
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ electrochemical synthesis of Pd aerogels as highly efficient anodic electrocatalysts for alkaline fuel cells†
Chen
Wang‡
a,
Wei
Gao‡
 a,
Xinhao
Wan‡
a,
Bin
Yao
a,
Wenjing
Mu
a,
Jie
Gao
*b,
Qiangang
Fu
*a and
Dan
Wen
*a
a,
Xinhao
Wan‡
a,
Bin
Yao
a,
Wenjing
Mu
a,
Jie
Gao
*b,
Qiangang
Fu
*a and
Dan
Wen
*a
aState Key Laboratory of Solidification Processing, School of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an, 710072, P. R. China. E-mail: dan.wen@nwpu.edu.cn; fuqiangang@nwpu.edu.cn
bSchool of Life Sciences, Northwestern Polytechnical University, Xi'an, 710072, P. R. China. E-mail: jgao@nwpu.edu.cn
First published on 11th November 2022
Abstract
Improving the utilization of noble metals is extremely urgent for fuel cell electrocatalysis, while three-dimensional hierarchical noble metal aerogels with abundant sites and channels are proposed to reinforce their electrocatalytic performances and decrease their amounts. Herein, novel Pd aerogels with tunable surface chemical states were prepared through a facile in situ electrochemical activation, starting with PdOx aerogels by the hydrolysis method. The hierarchical porous Pd aerogels showed unprecedented high activity towards the electrocatalytic oxidation of fuels including methanol (2.99 A mgPd−1), ethanol (8.81 A mgPd−1), and others in alkali, outperforming commercial catalysts (7.12- and 13.66-fold, corresponding to methanol and ethanol). Theoretical investigation unveiled the hybrid surface states with metallic and oxidized Pd species in Pd aerogels to regulate the adsorption of intermediates and facilitate the synergistic oxidation of adsorbed *CO, resulting in enhanced activity with the MOR as the model. Therefore, efficient Pd aerogels through the in situ electrochemical activation of PdOx aerogels were proposed and showed great potential for fuel cell anodic electrocatalysis.
Introduction
The fast development of electrochemical apparatuses like liquid fuel cells for the efficient conversion between renewable energy and sustainable electricity is contributing to the future goal of carbon neutralization.1–4 Noble metals, with their well-known properties such as extreme scarcity and high costs, are still irreplaceable as electrocatalysts for commercial electrochemical devices, and are seriously studied to improve their utilization via structure design and component regulation.5–8 Three-dimensional (3D) porous structures for electrocatalysts can supply more surface-exposed metal sites, facilitate the transportation of electrons, and accelerate the diffusion of liquid electrolytes and reactants/products, therefore they are conducive to building novel structures with decreased amounts of noble metals.9–11 Noble metal aerogels (NMAs) have been proposed recently as typical 3D metallic structures by the self-assembly of small-sized noble metal nanoparticles as building blocks into interconnected networks, according to pioneer research demonstrated by Eychmüller and co-workers.12 With the features of ultralow mass density and plentiful meso-/macropores originating from the aerogel structure, and the active intrinsic electrocatalytic properties inherited from noble metals, NMAs have been broadly explored as electrocatalysts for electrochemical reactions including oxygen reduction, methanol oxidation, and CO2 reduction, and outperformed their corresponding noble metal bulks and nanosized building blocks in electrocatalytic activity.13–20 Typically, Pd aerogels by the assembly of Pd nanoparticles exhibited abundant pore structures, large specific surface areas of 40–108 m2 g−1, and low densities of 0.025–0.059 g cm−3, which promoted the bioelectrocatalytic performance with 3 times higher activity compared to Pd nanoparticles.21Two different bottom-up approaches are generally proposed to realize the gelation of NMAs, namely, the one-step direct formation of NMAs by the reduction of corresponding metal salt solutions, and the two-step method of self-assembly of noble metal building blocks into NMAs with de-stabilizing agents or physical treatments.22 For instance, Zhu et al. reported the simple preparation of MCu (M = Pd, Pt, and Au) bimetallic aerogels by the one-step reduction of metal precursors with NaBH4 as the reductant,23 while Du et al. demonstrated a two-step freeze–thaw method to construct various NMAs as a result of the salting-out behavior and ice-templating effect of precursor solutions.24 In the one-step gelation method, chemicals with strong reducibility like sodium borohydride are generally employed to convert noble metal ions into metal ligaments, while in the two-step preparation of NMAs, organic ligands are adopted widely to prevent nanoparticle precursors from aggregating, and then interconnect with each other for the gelation of nanoparticles. Thus, mild synthetic conditions without organic ligands are much required to simplify the preparation of NMAs and further explore their applications. Meanwhile, NMAs with controllable and tunable surface properties are needed to comprehend the effects of chemical states and ligands on the activity from the viewpoint of electrocatalysis.25 The in situ electrochemical method is a facile pathway for regulating the surface properties of electrodes, and is adopted for electrocatalysis. Therefore, the in situ strategy to modulate NMAs with expected surface properties is desired for better understanding the catalytic mechanism and improving the performances.
Here in this work, we introduce a facile in situ electrochemical activation method to prepare Pd aerogels as efficient anodic electrocatalysts with PdOx aerogels as precursors. Using a mild aqueous hydrolysis method, 3D porous PdOx aerogels with high specific surface areas were easily prepared, and the effects of detailed synthetic conditions and parameters on the aerogel gelation are also discussed. The conversion of PdOx aerogels into Pd aerogels was realized by in situ electrochemical reduction in an alkaline electrolyte, with controllable surface oxidation states. The in situ-activated Pd aerogels showed excellent activity towards the electrocatalytic oxidation of different fuels in alkali, especially alcohols like methanol, ethanol, and ethylene glycol. Impressively, the mass activities for Pd aerogels are about 2.99 and 8.81 A mgPd−1 towards the electro-oxidation of methanol and ethanol, which are about 7.12 and 13.66 times higher than those of commercial Pd/C (0.42 and 0.64 A mgPd−1), respectively. Density functional theory (DFT) calculation further unveiled the change in the d-band center of Pd and the mechanism of the improved catalytic activity with methanol as the model. The hybrid metallic Pd and oxidized Pd species in Pd aerogels had lower adsorption energy for *OH and *CO intermediates, as well as smaller energy barriers for the conversion of *CO into *COOH, thus contributing to better activity. Therefore, a novel in situ activation strategy for the fabrication of Pd aerogels from PdOx aerogels was well explained. The excellent electrocatalytic properties of Pd aerogels verified from both experimental and theoretical results further broadened the synthesis of NMAs and their fuel cell applications.
Experimental
Reagents and materials
Sodium carbonate anhydrous (Na2CO3, 99.99%), palladium(II) chloride (PdCl2, anhydrous, 60% Pd basis), and palladium on carbon (10 wt% Pd) were purchased from Sigma-Aldrich. Potassium hydroxide (KOH, ≥85.0%), sodium chloride (NaCl, ≥99.5%), sodium sulphate (Na2SO4, ≥99.0%), sodium nitrate (NaNO3, ≥99.0%), isopropanol (≥99.7%), ethylene glycol (≥99.5%), glycerol (≥99.0%) and hydrochloric acid (HCl, 36.0–38.0%) were obtained from Sinopharm Chemical Reagent Co., Ltd. Ethanol absolute (C2H5OH) and acetone (≥99.8%) were bought from Merck. Methanol (CH3OH, 99.5%), and sodium hydroxide (NaOH) were purchased from Aladdin. All the water used in the experiments was Milli-Q water.Preparation of PdOx aerogels and Pd aerogels
The PdOx hydrogels were synthesized according to the following method. 120 mg Na2CO3 was dissolved in 9 mL Milli-Q water. Then 1 mL H2PdCl4 (1.2 mgPd mL−1, PdCl2 dissolved in dilute hydrochloric acid solution) was added to the above solution. After a slight shake for five minutes, the solution was placed at room temperature. The orange transparent film-like hydrogels completely settled down at the bottom after 6 h. The solution was washed 3 times by adding 12 times the amount of water. Then, the supernatant was carefully replaced with acetone. Finally, the hydrogels in acetone were transferred to a critical point dryer for supercritical CO2 drying to obtain the PdOx aerogels. Notably, besides Na2CO3, some other inorganic salts were also utilized to synthesize the PdOx hydrogels under the same conditions, including NaCl, Na2SO4, and NaNO3. Besides, the concentration of the inorganic salts and the pH of the reaction solution (controlled by the NaOH and HCl solutions) were adjusted to figure out the formation conditions of the PdOx hydrogels.In situ electrochemical activation
The in situ electrochemical activation of PdOx aerogels to obtain Pd aerogels was realized using the three-electrode setup, wherein PdOx dropped on a Ti plate (or a glassy carbon electrode) was employed as the working electrode, Ag/AgCl (3 M KCl) electrode was used as the reference electrode, and Pt wire functioned as the counter electrode. To activate PdOx aerogels, the chronoamperometry method was used at a potential of −1.0 V vs. Ag/AgCl for different periods in 1 M KOH solution. After reduction, the sample was washed with water, and then dried in vacuum for further characterization.Apparatus
A CO2 critical point dryer (13200JE-AB) was employed to obtain the aerogels. Scanning electron microscopy (SEM, NANOSEM450, US) and transmission electron microscopy (TEM, Tecnai G2 F30) instruments equipped with energy-dispersive X-ray spectroscopy (EDS) equipment were utilized to characterize the morphologies of the as-prepared materials. XPS data were collected by X-ray photoelectron spectroscopy (Kratos, Axis Supra). XRD patterns were obtained with a SHIMADZU XRD-7000 X-ray diffractometer. The samples for the XPS and XRD test were prepared by dropping a certain amount of material solution onto a Ti substrate (about 6 mg sample). UV-vis spectroscopy of the supernatant of the hydrogel solution was performed on a U-3900H UV/VIS spectrophotometer (2J2-0034). The N2 physisorption isotherms and the distribution of the pore size of the materials were obtained using an ASAP 2460 surface area and porosity analyzer (Micromeritics). The electrochemical activation of PdOx was conducted via the chronoamperometry method. The reduction degree of PdOx could be controlled by adjusting the potential and the treating time. The electrocatalytic performance of samples was tested by utilizing a CHI660E electrochemical workstation (Shanghai Chenhua Instruments Co., Ltd, China) with a standard three-electrode system at room temperature.Electrochemical quartz crystal microbalance (EQCM) test
The Autolab electrochemical quartz crystal microbalance (EQCM) module with equipment and electrodes (gold disks on quartz crystals) was utilized to in situ monitor the mass change during the electrochemical activation. The electrochemical activation was conducted via the chronoamperometry method in 1 M KOH solution with a potential of −1 V. The mass change during the electrochemical activation was calculated according to the Sauerbrey equation: Δf = −Cf × Δm, where Δf is the frequency change, Cf is the sensitivity factor of the crystal (0.0815 Hz ng−1 cm2 for 6 MHz at 20 °C), and Δm is the mass change per unit area (g cm−2).Catalysis toward the electrooxidation of different chemicals
The ink of PdOx aerogels was prepared by dissolving about 1.75 mg PdOx aerogels in 2 mL Milli-Q water. As a comparison, the ink of Pd/C (10 wt%) was prepared by mixing 10 mg Pd/C with 1 mL of 2-propanol and 3 mL of Milli-Q water. All inks were sonicated for at least 10 minutes before use. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was utilized to calculate the concentrations of Pd in inks. A certain amount of ink was dropped on glassy carbon electrodes (GCEs, 3 mm in diameter) and dried at room temperature to make sure the loading of Pd was 1 μg for each GCE. The electrocatalytic performance of the materials was tested in 1 M methanol + 1 M KOH aqueous solution for the MOR and in 1 M C2H5OH + 1 M KOH aqueous solution for the EOR. A Modified GCE, Ag/AgCl (3 M KCl), and Pt wire were employed as the working electrode, reference electrode, and counter electrode, respectively. The electrocatalytic stability of the samples was investigated via the chronoamperometry method. The corresponding potentials were set to −0.23 V for the MOR and −0.31 V for the EOR, respectively.DFT calculations
The density functional theory (DFT) calculations were performed in the Vienna Ab initio Simulation Package (VASP) code.26,27 The interactions between ion cores and valence electrons were calculated with the projector augmented wave (PAW) method.28,29 The exchanged correlation interactions were handled by the use of a generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE) function.30 The valence electronic states were expanded in the plane wave basis sets within a cutoff energy of 450 eV. A four-layer face centered cubic (4 × 4 × 4) structure of Pd(111) was adopted in which the lattice constant was 3.94 Å. Two or eight oxygen atoms were placed to simulate the state of Pd surface oxidation. A vacuum layer of 15 Å was adopted to avoid the periodic interactions and the bottom two layers were fixed in order to increase the calculation efficiency. The Brillouin zone was modeled using the gamma centered Monkhorst–Pack scheme, in which a 3 × 3 × 1 grid was adopted.Each electrochemical reaction step of methanol oxidation involves a H+ + e− pair transfer from the adsorbed species on the surface to the electrolyte. For the calculations of GH+ + Ge− under the normal conditions (pH = 0, U = 0), we employed the computational hydrogen electrode (CHE) model proposed by Nørskov to calculate the chemical potential of H+ + e−: GH+ + e− = ½GH2. The change in the Gibbs free energy of each (H+ + e−) pair transfer reaction was calculated by the computational hydrogen electrode (CHE) model: ΔG = ΔE + ΔZPE + ΔH(0 → 298.15 K) − TΔS, in which ΔE, ΔZPE, ΔH and ΔS are the change in potential energy, the change in the zero-point energy, the integrated heat capacity from 0 K to 298.15 K, and the change in the entropy. The zero-point energy was calculated by the summation of all vibrational frequencies: ZPE = ½∑ℏν, where ν corresponded to the vibrational frequency of each normal mode.
During the DFT calculation, the adsorption energies for intermediates at 298 K are calculated with respect to H2O (l), CO2 (g) and H2 (g) in the following way:
G CH3O = G*CH3O + (1/2)GH2 + GH2O, GCH2O = G*CH2O + (1/2)GH2 + GH2O, GCHOH = G*CHO + (2)GH2 + GH2O, GCOH = G*COH + (3/2)GH2 + GH2O, GCO = G*CO + 2GH2 + GH2O, GCOOH = G*COOH + (5/2)GH2, and GCOads = G*CO − GCO − G*.
Results and discussion
Preparation of PdOx aerogels
The overall process of preparing Pd aerogels includes the hydrolysis of precursors to synthesize PdOx aerogels and subsequent in situ electrochemical activation to obtain Pd aerogels, as illustrated in Scheme 1. Effects of various synthetic conditions and parameters on the gelation of PdOx aerogels and activation of Pd aerogels would be discussed and explored in detail.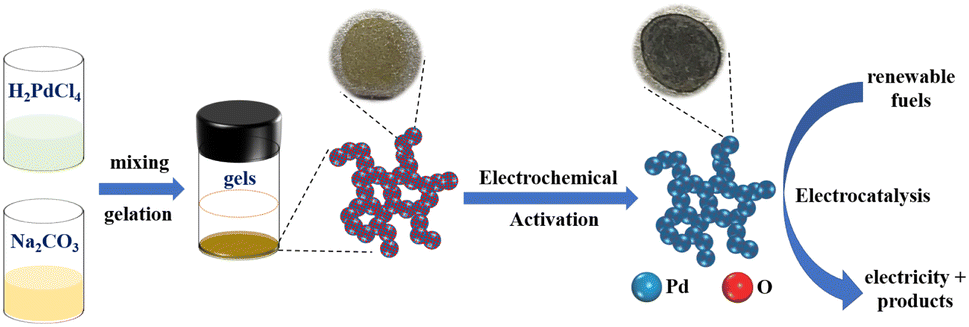 | ||
| Scheme 1 The schematic diagram for the in situ electrochemical activation of PdOx aerogels by the hydrolysis method to produce Pd aerogels as efficient electrocatalysts. | ||
PdOx aerogels as the precursors were firstly synthesized through a mild hydrolysis method. To prepare PdOx aerogels, a mixture solution of H2PdCl4 and Na2CO3 with the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 was shaken gently and then allowed to settle for 6 hours for the gelation. During the gelling process, significant changes in color from light yellow to dark-brown with prolonged time were observed from optical images (Fig. 1a). Meanwhile, the ultraviolet-visible (UV-vis) spectra of the supernatant solution with different reacting times were also monitored to study the reaction process (Fig. S1, ESI†). The characteristic peaks at 208, 234, 305, and 418 nm for H2PdCl4 disappeared quickly after adding Na2CO3, while a new peak around 270 nm emerged immediately, indicating the formation of new Pd(II) species (denoted as PdOx).31 With prolonged time, the intensity of the new peak at 270 nm increased within the initial 40 minutes and it decreased in the following time, suggesting the gradual gelation process of PdOx. After 6 hours, brown films were observed at the bottom of the vial (Fig. 1a), while the supernatant was clear with the complete vanishing of the UV-vis signal at 270 nm. The above phenomena suggested the successful preparation of the PdOx hydrogels through the hydrolysis of H2PdCl4 with Na2CO3 in solution at room temperature, without any reductants or surfactants. To further investigate the gelation process, transmission electron microscopy (TEM) images for the samples under different reacting times (0, 20, 40, and 120 minutes) were collected (Fig. S2, ESI†). After mixing H2PdCl4 and Na2CO3 (0 minute), large amounts of ultrafine nanoparticles of about 3 nm were formed (Fig. S2a, ESI†). In the initial 20 minutes, the assembly of the above nanoparticles happened quickly to form loosely interconnected frameworks (Fig. S2b, ESI†). With increase of the reacting time up to 40 and 120 minutes, the frameworks extended to larger networks, with the close assembly of ultrafine nanoparticles and clear macro-/mesoporous structures (Fig. S2c and d, ESI†). Therefore, the gelation process was believed to be the gradual self-assembly of hydrolyzed PdOx nanoparticles into 3D networks.
:100 was shaken gently and then allowed to settle for 6 hours for the gelation. During the gelling process, significant changes in color from light yellow to dark-brown with prolonged time were observed from optical images (Fig. 1a). Meanwhile, the ultraviolet-visible (UV-vis) spectra of the supernatant solution with different reacting times were also monitored to study the reaction process (Fig. S1, ESI†). The characteristic peaks at 208, 234, 305, and 418 nm for H2PdCl4 disappeared quickly after adding Na2CO3, while a new peak around 270 nm emerged immediately, indicating the formation of new Pd(II) species (denoted as PdOx).31 With prolonged time, the intensity of the new peak at 270 nm increased within the initial 40 minutes and it decreased in the following time, suggesting the gradual gelation process of PdOx. After 6 hours, brown films were observed at the bottom of the vial (Fig. 1a), while the supernatant was clear with the complete vanishing of the UV-vis signal at 270 nm. The above phenomena suggested the successful preparation of the PdOx hydrogels through the hydrolysis of H2PdCl4 with Na2CO3 in solution at room temperature, without any reductants or surfactants. To further investigate the gelation process, transmission electron microscopy (TEM) images for the samples under different reacting times (0, 20, 40, and 120 minutes) were collected (Fig. S2, ESI†). After mixing H2PdCl4 and Na2CO3 (0 minute), large amounts of ultrafine nanoparticles of about 3 nm were formed (Fig. S2a, ESI†). In the initial 20 minutes, the assembly of the above nanoparticles happened quickly to form loosely interconnected frameworks (Fig. S2b, ESI†). With increase of the reacting time up to 40 and 120 minutes, the frameworks extended to larger networks, with the close assembly of ultrafine nanoparticles and clear macro-/mesoporous structures (Fig. S2c and d, ESI†). Therefore, the gelation process was believed to be the gradual self-assembly of hydrolyzed PdOx nanoparticles into 3D networks.
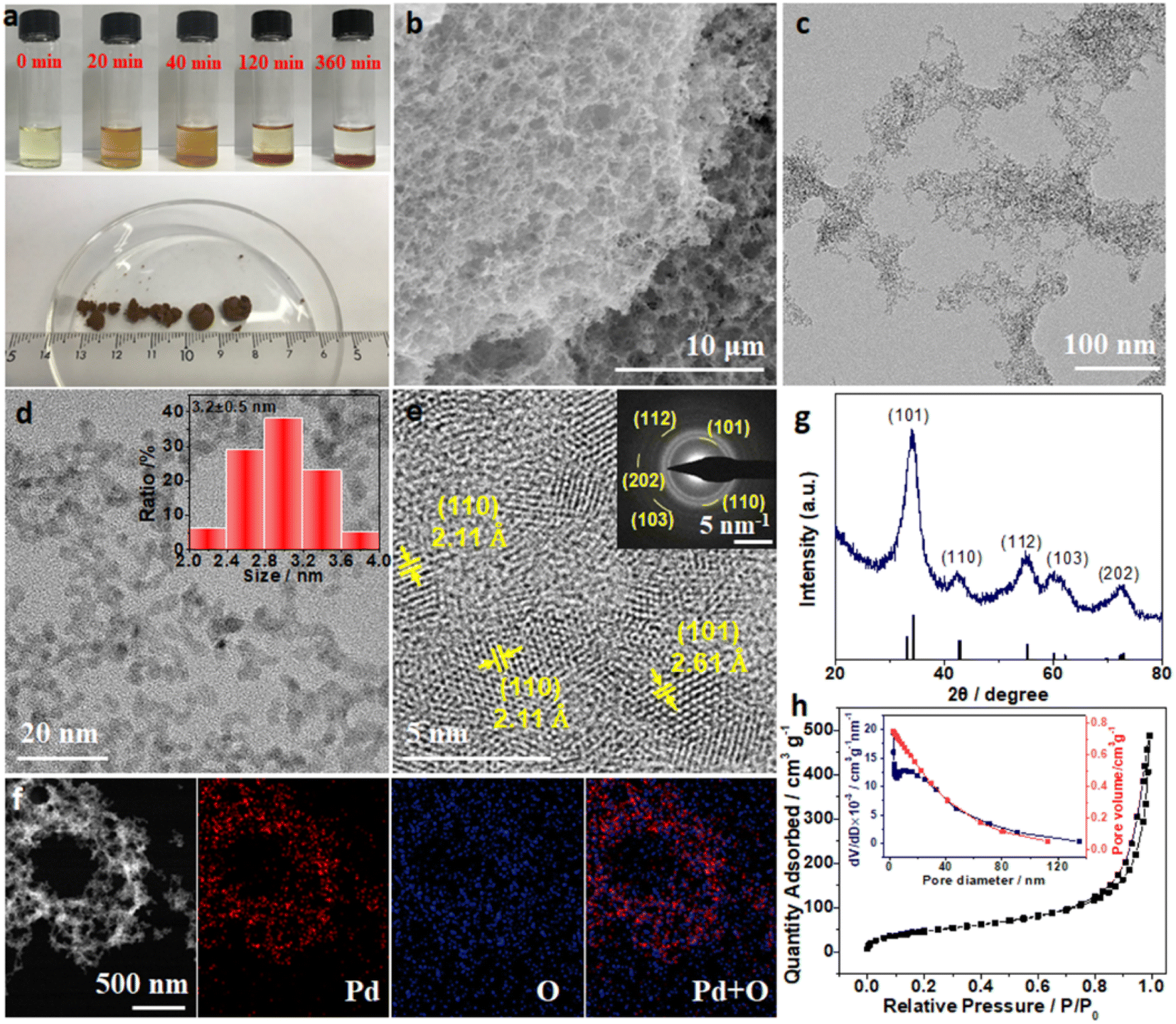 | ||
| Fig. 1 (a) Optical images of PdOx aerogels during synthesis and after CO2 supercritical drying. (b) SEM image, (c and d) TEM images; the inset of (d) shows the size distribution, and (e) HR-TEM image and SAED pattern for PdOx aerogels. (f) HAADF-STEM image and corresponding elemental distribution images of Pd and O for PdOx aerogels. (g) XRD pattern for PdOx aerogels. (h) N2 physisorption isotherm and corresponding pore size distribution for PdOx aerogels. | ||
The PdOx hydrogels completely settled down after 6 hours, and bulk PdOx aerogels with a diameter of about 1 cm were clearly obtained (Fig. 1a) after supercritical CO2 drying. The micro-structure of PdOx aerogels was explored by scanning electron microscopy (SEM) firstly, and Fig. 1b exhibits the interconnected porous structure built up from branches and pores. TEM images in Fig. 1c and d further revealed that the branches consisted of ultrafine nanoparticles with a uniform size of 3.2 ± 0.5 nm (the size distribution is shown in the inset of Fig. 1d) to interconnect and self-assemble with nearby nanoparticles. Moreover, the high-resolution TEM (HRTEM) image and corresponding selected-area electron diffraction (SAED) pattern shown in Fig. 1e not only demonstrated the polycrystalline nature of PdOx, but also matched well with various crystal faces of PdO. Specifically, the lattice spacings of 2.15 and 2.61 Å are indexed to the (110), and (101) facets of PdO, respectively. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and corresponding elemental mapping images in Fig. 1f also indicated the uniform distribution of Pd and O elements in the PdOx aerogels. Further analysis from the X-ray diffractometer (XRD) pattern (Fig. 1g) also confirmed the PdO structure, where diffraction peaks at about 34.02°, 42.32°, 55.02°, 60.18°, and 72.76° are ascribed to the (101), (110), (112), (103), and (202) reflection planes of tetragonal PdO (PDF# 88-2434) with the space group of I4/mmm (139), respectively. To understand the micro-structure and pore distribution of PdOx aerogels, the nitrogen physisorption isotherm was measured (Fig. 1h). The distribution of pore size calculated from the desorption branch in the isotherm using the Barrett–Joyner–Halenda (BJH) equation illustrated the hybrid microporous and mesoporous structures in PdOx aerogels (Fig. 1h). The combination of type II and IV curves further suggested the existence of macropores in PdOx aerogels.32 Due to the rich hybrid pores, the specific surface area of the as-prepared PdOx aerogels is calculated to be 177.7 m2 g−1, which is much larger than those of other NMAs (Table S1, ESI†). Therefore, the 3D porous structure of PdOx aerogels obtained by this method exhibited a large specific surface area and abundant pores, contributing to more active sites for intermediates and rich channels for mass transfer, which would be beneficial to electrocatalysis.
To better dig out the gelation process of PdOx aerogels, the systematic control experiments with various parameters were investigated and are discussed in the ESI and Fig. S3–S7,† including the ratios of H2PdCl4 to Na2CO3, the pH values of the solution, and various salts. The gelation mechanism for PdOx hydrogels may also be proposed from the above control experiments. The hydrolysis of Na2CO3 provided a weak basic condition (pH = 11) to realize the fast conversion of PdCl42− ions into hydrated PdO in the very beginning.33,34 Then the dehydration of precursors and salting out as well as the subsequent gelation process of PdOx nanoparticles occurred under a relatively high salt concentration to assemble 3D PdOx hydrogels.35 It is worth mentioning that only a basic environment with pH = 11 but without adding Na2CO3 cannot lead to the gelation of PdOx hydrogels. Na2CO3 played an irreplaceable role in this process by not only providing a basic environment via hydrolysis, but also maintaining a high salt concentration to drive the assembly of nanoparticles into hydrogels.
In situ electrochemical activation of PdOx aerogels into Pd aerogels
Pd-based materials have been widely developed as electrocatalysts for the oxidation of various fuels like methanol, ethanol, formic acid, etc. for liquid fuel cell applications.36–39 Here, electrocatalytic performances of PdOx aerogels with high porosity and surface area were also explored. However, when PdOx aerogels were used as the anodic materials towards electrocatalytic methanol oxidation, no significant activity was detected in alkaline solution (Fig. S8, ESI†) because the oxidized Pd species are inactive in PdOx aerogels. Since the in situ electrochemical activation method can quickly and exactly reduce oxidized metals into metallic states under appropriate voltage without any agents, a clean surface and tunable surface chemical states on electrodes are obtained for electrocatalysis.25 To activate PdOx aerogels, samples on the Ti plate were investigated as the proof of concept and in situ electrochemically activated in 1 M KOH solution. Under a bias potential of −1.0 V (vs. Ag/AgCl), the fast reduction of PdOx was observed by the change of sample color from yellow to black within 10 seconds, as shown in Fig. 2a and Video S1 (ESI).† Then Pd aerogels were obtained from the in situ reduction of PdOx aerogels. Such an in situ electrochemical activation process was further monitored and confirmed by the electrochemical quartz crystal microbalance (EQCM) measurement by testing the mass change, and the quick loss of mass of PdOx within several seconds demonstrated the loss of oxygen atoms and reduction of PdOx into Pd, as illustrated in Fig. S9 (ESI).† To estimate the electrochemically active surface areas (ECSAs) of Pd aerogels, their typical cyclic voltammetry (CV) curves were recorded in 1 M KOH solution. For Pd aerogels, the small peak at −0.3 V (vs. Ag/AgCl) is related to the oxidation of metallic Pd, while the reverse peak at −0.365 V (vs. Ag/AgCl) is ascribed to the reduction of newly oxidized Pd species.40 PdOx aerogels, in comparison, showed no apparent oxidation and reduction of metallic Pd species. Such electrochemical behaviors further suggested the high content of metallic Pd species in Pd aerogels after activation. From Fig. 2b, the ECSA for Pd aerogels is 102.9 m2 g−1, suggesting the good exposure of metallic Pd species due to the large surface area.41,42 The obtained Pd aerogels collected from the Ti plate were characterized to comprehend their structure and properties. The TEM image for Pd aerogels in Fig. 2c indicated the preservation of the interconnected network structure, and the average size of nanoparticles of about 3.6 ± 0.6 nm, which is close to that of the original PdOx aerogels. The X-ray energy dispersive spectrum (EDS) result of Pd aerogels (Fig. S10 and Table S2, ESI†) displayed a dramatically increased Pd/O atomic ratio (4.03) as compared to that (0.60) of PdOx aerogels, revealing the effective removal of oxygen and reduction of Pd during this in situ electrochemical reduction process. The interplanar distances of 2.28 and 2.06 Å in the HRTEM image (Fig. 2d) for Pd aerogels are ascribed to the (111) and (220) planes of metallic Pd, respectively. Meanwhile, corresponding SAED pattern of Pd aerogels matched well with various crystal faces of Pd, further identifying the formation of metallic Pd during reduction. The XRD pattern of Pd aerogels (onto a silicon substrate, Fig. 2e) presented the characteristic peaks at 40.06°, 46.86°, 68.14°, and 81.84°, which can be indexed to the (111), (200), (220), and (311) reflection planes of metallic Pd (PDF# 87-0643). Therefore, the above results suggested the successful formation of metallic Pd nanocrystals with a gel-like structure (i.e., Pd aerogels) from the in situ electrochemical reduction of PdOx aerogels.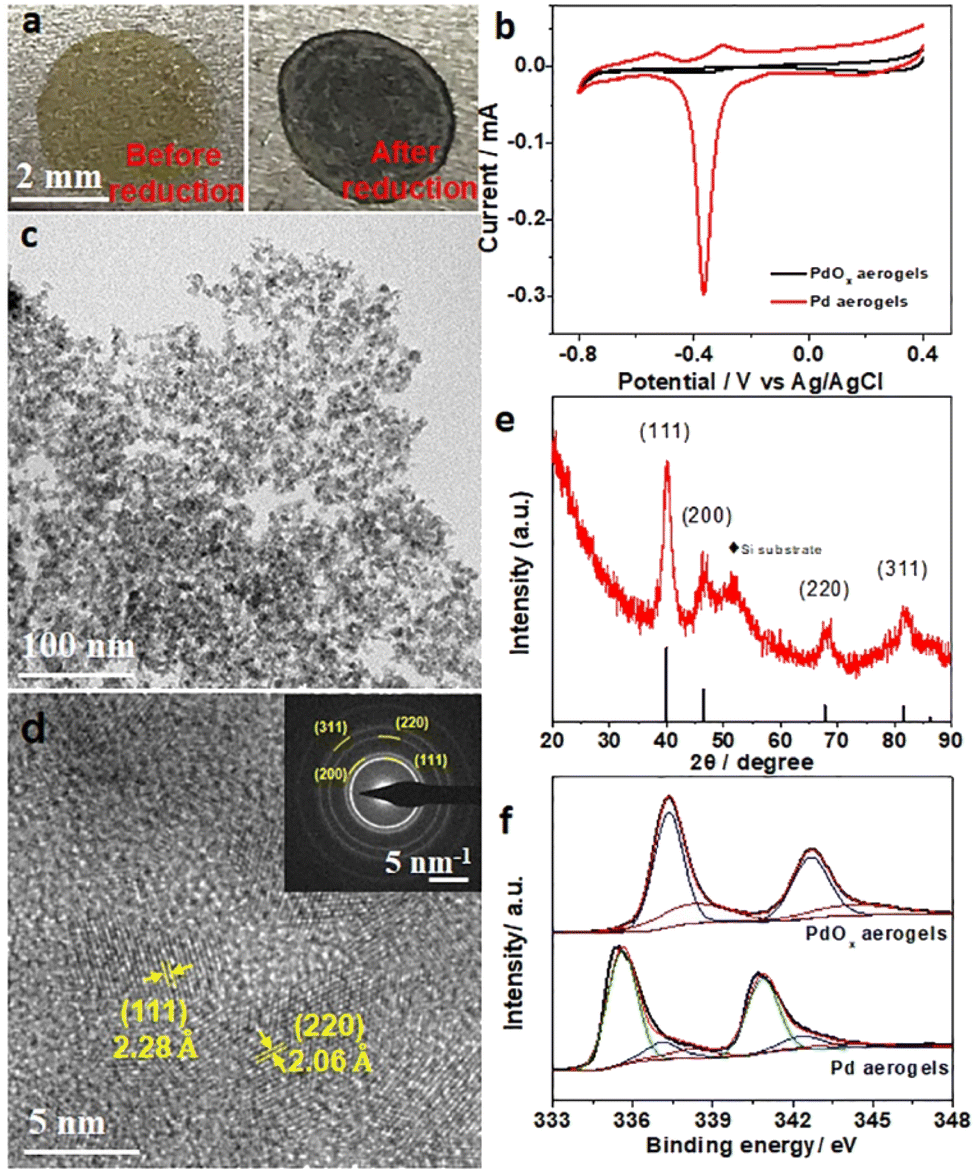 | ||
| Fig. 2 (a) Optical images for PdOx aerogels on a Ti plate before and after in situ electrochemical activation. (b) CV curves for PdOx aerogels and Pd aerogels in 1 M KOH solution with a scan rate of 50 mV s−1 (c) TEM image, (d) HR-TEM image and SAED pattern for Pd aerogels. (e) XRD pattern for Pd aerogels. (f) XPS spectra of Pd 3d orbitals before and after in situ electrochemical activation. | ||
To have a better understanding of the changes in the composition and surface chemical states, the Pd 2p orbitals of both PdOx aerogels and Pd aerogels were measured. The X-ray photoelectron spectroscopy (XPS) results in Fig. 2f can be deconvoluted into three doublets, where the subpeaks at 335.3, 336.9, and 338.0 eV for Pd 3d5/2 orbital are assigned to metallic Pd(0), Pd2+ and Pd4+ species, respectively.43,44 Both Pd2+ and Pd4+ species are mainly observed for PdOx aerogels, while the content of metallic Pd(0) is dominant (with a Pd(0) ratio of 0.635 in the Pd 3d spectrum, Table S3, ESI†), and the ratios of Pd2+ and Pd4+ species are significantly lower in Pd aerogels because of the reduction of Pd2+ and Pd4+ into metallic Pd(0). Thus, changes in XPS results obviously uncovered the efficient reduction process of PdOx aerogels into Pd aerogels with metallic Pd(0) species during the in situ electrochemical reduction. However, this incomplete conversion process with the existence of some oxidized Pd species accompanied by metallic Pd species on the surface should be carefully considered because this may affect the electrocatalytic properties of the as-obtained Pd aerogels.
The in situ electrochemical activation of materials provided a fast approach to convert pre-catalysts into catalytically active species, under simple conditions and with accurate regulation. Though fast activation within seconds of PdOx aerogels was proposed and proved above, the effect of reducing time should also be quantitatively investigated. Changes in surface chemical states and electrocatalytic activity along with different reducing times for PdOx aerogels were explored, where samples on a Ti plate were employed for better characterization and testing. Under various reducing times of 0, 5, 30, 300, and 1200 seconds, similar peaks indexed to Pd(0), Pd2+ and Pd4+ were analyzed from XPS data (Fig. 3a). The contents of metallic Pd(0) species and the ratios of Pd(0) to the oxidized Pd species increased (ratio of Pd(0)/oxidized Pd up to 3.63) along with longer activation times, suggesting that more oxidized Pd species were reduced into metallic Pd. Meanwhile, the electrocatalytic performances for the above samples were also obtained using methanol and ethanol oxidation reactions (MOR, and EOR) as the model reactions in 1 M KOH solution with 1 M methanol and 1 M ethanol, respectively. From CV curves in Fig. S11 (ESI)† for the MOR and EOR, the catalytic peak current densities improved apparently for samples with increased reduction times. Especially, the sample with 300 s reduction showed the highest peak current densities among all Pd aerogels with different activation times (Fig. 3b). However, prolonging the time to 1200 s contributed to insignificant improvement further but even deterioration, and high-valence PdOx species still remained in this sample to form a mixed surface chemical state. Furthermore, activating PdOx aerogels into Pd aerogels was also successfully applied on the glassy carbon (GC) electrode with an activation period of 300 s. As the anodic catalysts, Pd aerogels exhibited excellent mass activity towards both the MOR and EOR (Fig. 3c and d), where the peak current densities for the MOR and EOR are 2.99 and 8.81 A mgPd−1, respectively, which are much larger than those for pristine PdOx aerogels (0.14 and 0.14 A mgPd−1) and commercial Pd/C (0.42 and 0.64 A mgPd−1). The catalytic performances of Pd aerogels as anodic catalysts are better than those of recently reported Pd-based samples (Table S4, ESI†), which are among the best materials. The Tafel slope for Pd aerogels was apparently lower than those for PdOx aerogels and Pd/C, as obtained from the linear sweeping voltammperometry curves in Fig. S12 (ESI),† indicating the enhanced kinetics toward the oxidation of adsorbed *CO species. The smaller charge transfer resistance of Pd aerogels compared to PdOx aerogels and commercial Pd/C facilitated the electron transfer and better conductivity towards the MOR process (Fig. S13, ESI†). Also, Pd aerogels exhibited excellent durability towards the MOR and EOR with better retention of current densities under the identical conditions, as compared with commercial Pd/C (Fig. S14, ESI†). The morphology and structure of the used Pd aerogels were also well preserved after the durability test (Fig. S15, ESI†). Therefore, the structure and composition of the PdOx aerogels were facilely regulated through this in situ electrochemical activation method with well-controlled surface chemical states for Pd aerogels, and their effects on the anodic catalytic activity for both methanol and ethanol are also uncovered. Such an efficient yet simple in situ electrochemical activation strategy may also be proposed for other NMAs to further modulate their surface states and activity.
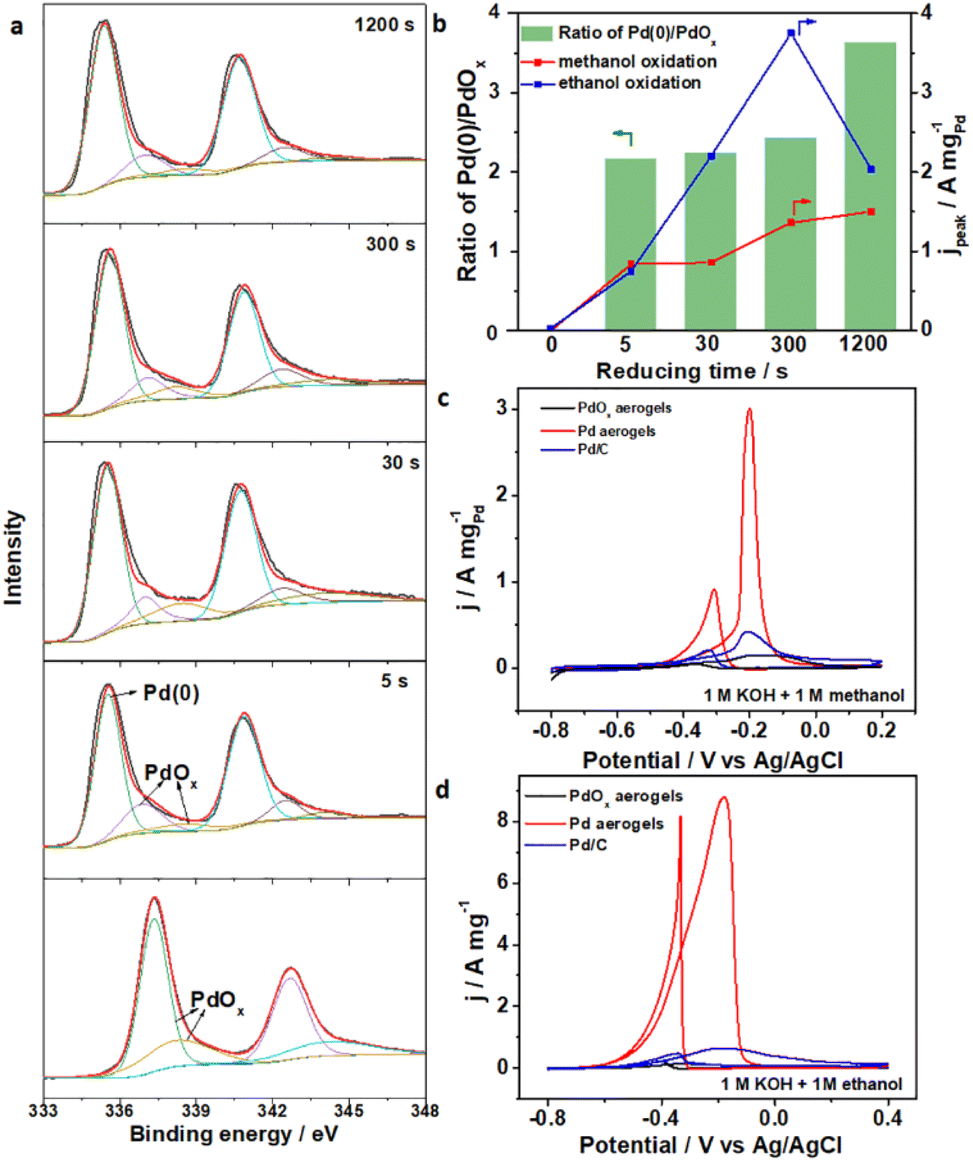 | ||
| Fig. 3 (a) XPS spectra of Pd 3d orbitals for samples with different electrochemical activation times. (b) Changes in the ratios of Pd(0)/PdOx, the MOR and the EOR forward peak current densities along with different reduction times. CV curves for commercial Pd/C, PdOx aerogels and Pd aerogels on a GC electrode in (c) 1 M KOH solution containing 1 M methanol, and (d) 1 M KOH solution containing 1 M ethanol with the scan rate of 50 mV s−1. | ||
Electrocatalytic mechanism and applications of Pd aerogels
Pd-based materials have been well investigated as MOR electrocatalysts due to their good intrinsic properties.45,46 Here, the in situ obtained Pd aerogels have a metallic phase structure with mixed surface composites of both metallic and oxidized species, which may lead to better catalytic activity. To comprehend the effect of the structure on the electrocatalytic properties, the mechanism of the MOR as the representative was unveiled for Pd aerogels using density functional theory (DFT) calculation. For bulk Pd, Pd aerogels, and PdOx aerogels, their surface composites based on Pd(111) are demonstrated in Fig. S16 (ESI),† wherein Pd aerogels had two oxygen atoms coordinated to neighboring Pd atoms (labelled Pd/Pd2O), while PdOx aerogels had all Pd atoms fully bonded with oxygen atoms (labelled Pd8O). The partial density of state (PDOS) of Pd d orbital results for different samples suggested the downshift in the d-band center with the more oxidized surface, which would weaken the adsorption of intermediates of *CO (Fig. S17, ESI†).47 According to previous experimental investigations, the MOR process followed the sequence: CH3OH(g) → *CH3O → *CH2O → *CHO → *CO → *COOH → CO2(g).48 The Gibbs free energy for the breaking of the O–H bond to generate *CH3O on different catalysts was calculated firstly. The Pd8O surface required a very high Gibbs free energy of 1.71 eV for the dehydrogenation of methanol to form the *CH3O intermediate, while the following step of dehydrogenation of *CH3O would produce gaseous HCHO(g) (which cannot be adsorbed on the surface) but not adsorbed *CH2O intermediate to discontinue the further oxidation (Fig. S18, ESI†). Thus, the fully oxidized Pd surface with this Pd8O model analyzed and unveiled the difficulty in the dehydrogenation of methanol, leading to the poor electrocatalytic activity. As a comparison, the Pd sites in pure metallic Pd and Pd/Pd2O had much lower energy barriers of 0.78 and 0.66 eV for *CH3O generation, respectively. Therefore, the Pd/Pd2O surface is more favored for the better dehydrogenation of methanol to initiate the oxidation process. Furthermore, in the overall MOR process, the strong adsorption of *CO on the Pd surface mainly led to the poisoning and deterioration of the activity, and its oxidation with adsorbed *OH on the surface sites is crucial.49 Compared to Pd with the adsorption energies of −1.59 eV for the *CO intermediate (Fig. S19a, ESI†), the smaller value of −1.45 eV demonstrated the moderate bonding of *CO on the Pd/Pd2O surface. Effective Pd-based catalysts usually possess good anti-CO poisoning ability, which means weak bonding of *CO on Pd-based catalysts is necessary.39 To further clarify the weaker bonding of *CO on Pd aerogels, the CO stripping test was performed50 (Fig. S20, ESI†). Significant negative shifts in onset and peak potentials for Pd aerogels suggested the weaker adsorption of CO on it compared to Pd/C,51 which is consistent with theoretical calculation. Additionally, the smaller adsorption energy (−3.03 eV) of active *OH from water dissociation on the Pd/Pd2O surface compared to it of −3.14 eV on the Pd surface (Fig. S19b, ESI†), also illustring the hybrid surface to facilitate the generation of *OH for the oxidation of adsorbed *CO. On the other hand, as the potential-determining step (PDS) of the MOR on Pd(111), the energy barrier for the oxidization of *CO into *COOH is also calculated. Pure Pd had a larger energy barrier of 1.45 eV, while that for Pd/Pd2O was 1.36 eV, suggesting the easier oxidation of *CO on the hybrid surface for Pd aerogels (Fig. 4). Thus, theoretical results revealed that the Pd/Pd2O surface can weaken the adsorption of *CH3O, *CO, and *OH intermediates, simultaneously reducing the energy barriers for the dehydrogenation of methanol and the oxidation of *CO into *COOH due to the synergetic effect of adsorption of *CO and *OH, as compared with the pure metallic Pd surface. Such a hybrid oxidized and metallic state for Pd in the in situ activated Pd aerogels is favored to contribute to better electrocatalytic MOR activity, which is also in good accordance with the experimental results.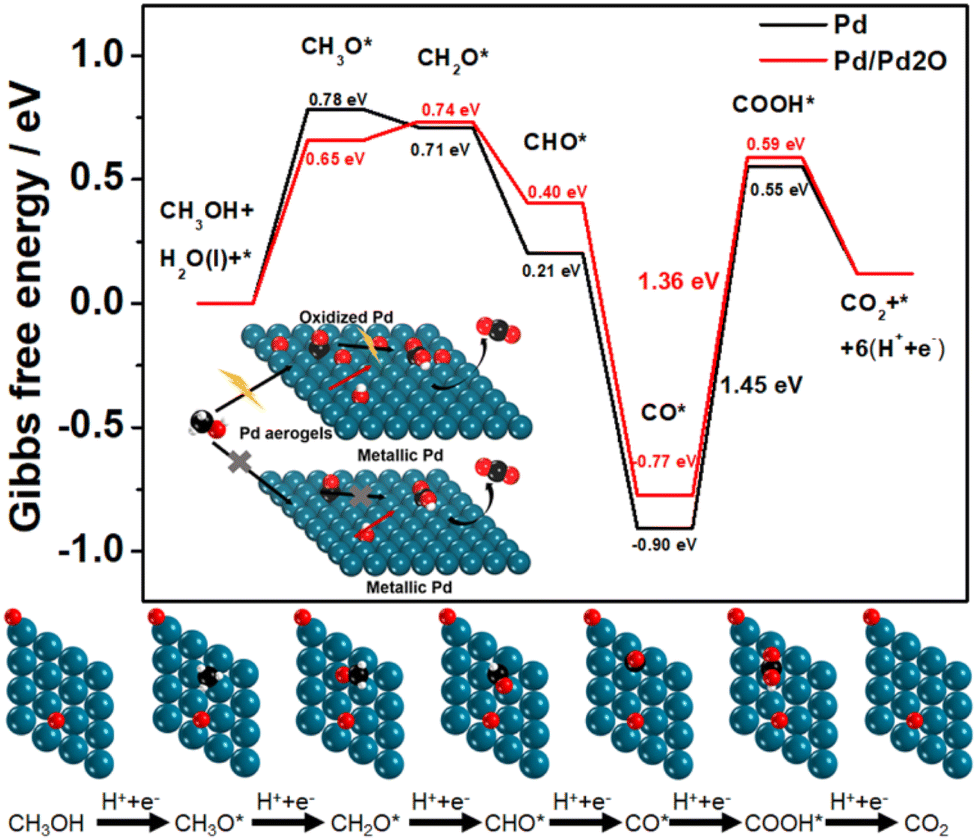 | ||
| Fig. 4 The Gibbs free energy profiles of MOR pathways on Pd/Pd2O for Pd aerogels and pure Pd as the comparison, and the models of intermediates. The balls with white, black, red, and blue represent H, C, O, and Pd atoms, respectively. Inset is the proposed mechanism for the MOR. | ||
Various fuel cells with alcohols (like ethanol, ethylene glycol, and glycerol), formic acid, and glucose as the chemical fuels have been proposed recently, and different Pd-based materials have been investigated as efficient anodic catalysts.52–54 Herein, the electrocatalytic performances towards isopropanol, ethylene glycol, glycerol, formic acid, and glucose for alkaline fuel cells on Pd aerogels as the anodic electrocatalysts were explored. Surprisingly, the Pd aerogels exhibited much higher activity towards the above fuels, showing lower onset potentials and larger peak current densities as anodic electrocatalysts towards various fuels for their fuel cell applications (Fig. S21, Table S5, ESI†), while the commercial Pd/C and PdOx aerogels as the control displayed lower activity towards the oxidation of alcohols including isopropanol, ethylene glycol, and glycerol (Fig. 5a–c), as well as formic acid and glucose (Fig. S22, ESI†) in alkali. Derived from the CV curves for the oxidation of isopropanol, ethylene glycol, and glycerol, the mass activities for various catalysts were obtained and are summarized in Fig. 5d. Notably, the mass activities for Pd aerogels are 3.8, 4.5, and 4.4 times larger than those for commercial Pd/C. Additionally, the electrocatalytic performances of Pd aerogels towards the oxidation of formic acid and glucose are also much better than those for PdOx aerogels and commercial Pt/C, as presented in Fig. S22 (ESI).† Thus, the above results confirmed the high electrocatalytic activity of Pd aerogels as efficient anodic catalysts towards different fuels. Two structural features of Pd aerogels are mainly important for the improved activity. On the one hand, the 3D Pd aerogels with high porosity and large surface area can provide rich active sites and more channels for better transfer of intermediates and electrons. On the other hand, the hybrid metallic and oxidized surface states for Pd aerogels may regulate the adsorption of intermediates and promote their dehydrogenation, and the oxidation of adsorbates, like the DFT calculation-revealed MOR process. Therefore, by in situ electrochemical activation of PdOx aerogels, the Pd aerogels with a preserved 3D structure and modified surface chemical states showed remarkable electrocatalytic activity towards the oxidation of different fuels, showing promise as novel and efficient anodic electrocatalysts for different fuel cell applications.
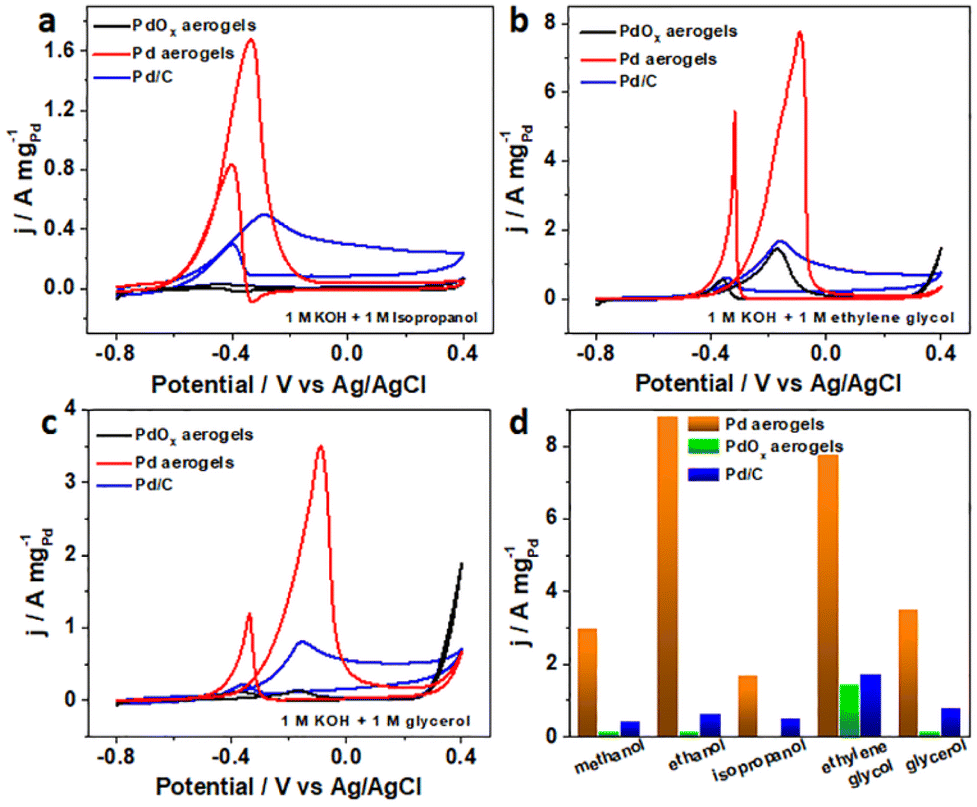 | ||
| Fig. 5 CV curves for commercial Pd/C, PdOx aerogels and Pd aerogels on the GC electrode in 1 M KOH solution containing (a) 1 M isopropanol, (b) 1 M ethylene glycol, and (c) 1 M glycerol with the scan rate of 50 mV s−1. (d) Mass activities for various Pd electrocatalysts towards alcohol oxidations. | ||
Conclusions
In summary, novel 3D Pd aerogels were synthesized through the in situ electrochemical activation of PdOx aerogels which were obtained by a simple hydrolysis method to construct a highly porous structure. After in situ activation, the hybrid surface states of metallic and oxidized Pd species on Pd aerogels were proposed which exhibited good synergy for electrocatalysis. Further theoretical analysis demonstrated the changes in the d-band center for Pd aerogels, smaller adsorption energies for the adsorption of various intermediates (*OH and *CO), and lower energy barriers for the conversion of intermediates on the mixed surface metallic and oxidized Pd species of Pd aerogels with the MOR process as the model. As a result, the in situ-activated Pd aerogels as the anodic electrocatalysts for the oxidation of various fuels (namely, methanol, ethanol, isopropanol, ethylene glycol, glycerol, formic acid, and glucose) showed remarkable activity compared to commercial Pd/C. Conclusively, Pd aerogels from the in situ activation of PdOx aerogels were facilely realized to construct a highly porous structure and hybrid surface chemical states and functioned as active electrocatalysts, which not only broadens the development of metal aerogels, but also shows promise for fuel cell applications.Data availability
All data is available in the main text or the ESI.†Author contributions
D. W., Q. F., and J. G. conceived and supervised the research. D. W., W. G., C. W., and Q. F. designed the experiments. C. W., W. G., B. Y., and W. M. performed most of the experiments, characterizations, electrochemical testing and data analysis. X. W. and J. G. performed DFT simulations. W. G., C. W., Q. F., and J. G. wrote the paper. C. W., W. G., and X. W. contributed equally. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21804108, 21902128), the 1000 Youth Talent Program of China, the China Postdoctoral Science Foundation (No. 2021M692620), the Fundamental Research Funds for the Central Universities (No. 3102019JC005), and the Research Fund of the State Key Laboratory of Solidification Processing (NPU), China (2021-QZ-01). We also thank the Analytical & Testing Center of Northwestern Polytechnical University for SEM, TEM and XRD characterization.Notes and references
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- D. Bruce, K. Haresh and T. Jean-Marie, Science, 2011, 334, 928–935 CrossRef PubMed.
- J. Liu, X. Sun, P. Song, Y. Zhang, W. Xing and W. Xu, Adv. Mater., 2013, 25, 6879–6883 CrossRef CAS PubMed.
- Q. W. Chang, Y. M. Hong, H. J. Lee, J. H. Lee, D. Ologunagba, Z. X. Liang, J. Kim, M. J. Kim, J. W. Hong, L. Song, S. Kattel, Z. Chen, J. G. Chen and S. I. Choi, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, 2112109119 CrossRef PubMed.
- V. Mantella, L. Castilla-Amorós and R. Buonsanti, Chem. Sci., 2020, 11, 11394–11403 RSC.
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J.-Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed.
- D. Göhl, A. Garg, P. Paciok, K. J. J. Mayrhofer, M. Heggan, Y. Shao-Horn, R. E. Dunin-Borkowski, Y. Román-Leshkov and M. Ledendecker, Nat. Mater., 2020, 19, 287–291 CrossRef PubMed.
- J. Kim, H. Kim, W. J. Lee, B. Ruqia, H. Baik, H. S. Oh, S. M. Paek, H. K. Lim, C. H. Choi and S. I. Choi, J. Am. Chem. Soc., 2019, 141, 18256–18263 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- C. Zhu, D. Du, A. Eychmüller and Y. Lin, Chem. Rev., 2015, 115, 8896–8943 CrossRef CAS PubMed.
- Y. Guo, J. Bae, Z. Fang, P. Li, F. Zhao and G. Yu, Chem. Rev., 2020, 120, 7642–7707 CrossRef CAS PubMed.
- N. Bigall, A.-K. Herrmann, M. Vogel, M. Rose, P. Simon, W. Carrillo-Cabrera, D. Dorfs, S. Kaskel, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2009, 48, 9731–9734 CrossRef CAS PubMed.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 57, 14149–14153 CrossRef CAS PubMed.
- M. Zareie Yazdan-Abad, M. Noroozifar, A. S. Douk, A. R. Modarresi-Alam and H. Saravani, Appl. Catal., B, 2019, 250, 242–249 CrossRef CAS.
- R. Du, J. Wang, Y. Wang, R. Hübner, X. Fan, I. Senkovska, Y. Hu, S. Kaskel and A. Eychmüller, Nat. Commun., 2020, 11, 1590 CrossRef CAS PubMed.
- R. Du, W. Jin, R. Hübner, L. Zhou, Y. Hu and A. Eychmüller, Adv. Energy Mater., 2020, 10, 1903857 CrossRef CAS.
- Q. Fang, H. Wang, X. Lv, X. Wei, X. Luo, J. Huang, L. Jiao, W. Gu, W. Song and C. Zhu, ACS Sustainable Chem. Eng., 2021, 9, 13039–13046 CrossRef CAS.
- H. Wang, L. Jiao, L. Zheng, Q. Fang, Y. Qin, X. Luo, X. Wei, L. Hu, W. Gu, J. Wen and C. Zhu, Adv. Funct. Mater., 2021, 31, 2103465 CrossRef CAS.
- Y. Zheng, J. Yang, X. Lu, H. Wang, A. A. Dubale, Y. Li, Z. Jin, D. Lou, N. K. Sethi, Y. Ye, J. Zhou, Y. Sun, Z. Zheng and W. Liu, Adv. Energy Mater., 2021, 11, 2002276 CrossRef CAS.
- R. Zhang, L. Zhu, X. Liu, J. Zhu and Y. Zhao, ACS Sustainable Chem. Eng., 2021, 9, 7837–7845 CrossRef CAS.
- D. Wen, A.-K. Herrmann, L. Borchardt, F. Simon, W. Liu, S. Kaskel and A. Eychmüller, J. Am. Chem. Soc., 2014, 136, 2727–2730 CrossRef CAS PubMed.
- W. Liu, A.-K. Herrmann, N. C. Bigall, P. Rodriguez, D. Wen, M. Oezaslan, T. J. Schmidt, N. Gaponik and A. Eychmüller, Acc. Chem. Res., 2015, 48, 154–162 CrossRef CAS PubMed.
- C. Z. Zhu, Q. R. Shi, S. F. Fu, J. H. Song, H. B. Xia, D. Du and Y. H. Lin, Adv. Mater., 2016, 28, 8779–8783 CrossRef CAS PubMed.
- R. Du, J.-O. Joswig, R. Hübner, L. Zhou, W. Wei, Y. Hu and A. Eychmüller, Angew. Chem., Int. Ed., 2020, 59, 8293–8300 CrossRef CAS PubMed.
- X. Shang, B. Dong, Y. M. Chai and C. G. Liu, Sci. Bull., 2018, 63, 853–876 CrossRef CAS.
- G. Kresse and J. Furthmuller, ACS Appl. Energy Mater., 1996, 6, 15–50 CAS.
- L. C. Grabow, A. A. Gokhale, S. T. Evans, J. A. Dumesic and M. Mavrikakis, J. Phys. Chem. C, 2008, 112, 4608–4617 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- P. J. Perdew, K. M. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef PubMed.
- C. D. Tait, D. R. Janecky and P. S. Z. Rogers, Geochim. Cosmochim. Acta, 1991, 55, 1253–1264 CrossRef CAS.
- A. A. Dubale, Y. Zheng, H. Wang, R. Hübner, Y. Li, J. Yang, J. Zhang, N. K. Sethi, L. He, Z. Zheng and W. Liu, Angew. Chem., Int. Ed., 2020, 59, 13891–13899 CrossRef CAS PubMed.
- B. W. Mountain and S. A. Wood, Econ. Geol., 1988, 83, 492–510 CrossRef CAS.
- J. M. van Middlesworth and S. A. Wood, Geochim. Cosmochim. Acta, 1999, 63, 1751–1765 CrossRef CAS.
- R. Du, Y. Hu, R. Hübner, J. O. Joswig, X. Fan, K. Schneider and A. Eychmüller, Sci. Adv., 2019, 5, eaaw4590 CrossRef PubMed.
- E. Antolini, Energy Environ. Sci., 2009, 2, 915–931 RSC.
- A. Janssen, V. Pawlik, A. D. von Rueden, L. Xu, C. Wang, M. Mavrikakis and Y. Xia, Adv. Mater., 2021, 33, 2103801 CrossRef CAS PubMed.
- W. Qiao, M. Zha, Y. Yang, G. Hu and L. Feng, Chem. Commun., 2022, 58, 10651–10654 RSC.
- J. X. Li, Y. J. Chang, D. Z. Li, L. G. Feng and B. G. Zhang, Chem. Commun., 2021, 57, 7035–7038 RSC.
- X. Lao, M. Yang, X. Sheng, J. Sun, Y. Wang, D. Zheng, M. Pang, A. Fu, H. Li and P. Guo, ACS Appl. Energy Mater., 2022, 5, 1282–1290 CrossRef CAS.
- L. Tao, D. Yu, J. S. Zhou, X. Lu, Y. X. Yang and F. M. Gao, Small, 2018, 14, 1704503 CrossRef PubMed.
- L. Yang, X. Wang, D. Liu, G. Cui, B. Dou and J. Wang, Appl. Catal., B, 2020, 263, 118304 CrossRef CAS.
- D.-J. Yang, I. Kamienchick, D. Y. Youn, A. Rothschild and I.-D. Kim, Adv. Funct. Mater., 2010, 20, 4258–4264 CrossRef CAS.
- E. Gracia-Espino, G. Hu, A. Shchukarev and T. Wågberg, J. Am. Chem. Soc., 2014, 136, 6626–6633 CrossRef CAS PubMed.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183–4206 CrossRef CAS PubMed.
- W. Qiao, X. D. Yang, M. Li and L. G. Feng, Nanoscale, 2021, 13, 6884–6889 RSC.
- T.-J. Wang, F.-M. Li, H. Huang, S.-W. Yin, P. Chen, P.-J. Jin and Y. Chen, Adv. Funct. Mater., 2020, 30, 2000534 CrossRef CAS.
- J. Yang, Y. Zhou, H. Su and S. Jiang, J. Electroanal. Chem., 2011, 662, 251–256 CrossRef CAS.
- Y. Lu and W. Chen, ACS Catal., 2012, 2, 84–90 CrossRef CAS.
- M. K. Kabiraz, J. Kim and S. I. Choi, Bull. Korean Chem. Soc., 2021, 42, 802–805 CrossRef CAS.
- Y. F. Bao and L. G. Feng, Acta Phys. Sin., 2021, 37, 2008031 Search PubMed.
- H. Xu, H. Shang, C. Wang and Y. Du, Small, 2021, 17, 2005092 CrossRef CAS PubMed.
- S. Han, C. He, Q. Yun, M. Li, W. Chen, W. Cao and Q. Lu, Coord. Chem. Rev., 2021, 445, 214085 CrossRef CAS.
- H. Lv, L. Sun, L. Zou, D. Xu, H. Yao and B. Liu, Chem. Sci., 2019, 10, 1986–1993 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM, TEM images, UV-vis spectra, optical images, electrochemical tests, models for DFT calculations, additional discussions, and supplementary video. See DOI: https://doi.org/10.1039/d2sc05425f |
| ‡ Chen Wang, Wei Gao, and Xinhao Wan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |