
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly sensitive urine glucose detection with graphene field-effect transistors functionalized with electropolymerized nanofilms†
Gonzalo E.
Fenoy
 a,
Waldemar A.
Marmisollé
*a,
Wolfgang
Knoll
bc and
Omar
Azzaroni
*ad
a,
Waldemar A.
Marmisollé
*a,
Wolfgang
Knoll
bc and
Omar
Azzaroni
*ad
aInstituto de Investigaciones Fisicoquímicas Teóricas y Aplicadas (INIFTA), Departamento de Química, Facultad de Ciencias Exactas, Universidad Nacional de La Plata (UNLP), CONICET, 64 and 113, La Plata (1900), Argentina. E-mail: wmarmi@inifta.unlp.edu.ar; azzaroni@inifta.unlp.edu.ar
bAIT Austrian Institute of Technology, Biosensor Technologies, Tulln, Austria
cDepartment of Scientific Coordination and Management, Danube Private University, Krems, Austria
dCEST-UNLP Partner Lab for Bioelectronics (INIFTA), Diagonal 64 y 113, La Plata (1900), Argentina
First published on 13th October 2021
Abstract
We introduce a new approach for glucose oxidase (GOx) immobilization on graphene field-effect transistors (gFETs) to fabricate highly sensitive glucose sensors. The strategy relies on the electropolymerization of a layer of the copolymer poly(3-amino-benzylamine-co-aniline) (PABA) on graphene-based transistors. The synthesized polymer film provides the suitable electrostatic charge and non-denaturing environment for enzyme immobilization without the need of any chemical primer. Then, the local pH changes triggered by the enzyme-catalyzed oxidation produce a shift in the Dirac potential of the gFETs to more negative values, which is evidenced by the differences in the graphene channel conductivity and constitutes the signal transduction mechanism of the sensing devices. The assembled biosensors revealed a low LOD of 4.1 μM and were capable of detecting glucose in the range from 10 to 1000 μM in a flow configuration. Moreover, they showed sensitivity of −24.9 μA per decade of glucose concentration and a fast response time, with an average value of 190 seconds, while allowing the operation at small gate-source and drain-source voltages. Finally, the biosensors were able to successfully monitor the analyte in urine samples, showing their potential towards the fabrication of point-of-care glucose testing devices.
Introduction
Diabetes mellitus is a group of diseases characterized by hyperglycemia that affects millions of people worldwide.1,2 When not treated properly, diabetes can provoke serious complications such as blindness, kidney failure, and heart attack, among others.3,4 Furthermore, other glucose metabolism anomalies may also imply severe problems and diseases.2,5 Therefore, regular monitoring of the glucose levels is crucial for the optimal supervision of the progression of those diseases and conditions. In this regard, point-of-care (POC) biosensors allow patients (as well as healthy individuals) to perform the tracking of their glucose concentration easily and accurately, without the need of specific staff or a visit to the hospital.6 While the POC testing of blood glucose can present some inconveniences related to blood drawing (either intravenously or by finger pricking), the detection of glucose in urine can be easily performed.7 Moreover, the presence of glucose in urine (glycosuria) is a dangerous condition which indicates a worsening of diabetes.8In this regard, electrochemical biosensing is one of the most efficient and employed methods to detect biomolecules for the diagnosis of different conditions and diseases.13,14 Particularly, the biosensing field has shown great growth upon the appearance of graphene field-effect transistors (gFETs), as these devices show an attractive option for highly sensitive and reliable detection, which also allows real-time monitoring and miniaturization.9 Briefly, the sensing mechanism of gFETs lies in the variation of the electrical resistance of the graphene channel upon adsorption of ions or molecules.10,11 In this way, the devices have demonstrated the effective detection of ions, gaseous molecules, and various biologically relevant entities.10,12,13 Particularly, sensing with gFETs coupled with enzymatic reactions has shown high specificity and selectivity, achieving the effective detection of glucose, acetylcholine, and urea, among others biomarkers.11,14,15 However, in order to fabricate enzymatic gFET biosensors, the graphene channel must be functionalized to introduce specific recognition moieties. In this regard, adequate enzyme immobilization is a key factor in order to develop sensitive and reliable devices, as most of the figures of merit of biosensors are crucially affected by enzyme stability.16 Then, upon anchoring, the enzyme must preserve its activity and accessibility to its active sites without detaching, while the immobilization method must also maintain the functionality of the transducing element.17
Particularly, when considering enzymatic glucose sensors, glucose oxidase (GOx) has been immobilized on diverse surfaces through a vast amount of strategies, such as physical adsorption, covalent attachment, and physical entrapment or encapsulation.18–20 While GOx immobilization via covalent attachment has been reported as a useful strategy, it is well-known that this approach can exhibit some problems, such as non-adequate reproducibility and the risk of altering the structure and functionality of the enzyme, as key groups could be involved in the immobilization process.18,21,22 Moreover, and particularly when considering enzyme immobilization on graphene surfaces (as that occurring in gFETs), the direct covalent attachment to graphene has been reported to cause damage to the sp2 structure of the material, which could produce the deterioration of the signal transduction of the biosensors.23 On the other hand, physical adsorption (for example via electrostatic immobilization) makes use of the interactions between surface functional groups and enzymes, and the binding, usually, does not change the native structure of the enzyme, preventing the alteration of the active sites of the enzyme and retaining its catalytic activity upon immobilization.24 Particularly considering GOx electrostatic immobilization, it has been reported that this enzyme strongly interacts in the presence of molecules that have a high number of reactive amino groups, such as polyethyleneimine (PEI).25
In this regard, the functionalization of gFETs with polyamines has been shown useful in order to perform the electrostatic immobilization of different enzymes such as urease, arginase, and acetylcholinesterase.11,14,26 Moreover, we have recently demonstrated the electrodeposition of polymers bearing multiple amino groups (poly(3-amino-benzylamine-co-aniline)) (PABA) on gFETs as an effective approach for the generation of platforms capable of immobilizing enzymes by electrostatic interactions with high stability and without altering their functionality. The electropolymerization technique brings numerous benefits as a functionalization approach, yielding highly stable films with an accurate control of the thickness.11 Moreover, it has been recently reported that the functionalization of gFETs with amino-moiety-bearing polymers improves the pH response of the transistors, as the variation in the degree of protonation of the polymeric layer in the vicinity of the graphene channel causes a change in the Fermi level.11,14,27 In this regard, PABA possesses different kinds of amine–imine moieties, which are capable of transducing pH changes in a wide pH range. Then, the electropolymerized layer not only offers an appropriate environment for the enzyme immobilization, but it also enhances the pH sensitivity of the graphene-based transistors.11
Finally, regarding gFET-based glucose sensors, the most reported GOx immobilization method implies the covalent attachment employing a linker molecule (pyrene-like),15,28 while also physical entrapping29,30 has been used as a useful strategy. However, as far as we know, there are no reports on biosensors based on the electrostatic anchoring of GOx on gFETs. Hence, in this work we carried out the electropolymerization of PABA on gFETs and the subsequent electrostatic GOx anchoring as an alternative strategy for the fabrication of glucose biosensors. The PABA layer provides a suitable environment for the enzyme immobilization by making use of the extra amino moieties. Later, the local pH changes caused by the enzyme-catalyzed oxidation are chemically transduced by the variety of amine groups in PABA into changes in the surface charge density, which ultimately modify the gFET conductivity. This chemically amplified sensing mechanism allows the glucose quantification in a flow measurement setup, as well as the monitoring of the analyte in diluted urine, showing their potential towards the fabrication of point-of-care biosensors. It is worth noting that, contrary to traditional GOx-based glucose biosensors that rely on electrochemical transduction via redox wiring of GOx, the present strategy does not rely on the redox wiring of the enzyme, but on the highly reproducible pH shift resulting from the enzymatic activity of GOx.
Materials and methods
Chemicals and reagents
Cysteamine was obtained from Acros Organics. Glucose oxidase (GOx) (200–250 U mg−1, from Aspergillus brasiliensis) was obtained from Calzyme. Glucose, aniline (≥99.5%) (ANI), 3-aminobenzylamine (99%) (ABA), 4-(2-hydroxyethyl)-1piperazineethanesulfonic acid (≥99.5%) (HEPES), KCl, ethanol, KOH, HCl, H2SO4, hydrazine monohydrate (99%), (3-aminopropyl)-triethoxysilane (97%) (APTES), and N,N-dimethylformamide (≥99.8%) (DMF) were purchased from Sigma Aldrich. All aqueous solutions were prepared with Milli-Q water.Instrumentation
Scanning electron microscopy (SEM) images were recorded using a SUPRA 40 field emission scanning electron microscope (Zeiss). Surface plasmon resonance (SPR) spectroscopy was performed with an SPR Navi 210 A instrument (BioNavis Ltd, Tampere, Finland). Electrical measurements were performed using a probe station source meter unit U2422A (Keysight Technologies, USA). Cyclic voltammetry experiments were performed with a μAutolabIII/FRA2 potentiostat.gFET fabrication
In order to fabricate the gFETs, a previously reported protocol was followed.11 Briefly, to obtain the channel of the transistors, the glass area between the Au interdigitated gold electrodes (IDEs) (Micrux Technologies) was functionalized with graphene oxide (GO), which was subsequently reduced (rGO) with hydrazine. The gFETs were stored under vacuum.PABA electrosynthesis
The functionalization of the gFETs with the copolymer bearing pendant amino moieties, poly(3-amino-benzylamine-co-aniline) (PABA), was performed by cyclic voltammetry (CV) in acidic solution, as previously described.31,32 The electrochemical cell consisted of a batch add-on obtained from Micrux Technologies, using a Pt wire as the counter electrode and Ag/AgCl (3 M KCl) as the reference electrode, while both source and drain electrodes were connected as the WE. Later, the electrosynthesis of PABA on the gFETs was performed by CV in a solution containing 0.07 M ABA and 0.03 M ANI in 0.5 M H2SO4, from −0.15 to 1 V vs. Ag/AgCl at 50 mV s−1 up to 20 voltammetric cycles. By employing this feed ratio (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), a film composition of approximately 30% in ABA is expected based on previous studies.33 The polymer-modified transistors (PABA-gFETs) were finally rinsed with Milli-Q and a buffer (0.1 mM HEPES, 10 mM KCl, pH 7) solution.
:3), a film composition of approximately 30% in ABA is expected based on previous studies.33 The polymer-modified transistors (PABA-gFETs) were finally rinsed with Milli-Q and a buffer (0.1 mM HEPES, 10 mM KCl, pH 7) solution.
GOx electrostatic immobilization
In order to instigate the electrostatic anchoring of GOx on the PABA-gFETs, a 1 mg mL−1 solution of the enzyme in buffer (0.1 mM HEPES, 10 mM KCl) was prepared and adjusted to pH 6 (the isoelectric point of GOx is known to be 4.2 (ref. 34)). Afterwards, the array area of the PABA-gFETs was immersed in the GOx solution for 30 minutes and rinsed with the buffer solution. The modified transistors were stored at 4 °C.Surface Plasmon resonance spectroscopy
SPR Au substrates (SPR102 AU, BioNavis) (and Au-coated glass slides for the SEM image) were functionalized with an amino-terminated self-assembled monolayer (SAM) by incubating the substrates in 5 mM cysteamine ethanolic solution for 8 h. Then, the drop casting of the aqueous dispersion of GO flakes and subsequent chemical reduction were performed. Next, the PABA electro-polymerization, was performed as described earlier, yielding PABA–rGO-Au substrates. Finally, the PABA–rGO-Au substrates were mounted on the SPR apparatus and GOx immobilization was performed as reported for the transistors. The amount of immobilized enzyme was calculated according to:where k·dp is 1.09.10−7 cm/° (at 670 nm) and dn/dc was considered as 0.182 cm3 g−1 for globular proteins.
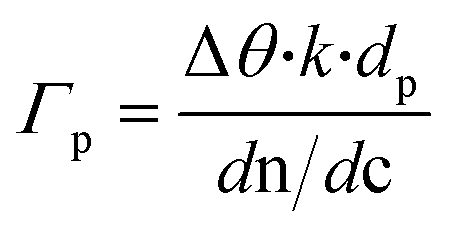
Electrical characterization and a sensing setup
In order to perform the electrical characterization of the devices, the interdigitated Au microelectrodes were used as source and drain electrodes, respectively, while the gate electrode was a Ag/AgCl reference electrode. For evaluating the transfer characteristics of the gFETs, a PEEK batch cell purchased from Micrux was used, while an PMMA flow-cell (also from Micrux) was employed for the continuous flow measurements. In order to perform the flow measurements, automatic sample collection was performed at a constant flow rate of 300 μL min−1 using a peristaltic pump (Ismatec) coupled to a switching valve (Rheodyne). A source–drain bias of 100 mV (VDS) was employed for all the measurements. For the gFET pH response experiments, different pH solutions were prepared by dropwise addition of HCl or KOH to the buffer solution (0.1 mM HEPES, 10 mM KCl). For the glucose sensing experiments, solutions with different concentrations of the analyte were prepared in 0.1 mM HEPES–10 mM KCl buffer and adjusted to pH 6. For the interference measurements, 20 μL of different interferent solutions were added to the batch cell (250 μL initial volume) in order to reach the final concentration. For the experiments of glucose sensing in real samples, urine from healthy individuals was collected, filtered (0.2 μm), and diluted with Milli-Q water (1:5), and the pH was corrected to 6. This sample (250 μL) was injected in the cell as the initial solution, while the same sample was spiked with glucose and different volumes of it were added to the cell in order to reach the desired spiked glucose concentration values.
Results and discussion
gFET fabrication and characterization
The first step of the biosensor production involved the fabrication of the gFETs (Fig. 1(A)). The SEM image in Fig. 1(B) shows the effective deposition of the reduced graphene oxide on the glass area, which yields the bridging of the Au electrodes and forms the channel of the transistors (further SEM images are presented in Fig. S1†). It has been reported that the use of IDEs enables a stable response as well as high device-to-device reproducibility (the inset of Fig. 1(B) shows a picture of a commercial IDE).11 In this regard, the reproducibility of the employed fabrication protocol was evaluated by recording the resistance between both source and drain electrodes for a batch of 30 gFETs, showing a very low RSD (Fig. S2†), similar to previously reported studies employing similar approaches.11,14,35 Moreover, and in line with recent studies,11 the gFET fabrication protocol resulted in very high device transconductance values, which reached values up to 1500 μS (Fig. S3†). The transconductance is one of the key parameters for the fabrication of highly sensitive biosensors.29 Then, the charge carrier mobility of the transistors was calculated according to eqn (1):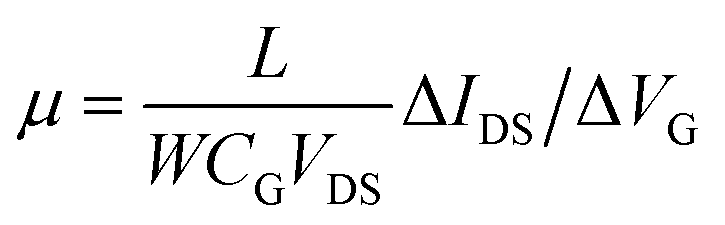 | (1) |
 | ||
| Fig. 1 Scheme of the gFET fabrication process (A). SEM image of the interdigitated electrode area of a gFET (B). Transfer characteristics of a gFET at different pH values (10 mM KCl, 0.1 mM HEPES, VDS = 100 mV) (C). Linear fitting of the changes in the Dirac point voltage as a function of the solution pH (pH-response) of the bare gFET (D). | ||
In order to study the performance of the gFETs, an electrolyte-gated setup was employed for the measurements, using a Ag/AgCl as the gate electrode (VG). The change of the VG under a constant drain–source voltage (VDS) modifies the resistance of the reduced graphene oxide channel, which results in a V-shaped transfer curve. The point of minimum drain–source current is known as the Dirac point observed at the Dirac point voltage (Vi). If the voltage is more positive than Vi, the charge carriers are mostly electrons, whereas holes are the carriers for more negative voltages. Firstly, the pH response of the bare gFETs was evaluated. From Fig. 1(C), it can be observed that, as the pH of the solution decreases, the Dirac point of the devices shifts to more negative potentials due to H3O+ adsorption (n-type doping). Likewise, at higher pH values, the OH− adsorption (p-type doping) causes a shift in the Dirac point to more positive values.11,36 Then, from Fig. 1(D), it can be observed that the devices showed a pH-sensitivity of 33 ± 1 mV pH−1 unit in terms of the changes in the Dirac point voltage in the pH range from 3.9 to 7.8.
PABA functionalization
Then, the electropolymerization of PABA in acidic medium was performed in order to functionalize the gFETs with a suitable surface to immobilize GOx, according to a procedure recently described.31 The copolymer of ANI and ABA carries pendant amino groups that enable the incorporation of negative biomolecules through electrostatic interactions. A schematic representation of the experimental configuration employed to perform the electropolymerization is shown in Fig. 2(A), together with the structure of the polymer. Briefly, both drain and source electrodes were connected as the working electrode of a three-electrode cell, while a Pt wire was used as a counter electrode and a Ag/AgCl as the reference electrode. In order to study the stability of the gFETs upon the polymerization procedure, the cycling of the gFETs in the highly acidic medium was performed. The transistor performance showed no changes after 20 voltammetric cycles (Fig. S4†).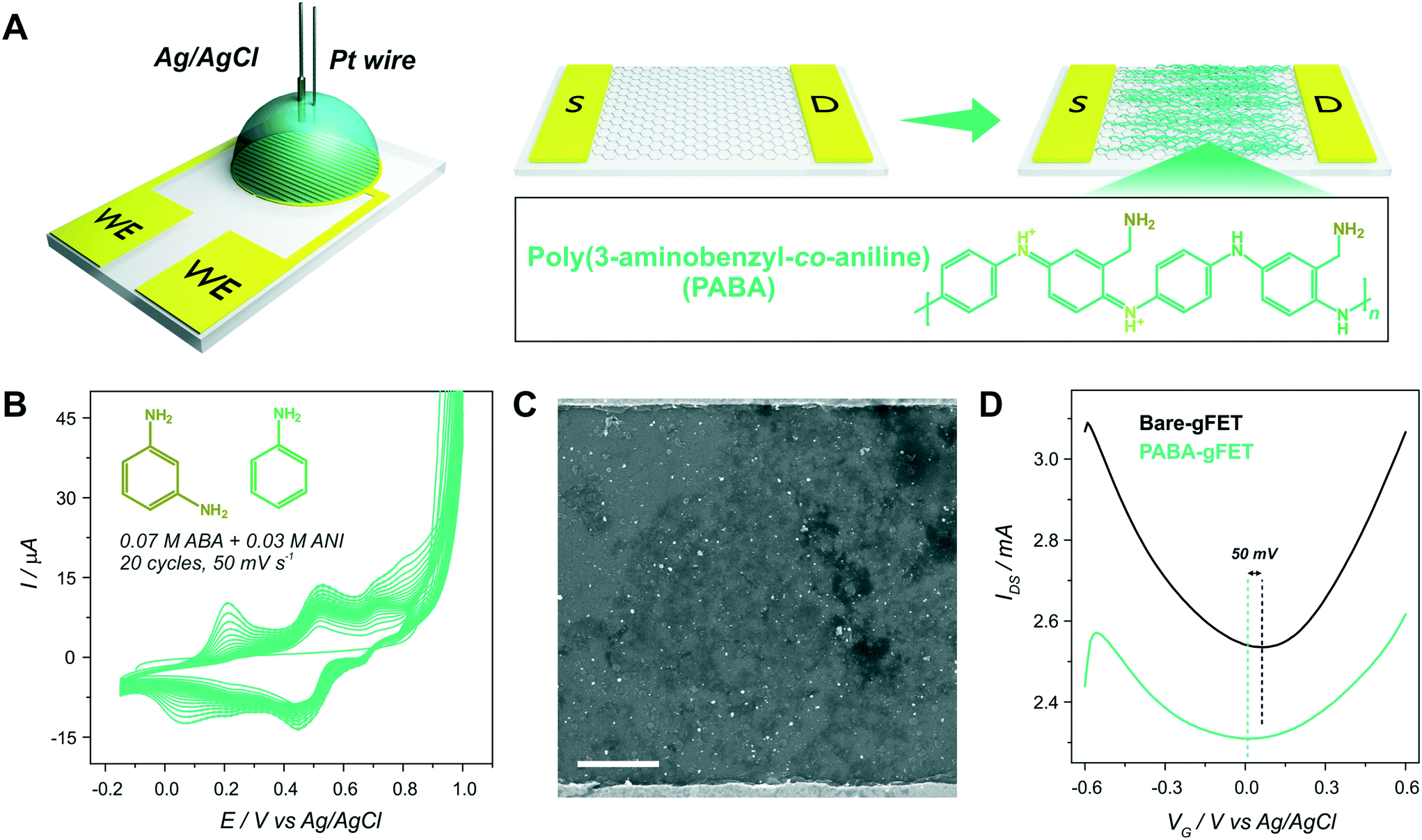 | ||
| Fig. 2 Scheme of the setup employed to perform PABA electropolymerization on the gFETs and structure of the polymer (A). Voltammograms for the electropolymerization of PABA on a gFET (B). SEM image of the channel area of a PABA-gFET; the scale bar corresponds to 2 μm (C). Transfer characteristics of a gFET before and after PABA electropolymerization (10 mM KCl, 0.1 mM HEPES, pH 7, VDS = 100 mV) (D). | ||
Voltammograms for the PABA electrosynthesis on a gFET are shown in Fig. 2(B). The characteristics of the voltammetric response are comparable to those previously reported.11,33 Briefly, the peak observed at approximately 0.2 V for the anodic sweep (and the corresponding one in the cathodic sweep) is ascribed to the convolution of the first redox couple for both pure poly(3-aminobenzylamine) and PANI (leuco-emeraldine to emeraldine transition).32,33 A similar behavior is observed for the peak appearing at about 0.5 V, which is attributed to the overlapping of the pure poly(3-aminobenzylamine) and the so-called “middle peak” of PANI.31 Finally, the high anodic current values starting from about 0.8 V are ascribed to the oxidative electropolymerization.32 The thickness of the PABA film is estimated to be approximately 7 nm from previously reported results.11,32Fig. 2(C) shows the SEM image of the channel area of a PABA-gFET. The effective deposition of the PABA polymer film can be observed.
The transfer characteristics at pH 7 of a gFET before and after PABA electropolymerization are shown in Fig. 2(D). It can be observed that, after the electropolymerization procedure, the Dirac point of the transistors shifts 50 mV toward more negative values. This result is ascribed to the electrostatic gating effect caused by the positively charged film on the graphene transistors, and it is in line with previous reports.11,37 Moreover, the devices maintain high transconductance values even after PABA electropolymerization, revealing that no deterioration of the gFETs happens during this functionalization procedure.
Moreover, it has been previously reported that, due to the multiple protonation states of the electrodeposited copolymer, the functionalized devices show an improved pH response compared with the bare gFETs. It has been stated that the variation of the protonation degree of polymers induces a shift in the Fermi level of the graphene channel, affecting the pH response of the transistors.11,27
GOx immobilization
After PABA electrosynthesis, the amino moieties of the copolymer were used as anchoring sites for GOx immobilization (Fig. 3(A)). From Fig. 3(B), it can be observed that the anchoring of the enzyme in the FET response shows a noticeable shift of the Dirac point to more positive potentials, consistent with the adsorption of negatively charged species (i.e., a p-doping effect).11,37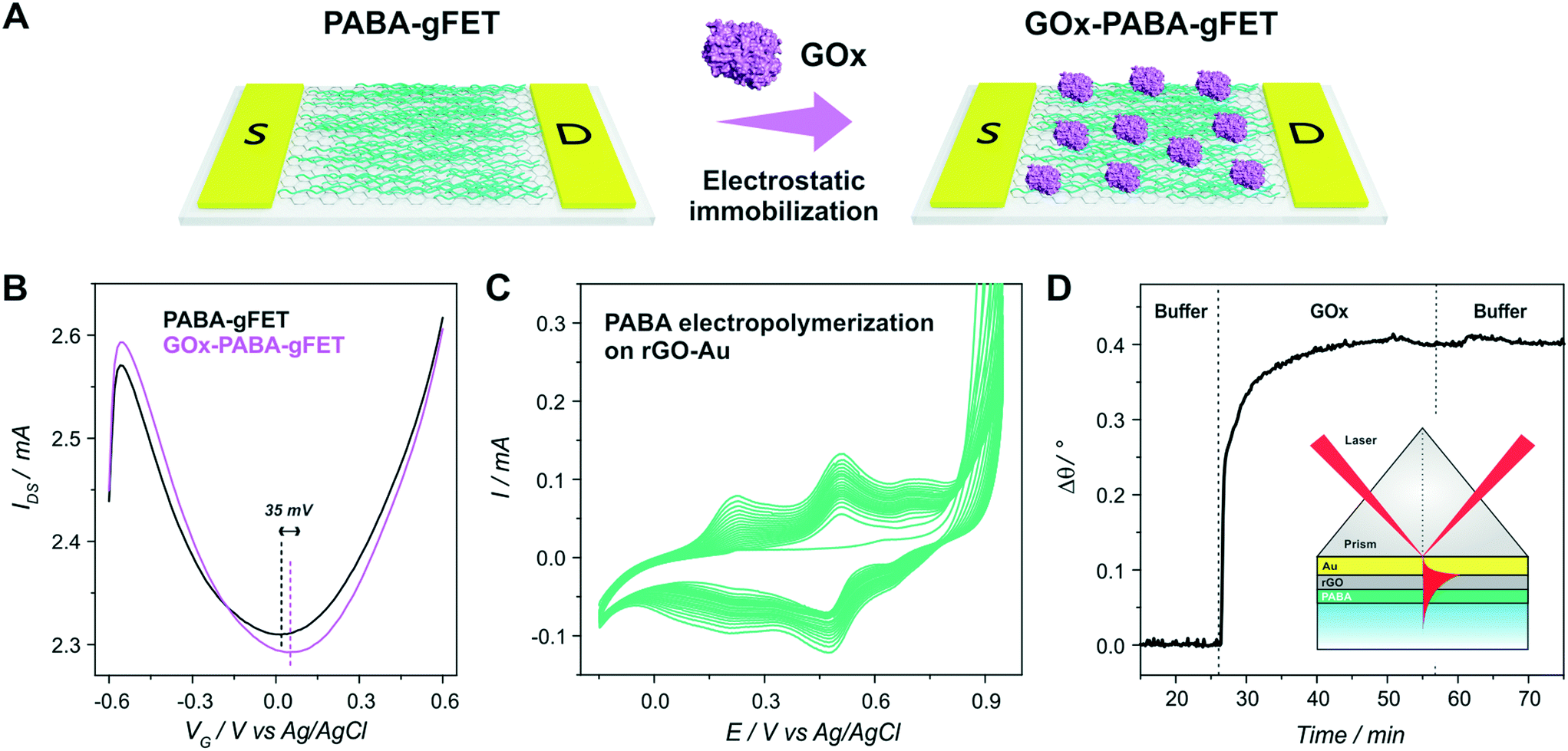 | ||
| Fig. 3 Scheme of the GOx immobilization on the PABA-gFETs (A). Transfer characteristics of a PABA-gFET before and after GOx immobilization (10 mM KCl, 0.1 mM HEPES, pH 7) (B). CV curves for the electropolymerization of PABA on a rGO-modified Au substrate (C). Change in the SPR minimum reflectivity angle of a PABA–rGO-Au substrate upon GOx adsorption (D). | ||
Moreover, with the aim of quantifying the mass of the GOx layer immobilized on the PABA-gFETs, the electrostatic anchoring of GOx was monitored by the SPR measurements of the previously modified PABA–rGO-Au substrates. Briefly, rGO-Au substrates were fabricated by functionalizing the commercial SPR Au sensor surface with a self-assembled monolayer (SAM) of cysteamine. Then, the substrate was modified by the deposition of rGO and finally the PABA electropolymerization was performed on the rGO–Au substrates which are then used in the SPR measurements.
A SEM image of the rGO-Au substrate is presented in Fig. S5,† showing the rGO deposition and distribution on the Au surface, which is similar to that obtained on the gFETs. Furthermore, the cyclic voltammetry curves for PABA electropolymerization on the rGO–Au substrates are shown in Fig. 3(C). It can be seen that the curves exhibit the same features as those previously observed for the electropolymerization on gFETs. Then, the reproducibility of the modification steps on the SPR sensors reinforces the validity of the estimation of the amount of GOx immobilized on the IDEs by SPR. The GOx immobilization on PABA was studied by monitoring the change in the minimum reflectivity angle (θ) while the different solutions were flowed through the cell (Fig. 3(D)). The obtained Δθ value yielded a mass surface coverage of 240 ng cm−2, equivalent to 1.5 pmol cm−2. This value agrees with typical GOx surface coverages on amino-functionalized surfaces.38
Finally, it becomes important to highlight that, due to the electrochemical functionalization procedure, no anchoring primers (as pyrene-like molecules) were required for the enzyme integration, which constitutes a very interesting advantage of this functionalization strategy.
Glucose sensing
Finally, the GOx-modified transistors were evaluated as glucose sensors. First of all, the effect of glucose addition on the static response of the devices was studied in order to investigate the signal transduction mechanism of the biosensors. Fig. 4(A) shows a scheme of the glucose oxidation on the biosensors, while Fig. 4(B) shows the transfer characteristics of a GOx–PABA-gFET in a buffer solution with and without 50 μM glucose (the same experimental results for a PABA-modified transistor are shown in Fig. S6,† exhibiting no significant change in the Dirac point of the transistor). It can be observed that, upon the addition of glucose to the electrolyte, the Dirac point of the transistors shifts to more negative potentials, in line with the shift obtained for a decrease in the solution pH. This shift could be related to the generation of gluconic acid, whose dissociation releases protons causing a diminution of the local pH in the vicinity of the graphene channel. However, and while this signal transduction mechanism has been widely theoretically and experimentally proven39,40 and the local pH decrease in the surroundings of graphene channel has been stated as the signal transduction mechanism for numerous gFET-based biosensors,11,41,42 it has been also reported for glucose gFET sensors that this shift could also be ascribed to the generation of H2O2, which is also produced upon glucose-catalyzed oxidation.15 In this regard, in order to elucidate the signal transduction mechanism of the biosensor, the transfer characteristics of a biosensor were studied in the absence and presence of 50 μM H2O2, showing no variation in the position of the Dirac point voltage (Fig. S7†). Moreover, an analogous experiment was performed in a flow configuration (Fig. S8†), yielding no significant variation in IDS while the H2O2 solution was flowed. Therefore, both experiments support the idea that the sensor signal transduction mechanism is driven by the change in pH caused by the generation of gluconic acid upon glucose-catalyzed oxidation. In this respect, it has been reported that PABA improves the pH response of the transistors especially towards lower pH solutions, as the amine and imine moieties of both monomers present in the copolymer can change their protonation states and, therefore, enhance pH sensitivity of the graphene channel.11 Consequently, it can be stated that the immobilized GOx on the PABA-modified transistors effectively catalyzes the oxidation of glucose, yielding gluconic acid and H2O2, and thus, triggering a diminution in the local pH, involving the signal transduction mechanism of the biosensors. | ||
| Fig. 4 Scheme of the glucose oxidation reaction on the transistors (A). Transfer characteristics of an GOx–PABA-gFET in the absence and presence of 0.1 mM glucose (VDS = 0.1 V, 10 mM KCl, 0.1 mM HEPES, pH 6) (B). Flow-response of a GOx–PABA-gFET (300 μL min−1, VG = −0.2 V, VDS = 0.1 V) at different analyte concentrations (C). Linear fitting for the GOx–PABA-gFETs showing the results obtained for two different electrodes (D). Response of a GOx–PABA-gFET to different potential interference injections (VG = −0.2 V, VDS = 0.1 V, 10 mM KCl, 0.1 mM HEPES, pH 6) (n = 2) (E). Response of a GOx–PABA-gFET to the injection of spiked urine samples (VG = −0.2 V, VDS = 0.1 V) (F) and linear fitting of the response (results for two measurements) (G). | ||
It is relevant to note here that, since the signal transduction mechanism of the biosensors is rooted in the pH change caused upon glucose oxidation, it is not necessary to incorporate redox-active species that operate as mediators (such as ferrocene, quinone, ruthenium complexes, or even redox polymers) in the film architecture in order to establish an efficient electron transfer between enzyme and electrodes. This mechanism avoids usual problems that appear upon mediator integration, such as response to interferences and the toxicity of the mediators (limiting the biosensor's in vivo application), together with the increased sensor fabrication complexity added by this step. Furthermore, the presence of dissolved oxygen is frequently an issue for redox-based glucose sensors as it competes with redox mediators causing a decrease in the amperometric response.43 This is not a problem for a biosensor device based on the present strategy.
Afterwards, the response of the devices to different flowing glucose concentration solutions was evaluated in a continuous mode as shown in Fig. 4(C) and S9.† As it was explained above, the catalysis of the enzyme substrate causes a decrease in the local pH, shifting the Dirac point of the biosensors to more negative values. Thus, if the gate voltage is set to a more negative value than the Dirac point (that is, the hole regime, VG < Vi), a decrease in the registered drain–source current can be observed upon glucose addition (at constant VG and VDS). From Fig. 4(C) and (D), it can be observed that the flow response of the biosensors exhibits logarithmic dependence on the glucose concentration, which can be understood by recalling the linear response of the pH (shown in section 3.1) and assuming that the proton concentration is proportional to the glucose concentration.11,27 Furthermore, this logarithmic dependence of the glucose response supports the idea of the pH-based signal transduction mechanism of the biosensors. The sensitivity of the GOx–PABA-gFETs in terms of the changes in the drain–source current (ΔIDS) reached −24.9 ± 0.5 μA per decade of glucose concentration (R2 = 0.9976). Furthermore, the stable flow response of the transistors to the continuous flow of the analyte indicated that they could be employed for the real-time sensing of glucose. Moreover, it can be observed that the flow of the buffer solution after the highest glucose concentration recovered the drain–source current to almost the original value (ΔIDS ∼ 0 ). This result implies that the biosensor response to glucose is totally reversible, and the devices could be reused for further measurements. A diminution of the registered drain–source current of ∼57 μA was observed when a solution of glucose concentration of 1 mM was flowed through the cell. Then, by making use of the sensitivity values obtained for the pH response of the sensors for the flow pH response reported before,11 the change in IDS for the highest glucose concentration can be associated with a decrease in the local pH of approximately 1.0 pH unit, i.e. from 6.0 to 5.0 under the steady-state conditions reached by continuous flowing.
Moreover, the flow response of the biosensors showed an average value for the time response of 190 seconds for 95% of the maximum response, enabling the fast real-time detection of the analyte by the devices. The device-to-device reproducibility was also assessed, showing an average relative standard deviation (RSD) for the 8 different evaluated glucose concentrations of 7% (n = 2). Furthermore, the repeatability of the devices showed a relative standard deviation (RSD) of 7.2% for three successive measurements of 50 μM glucose (static measurement). The stability of the biosensors was also investigated, showing that the devices maintained 92% of the original response after 3 days of storage at 4 °C in buffer HEPES solution (Fig. S10†).
The limit of detection (LOD) of the GOx–PABA-gFET sensor was determined to be 3 times the standard deviation for the buffer injection (blank), yielding a value of 4.1 μM. This low LOD would allow for the determination of glucose in a variety of body fluids, as it is known that the glucose concentration ranges in body fluids are 2–30 mM, 100–600 μM, 30–80 μM and 20–600 μM for blood, tears, saliva and sweat, respectively.44 Moreover, the low LOD would even enable the sample dilution, minimizing possible interferences present in the complex matrix. In order to gain further insight regarding this aspect, the response of the transistors to different potential interferences was analyzed (Fig. 4(E)). It can be seen that the biosensors show no appreciable variation in IDS upon injection of ascorbic acid (AA), dopamine (DA) and urea, which are usual analytes present in biological fluids as blood, cerebrospinal fluid and urine, respectively.45–47
Table 1 exhibits the relevant analytical figures of merit of other recently reported FET-based sensors for glucose detection, together with those obtained in this work. From the comparison, it can be concluded that the biosensors here presented show a very low LOD and wide detection range compared to other devices. Moreover, the operation in the electrolyte gating setup enables the use of low gate-source and drain-source potentials, minimizing the alteration of the biomolecules attached to the devices, and, therefore, ensuring enzyme functionality and guaranteeing a stable response. The employed setup also minimizes the occurrence of parasitic electrochemical reactions while showing very high sensitivity.11,50
| System | LOD | Dynamic range | Measurement setup | Ref. |
|---|---|---|---|---|
| Graphene transistor with AuNP-graphene gate | 4 μM | 10–400 μM | V GS = 0.5 V | 51 |
| V DS = 0.05 V | ||||
| Flexible CVD-graphene FET | 3.3 mM | 3.3–10.9 mM | V GS = 1 V | 15 |
| V DS = 0.2 V | ||||
| Silk-CVD-graphene FET | 0.1 mM | 0.1–10 mM | V GS = 0 V | 30 |
| V DS = 0.1 V | ||||
| CVD-graphene FET | 0.1 mM | 0.1–10 mM | V GS = 0 V | 28 |
| V DS = 0.1 V | ||||
| PEDOT:PSS organic thin-film transistor | 1.1 mM | 1.1–16.5 mM | V GS = 0 V | 52 |
| V DS = −1.5 V | ||||
| PEDOT:PSS organic thin-film transistor | 0.1 mM | 0.1–2 mM | V GS = 0.6 V | 53 |
| V DS = 0.2 V | ||||
| Charge transfer technique FET | 0.1 mM | 0.1–200 mM | V REF = 2.84 V | 54 |
| PABA-graphene FET | 4.1 μM | 10 μM–1 mM | V GS = −0.2 V | This work |
| V DS = 0.1 V |
Furthermore, in order to study the response of the biosensors in a complex biological fluid, we performed glucose sensing measurements in diluted urine samples (Fig. 4(F) and S11†). The cell was filled with the diluted real sample and, in order to determine its glucose concentration, injections of the same spiked sample were subsequently performed. Next, the linear fitting of the response of the biosensor in the biological fluid yielded a sensitivity of −13 ± 2 μA per decade of glucose concentration (R2 = 0.9462, n = 2) (Fig. 4(G)). By employing this sensitivity, a glucose concentration in the original sample of 40 ± 5 μM was found, consistent with the reported analyte concentration found in healthy individuals.48 These results show that the fabricated biosensors can be used for the determination of glucose in complex biological samples without the requirement of arduous sample processing steps, paving the way for the development of point-of-care gFET-based glucose biosensors.
Table S1† shows the comparison of the performance of the sensor developed in this work in biological samples with those of other recently developed FET-based glucose sensors. It can be noted that the present biosensors show a relatively low LOD, wide dynamic range and low RSD for the determination of glucose in the given biological sample. Moreover, it is worth noting that the developed sensor presents several advantages when compared with traditional methods for the detection of glucose in urine, such as the widely employed glucose strips (Table S2†), such as a low response to interferents, lower assay time and quantitative determination of the analyte, among others. Additionally, it has been recently concluded that dipstick testing for urinary glucose is insufficiently sensitive to be used as a screening tool for diabetes mellitus, which emphasizes the need for an accurate device for the detection of glucose in urine.49
Conclusions
We have reported a novel approach for the fabrication of glucose enzymatic gFET biosensors employing electropolymerization as the strategy for the graphene channel functionalization. Due to the advantages of voltammetric electrosynthesis, the PABA film thickness could be precisely controlled, and it showed excellent stability. Moreover, the presence of diverse amine–imine moieties not only allowed for PABA to transduce pH changes in a wide pH range, but it also allowed the subsequent electrostatic immobilization of GOx, granting a straightforward anchoring method without the need of any chemical primer that could affect the transistor performance.The GOx–PABA-gFETs displayed a shift in the Dirac potential to more negative values in the presence of the enzyme substrate due to GOx-catalyzed oxidation, allowing the real-time sensing of glucose in the range from 10 to 1000 μM. Moreover, the devices showed appropriate reproducibility and a fast response time. Compared with previously reported gFET sensors, the developed transistors showed a low LOD and wide detection range, while allowing the operation at small gate-source and drain-source potentials. Furthermore, the biosensors were able to successfully detect the analyte in a complex biological fluid such as diluted urine.
Further work could be directed towards the integration of different gFET-based biosensors with the aim of fabricating a simple platform for the simultaneous detection of multiple biorelevant analytes such as glucose, acetylcholine, and urea in biological fluids.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by ANPCyT (PICT-2017-1523), the CEST-Competence Center for Electrochemical Surface Technologies (CEST−UNLP Partner Lab for Bioelectronics) and the Universidad Nacional de La Plata (PID-X867). G. E. F. acknowledges CONICET for a doctoral fellowship and thanks the Biosensors Technologies Group for the support provided during his stay at AIT. Dr. Patrik Aspermair is greatly acknowledged for SEM measurements.References
- E. Jean-Marie, Diabetes Care, 2010, 33, S62–S69 CrossRef PubMed.
- E. Sehit and Z. Altintas, Biosens. Bioelectron., 2020, 159, 112165 CrossRef CAS PubMed.
- E. W. Gregg, N. Sattar and M. K. Ali, Lancet Diabetes Endocrinol., 2016, 4, 537–547 CrossRef PubMed.
- D. M. Nathan, N. Engl. J. Med., 1993, 328, 1676–1685 CrossRef CAS PubMed.
- A. K. Lee, B. Warren, C. J. Lee, J. W. McEvoy, K. Matsushita, E. S. Huang, A. R. Sharrett, J. Coresh and E. Selvin, Diabetes Care, 2018, 41, 104–111 CrossRef PubMed.
- V. Gubala, L. F. Harris, A. J. Ricco, M. X. Tan and D. E. Williams, Anal. Chem., 2012, 84, 487–515 CrossRef CAS PubMed.
- C. Radhakumary and K. Sreenivasan, Anal. Chem., 2011, 83, 2829–2833 CrossRef CAS PubMed.
- H. G. Rennke and B. M. Denker, Renal pathophysiology: the essentials, Lippincott Williams & Wilkins, 2019 Search PubMed.
- W. Fu, L. Jiang, E. P. van Geest, L. M. C. Lima and G. F. Schneider, Adv. Mater., 2017, 29, 1603610 CrossRef PubMed.
- B. Zhan, C. Li, J. Yang, G. Jenkins, W. Huang and X. Dong, Small, 2014, 10, 4042–4065 CrossRef CAS PubMed.
- G. E. Fenoy, W. A. Marmisollé, O. Azzaroni and W. Knoll, Biosens. Bioelectron., 2020, 148, 111796 CrossRef CAS PubMed.
- X. Zhang, Q. Jing, S. Ao, G. F. Schneider, D. Kireev, Z. Zhang and W. Fu, Small, 2020, 16, 1–24 Search PubMed.
- G. Wu, Z. Dai, X. Tang, Z. Lin, P. K. Lo, M. Meyyappan and K. W. C. Lai, Adv. Healthcare Mater., 2017, 6, 1700736 CrossRef PubMed.
- E. Piccinini, C. Bliem, C. Reiner-Rozman, F. Battaglini, O. Azzaroni and W. Knoll, Biosens. Bioelectron., 2017, 92, 661–667 CrossRef CAS PubMed.
- Y. H. Kwak, D. S. Choi, Y. N. Kim, H. Kim, D. H. Yoon, S. S. Ahn, J. W. Yang, W. S. Yang and S. Seo, Biosens. Bioelectron., 2012, 37, 82–87 CrossRef CAS PubMed.
- G. E. Fenoy, C. Bilderling, W. Knoll, O. Azzaroni and W. A. Marmisollé, Adv. Electron. Mater., 2021, 2100059 CrossRef CAS.
- A. Sassolas, L. J. Blum and B. D. Leca-Bouvier, Biotechnol. Adv., 2012, 30, 489–511 CrossRef CAS PubMed.
- C. Chen, Q. Xie, D. Yang, H. Xiao, Y. Fu, Y. Tan and S. Yao, RSC Adv., 2013, 3, 4473–4491 RSC.
- M. L. Cortez, W. Marmisollé, D. Pallarola, L. I. Pietrasanta, D. H. Murgida, M. Ceolín, O. Azzaroni and F. Battaglini, Chem. – Eur. J., 2014, 20, 13366–13374 CrossRef CAS PubMed.
- D. Pallarola, C. von Bildering, L. I. Pietrasanta, N. Queralto, W. Knoll, F. Battaglini and O. Azzaroni, Phys. Chem. Chem. Phys., 2012, 14, 11027–11039 RSC.
- A. Vakurov, C. E. Simpson, C. L. Daly, T. D. Gibson and P. A. Millner, Biosens. Bioelectron., 2005, 20, 2324–2329 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- S. Niyogi, E. Bekyarova, M. E. Itkis, H. Zhang, K. Shepperd, J. Hicks, M. Sprinkle, C. Berger, C. N. Lau, W. A. Deheer, E. H. Conrad and R. C. Haddon, Nano Lett., 2010, 10, 4061–4066 CrossRef CAS PubMed.
- T. Jesionowski, J. Zdarta and B. Krajewska, Adsorption, 2014, 20, 801–821 CrossRef CAS.
- S. B. Bankar, M. V. Bule, R. S. Singhal and L. Ananthanarayan, Biotechnol. Adv., 2009, 27, 489–501 CrossRef CAS PubMed.
- T. Berninger, C. Bliem, E. Piccinini, O. Azzaroni and W. Knoll, Biosens. Bioelectron., 2018, 115, 104–110 CrossRef CAS PubMed.
- L. H. Hess, A. Lyuleeva, B. M. Blaschke, M. Sachsenhauser, M. Seifert, J. A. Garrido and F. Deubel, ACS Appl. Mater. Interfaces, 2014, 6, 9705–9710 CrossRef CAS PubMed.
- Y. Huang, X. Dong, Y. Shi, C. M. Li, L.-J. Li and P. Chen, Nanoscale, 2010, 2, 1485 RSC.
- M. Zhang, C. Liao, C. H. Mak, P. You, C. L. Mak and F. Yan, Sci. Rep., 2015, 5, 1–6 CAS.
- X. You and J. J. Pak, Sens. Actuators, B, 2014, 202, 1357–1365 CrossRef CAS.
- G. E. Fenoy, J. M. Giussi, C. von Bilderling, E. M. Maza, L. I. Pietrasanta, W. Knoll, W. A. Marmisollé and O. Azzaroni, J. Colloid Interface Sci., 2018, 518, 92–101 CrossRef CAS PubMed.
- G. E. Fenoy, J. Scotto, J. Azcárate, M. Rafti, W. A. Marmisollé and O. Azzaroni, ACS Appl. Energy Mater., 2018, 1, acsaem.8b01021 CrossRef.
- W. A. Marmisollé, D. Gregurec, S. Moya and O. Azzaroni, ChemElectroChem, 2015, 2, 2011–2019 CrossRef.
- R. Wilson and A. P. F. Turner, Biosens. Bioelectron., 1992, 7, 165–185 CrossRef CAS.
- C. Reiner-Rozman, M. Larisika, C. Nowak and W. Knoll, Biosens. Bioelectron., 2015, 70, 21–27 CrossRef CAS PubMed.
- P. Salvo, B. Melai, N. Calisi, C. Paoletti, F. Bellagambi, A. Kirchhain, M. G. Trivella, R. Fuoco and F. Di Francesco, Sens. Actuators, B, 2018, 256, 976–991 CrossRef CAS.
- Y. Y. Wang and P. J. Burke, Nano Res., 2014, 7, 1650–1658 CrossRef CAS PubMed.
- J. Hodak, R. Etchenique, E. J. Calvo, K. Singhal and P. N. Bartlett, Langmuir, 1997, 13, 2708–2716 CrossRef CAS.
- S. D. Caras, D. Petelenz and J. Janata, Anal. Chem., 1985, 57, 1920–1923 CrossRef CAS PubMed.
- A. B. Kharitonov, M. Zayats, A. Lichtenstein, E. Katz and I. Willner, Sens. Actuators, B, 2000, 70, 222–231 CrossRef CAS.
- Y. Ohno, K. Maehashi, Y. Yamashiro and K. Matsumoto, Nano Lett., 2009, 9, 3318–3322 CrossRef CAS PubMed.
- T. Ono, Y. Kanai, K. Inoue, Y. Watanabe, S. I. Nakakita, T. Kawahara, Y. Suzuki and K. Matsumoto, Nano Lett., 2019, 19, 4004–4009 CrossRef CAS PubMed.
- P. Wang, S. Amarasinghe, J. Leddy, M. Arnold and J. S. Dordick, Polymers, 1998, 39, 123–127 CrossRef CAS.
- E. W. Nery, M. Kundys, P. S. Jeleń and M. Jönsson-Niedziólka, Anal. Chem., 2016, 88, 11271–11282 CrossRef PubMed.
- K. M. Mitchell, Anal. Chem., 2004, 76, 1098–1106 CrossRef CAS PubMed.
- G. Block, M. Dietrich, E. Norkus, C. Jensen, N. L. Benowitz, J. D. Morrow, M. Hudes and L. Packer, Epidemiology, 2006, 17, 404–412 CrossRef PubMed.
- S. Bouatra, F. Aziat, R. Mandal, A. C. Guo, M. R. Wilson, C. Knox, T. C. Bjorndahl, R. Krishnamurthy, F. Saleem, P. Liu, Z. T. Dame, J. Poelzer, J. Huynh, F. S. Yallou, N. Psychogios, E. Dong, R. Bogumil, C. Roehring and D. S. Wishart, PLoS One, 2013, 8, e73076 CrossRef CAS PubMed.
- E. Witkowska Nery, M. Kundys, P. S. Jeleń and M. Jönsson-Niedziółka, Anal. Chem., 2016, 88, 11271–11282 CrossRef CAS PubMed.
- Y. W. Ooi and S. Teece, Zhonghua Jizhen Yixue Zazhi, 2006, 23, 139–140 Search PubMed.
- Q. He, H. G. Sudibya, Z. Yin, S. Wu, H. Li, F. Boey, W. Huang, P. Chen and H. Zhang, ACS Nano, 2010, 4, 3201–3208 CrossRef CAS PubMed.
- M. Ma, Y. Zhou, J. Li, Z. Ge, H. He, T. Tao, Z. Cai, X. Wang, G. Chang and Y. He, Analyst, 2020, 145, 887–896 RSC.
- J. Liu, M. Agarwal and K. Varahramyan, Sens. Actuators, B, 2008, 135, 195–199 CrossRef CAS.
- Z.-T. Zhu, J. T. Mabeck, C. Zhu, N. C. Cady, C. A. Batt and G. G. Malliaras, Chem. Commun., 2004, 1556 RSC.
- S. R. Lee, K. Sawada, H. Takao and M. Ishida, Biosens. Bioelectron., 2008, 24, 650–656 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sd00007a |
| This journal is © The Royal Society of Chemistry 2022 |