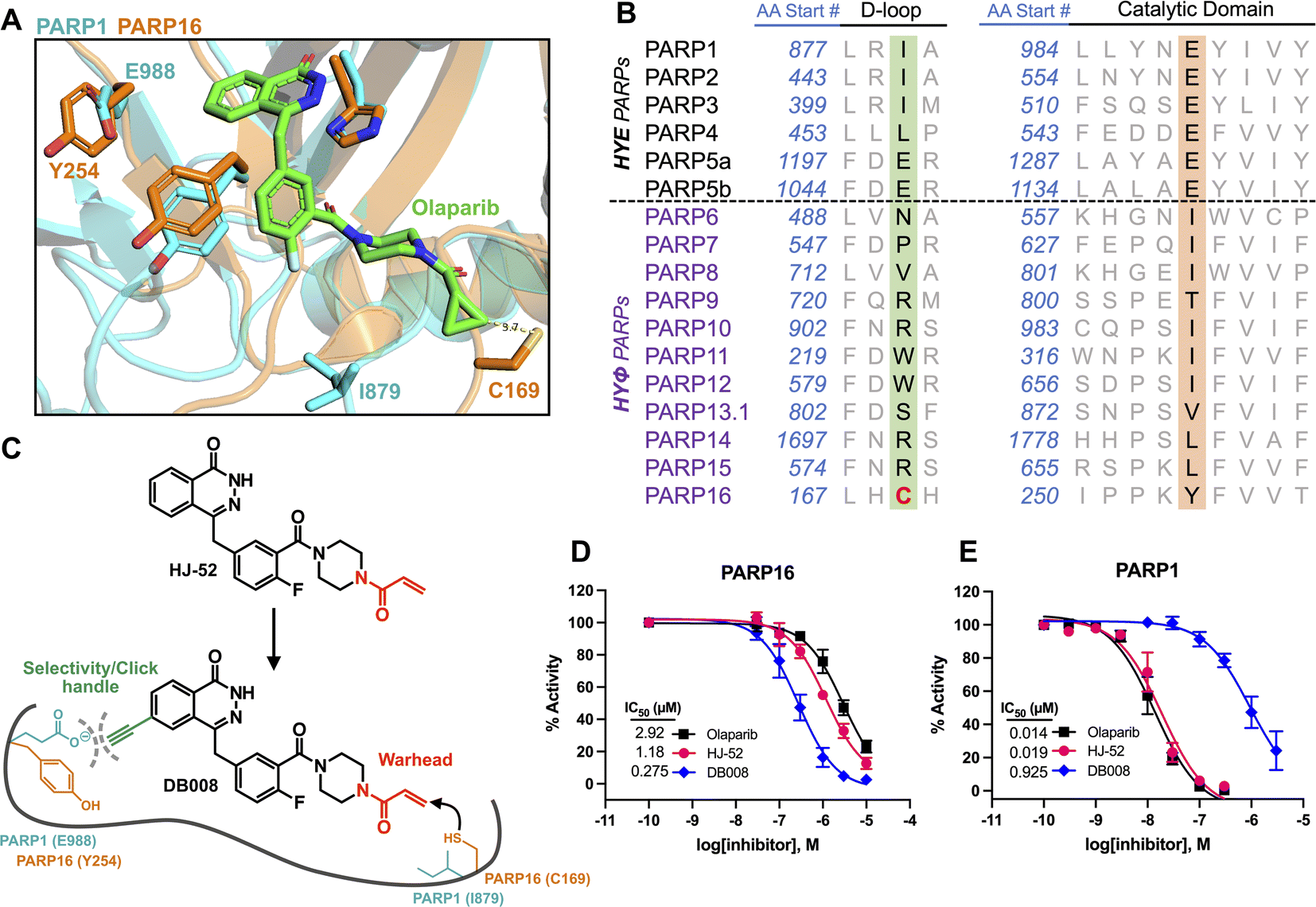 | ||
| Fig. 1 Structure-based design leads to an electrophilic PARP16 inhibitor that covalently modifies a non-conserved cysteine. (A) Crystal structure of the active site of PARP16 (orange, PDB: 4F0D) overlaid with PARP1 (cyan, PDB: 5DS3) bound to olaparib (green). (B) PARP family sequence alignment generated with T-Coffee multiple sequence alignment algorithm. The non-conserved D-loop cysteine (Cys169) of PARP16 is highlighted in red. (C) Structure of HJ-52 and DB008, with the acrylamide selectivity element shown in red, and the dual selectivity element/clickable alkyne handle shown in green. Biochemical activity assay to assess potency of olaparib, HJ-52, and DB008 against PARP16 (D) and PARP1 (E); n ≥ 3 biological replicates. | ||
As a proof-of-concept, we synthesized HJ-52 (Fig. 1C), an olaparib analog in which the cyclopropyl amide was replaced with an acrylamide warhead designed to react with Cys169 of the D-loop in PARP16. To determine if HJ-52 is able to covalently modify PARP16, we performed whole-protein electrospray ionization (ESI) mass spectrometry. Gratifyingly, incubation of recombinant SUMO-tagged PARP16 that is devoid of the transmembrane domain (SUMO-PARP16ΔTM) with a 50- or 100-fold excess of HJ-52 led to the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 covalent complex between SUMO-PARP16ΔTM and HJ-52 (Fig. S1†). Using an in vitro MARylation assay, we found that HJ-52 was about 2.5-fold more potent against PARP16 than olaparib (Fig. 1D); however, when we tested HJ-52 against PARP1, HJ-52 was equipotent to olaparib (Fig. 1E). Nevertheless, these results show that Cys169 of PARP16 can be targeted by an appropriately positioned electrophile.
:1 covalent complex between SUMO-PARP16ΔTM and HJ-52 (Fig. S1†). Using an in vitro MARylation assay, we found that HJ-52 was about 2.5-fold more potent against PARP16 than olaparib (Fig. 1D); however, when we tested HJ-52 against PARP1, HJ-52 was equipotent to olaparib (Fig. 1E). Nevertheless, these results show that Cys169 of PARP16 can be targeted by an appropriately positioned electrophile.
To improve the selectivity of HJ-52, we turned to a strategy that we have used successfully to transform PARP inhibitors with high affinity for PARylating PARPs, like PARP1, into selective inhibitors of MARylating PARPs. Key to this strategy is exploiting the differences in the identify of an amino acid in the third position of a structurally conserved triad (histidine-tyrosine-variable amino acid, HYX). MARylating PARPs, like PARP16, contain a hydrophobic amino acid (Φ) in the third position of the triad whereas PARylating PARPs, like PARP1, contain a glutamate (E) in this position (Fig. 1B). The amino acid at this position is juxtaposed to the nicotinamide sub-pocket of the NAD+ binding site.18 Our mutagenesis and structural studies revealed that the amino acid at this position can act as a “gatekeeper” by controlling access to a hydrophobic cavity adjacent to the nicotinamide sub-pocket of the NAD+ binding site.6 Structural analysis of PARPs show that a Φ-gatekeeper allows access to this hydrophobic cavity, whereas an E-gatekeeper occludes it. Indeed, we and others have shown that PARP inhibitors containing strategically positioned hydrophobic groups (e.g., propynyl, substituted alkyl and aromatic groups) access the hydrophobic cavity and interact favorably with HYΦ PARPs, resulting in selective inhibition of HYΦ PARPs over HYE PARPs.6,19,20
PARP16 contains a tyrosine (Tyr254, human PARP16 numbering) in the gatekeeper position, which is unique among the MARylating PARPs. Structural analysis shows that Tyr254 is orientated away from the nicotinamide sub-pocket, allowing access to its hydrophobic cavity. Guided by structural analysis, we designed and synthesized a HJ-52 analog (DB008, Fig. 1C) that contains an ethynyl group at the C-6 position of the phthalazinone scaffold. We envisioned this terminal alkyne serving two roles: (i) promoting selective binding to PARP16 over PARP1 and other HYE PARPs by occupying the hydrophobic cavity and (ii) providing a clickable handle for assessment of target engagement using copper-catalyzed azide–alkyne cycloaddition (CuAAC).21 We found that DB008 was 4.3-fold more potent against PARP16 compared to HJ-52 (IC50 = 275 nM versus 1180 nM) (Fig. 1D). Although this increase in potency against PARP16 was modest, inhibition of PARP1 decreased by ∼50-fold compared to HJ-52 (IC50 = 925 nM versus 19 nM) (Fig. 1E). A similar trend, although not as pronounced, was also observed with PARP2 (Fig. S2†). Together these results demonstrate that the addition of the ethynyl group at the C-6 position of the phthalazinone scaffold of HJ-52 resulted in an inhibitor with improved potency and selectivity for PARP16.
Off-target PARP engagement by DB008 is reversible
We next sought to assess the selectivity of DB008 across the PARP family using our PASTA (PARP activity screening and inhibitor testing assay) biochemical plate assay22 (Fig. 2A). We were encouraged to see that DB008 displayed an excellent selectivity profile among the MARylating PARPs. One of the prominent off-targets of DB008 is PARP2 (IC50 = 139 nM), which was not surprising since all of the approved PARP1 inhibitors (olaparib, rucaparib, niraparib, talazoparib) and the recent clinical PARP7 candidate (RBN-2397) potently inhibit PARP2.7,23 Nevertheless, we wanted to determine if the inhibition of PARP1/2 by DB008 was a result of reversible or irreversible binding. For these studies, we evaluated the effects of DB008 and other PARP inhibitors on endogenous PARylation in HEK 293T cells. In HEK 293T cells, as is the case in many other mammalian cells, endogenous PARylation is predominately mediated by PARP1/2. We first confirmed inhibition of PARylation in a cellular context by treating HEK 293T cells with DB008, HJ-52, and olaparib as a positive control, followed by treatment with a poly(ADP-ribose) glycohydrolase (PARG) inhibitor24 to boost the endogenous PARylation signal for detection by western blot (Fig. 2B). Our cellular PARylation assay recapitulated the results from our in vitro PASTA assay, with olaparib and HJ-52 displaying low nanomolar potency against PARP1, and DB008 inhibiting PARP1 with an IC50 ∼ 1 μM (Fig. 2B and C). We did not detect auto-PARylation of PARP2 (molecular weight = 66 kDa), which is likely due to the relatively low expression levels of PARP2 (170 nM) compared to PARP1 (4900 nM) in HEK 293T cells.25 To determine if inhibition of PARylation is reversible or irreversible, we performed a washout experiment in which HEK 293T cells were treated with DB008, followed by a series of washouts (replacing media with fresh media lacking DB008). We observed a complete rescue of PARylation after the washout conditions, demonstrating that DB008 does not inhibit PARP1/2 in a covalent manner (Fig. 2D).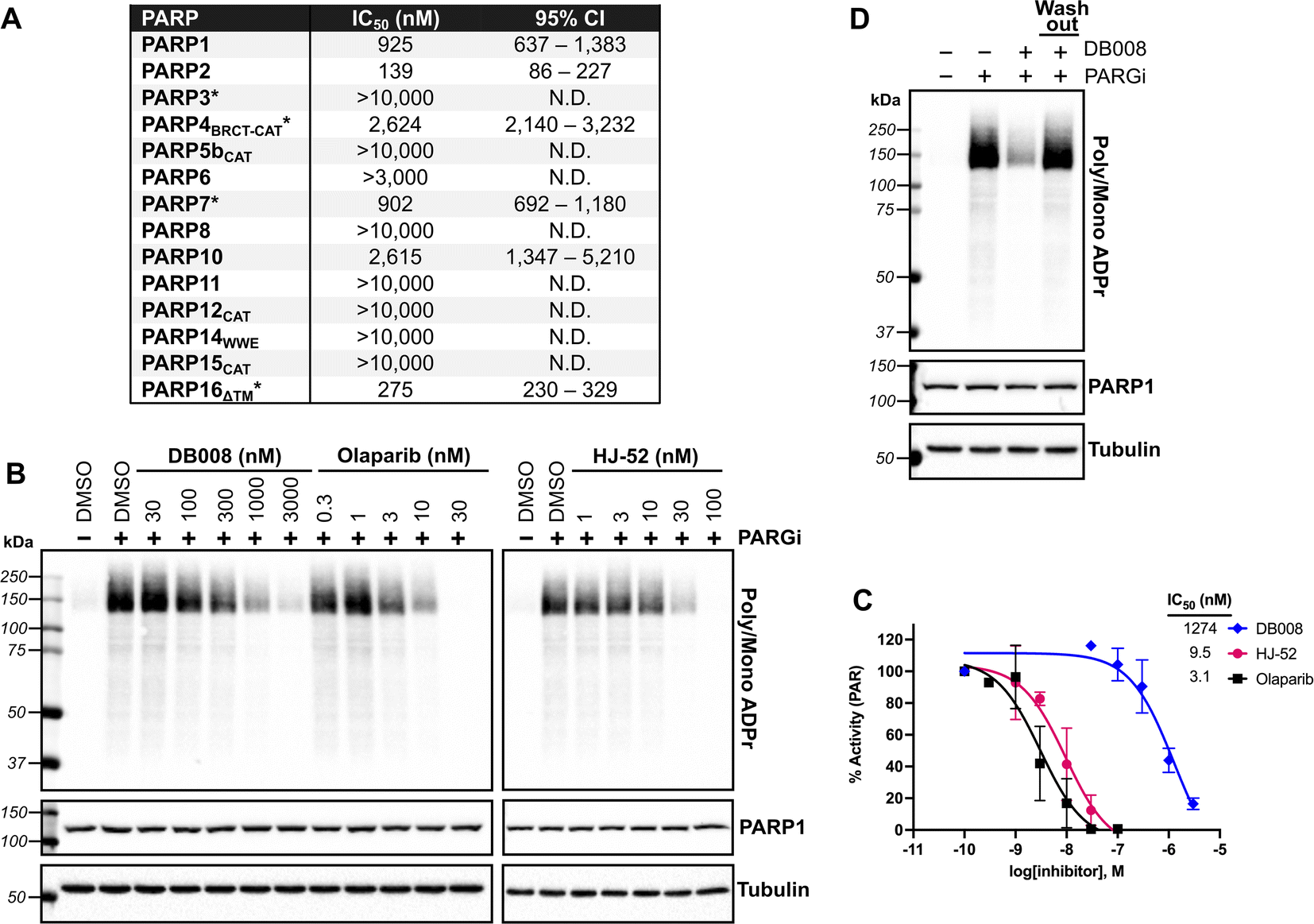 | ||
| Fig. 2 Biochemical and cellular assessment of DB008 selectivity across the PARP family. (A) DB008 potency against the PARP family determined using our PASTA assay (trans-modification) unless otherwise indicated: *auto-modification assay format using native NAD+ (100 μM for PARP3, PARP4, and PARP7; 400 μM for PARP16). N.D. = not determined. Individual dose–response curves can be found in Fig. S5.† (B) Cellular inhibition of PARP1 was determined by treating HEK 293T cells with a dose response of PARP inhibitors (30 min) followed by PARG inhibitor (15 min) to amplify the PARylation signal. Western blotting for PARylation was done using a mono/poly ADPr antibody from cell signaling technology. (C) Quantification of inhibition from (B); n = 2 biological replicates. (D) DB008 is not a covalent inhibitor of PARP1/2 in cells. Cellular PARP1/2 inhibition assay performed as in (B), except a washout condition before PARG inhibitor treatment was included; n = 2 biological replicates. | ||
DB008 selectivity labels PARP16 in cells
Next, we wanted to confirm that DB008 forms a covalent bond with Cys169 of PARP16 and profile its proteome-wide selectivity using CuAAC. Using DOCKovalent, we generated a model of DB008 bound to PARP16.26 In this model, the acrylamide emanating from the piperazine linker formed a covalent bond with Cys169 (Fig. 3A). Additionally, the ethynyl at C-6 position of the core phthalazinone scaffold occupied the hydrophobic cavity gated by Tyr254 (Fig. 3A). To confirm that DB008 reacts covalently with Cys169 of PARP16, we transiently expressed Myc2x-tagged PARP16 in HEK 293T cells and treated cells with increasing concentrations of DB008 for 2 hours. CuAAC with TAMRA-azide in cell lysates followed by SDS-PAGE and in-gel fluorescence imaging revealed that DB008 selectively labels Myc2x-PARP16 in a dose-dependent manner (Fig. 3B), with saturation occurring at ∼300 nM DB008 [apparent Kd = 90.0 nM (95% confidence interval: 52.5–231 nM)] (Fig. 3C). Critically, when we mutated Cys169 of Myc2x-PARP16 to a serine (C169S), we observed a near complete reduction in labeling by DB008 (Fig. 3B). We also performed a time course using the 300 nM saturating dose of DB008, which demonstrated that covalent labeling of PARP16 occurs in a time-dependent manner and that saturation occurs at approximately 120 minutes (Fig. 3D and E). Together, these results show that DB008 is membrane-permeable, that the ethynyl group of DB008 can be used to monitor covalent labeling of PARP16 in cells, and that Cys169 in the D-loop of PARP16 is the major site of covalent modification.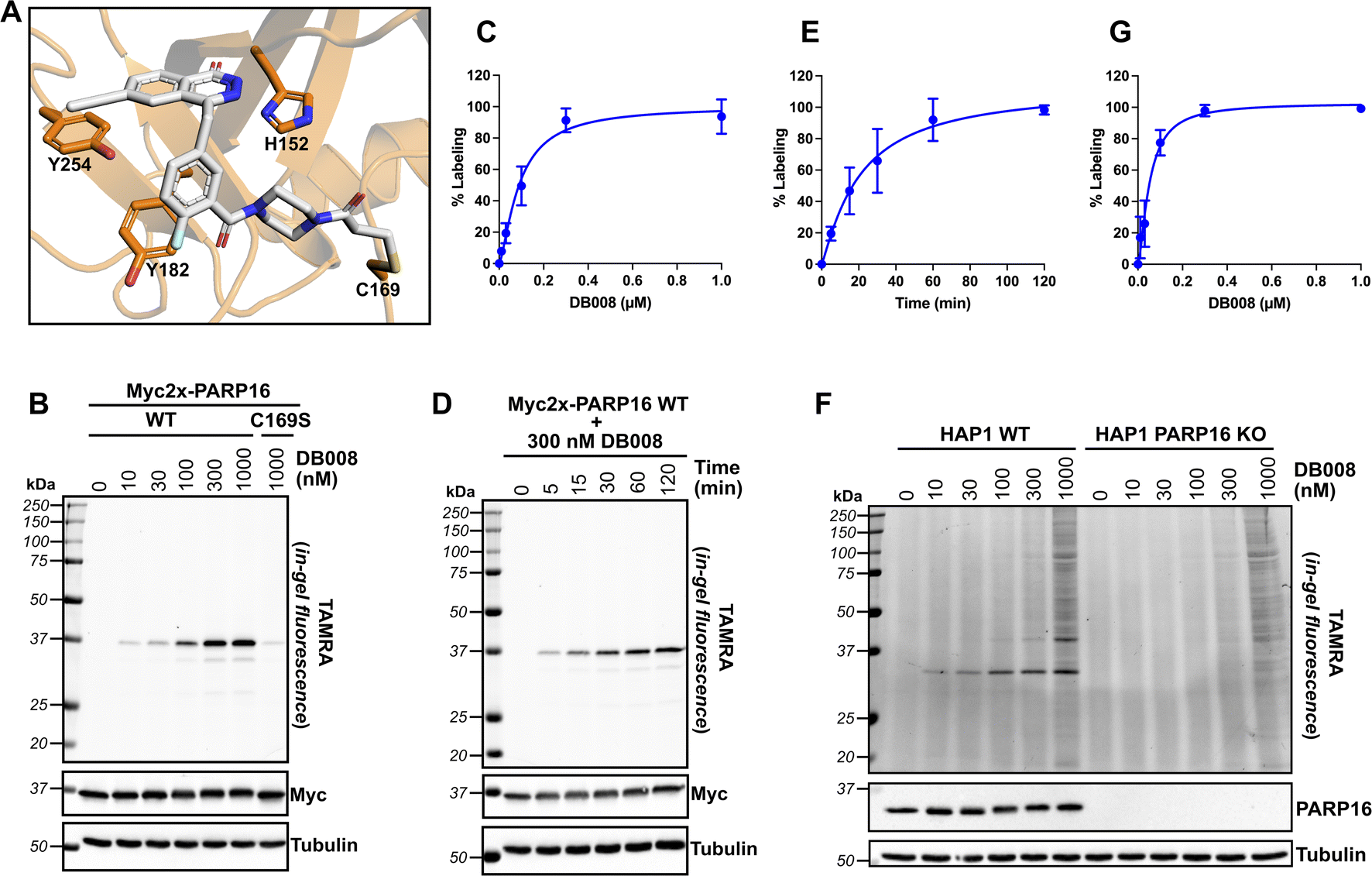 | ||
| Fig. 3 DB008 covalently modifies Cys169 of PARP16 and exhibits excellent proteome-wide selectivity in the irreversible binding mode. (A) Model of DB008 covalently bound to C169 of PARP16. (B) HEK 293T cells were transfected with Myc2x-tagged PARP16 WT or the C169S mutant, treated with a DB008 dose response for 2 h, followed by lysis and clicking to TAMRA-azide for in-gel fluorescence detection of PARP16 labeling. (C) Quantification of TAMRA signal from (B); n = 3 biological replicates. (D) HEK 293T cells were transfected with Myc2x-tagged PARP16 WT, treated with a 300 nM DB008 on a time course, followed by lysis and clicking to TAMRA-azide for in-gel fluorescence detection of PARP16 labeling. (E) Quantification of TAMRA signal from (D); n = 3 biological replicates. (F) DB008 proteome-wide labeling in HAP1 cells. HAP1 WT and HAP1 PARP16 KO cells were treated with a DB008 dose response for 2 h, followed by lysis and clicking to TAMRA-azide for in-gel fluorescence detection of endogenous PARP16 labeling. (G) Quantification of TAMRA signal from (F); n = 2 biological replicates. | ||
We next examined the proteome-wide selectivity of DB008. We treated HAP1 wild-type (WT) cells with increasing concentrations of DB008, followed by CuAAC with TAMRA-azide in cell lysates. Strikingly, we observed selective labeling (up to the saturating dose of 300 nM) of a ∼32 kDa band which correlated with the molecular weight of endogenous PARP16 [apparent Kd of 51.6 nM (95% confidence interval: 29.5–129 nM)] (Fig. 3F and G). To determine if the ∼32 kDa band was indeed PARP16, we treated HAP1 PARP16 knock-out (KO) cells with DB008 and processed samples as described above. We did not observe labeling of a ∼32 kDa band in PARP16 KO cells, demonstrating that PARP16 is the prominent target of DB008 in cells. Together these results show that PARP16 is the major target of DB008 in cells and highlight the excellent proteome-wide selectivity of DB008 under conditions of saturable PARP16 labeling.
It is worth noting that despite robust expression and DB008-mediated labeling of exogenous PARP16, we have not been able to detect PARP16-dependent auto-MARylation or trans-MARylation activity in cells (data not shown). We do see auto-MARylation in vitro using a modified version of our PASTA assay (illustrated in Fig. 1D), however, only at high concentrations of NAD+ (400 μM; 4–40-fold the amount used for the other PARPs) and PARP16 (1 μM added to the plate), conditions that force self-modification similar to assays developed by others.17,27 We are therefore unable to determine a cellular IC50 value for DB008 against PARP16. However, the IC50 is not the best measure of potency for covalent inhibitors because incubation time can dramatically shift IC50 values due to the time-dependent nature of their inhibition. Instead, the more appropriate parameter for measuring potency of covalent inhibitors is Kinact/KI, a second-order rate constant that describes the efficiency of covalent bond formation.28 To calculate Kinact/KI, we conducted time-dependent labeling of Myc2x-PARP16 expressed in HEK 293T cells, with increasing concentrations of DB008, and determined the Kinact/KI of DB008 to be 5.95 × 103 M−1 s−1 (Fig. S3†). Importantly, the relationship between inhibitor concentration and Kobs resulted in a saturation binding curve (Fig. S3C†), indicative of a two-step, specific binding model wherein the inhibitor binds the protein first, forming a reversible protein–inhibitor complex, followed by covalent bond formation between the nucleophilic residue and electrophilic inhibitor.
DB008 as a cellular PARP16 target engagement probe
The lack of detectable cellular PARP16 MARylation activity makes it difficult to assess the potency of inhibitors against PARP16 in cells. We were thus encouraged to use DB008 and CuAAC to profile PARP16 inhibitors using competition-based assays. In this way, we could directly assess PARP16 active site engagement by putative PARP16 inhibitors. We chose to focus on talazoparib and epigallocatechin gallate (EGCG) (Fig. 4A), two recently reported PARP16 inhibitors10,12 that are commercially available and have been shown to induce cancer cell death and/or mitigate the unfolded protein response in the context of vascular stress. Talazoparib, a potent PARP1/2 inhibitor, was found to bind PARP16 in SCLC cells using inhibitor-bead conjugates in a chemical proteomics-based approach.10 Using our modified PASTA assay, we determined talazoparib inhibition of PARP16 catalytic activity with an IC50 in the 100–300 nM range,10 supporting the notion that PARP16 may be a relevant off-target of talazoparib in cells. EGCG, a major catechin found in tea, was identified as a binder of recombinant GST-PARP16 from a high-throughput optical-based microarray screen. An in vitro ADP-ribosylation assay using biotinylated-NAD+ was used to calculate an IC50 of 14.52 μM for EGCG against GST-PARP16.12 EGCG, used at 100 μM, was shown to suppress PERK-mediated UPR in response to ER-stress in QGY-7703 and HeLa cells. EGCG was also used at 30–100 μM to reduce PARP16-dependent ER stress in models of vascular aging29 and neointimal hyperplasia.30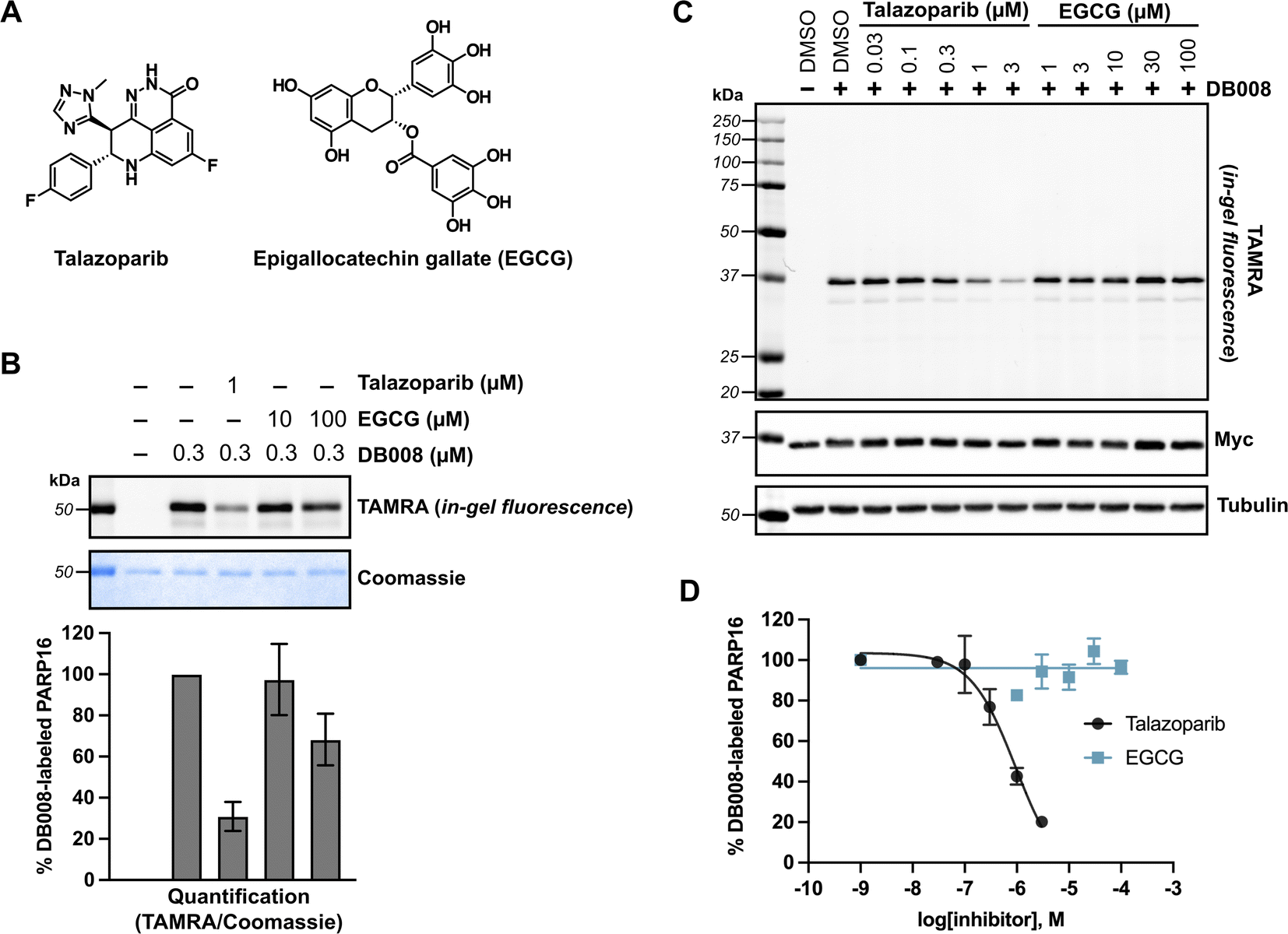 | ||
| Fig. 4 Competition labeling experiments using DB008 enables validation of previously reported PARP16 inhibitors. (A) Chemical structures of talazoparib (Tal) and epigallocatechin gallate (EGCG). (B) Tal, but not EGCG, effectively blocks covalent labeling of PARP16 by DB008. In vitro competition assay wherein talazoparib and EGCG were incubated with recombinant PARP16 (40 min) followed by treatment with DB008 (20 min) and clicking to TAMRA-azide for in gel-fluorescence detection of PARP16 labeling; n = 2 biological replicates. (C) Tal, but not EGCG, inhibits PARP16 labeling by DB008 in cells. Cellular competition assay wherein Myc2x-PARP16 expressing HEK 293T cells were dosed with Tal and EGCG for 1 h, then treated with DB008 (0.3 μM) for 30 min, followed by lysis and clicking to TAMRA-azide for in gel-fluorescence detection of PARP16 labeling. (D) Quantification of TAMRA signal from (C); n = 2 biological replicates. | ||
We first performed an in vitro competition assay wherein EGCG and talazoparib were pre-incubated with recombinant PARP16, followed by incubation with DB008 and subsequent CuAAC with TAMRA-azide for gel-fluorescence imaging (Fig. 4B). While 1 μM talazoparib competed DB008 labeling effectively (∼70%)—consistent with previous studies using in vitro PARP16 catalytic activity assays10,17—100 μM EGCG only partially competed (∼30%) labeling. We then wanted to evaluate how effectively talazoparib and EGCG compete labeling of PARP16 by DB008 in HEK 293T cells. While talazoparib competed labeling of PARP16 by DB008 with an IC50 of 949 nM, we observed no competition of DB008 labeling with EGCG up to 100 μM, suggesting that EGCG does not bind the active site of PARP16 in cells (Fig. 4C and D). Thus, the effects of EGCG on the UPR are not likely due to direct inhibition of PARP16. However, we cannot rule out that EGCG binds to another, non-DB008 competed site on PARP16. In any event, we caution the use of EGCG as a cellular probe for PARP16.
PARP16 and its catalytic activity is not required for the UPR in HAP1 cells
Previous studies suggest that PARP16, and in particular its catalytic activity, regulates the UPR in response to ER stress.8 We therefore sought to determine if inhibition of PARP16 by DB008 impacts the UPR in response to the ER stressor, tunicamycin. Unlike previous studies in HeLa cells, we did not observe any differences in tunicamycin-induced ATF4 and phosphorylated eIF2α levels, two commonly used markers of ER stress, between HAP1 WT and PARP16 KO cells (Fig. S4†). Moreover, treatment with DB008 had no effect on tunicamycin-induced ATF4 and phosphorylated eIF2α levels in HAP1 WT and PARP16 KO cells (Fig. S4†). Our results are consistent with a recent study that showed that siRNA-mediated knockdown of PARP16 had no effect on tunicamycin-induced ER stress in SCLC and Ewing sarcoma cells.10 Taken together, these data suggest that PARP16 may not be involved in the UPR as previously described, or perhaps the role of PARP16 in the UPR is cell-line specific.DB008 stabilizes PARP16 levels during nutrient starvation
In drosophila (S2 cells), PARP16 catalytic activity has been linked to the formation of a novel stress assembly, known as Sec bodies, in response to amino acid starvation.31 Sec bodies are membraneless organelles that form at ER-exit sites (ERES) to protect components of the early secretory pathway from degradation and to promote cell survival during periods of amino acid deprivation.32 It was shown that upon amino acid starvation in S2 cells, the loss of PARP16 prevented the formation of Sec bodies.31 It was proposed that amino acid starvation stimulated PARP16 activity, leading to MARylation of Sec16, a key component of Sec bodies, though direct evidence was lacking. This study motivated us to determine if human PARP16 is regulated in response to nutrient stress. In mammalian cells, replacement of complete growth medium with Hanks' balanced salt solution (HBSS), which lacks amino acids and serum, induces nutrient stress.33 As expected, we found that incubation of HAP1 cells with HBSS for 16 hours induced phosphorylation of eIF2α and shut down global protein synthesis, measured using a puromycin incorporation assay34 (Fig. 5A). Neither knockout of PARP16 nor treatment with DB008 impacted phosphorylated eIF2α or global protein synthesis levels (Fig. 5A). Surprisingly, however, we observed that PARP16 levels drastically reduce during amino acid and serum starvation, which was rescued by treatment with 100 nM DB008 (Fig. 5A). This suggests that PARP16 catalytic activity may regulate its stability during nutrient stress. | ||
| Fig. 5 DB008 and talazoparib treatment rescue nutrient starvation-induced loss of soluble PARP16. (A) Amino acid (aa) and serum starvation decreases PARP16 levels which is prevented by DB008 treatment. HAP1 WT and HAP1 PARP16 KO cells were incubated in complete media (IMDM + 10% FBS) or starvation media (1× Hanks' balanced salt solution, HBSS) in the presence or absence of 100 nM DB008 for 16 h, then dosed with 5 μg ml−1 puromycin for 5 minutes to capture the state of translation, followed by western blotting. (B) In contrast to inhibition of PARP16, neither inhibition of the proteasome nor lysosome prevent aa starvation-mediated decreases in PARP16 levels. HAP1 WT cells were incubated in complete media or starvation media (1× HBSS) in the presence of 300 nM DB008, 10 μM MG132 (proteosome inhibitor), or the lysosomal inhibitors bafilomycin A1 (Baf-A1; 1 μM), or chloroquine (CLQ; 50 μM) for 16 h, followed by western blotting. (C) aa and serum starvation leads to a loss of soluble PARP16, which is rescued by the PARP16 inhibitors, DB008 and talazoparib (Tal). HAP1 WT cells were incubated in complete media or starvation media and dosed with either DB008 or Tal for 16 h, followed by western blotting. Cells were lysed in RIPA buffer and after clarification, western blotting was performed on the RIPA-soluble fraction (supernatant) and RIPA-insoluble fraction (pellet). | ||
We wondered if nutrient stress induces PARP16 degradation, either through a proteasomal or lysosomal pathway. We therefore incubated HAP1 WT cells in complete media or HBSS in the presence or absence of DB008, the proteasome inhibitor MG132, or two structurally (and mechanistically) distinct lysosomal inhibitors, bafilomycin A1 and chloroquine. Antibodies against poly-ubiquitin and p62, markers for proteasome and lysosome inhibition, respectively, confirmed the activity of MG132, bafilomycin A1 and chloroquine in complete media (Fig. 5B). Nevertheless, unlike DB008, these degradation inhibitors did not impact PARP16 levels under amino acid and serum starvation (Fig. 5B). These results show that PARP16 is not degraded via proteasomal or lysosomal pathways during nutrient stress.
What, then, is the fate of PARP16 under nutrient stress? Perhaps nutrient stress induces PARP16 aggregation in an activity-dependent manner, leading to a loss of soluble PARP16. To test this hypothesis, we isolated a RIPA-soluble fraction (supernatant) and a RIPA-insoluble fraction (pellet) after centrifugation of the lysates derived from amino acid and serum starved cells treated with either DB008 or talazoparib. We found that amino acid and serum starvation dramatically increased PARP16 levels in the RIPA-insoluble fraction, which was counteracted in a dose-dependent manner by treatment with either DB008 or talazoparib (Fig. 5C). Concurrently, DB008 and talazoparib treatment increased, dose dependently, PARP16 levels in the RIPA-soluble fraction (Fig. 5C). Together, these results support the hypothesis that nutrient stress induces PARP16 aggregation, which is dependent on its catalytic activity.
Conclusions
We have described the design, synthesis, and characterization of DB008, a selective, covalent inhibitor of PARP16 that contains an acrylamide electrophile. Mutagenesis studies demonstrate that Cys169 is the major site of covalent modification by DB008. While covalent inhibitors of other PARP family members have been described (e.g., PARP1),35 DB008 is the first example of a cysteine-targeted covalent PARP inhibitor. The ethynyl group at the C-6 position of the phthalazinone scaffold of DB008 imparts enhanced PARP family-wide selectively compared to its precursor HJ-52 and can be used as a click handle for CuAAC-mediated labeling of PARP16 in cell lysates. DB008 irreversibly inhibits PARP16, but not other PARP family members. Proteome-wide profiling using click chemistry demonstrated that DB008 is highly selective for PARP16 in the irreversible binding mode. PARP family-wide, activity-based screening showed that even in the reversible binding mode, DB008 exhibits good selectivity for PARP16. Indeed, PARP2 is the only other PARP that is appreciably inhibited by DB008. Because PARP16, but not PARP2, is irreversibly inhibited by DB008, cell-based studies using washout experiments will ensure that only PARP16 is inhibited. In future studies, it will be interesting to explore how modifications at the C-6 position impact enhanced PARP family-wide selectivity and potency against PARP16. Indeed, in other studies, we have seen how the hydrophobic cavity-directed substituent can influence PARP family-wide selectivity.6,19,20Our finding that nutrient stress causes PARP16 to be sequestered in a detergent-insoluble fraction is reminiscent of what has been reported for PARP2 when cells are deprived of serum.36 It will be interesting to determine if PARP2 sequestration is also dependent on its catalytic activity, similar to what we report here for PARP16. Whether the effects of DB008 and talazoparib on PARP16 solubility are due to changes in PARP16 auto-MARylation or trans-MARylation of targets that regulate PARP16 solubility is not clear. One challenge to address this question has to do with our inability to detect PARP16 MARylation in cells. This could be due to a multitude of reasons, including the potential that PARP16 modifies its targets on uncharacterized sites that are enzymatically or chemically labile. However, it is also entirely possible that the effects of DB008 and talazoparib are independent of PARP16 catalytic activity. For example, binding of DB008 or talazoparib may prevent PARP16 from unfolding, or perhaps may alter protein–protein interactions that prevent PARP16 sequestration into the pellet during starvation. Therefore, more studies need to be done to validate the role of ADP-ribosylation on protein stability or trafficking during nutrient deprivation. Additionally, the biological make-up of the RIPA-insoluble fraction needs to be explored, as it may reveal similarities to the Sec bodies discovered in drosophila.
Author contributions
D. S. B. conceptualization, investigation, methodology, validation, writing – original draft, visualization. S. S. resources (synthetic intermediates). H. J. resources (synthesized HJ-52). R. K. M. investigation (HJ-52-PARP16 whole protein ESI mass spectrometry). I. T. K. and I. R. S. resources (expression of PARPs for in vitro assays). B. T. and N. L. investigation (generated model of DB008 covalently bound to PARP16). M. S. C. conceptualization, supervision, writing – review & editing, funding acquisition.Data availability
All data have been provided in the main text and ESI.† We do not have addition data to provide.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank current and former Cohen lab members for many helpful discussions regarding experimental design, data analysis, and general advice for this project. We also thank Larry David and the OHSU proteomics core for analysis of HJ-52 covalent binding to PARP16. This work was supported by an Achievement Rewards for College Scientists (ARCS; to D. S. B.) and by the National Institutes of Neurological Disorders and Stroke 2R01NS088629 (to M. S. C.).Notes and references
- M. S. Cohen and P. Chang, Nat. Chem. Biol., 2018, 14, 236–243 CrossRef CAS PubMed.
- D. J. Sanderson and M. S. Cohen, Crit. Rev. Biochem. Mol. Biol., 2020, 55, 1–14 CrossRef PubMed.
- J. Mateo, C. J. Lord, V. Serra, A. Tutt, J. Balmaña, M. Castroviejo-Bermejo, C. Cruz, A. Oaknin, S. B. Kaye and J. S. de Bono, Ann. Oncol., 2019, 30, 1437–1447 CrossRef CAS PubMed.
- M. G. Nizi, M. M. Maksimainen, L. Lehtiö and O. Tabarrini, J. Med. Chem., 2022, 65, 7532–7560 CrossRef CAS PubMed.
- H. Zhang, P. Yu, V. S. Tomar, X. Chen, M. J. Atherton, Z. Lu, H.-G. Zhang, S. Li, A. Ortiz, J. Gui, N. A. Leu, F. Yan, A. Blanco, M. L. Meyer-Ficca, R. G. Meyer, D. P. Beiting, J. Li, S. Nunez-Cruz, R. S. O'Connor, L. R. Johnson, A. J. Minn, S. S. George, C. Koumenis, J. A. Diehl, M. C. Milone, H. Zheng and S. Y. Fuchs, Nat. Cancer, 2022, 3, 808–820 CrossRef CAS PubMed.
- I. T. Kirby, A. Kojic, M. R. Arnold, A.-G. Thorsell, T. Karlberg, A. Vermehren-Schmaedick, R. Sreenivasan, C. Schultz, H. Schüler and M. S. Cohen, Cell Chem. Biol., 2018, 25, 1547–1553.e12 CrossRef CAS PubMed.
- J. M. Gozgit, M. M. Vasbinder, R. P. Abo, K. Kunii, K. G. Kuplast-Barr, B. Gui, A. Z. Lu, J. R. Molina, E. Minissale, K. K. Swinger, T. J. Wigle, D. J. Blackwell, C. R. Majer, Y. Ren, M. Niepel, Z. A. Varsamis, S. P. Nayak, E. Bamberg, J.-R. Mo, W. D. Church, A. S. A. Mady, J. Song, L. Utley, P. E. Rao, T. J. Mitchison, K. W. Kuntz, V. M. Richon and H. Keilhack, Cancer Cell, 2021, 39, 1214–1226.e10 CrossRef CAS PubMed.
- M. Jwa and P. Chang, Nat. Cell Biol., 2012, 14, 1223–1230 CrossRef CAS PubMed.
- C. Hetz, E. Chevet and H. P. Harding, Nat. Rev. Drug Discovery, 2013, 12, 703–719 CrossRef CAS PubMed.
- V. Palve, C. E. Knezevic, D. S. Bejan, Y. Luo, X. Li, S. Novakova, E. A. Welsh, B. Fang, F. Kinose, E. B. Haura, A. N. Monteiro, J. M. Koomen, M. S. Cohen, H. R. Lawrence and U. Rix, Cell Chem. Biol., 2022, 29, 202–214.e7 CrossRef CAS PubMed.
- S. Challa, B. R. Khulpateea, T. Nandu, C. V. Camacho, K. W. Ryu, H. Chen, Y. Peng, J. S. Lea and W. L. Kraus, Cell, 2021, 184, 4531–4546.e26 CrossRef CAS PubMed.
- J. Wang, C. Zhu, D. Song, R. Xia, W. Yu, Y. Dang, Y. Fei, L. Yu and J. Wu, Cell Death Discovery, 2017, 3, 17034 CrossRef CAS PubMed.
- R. M. Centko, G. W. Carlile, I. Barne, B. O. Patrick, P. Blagojevic, D. Y. Thomas and R. J. Andersen, ACS Omega, 2020, 5, 25593–25604 CrossRef CAS PubMed.
- K. Hallenbeck, D. Turner, A. Renslo and M. Arkin, Curr. Top. Med. Chem., 2016, 17, 4–15 CrossRef PubMed.
- J. Singh, J. Med. Chem., 2022, 65, 5886–5901 CrossRef CAS PubMed.
- A.-G. Thorsell, T. Ekblad, T. Karlberg, M. Löw, A. F. Pinto, L. Trésaugues, M. Moche, M. S. Cohen and H. Schüler, J. Med. Chem., 2017, 60, 1262–1271 CrossRef CAS PubMed.
- T. J. Wigle, W. D. Church, C. R. Majer, K. K. Swinger, D. Aybar, L. B. Schenkel, M. M. Vasbinder, A. Brendes, C. Beck, M. Prahm, D. Wegener, P. Chang and K. W. Kuntz, SLAS Discovery, 2020, 25, 241–252 CrossRef CAS PubMed.
- I. T. Kirby and M. S. Cohen, Curr. Top. Microbiol., 2018, 211–231 Search PubMed.
- L. B. Schenkel, J. R. Molina, K. K. Swinger, R. Abo, D. J. Blackwell, A. Z. Lu, A. E. Cheung, W. D. Church, K. Kunii, K. G. Kuplast-Barr, C. R. Majer, E. Minissale, J.-R. Mo, M. Niepel, C. Reik, Y. Ren, M. M. Vasbinder, T. J. Wigle, V. M. Richon, H. Keilhack and K. W. Kuntz, Cell Chem. Biol., 2021, 28, 1158–1168.e13 CrossRef CAS PubMed.
- R. K. Morgan, I. T. Kirby, A. Vermehren-Schmaedick, K. Rodriguez and M. S. Cohen, ACS Med. Chem. Lett., 2018, 10, 74–79 CrossRef PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS PubMed.
- I. T. Kirby, D. J. Sanderson and M. S. Cohen, STAR Protoc., 2021, 2, 100344 CrossRef CAS PubMed.
- A.-G. Thorsell, T. Ekblad, T. Karlberg, M. Löw, A. F. Pinto, L. Trésaugues, M. Moche, M. S. Cohen and H. Schüler, J. Med. Chem., 2016, 60, 1262–1271 CrossRef PubMed.
- D. I. James, K. M. Smith, A. M. Jordan, E. E. Fairweather, L. A. Griffiths, N. S. Hamilton, J. R. Hitchin, C. P. Hutton, S. Jones, P. Kelly, A. E. McGonagle, H. Small, A. I. J. Stowell, J. Tucker, I. D. Waddell, B. Waszkowycz and D. J. Ogilvie, ACS Chem. Biol., 2016, 11, 3179–3190 CrossRef CAS PubMed.
- N. H. Cho, K. C. Cheveralls, A.-D. Brunner, K. Kim, A. C. Michaelis, P. Raghavan, H. Kobayashi, L. Savy, J. Y. Li, H. Canaj, J. Y. S. Kim, E. M. Stewart, C. Gnann, F. McCarthy, J. P. Cabrera, R. M. Brunetti, B. B. Chhun, G. Dingle, M. Y. Hein, B. Huang, S. B. Mehta, J. S. Weissman, R. Gómez-Sjöberg, D. N. Itzhak, L. A. Royer, M. Mann and M. D. Leonetti, Science, 2022, 375, eabi6983 CrossRef CAS PubMed.
- N. London, R. M. Miller, S. Krishnan, K. Uchida, J. J. Irwin, O. Eidam, L. Gibold, P. Cimermančič, R. Bonnet, B. K. Shoichet and J. Taunton, Nat. Chem. Biol., 2014, 10, 1066–1072 CrossRef CAS PubMed.
- A. Galera-Prat, J. Alaviuhkola, H. I. Alanen and L. Lehtiö, Protein Eng., Des. Sel., 2022, 35, gzac006 CrossRef PubMed.
- J. M. Strelow, J. Biomol. Screening, 2016, 22, 3–20 CrossRef PubMed.
- D. Yang, Q. Wang, G. Wei, J. Wu, Y. C. Zhu, Q. Zhu, T. Ni, X. Liu and Y. Z. Zhu, Aging, 2020, 12, 21423–21445 CrossRef CAS PubMed.
- F. Long, D. Yang, J. Wang, Q. Wang, T. Ni, G. Wei, Y. Zhu and X. Liu, Acta Pharm. Sin. B, 2021, 11, 1261–1273 CrossRef CAS PubMed.
- A. Aguilera-Gomez, M. M. van Oorschot, T. Veenendaal and C. Rabouille, Elife, 2016, 5, e21475 CrossRef PubMed.
- M. Zacharogianni, A. Aguilera-Gomez, T. Veenendaal, J. Smout and C. Rabouille, Elife, 2014, 3, e04132 CrossRef PubMed.
- J. Mejlvang, H. Olsvik, S. Svenning, J.-A. Bruun, Y. P. Abudu, K. B. Larsen, A. Brech, T. E. Hansen, H. Brenne, T. Hansen, H. Stenmark and T. Johansen, J. Cell Biol., 2018, 217, 3640–3655 CrossRef CAS PubMed.
- E. K. Schmidt, G. Clavarino, M. Ceppi and P. Pierre, Nat. Methods, 2009, 6, 275–277 CrossRef CAS PubMed.
- G. J. Brighty, R. C. Botham, S. Li, L. Nelson, D. E. Mortenson, G. Li, C. Morisseau, H. Wang, B. D. Hammock, K. B. Sharpless and J. W. Kelly, Nat. Chem., 2020, 12, 906–913 CrossRef CAS PubMed.
- Q. Sun, M. I. Gatie and G. M. Kelly, Biochem. Cell Biol., 2019, 97, 600–611 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures, methods, and detailed synthetic methods. See DOI: https://doi.org/10.1039/d2sc04820e |
| This journal is © The Royal Society of Chemistry 2022 |