
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTen-step asymmetric total syntheses of potent antibiotics anthracimycin and anthracimycin B†
Peilin
Tian
a,
Wenkang
Ye
b,
Xiayan
Zhang
a,
Yi
Tong
a,
Pei-Yuan
Qian
 *bc and
Rongbiao
Tong
*ac
*bc and
Rongbiao
Tong
*ac
aDepartment of Chemistry, The Hong Kong University of Science and Technology, Clearwater Bay, Kowloon, Hong Kong, China. E-mail: rtong@ust.hk; Fax: +86 23581594; Tel: +86 23587357
bDepartment of Ocean Science, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: boqianpy@ust.hk
cThe Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), Nansha, Guangzhou 511458, China
First published on 15th October 2022
Abstract
The increase in antibiotic resistance calls for the development of novel antibiotics with new molecular structures and new modes of action. However, in the past few decades only a few novel antibiotics have been discovered and progressed into clinically used drugs. The discovery of a potent anthracimycin antibiotic represents a major advance in the field of antibiotics. Anthracimycin is a structurally novel macrolide natural product with an excellent biological activity profile: (i) potent in vitro antibacterial activity (MIC 0.03–1.0 μg mL−1) against many methicillin-resistant Staphylococcus aureus (MRSA) strains, Bacillus anthracis (anthrax), and Mycobacterium tuberculosis; (ii) low toxicity to human cells (IC50 > 30 μM); (iii) a novel mechanism of action (inhibiting DNA/RNA synthesis). While the first total synthesis of anthracimycin was elegantly accomplished by Brimble et al. with 20 steps, we report a 10-step asymmetric total synthesis of anthracimycin and anthracimycin B (first total synthesis). Our convergent strategy features (i) one-pot sequential Mukaiyama vinylogous aldol/intramolecular Diels–Alder reaction to construct trans-decalin with high yield and excellent endo/exo selectivity and (ii) Z-selective ring-closing metathesis to forge the 14-membered ring. In vitro antibacterial evaluation suggested that our synthetic samples exhibited similar antibacterial potency to the naturally occurring anthracimycins against Gram-positive strains. Our short and reliable synthetic route provides a supply of anthracimycins for further in-depth studies and allows medicinal chemists to prepare a library of analogues for establishing structure–activity relationships.
Introduction
The alarming prevalence of bacteria resistant to multiple antibiotics raises a global concern on the availability of new antibiotics effective against such “superbugs” that are resistant to most or all known antibiotics.1,2 The search for new antibiotics with different strategies including bioassay-guided isolation of new antibacterial natural products, genome mining,3–8 and chemical synthesis/modifications of existing antibiotics9,10 continues to play an important role in addressing antibiotic resistance.11–14 However, only a few structurally novel antibacterial compounds have been discovered in the past five decades, and anthracimycin15 was one of these new antibiotics with a novel molecular structure and high antibacterial potency. Anthracimycin was discovered in 2013 by Fenical and co-worker from marine microorganism Streptomyces species (CNH365) to be a structurally novel potent antibiotic against Gram-positive bacteria (MIC 0.03–0.5 μg mL−1) including notorious methicillin-resistant Staphylococcus aureus (MRSA)16 and the bacterium Bacillus anthracis that has been used as a bioterrorism weapon to cause human-infectious anthrax (Fig. 1). In 2018, Reyes et al.17 identified 2-demethyl anthracimycin (anthracimycin B) from cultures of the deep-sea actinomycete Streptomyces cyaneofuscantus M-169 and found that it exhibited similar but less potent antibacterial activity against Gram-positive bacteria. Another anthramycin analogue (anthracimycin BII-2619) was also identified in 2018 by Sirota et al.18 through a genome mining strategy to display Gram-positive antibacterial activity with lower mammalian cytotoxicity compared to that of anthracimycin. In 2020, Fukuda19 and co-workers isolated 2-epi-anthracimycin along with anthracimycin and evaluated its cytotoxicity against Jurkat cells (IC50 = 50.5 μM). While most macrolide antibiotics bind to the 50S ribosomal subunit to inhibit protein biosynthesis,20 it was suggested that anthracimycin exerts its bactericidal effect through inhibiting DNA and/or RNA synthesis in the absence of DNA intercalation.21 The high antibacterial potency, new molecular structure, and novel mechanism of action make anthracimycin an ideal target for chemical synthesis and development of novel antibiotics.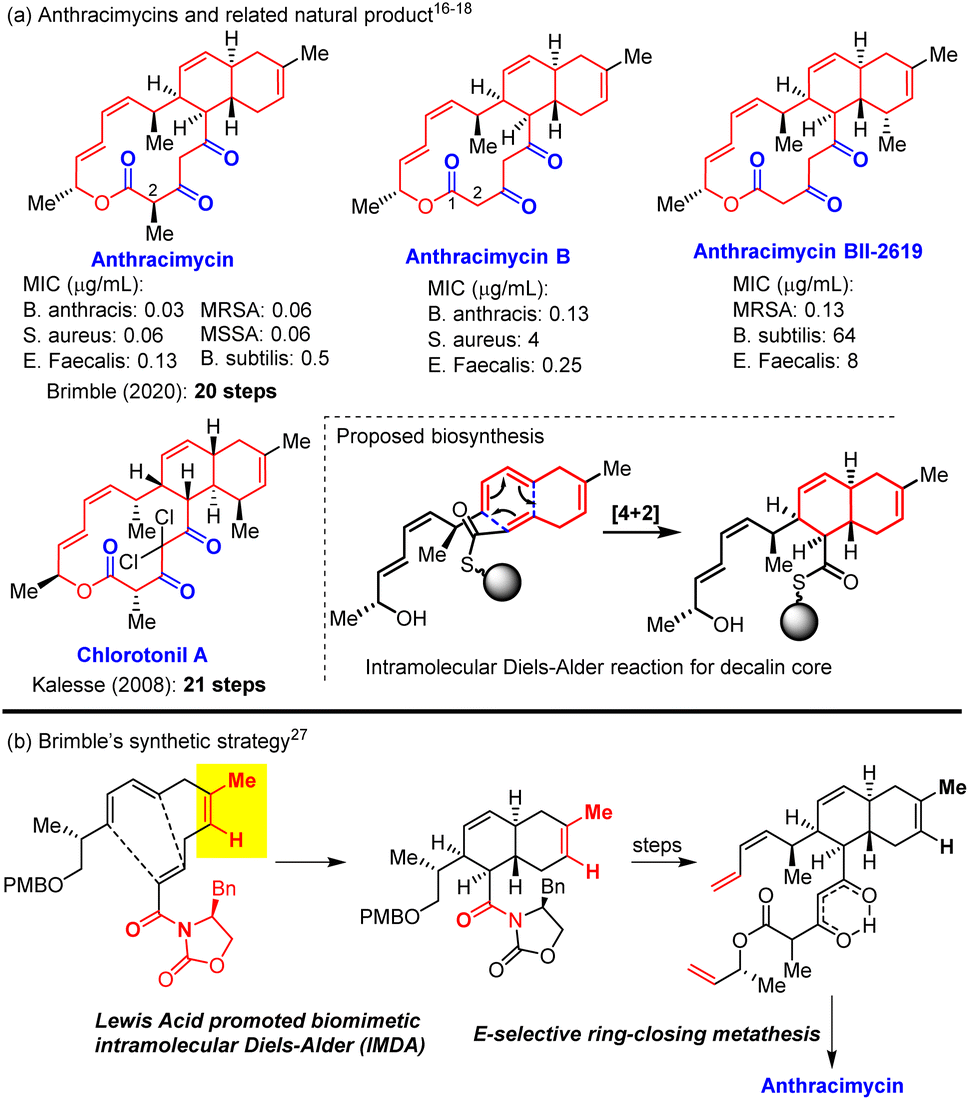 | ||
| Fig. 1 Anthracimycin natural products. | ||
In 2019, two PhD theses (J. L. Freeman22,23 and G. Lodovici24) disclosed detailed synthetic studies towards anthracimycin and presented a significant synthetic challenge of using the biomimetic intramolecular Diels–Alder (IMDA) reaction of the tetraene substrate (Scheme 1b)25,26 to construct the decalin core. Further optimization of Freeman's thesis work enabled Brimble27 and co-workers to achieve the first total synthesis of anthracimycin in 20 steps with 0.4 mg pure anthracimycin. It was noted that Kalesse et al.28 accomplished the total synthesis of structurally related chlorotonil A29 in 21 steps by using the halogen-directed IMDA reaction and macrolactonization as the key steps. In this paper, we report a 10-step asymmetric total synthesis of anthracimycin and anthracimycin B (the first total synthesis) with substantially improved overall yield (>3% overall yield, 25 mg and 14 mg obtained, respectively).
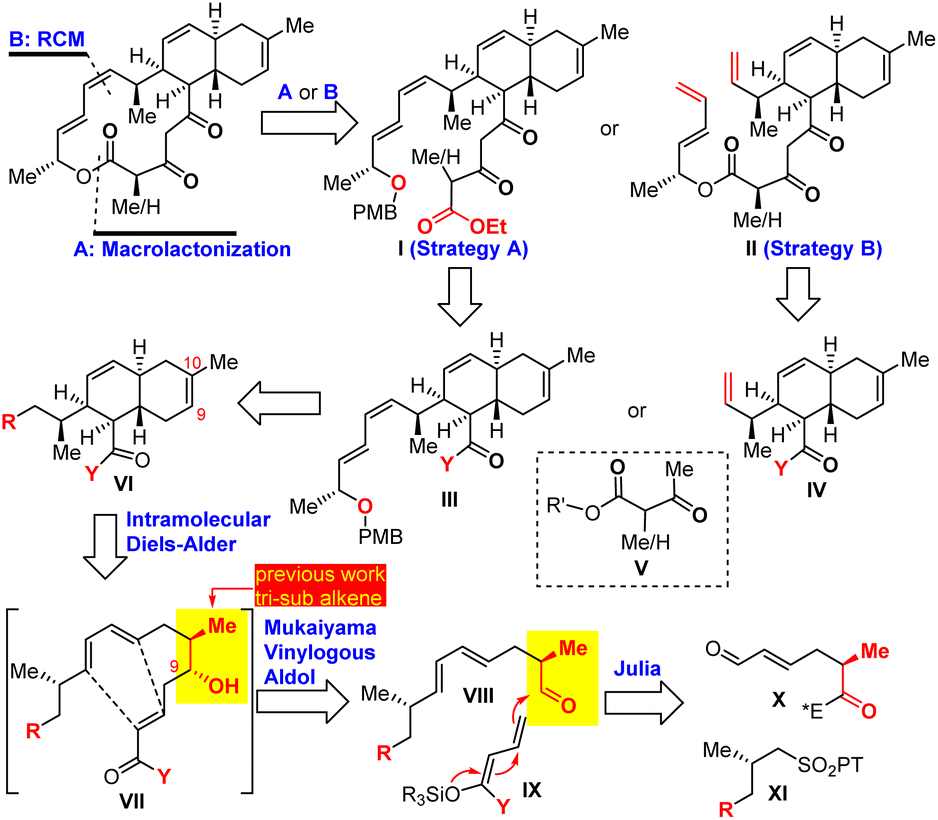 | ||
| Scheme 1 Retrosynthetic analysis of anthracimycins. | ||
Results and discussion
The key synthetic challenge of anthracimycin in the previous synthetic studies22–24,27 was the inefficient construction of the trans-decalin core through the biomimetic intramolecular Diels–Alder (IMDA) reaction of the tetraene substrate. To address this challenge, we proposed an IMDA reaction of an “unusual” triene substrate VII with a C9 secondary alcohol (Scheme 1), which served (1) as the precursor of the trisubstituted Z-9,10Δ alkene, (2) to enhance the endo/exo selectivity and diastereoselectivity, and (3) to improve the convergency of the synthesis because the presence of the alcohol would allow the fragment union through C–C bond formation with many well-established transformations (i.e., aldol, allylation, Grignard addition, etc.). With this central ideal in mind, our retrosynthetic analysis was conceived as depicted in Scheme 1. The 14-membered macrocycle would be forged in the last step by either macrolactonization28 of I or Z-selective ring-closing metathesis (RCM) of II. Notably, Z-selective RCM might be advantageous since the preparation of the more challenging Z-configured alkene substrate at an early stage of synthesis was eliminated. Both I and II can be obtained by condensation of 1,3-dicarbonyl V with III and IV, respectively. The designed triene VII substrate for the IMDA reaction could be easily synthesized by Julia–Kocienski olefination30 of aldehyde X and tetrazole XI followed by the Mukaiyama vinylogous aldol (MVA)31–35 reaction of aldehyde VIII and O,O-silyl ketene acetal IX.36Our synthesis consisted of two stages: trans-decalin synthesis and macrolactone synthesis as depicted in Scheme 2. We started with the preparation of known compound 2 (ref. 37 and 38) in 86% yield by Evans asymmetric alkylation39 of commercially available (S)-4-benzyl-3-propionyl-2-oxazolidinone (1). Olefin cross metathesis of 2 with crotonaldehyde delivered 3 as an exclusive E-configuration in 85% yield. Julia–Kocienski olefination of N-phenyl tetrazole sulfone 4a (ref. 22 and 40) with aldehyde 3 followed by Dess–Martin periodinane (DMP) oxidation provided (E,E)-diene 5a (E/Z > 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which was subjected to our planned Lewis acid-promoted Mukaiyama vinylogous aldol (MVA) reaction with the known silyl ketene acetal 6.36,41 Triarylborane [B(C6F5)3]42 was found to effectively promote MVA to provide triene 7 in 71% yield with a 6:1 diastereomeric ratio. The thermal IMDA reaction of triene 7 was first attempted at 165 °C in dichlorobenzene to produce a mixture of endo/exo products (endo/exo: 2.3:1) with 82% combined yield. To improve the endo/exo selectivity, we used BF3–Et2O to promote the IMDA reaction,29 which resulted in decomposition. Fortunately, we found that Et2AlCl (4.0 equiv.) was an effective promoter for the IMDA reaction to provide the desired trans-decalin as a mixture of 8a and 8b, which upon treatment with 37% HCl for removal of the TBS protecting group as well as lactonization generated tricyclic lactone 9 (dr > 15:1) in 87% yield. It was noted that Et2AlCl removed the PMB protecting group at −78 °C as evidenced by TLC analysis and then the IMDA reaction occurred at room temperature. Therefore, we believe that the excellent selectivity and the high yield of this transformation stem from the pre-formation of an aluminum-coordinated macrocycle intermediate (or transition state), which then undergoes an effective and endo-selective transannular Diels–Alder reaction. This result was remarkable regarding the yield, endo/exo selectivity and diastereoselectivity as compared to the previous IMDA reaction of tetraene substrates.22–24,27 Regioselective dehydration of 9 was achieved by mesylation (MsCl/Et3N) and elimination with LiBr/Li2CO3/DMF43,44 to afford trans-decalin 10 (92% yield), whose structure was confirmed by single crystal X-ray diffraction. We accomplished the first stage of the efficient trans-decalin synthesis and turned our attention to the second stage of the macrolactone synthesis. We first explored BF3–Et2O-mediated macrolactonization.29 The lactone ring of decalin 10 was hydrolyzed with KOH and the resulting unstable hydroxyl carboxylic acid was treated with TMSCHN2 to afford the corresponding ester 10′, which was noticeably unstable under either mild acidic or basic conditions and should be oxidized immediately with DMP to provide aldehyde 11. While attempting to elaborate aldehyde 11 into (E,Z)-diene 14a by Stork–Zhao olefination and Stille coupling,29 we encountered an unexpected difficulty in the formation of (Z)-vinyl iodide. Stork–Zhao olefination45 of aldehyde 11 under various conditions led to a E/Z mixture of vinyl iodide (E/Z 1:1). Alternatively, we attempted Z-selective Still–Gennari olefination46 of aldehyde 11 without success because (E,Z)-14a was obtained as a minor isomer [(E,Z)/(E,E): 1:2.9 to 1:10]. To solve the poor Z-selectivity in the 1,3-diene synthesis, we decided to explore Sonogashira coupling and partial hydrogenation with the Lindlar catalyst. Aldehyde 11 was converted into alkyne 12a with the Bestmann reagent, and then Sonogashira coupling with vinyl iodide 13a followed by Lindlar hydrogenation provided the desired (E,Z)-diene 14a in 64% yield over two steps. The installation of 1,3,5-tricarbonyls (14a → 16a) was challenging: Claisen condensation of ester 14a with 1,3-dicarbonyl 15a was not successful, while a four-step sequence (DIBAL-H reduction, DMP oxidation, Aldol reaction, and DMP oxidation) delivered 16a with 27% yield. Nevertheless, with tens of milligrams of 16a in hand, we explored the BF3·Et2O-mediated macrolactonization, but we were not able to identify any desired product from many attempted reactions under various conditions (temperature and equivalent of BF3·Et2O). TLC and NMR analysis clearly suggested that 16a decomposed in the presence of BF3·Et2O. Then, we investigated the second macrocyclization strategy: RCM. Because Z-selective Stork–Zhao olefination failed to produce Z-vinyl iodide for subsequent Stille coupling, we decided to take a risk: Z-alkene formation by RCM as the last step, which contrasts with the previous RCM for E-alkene formation.27 To verify this macrocyclization method, we prepared two substrates 16b and 16c, corresponding to anthracimycin and anthracimycin B, respectively. Wittig olefination of 11 followed by ester reduction with DIBAL-H and DMP oxidation afforded aldehyde 13b with 46% yield over 3 steps. The subsequent aldol reaction of 13b with dienolate of 1,3-dicarbonyl 14b/14c followed by DMP oxidation provided the key macrocyclization substrates 16b and 16c in 42% and 44% yield over two steps, respectively. The planned RCM of 16b and 16c proceeded smoothly with the Hoveyda–Grubbs II catalyst to furnish anthracimycin and anthracimycin B (exclusive Z-double bond), respectively, with >60% yield based on the crude NMR analysis (42–43% isolated yield). The NMR data of our synthetic samples were consistent with those reported for natural products (see ESI Tables S1–S4†). Additionally, single crystals of anthracimycin were obtained for X-ray diffraction analysis, which further confirmed the structure of our synthetic anthracimycin.
:1), which was subjected to our planned Lewis acid-promoted Mukaiyama vinylogous aldol (MVA) reaction with the known silyl ketene acetal 6.36,41 Triarylborane [B(C6F5)3]42 was found to effectively promote MVA to provide triene 7 in 71% yield with a 6:1 diastereomeric ratio. The thermal IMDA reaction of triene 7 was first attempted at 165 °C in dichlorobenzene to produce a mixture of endo/exo products (endo/exo: 2.3:1) with 82% combined yield. To improve the endo/exo selectivity, we used BF3–Et2O to promote the IMDA reaction,29 which resulted in decomposition. Fortunately, we found that Et2AlCl (4.0 equiv.) was an effective promoter for the IMDA reaction to provide the desired trans-decalin as a mixture of 8a and 8b, which upon treatment with 37% HCl for removal of the TBS protecting group as well as lactonization generated tricyclic lactone 9 (dr > 15:1) in 87% yield. It was noted that Et2AlCl removed the PMB protecting group at −78 °C as evidenced by TLC analysis and then the IMDA reaction occurred at room temperature. Therefore, we believe that the excellent selectivity and the high yield of this transformation stem from the pre-formation of an aluminum-coordinated macrocycle intermediate (or transition state), which then undergoes an effective and endo-selective transannular Diels–Alder reaction. This result was remarkable regarding the yield, endo/exo selectivity and diastereoselectivity as compared to the previous IMDA reaction of tetraene substrates.22–24,27 Regioselective dehydration of 9 was achieved by mesylation (MsCl/Et3N) and elimination with LiBr/Li2CO3/DMF43,44 to afford trans-decalin 10 (92% yield), whose structure was confirmed by single crystal X-ray diffraction. We accomplished the first stage of the efficient trans-decalin synthesis and turned our attention to the second stage of the macrolactone synthesis. We first explored BF3–Et2O-mediated macrolactonization.29 The lactone ring of decalin 10 was hydrolyzed with KOH and the resulting unstable hydroxyl carboxylic acid was treated with TMSCHN2 to afford the corresponding ester 10′, which was noticeably unstable under either mild acidic or basic conditions and should be oxidized immediately with DMP to provide aldehyde 11. While attempting to elaborate aldehyde 11 into (E,Z)-diene 14a by Stork–Zhao olefination and Stille coupling,29 we encountered an unexpected difficulty in the formation of (Z)-vinyl iodide. Stork–Zhao olefination45 of aldehyde 11 under various conditions led to a E/Z mixture of vinyl iodide (E/Z 1:1). Alternatively, we attempted Z-selective Still–Gennari olefination46 of aldehyde 11 without success because (E,Z)-14a was obtained as a minor isomer [(E,Z)/(E,E): 1:2.9 to 1:10]. To solve the poor Z-selectivity in the 1,3-diene synthesis, we decided to explore Sonogashira coupling and partial hydrogenation with the Lindlar catalyst. Aldehyde 11 was converted into alkyne 12a with the Bestmann reagent, and then Sonogashira coupling with vinyl iodide 13a followed by Lindlar hydrogenation provided the desired (E,Z)-diene 14a in 64% yield over two steps. The installation of 1,3,5-tricarbonyls (14a → 16a) was challenging: Claisen condensation of ester 14a with 1,3-dicarbonyl 15a was not successful, while a four-step sequence (DIBAL-H reduction, DMP oxidation, Aldol reaction, and DMP oxidation) delivered 16a with 27% yield. Nevertheless, with tens of milligrams of 16a in hand, we explored the BF3·Et2O-mediated macrolactonization, but we were not able to identify any desired product from many attempted reactions under various conditions (temperature and equivalent of BF3·Et2O). TLC and NMR analysis clearly suggested that 16a decomposed in the presence of BF3·Et2O. Then, we investigated the second macrocyclization strategy: RCM. Because Z-selective Stork–Zhao olefination failed to produce Z-vinyl iodide for subsequent Stille coupling, we decided to take a risk: Z-alkene formation by RCM as the last step, which contrasts with the previous RCM for E-alkene formation.27 To verify this macrocyclization method, we prepared two substrates 16b and 16c, corresponding to anthracimycin and anthracimycin B, respectively. Wittig olefination of 11 followed by ester reduction with DIBAL-H and DMP oxidation afforded aldehyde 13b with 46% yield over 3 steps. The subsequent aldol reaction of 13b with dienolate of 1,3-dicarbonyl 14b/14c followed by DMP oxidation provided the key macrocyclization substrates 16b and 16c in 42% and 44% yield over two steps, respectively. The planned RCM of 16b and 16c proceeded smoothly with the Hoveyda–Grubbs II catalyst to furnish anthracimycin and anthracimycin B (exclusive Z-double bond), respectively, with >60% yield based on the crude NMR analysis (42–43% isolated yield). The NMR data of our synthetic samples were consistent with those reported for natural products (see ESI Tables S1–S4†). Additionally, single crystals of anthracimycin were obtained for X-ray diffraction analysis, which further confirmed the structure of our synthetic anthracimycin.
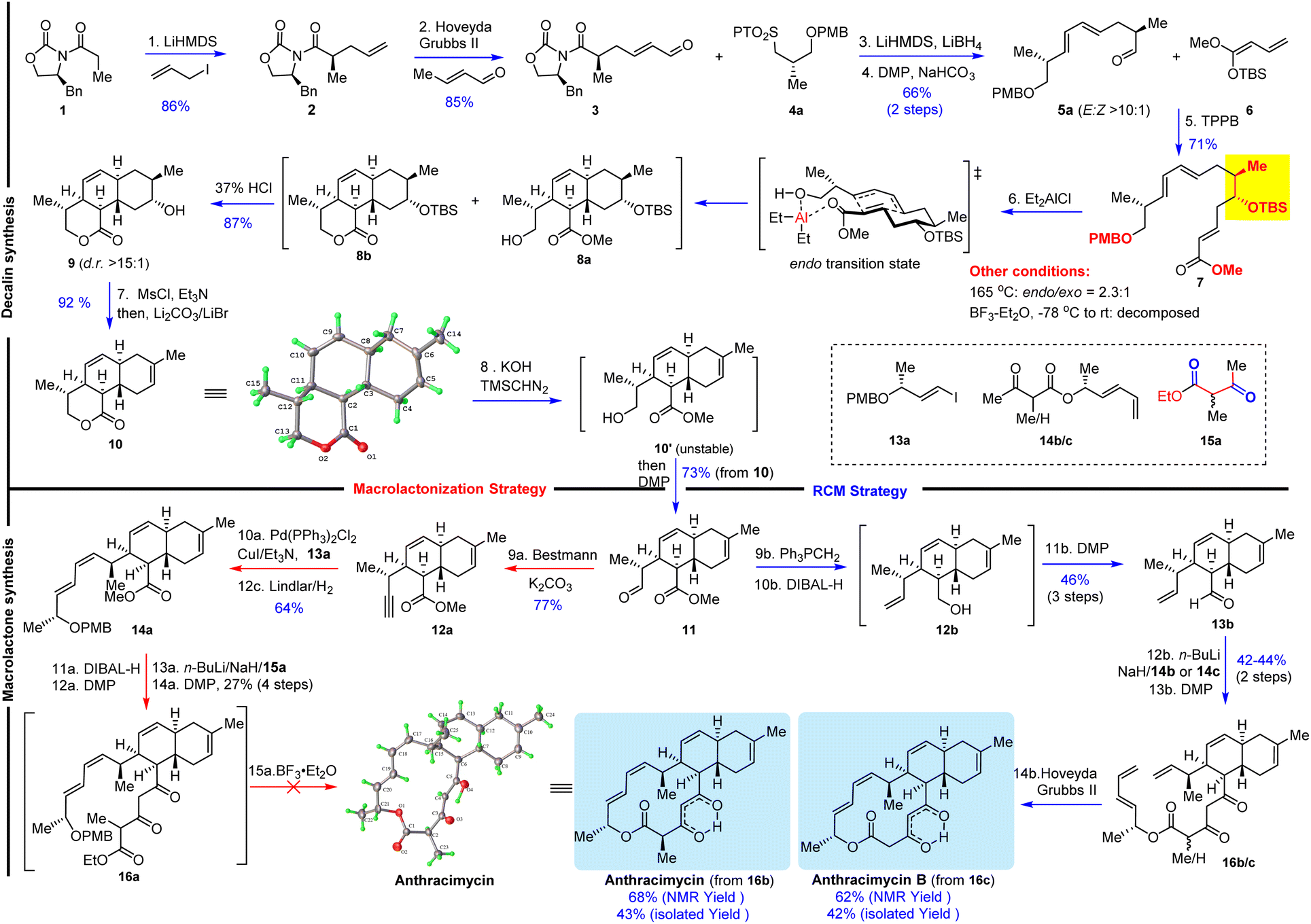 | ||
| Scheme 2 Asymmetric total synthesis of anthracimycin and anthracimycin B. | ||
Since our total synthesis required 14 steps, we set out to further optimize our synthetic route (Scheme 3). First, we employed N-phenyl tetrazole sulfone 4b,47 instead of 4a (Scheme 2), for Julia–Kocienski olefination with aldehyde 3 so that the late-stage conversion of lactone to vinyl ester (9 → 12b, Scheme 2) was unnecessary (and thus saving us 4 steps). Reductive removal of the Evans chiral auxiliary and DMP oxidation of the resulting alcohol afforded aldehyde 5b (62% yield) with 6:1 E/Z selectivity for the newly-formed alkene. Next, we explored the possibility of one-pot sequential Mukaiyama vinylogous aldol (MVA)/intramolecular Diels–Alder (IMDA) reaction since both involved a Lewis acid as a promoter. After examining several acid promoters including TiCl4, BF3–Et2O, CBS-HOTf, proline, Rh(I) and B(C6F5)3, we found that the sequential MVA and IMDA reactions could be achieved in one pot by treating the reaction mixture first with B(C6F5)3 and then with Et2AlCl to provide a mixture of trans-decalins 8c and 8d (63% combined yield on a 1.1 g scale) with a 3:1 diastereomeric ratio in favor of the desired 8c. Although the diastereoselectivity of the IMDA reaction was moderate as compared to substrate 7 probably due to the lack of a macrocyclic intermediate (or a transition state) for the highly diastereoselective transannular Diels–Alder reaction, the overall efficiency was much higher. Dehydration with the Burgess reagent allowed DIBAL-H reduction of the ester to be carried out in one pot. DMP oxidation of the resulting alcohol provided aldehyde 13b (66% yield over two steps), which was elaborated to anthracimycin and anthracimycin B following the same protocol as in Scheme 2. The optimized route for 13b allowed us to accomplish the total synthesis of anthracimycin and anthracimycin B in 10 steps with 3.3–3.6% isolated overall yield from commercially available Evans chiral auxiliary 1 without using protecting groups and with only 7 isolated intermediates. The concise and efficient route enabled us to prepare 25 mg of anthracimycin and 14 mg of anthracimycin B, which allowed us to evaluate their antibacterial activity.
 | ||
| Scheme 3 Optimized route for the synthesis of trans-decalin 13b. | ||
It is well recognized that the bioactivity of newly isolated natural products might be different from that of the corresponding synthetic samples (possibly due to contamination of natural samples). Therefore, it is imperative to verify their bioactivity using synthetic samples. The antibacterial activity of our synthetic anthracimycin and anthracimycin B was evaluated against several pathogenic strains, including Gram-negative strains: A. baumannii B-65371, E. cloacae NRRL-B-425, E. coli k12, and K. pneumoniae NRRL-B-408, and Gram-positive strains: MRSA ATCC 43300, S. aureus ATCC 25923, B. subtilis zk31 and M. luteus ATCC 10040. As shown in Table 1, synthetic anthracimycin and anthracimycin B were not effective against Gram-negative strains but they were potent against the Gram-positive bacteria (MIC: 0.03–0.8 μg mL−1), which is consistent with MIC values reported for natural anthracimycins.15,17,18
| Strains/cells | Ant | Ant B | Van |
|---|---|---|---|
| a The experimental results are expressed as the MIC, MBIC and IC50 (μg mL−1). All the bioactivity assays of compounds were performed three times. In antimicrobial experiments, kanamycin and vancomycin were used as positive controls. Ant: anthracimycin; Ant B: anthracimycin B; Van: vancomycin; HaCaT cells are human keratinocytes. | |||
| A. baumannii: B65371 | >40 | >40 | >40 |
| E. cloacae: NRRL-B-425 | >40 | >40 | >40 |
| E. coli: K12 | >40 | >40 | >40 |
| K. pneumoniae: NRRL-B-408 | >40 | >40 | >40 |
| MRSA: ATCC43300 | 0.03 ± 0.001 | 0.7 ± 0.13 | 0.8 ± 0.04 |
| S. aureus: ATCC 25923 | 0.04 ± 0.004 | 0.3 ± 0.023 | 0.8 ± 0.002 |
| MRSA ATCC29213 | 0.04 ± 0.004 | 1.0 ± 0.11 | 0.4 ± 0.045 |
| MRSA ATCC29213 Biofilm (MBIC) | 0.02 ± 0.006 | 0.6 ± 0.19 | 1.2 ± 0.094 |
| MRSA Sa115 | 0.04 ± 0.005 | 1.0 ± 0.094 | 0.4 ± 0.038 |
| S. aureus R2952 | 0.04 ± 0.047 | 1.0 ± 0.094 | 0.4 ± 0.023 |
| B. subtilis: zk31 | 0.04 ± 0.005 | 0.5 ± 0.075 | 0.07 ± 0.007 |
| M. luteus: ATCC 10040 | 0.02 ± 0.009 | 0.8 ± 0.024 | 1.6 ± 0.094 |
| HaCaT (IC50) | 14.74 ± 1.32 | 14.90 ± 1.93 | |
A cytotoxicity study using HaCaT (cell line from human skin) suggested that both synthetic anthracimycins possessed low cytotoxicity (IC50 > 14 μg mL−1), which holds great promise for further drug development. More interestingly, we found that both anthracimycins significantly inhibited the MRSA biofilm formation (MRSA ATCC29213) with MBIC values (MBIC 0.02 μg mL−1 and 0.6 μg mL−1, respectively) even lower than the respective MIC values. As compared to vancomycin, a frontline antibiotic for treatment of MRSA infections, anthracimycin was 60 times more potent. This result is significant because MRSA biofilm formation48–51 is primarily responsible for the reduced efficacy or inefficacy of many clinically used antibiotics for MRSA infections, and anthracimycin demonstrated potential as a new effective anti-MRSA antibiotic.
Conclusion
In summary, we have accomplished a 10-step asymmetric total synthesis of anthracimycin and anthracimycin B (the first total synthesis) with 3.3–3.6% overall isolated yields. The key strategy is the use of alcohol as the latent alkene to enable convergent fragment coupling (vinylogous Mukaiyama aldol) and to enhance the endo/exo selectivity and yield of the intramolecular Diels–Alder reaction. Additionally, the ring-closing metathesis using the Hoveyda–Grubbs II catalyst allowed the efficient construction of a 14-membered macrolide with exclusive Z-alkene selectivity. Our short and efficient synthetic route enabled us to prepare 25 mg of anthracimycin and 14 mg of anthracimycin B and allowed us to evaluate their antibacterial activities using our synthetic sample, which were consistent with those reported for the natural anthracimycins. Finally, we found that our synthetic anthracimycin significantly inhibited the MRSA biofilm formation with great promise to be a new effective anti-MRSA antibiotic.Data availability
Experimental procedures and characterization data are available within this article and its ESI.† Data are also available from the corresponding author on request.Author contributions
P. T., X. Z. and Y. T. performed the synthetic experiments. W. Y. performed the biological experiments. P. Q. and R. T. conceptualized and directed the project, and drafted the manuscript with the assistance from all co-authors.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research was financially supported by the Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (SMSEGL20Sc01-B) and Research Grant Council of Hong Kong (C6026-19G, 16307219, 16304618, and 16303617). Prof. Wei Shen Aik and Prof. Jun (Joelle) Wang from Hong Kong Baptist University are gratefully acknowledged for X-ray crystallographic analysis.Notes and references
- C. L. Ventola, Pharm. Ther., 2015, 40, 277–283 Search PubMed.
- S. Baker, Science, 2015, 347, 1064–1066 CrossRef CAS.
- E. J. Culp, N. Waglechner, W. Wang, A. A. Fiebig-Comyn, Y.-P. Hsu, K. Koteva, D. Sychantha, B. K. Coombes, M. S. Van Nieuwenhze, Y. V. Brun and G. D. Wright, Nature, 2020, 578, 582–587 CrossRef CAS.
- G. L. Challis, J. Med. Chem., 2008, 51, 2618–2628 CrossRef CAS PubMed.
- Y.-X. Li, Z. Zhong, W.-P. Zhang and P.-Y. Qian, Nat. Commun., 2018, 9, 3273 CrossRef.
- E. C. O'Neill, M. Schorn, C. B. Larson and N. Millán-Aguiñaga, Crit. Rev. Microbiol., 2019, 45, 255–277 CrossRef.
- L. Foulston, Curr. Opin. Microbiol., 2019, 51, 1–8 CrossRef CAS PubMed.
- N. Ziemert, M. Alanjary and T. Weber, Nat. Prod. Rep., 2016, 33, 988–1005 RSC.
- P. M. Wright, I. B. Seiple and A. G. Myers, Angew. Chem., Int. Ed., 2014, 53, 8840–8869 CrossRef CAS PubMed.
- S. E. Rossiter, M. H. Fletcher and W. M. Wuest, Chem. Rev., 2017, 117, 12415–12474 CrossRef CAS.
- A. Luther, M. Urfer, M. Zahn, M. Müller, S.-Y. Wang, M. Mondal, A. Vitale, J.-B. Hartmann, T. Sharpe, F. L. Monte, H. Kocherla, E. Cline, G. Pessi, P. Rath, S. M. Modaresi, P. Chiquet, S. Stiegeler, C. Verbree, T. Remus, M. Schmitt, C. Kolopp, M.-A. Westwood, N. Desjonquères, E. Brabet, S. Hell, K. LePoupon, A. Vermeulen, R. Jaisson, V. Rithié, G. Upert, A. Lederer, P. Zbinden, A. Wach, K. Moehle, K. Zerbe, H. H. Locher, F. Bernardini, G. E. Dale, L. Eberl, B. Wollscheid, S. Hiller, J. A. Robinson and D. Obrecht, Nature, 2019, 576, 452–458 CrossRef CAS PubMed.
- T. F. Durand-Reville, A. A. Miller, J. P. O'Donnell, X. Wu, M. A. Sylvester, S. Guler, R. Iyer, A. B. Shapiro, N. M. Carter, C. Velez-Vega, S. H. Moussa, S. M. McLeod, A. Chen, A. M. Tanudra, J. Zhang, J. Comita-Prevoir, J. A. Romero, H. Huynh, A. D. Ferguson, P. S. Horanyi, S. J. Mayclin, H. S. Heine, G. L. Drusano, J. E. Cummings, R. A. Slayden and R. A. Tommasi, Nature, 2021, 597, 698–702 CrossRef CAS PubMed.
- M. J. Mitcheltree, A. Pisipati, E. A. Syroegin, K. J. Silvestre, D. Klepacki, J. D. Mason, D. W. Terwilliger, G. Testolin, A. R. Pote, K. J. Y. Wu, R. P. Ladley, K. Chatman, A. S. Mankin, Y. S. Polikanov and A. G. Myers, Nature, 2021, 599, 507–512 CrossRef CAS PubMed.
- Z. Wang, B. Koirala, Y. Hernandez, M. Zimmerman, S. Park, D. S. Perlin and S. F. Brady, Nature, 2022, 601, 606–611 CrossRef CAS PubMed.
- K. H. Jang, S.-J. Nam, J. B. Locke, C. A. Kauffman, D. S. Beatty, L. A. Paul and W. Fenical, Angew. Chem. Int. Ed., 2013, 52, 7822–7824 CrossRef CAS.
- J. Larsen, C. L. Raisen, X. Ba, N. J. Sadgrove, G. F. Padilla-González, M. S. J. Simmonds, I. Loncaric, H. Kerschner, P. Apfalter, R. Hartl, A. Deplano, S. Vandendriessche, B. Černá Bolfíková, P. Hulva, M. C. Arendrup, R. K. Hare, C. Barnadas, M. Stegger, R. N. Sieber, R. L. Skov, A. Petersen, Ø. Angen, S. L. Rasmussen, C. Espinosa-Gongora, F. M. Aarestrup, L. J. Lindholm, S. M. Nykäsenoja, F. Laurent, K. Becker, B. Walther, C. Kehrenberg, C. Cuny, F. Layer, G. Werner, W. Witte, I. Stamm, P. Moroni, H. J. Jørgensen, H. de Lencastre, E. Cercenado, F. García-Garrote, S. Börjesson, S. Hæggman, V. Perreten, C. J. Teale, A. S. Waller, B. Pichon, M. D. Curran, M. J. Ellington, J. J. Welch, S. J. Peacock, D. J. Seilly, F. J. E. Morgan, J. Parkhill, N. F. Hadjirin, J. A. Lindsay, M. T. G. Holden, G. F. Edwards, G. Foster, G. K. Paterson, X. Didelot, M. A. Holmes, E. M. Harrison and A. R. Larsen, Nature, 2022, 602, 135–141 CrossRef CAS PubMed.
- V. Rodríguez, J. Martín, A. Sarmiento-Vizcaíno, M. De La Cruz, L. García, G. Blanco and F. Reyes, Mar. Drugs, 2018, 16, 406 CrossRef PubMed.
- F. L. Sirota, F. Goh, K. N. Low, L. K. Yang, S. C. Crasta, B. Eisenhaber, F. Eisenhaber, Y. Kanagasundaram and S. B. Ng, J. Genet. Genomicsics, 2018, 6, 63–73 Search PubMed.
- T. Fukuda, K. Nagai, A. Kanamoto and H. Tomoda, J. Antibiot., 2020, 73, 548–553 CrossRef CAS PubMed.
- J. W. Corcoran, Mode of action and resistance mechanisms of macrolides, Academic Press, Orlando, FL, 1984 Search PubMed.
- M. E. Hensler, K. H. Jang, W. Thienphrapa, L. Vuong, D. N. Tran, E. Soubih, L. Lin, N. M. Haste, M. L. Cunningham, B. P. Kwan, K. J. Shaw, W. Fenical and V. Nizet, J. Antibiot., 2014, 67, 549–553 CrossRef CAS.
- J. L. Freeman, M. A. Brimble and D. P. Furkert, Org. Chem. Front., 2019, 6, 2954–2963 RSC.
- J. L. Freeman, PhD, University of Auckland, 2019.
- G. Lodovici, PhD, University of York, 2019.
- E. Harunari, H. Komaki and Y. Igarashi, J. Antibiot., 2016, 69, 403–405 CrossRef CAS.
- S. Alt and B. Wilkinson, ACS Chem. Biol., 2015, 10, 2468–2479 CrossRef CAS.
- E. K. Davison, J. L. Freeman, W. Zhang, W. M. Wuest, D. P. Furkert and M. A. Brimble, Org. Lett., 2020, 22, 5550–5554 CrossRef CAS.
- N. Rahn and M. Kalesse, Angew. Chem., Int. Ed., 2008, 47, 597–599 CrossRef CAS.
- K. Gerth, H. Steinmetz, G. Höfle and R. Jansen, Angew. Chem., Int. Ed., 2008, 47, 600–602 CrossRef CAS.
- B. Chatterjee, S. Bera and D. Mondal, Tetrahedron: Asymmetry, 2014, 25, 1–55 CrossRef CAS.
- G. Casiraghi, F. Zanardi, G. Appendino and G. Rassu, Chem. Rev., 2000, 100, 1929–1972 CrossRef CAS PubMed.
- G. Casiraghi, L. Battistini, C. Curti, G. Rassu and F. Zanardi, Chem. Rev., 2011, 111, 3076–3154 CrossRef CAS PubMed.
- S. E. Denmark, J. R. Heemstra Jr and G. L. Beutner, Angew. Chem., Int. Ed., 2005, 44, 4682–4698 CrossRef CAS.
- M. Cordes and M. Kalesse, Molecules, 2019, 24, 3040 CrossRef.
- M. Kalesse, M. Cordes, G. Symkenberg and H.-H. Lu, Nat. Prod. Rep., 2014, 31, 563–594 RSC.
- R. V. Hoffman and H. O. Kim, J. Org. Chem., 1991, 56, 1014–1019 CrossRef CAS.
- X. Xiong, Y. Wu and B. Liu, Eur. J. Org. Chem., 2020, 2020, 948–960 CrossRef CAS.
- C. W. Reed, M. G. Fulton, K. D. Nance and C. W. Lindsley, Tetrahedron Lett., 2019, 60, 743–745 CrossRef CAS.
- D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 1737–1739 CrossRef CAS.
- A. Fürstner, B. Fasching, G. W. O'Neil, M. D. B. Fenster, C. Godbout and J. Ceccon, Chem. Commun., 2007, 3045–3047, 10.1039/b707835h.
- S. Simsek, M. Horzella and M. Kalesse, Org. Lett., 2007, 9, 5637–5639 CrossRef CAS.
- M. Christmann and M. Kalesse, Tetrahedron Lett., 2001, 42, 1269–1271 CrossRef CAS.
- V. Koch, M. Nieger and S. Brase, Org. Chem. Front., 2020, 7, 2670–2681 RSC.
- S. Poigny, S. Nouri, A. Chiaroni, M. Guyot and M. Samadi, J. Org. Chem., 2001, 66, 7263–7269 CrossRef CAS PubMed.
- G. Stork and K. Zhao, Tetrahedron Lett., 1989, 30, 2173–2174 CrossRef CAS.
- I. Janicki and P. Kielbasinski, Adv. Synth. Catal., 2020, 362, 2552–2596 CrossRef CAS.
- C. Jasper, R. Wittenberg, M. Quitschalle, J. Jakupovic and A. Kirschning, Org. Lett., 2005, 7, 479–482 CrossRef CAS PubMed.
- J.-O. Cha, J. I. Yoo, J. S. Yoo, H.-S. Chung, S.-H. Park, H. S. Kim, Y. S. Lee and G. T. Chung, Osong Public Health Res. Perspect., 2013, 4, 225–232 CrossRef PubMed.
- A. Boudet, P. Sorlin, C. Pouget, R. Chiron, J.-P. Lavigne, C. Dunyach-Remy and H. Marchandin, Front. Microbiol., 2021, 12, 750489 CrossRef.
- K. M. Craft, J. M. Nguyen, L. J. Berg and S. D. Townsend, MedChemComm, 2019, 10, 1231–1241 RSC.
- S. Cascioferro, D. Carbone, B. Parrino, C. Pecoraro, E. Giovannetti, G. Cirrincione and P. Diana, ChemMedChem, 2021, 16, 65–80 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2175241 and 2194803. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05049h |
| This journal is © The Royal Society of Chemistry 2022 |