
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntercalating methylene blue in molecular beacon for sensitive detection of salivary TNF-α towards early diagnosis of oral cancer†
Siyi
Zou
 a,
Hui
Wei
a,
Xiaolin
Cui
b,
Wing Cheung
Mak
c,
Xuejin
Li
d and
Guozhen
Liu
*a
a,
Hui
Wei
a,
Xiaolin
Cui
b,
Wing Cheung
Mak
c,
Xuejin
Li
d and
Guozhen
Liu
*a
aSchool of Medicine, The Chinese University of Hong Kong, Shenzhen 518172, China. E-mail: Liuguozhen@cuhk.edu.cn
bChristchurch Regenerative Medicine and Tissue Engineering (CReaTE) group, Department of Orthopaedic Surgery and Musculoskeletal Medicine, University of Otago, Christchurch, 8011, New Zealand
cDepartment of Biomedical Engineering, The Chinese University of Hong Kong, Hong Kong SAR, China
dSchool of Science and Engineering, The Chinese University of Hong Kong, Shenzhen 518172, China
First published on 29th April 2022
Abstract
Oral cancer is the sixth most common cancer globally, and pro-inflammatory cytokines are associated with oral cancer development and progression. Thus, monitoring salivary pro-inflammatory cytokines is essential for the early diagnosis of oral cancer. Electrochemical molecular beacons are popular in the design of biosensors for the real-time monitoring of analytes. Labelling redox probes on molecular beacons reporting electrochemical signals is critical to enhance their sensitivity. Herein, we designed an electrochemical biosensor, denoted as screen-printed carbon electrode (SPCE)-graphene oxidize-aptamer conjugated with methylene blue (MB) (SPCE-GO-aptamer (MB)), by intercalating methylene blue (i.e., the redox probe) in the hairpin structure of a molecular beacon having specific affinity to TNF-α on a graphene oxide (GO)-modified glass screen-printed carbon electrode for the quantitative detection of TNF-α. For comparison, another sensing interface termed SPCE-GO-aptamer-MB, on which methylene blue was covalently attached to the end of the molecular beacon on a GO-modified SPCE was employed for the quantitative detection of TNF-α. Techniques including electrochemistry, XPS, and SEM were used to characterize the surface of the functionalized sensor. Because a larger amount of MB could be intercalated in the hairpin of the molecular beacon, a relatively larger MB signal was observed for SPCE-GO-aptamer (MB) than that on SPCE-GO-aptamer-MB when both biosensing interfaces were exposed to the same concentration of TNF-α. The linear range of SPCE-GO-aptamer (MB) and SPCE-GO-aptamer-MB for the detection of TNF-α was 1–400 pg mL−1 with a detection limit of 1 pg mL−1 and 10–300 pg mL−1 with a detection limit of 10 pg mL−1, respectively. The SPCE-GO-aptamer(MB) sensing interface was validated by detecting TNF-α in saliva with a desirable performance, which promises a non-invasive method for monitoring the dynamic cytokine concentration in oral cancer.
Introduction
Oral cancer is generally preceded by potentially malignant oral disorders with the malignant neoplasm of the lip and oral cavity.1 Oral cancer is the sixth most frequent cancer globally and has an overall 5 year survival rate of less than 50%. The prevalent oral habits of betel quid chewing, smoking, alcohol consumption, and oral-genital sex can cause oral cancer. Oral cancer can be life-threatening, and thus early diagnostic tools are in high demand.2 The standard methods used to detect oral cancers are still comprehensive clinical examinations, expensive biochemical investigations, and invasive biopsies. Saliva carries molecules and cells originating in the aerodigestive tract, as well as nucleic acids, peptides, electrolytes, organic and inorganic salts and proteins, which are passively and actively transported from the circulatory system into the salivary glands.3 Thus saliva, as a non-invasive and relatively stress-free diagnostic alternative to blood, is the mirror of our body, reflecting its physiological and pathological status.Cytokines are signalling molecules between the cells in the immune system, which are present in various body fluids (blood, sweat, saliva, interstitial fluids, cerebrospinal fluids, etc.) at the pM concentration level.4 They are among the core indicators of the functional status of the body, where elevated concentrations of cytokines in body fluids are associated with a range of unhealthy conditions.5 Pro-inflammatory cytokines including TNF-α, IL-6, and IL-8 have been observed to be elevated in brain disorder diseases.6,7 Studies demonstrated that the tumour microenvironment generates chronic inflammation and promotes the secretion of diverse cytokines in different types and stages of cancers.8 It has been reported that pro-inflammatory cytokines including IL-1β, IL-2, IL-6, IL-1α and TNF-α are associated with the severity of oral mucosal tissue damage in patients with cancer,9 and their salivary levels are significantly higher in oral cancer patients compared to healthy controls.10 Thus, salivary cytokines can be potential biomarkers for the diagnosis of oral cancer.
TNF-α is a small 157-amino-acid pro-inflammatory cytokine implicated in nociceptive signalling. It functions as a “master regulator” cytokine, which initiates inflammation and drives pro-inflammatory cytokine cascades. Previous studies have identified the important role of TNF-α and TNF receptor 1 (TNFR1) in the invasiveness of oral cancer cell lines.11 In addition, the level of TNF-α becomes elevated during the progression of potentially malignant oral disorders, given that TNF-α regulated malignant transformation in an oral carcinogenesis model.12 Therefore, TNF-α is an ideal biomarker for the early diagnosis of oral cancer. Currently, enzyme-linked immunosorbent assays (ELISA), enzyme-linked immunospot assays (ELISPOT), and reverse transcriptase-polymerase chain reaction (RT-PCR)13 are the dominant methods to detect TNF-α in the clinical laboratory. However, these methods are time-consuming, expensive, and unsuitable for real-time detection.14 Thus, to overcome these limitations, numerous advanced biosensing technologies, especially electrochemical biosensors, have become attractive for the detection of TNF-α due to their high sensitivity, short assay time and device miniaturization.15,16 Previously, we developed a nanofabricated amperometric immunosensor to detect TNF-α in the range of 0.1–150 pg mL−1.17 The fabricated immunosensor provided high selectivity and stability and could be used to detect TNF-α secreted by live BV-2 cells with comparable accuracy to ELISA. Furthermore, an impedimetric immunosensor was designed to measure the TNF-α levels in artificial saliva and real human saliva in the range of 1–100 pg mL−1, which was applied for the early diagnosis of heart failure.18
Electrochemical molecular beacons, as the key component of immunosensors, have demonstrated advantages in biosensing because they can perform analyte-triggered structure switching as a result of analyte binding, which creates a readily detectable output in real time.19 To date, numerous reports have been published on labeling the redox probe molecules methylene blue (MB), ferrocene (Fc) and anthraquinone (AQ) at the end of aptamers to construct aptamer-based electrochemical biosensors for the detection of various analytes ranging from small molecules to large proteins. For example, Crulhas et al.20 developed MB-labeled IFN-γ aptamers for immobilization on the surface of a gold electrode, which was further fabricated into an electrochemical sensor for the detection of bovine interferon gamma (BoIFN-γ). The binding of BoIFN-γ led to the development of an aptamer hairpin structure, repelling the MB redox molecules from the electrode and reducing the electron transfer efficiency. The detection limit of this sensor was 0.1 nM in pristine buffer and 0.9 nM in blood. In another study, Zhang et al.21 constructed a ferrocene-labeled aptamer-equipped sensor for the detection of PDGF-BB with a detection limit of 10 pg mL−1. In addition, our group developed an electrochemical molecular beacon-based biosensor for the simultaneous detection of VEGF, IFN-γ, and TNF-α.22 These assays were based on an aptamer labeled with a single redox probe molecule.23 Redox probes can also be intercalated in the hairpin part based on static electric adsorption or π–π stacking. Several positively charged redox probes such as ruthenium hexamine were successfully intercalated in the negatively charged hairpin part of a molecular beacon, which was successfully used for the detection of IFN-γ with an improved sensitivity of 1.3 pg mL−1.24 However, ruthenium hexamine provided relatively weak voltammetric behavior compared to the other cationic redox probes such as methylene blue and paraquat.25 In addition, methylene blue has been commercially used as a label for electrochemical molecular beacons and aptamers. Thus, this study is interested in studying if methylene blue can be intercalated into a molecular beacon for biosensing.
Herein, we demonstrate the preparation of an electrochemical molecular beacon-based biosensor for the detection of salivary TNF-α. Firstly, graphene oxide (GO) was used to load streptavidin on the sensing interface due to its large theoretical surface area, high negative charge density, surface π–π interaction and excellent electrochemical properties, which can be modified with biomolecules and improve the electron transfer.26 Then, a nanocomposite containing the streptavidin-incorporated GO was prepared, which was deposited on a screen-printed carbon electrode (SPCE). MB was intercalated in the hairpin part of a molecular beacon through π–π stacking, which was then modified on the GO-modified SPCE to generate the SPCE-GO-Apt (MB) sensing interface through streptavidin and biotin binding. For comparison, a GO-modified SPCE was fabricated with a methylene blue (MB)-labelled molecular beacon against TNF-α to achieve an SPCE-GO-Apt-MB sensing interface through streptavidin and biotin binding. As shown in Scheme 1, at both sensing interfaces, amino group-terminated poly(ethyl glycol) (PEG) was fabricated as a mixed layer to help minimize the non-specific protein binding.27 Before the addition of TNF-α, the molecular beacon showed the natural configuration of having the MB close to the electrode surface for SPCE-GO-Apt-MB, producing an obvious electrochemical signal for MB. After the addition of TNF-α, the hairpin structure of the molecular beacon opened, causing the covalently labeled MB to move away from the electrode, which was expressed as a decrease in the electrochemical signal of MB. However, in the absence of TNF-α, the SPCE-GO-Apt (MB) presented a negligible electrochemical signal because MB was hidden in the hairpin to switch off the electron communication with the electrode. The electrochemical signal of MB recovered after the binding of TNF-α to the molecular beacon, which released MB. Both SPCE-GO-Apt-MB and the SPCE-GO-Apt (MB) were successfully used for the detection of salivary TNF-α, where the latter exhibited a better performance. This study paves the way for a point-of-care and non-invasive strategy to detect pro-inflammatory cytokines for the early diagnosis of oral cancer.
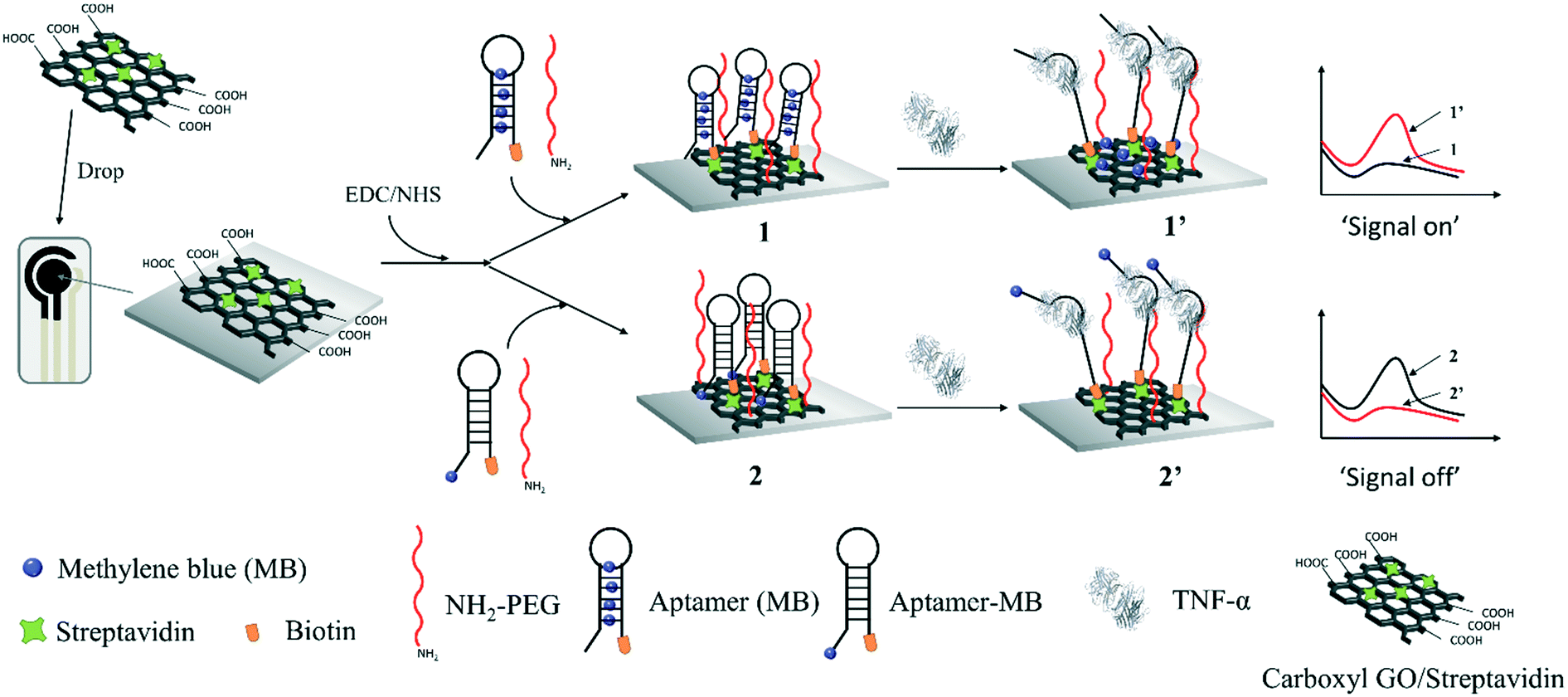 | ||
| Scheme 1 Schematic of the electrochemical molecular beacon-based biosensor for the detection of TNF-α based on intercalating redox probe or covalent binding MB to the hairpin of a molecular beacon. | ||
Materials and methods
Chemical and materials
Carboxyl graphene oxide (carboxyl GO), tris(hydroxymethyl) aminomethane (tris), methylene blue (MB), potassium chloride, hydrochloric acid, potassium ferricyanide, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl), magnesium chloride, 2-(morpholino) ethanesulfonic acid (MES), N-hydroxysuccinimide (NHS), TNF alpha Human ELISA Kit (Catalog #RAB1089) and NH2-PEG were purchased from Sigma-Aldrich. All aqueous solutions were prepared using deionized water, and pH = 7.4 PBS solution (containing 0.05 M K2HPO4/KH2PO4 and 0.05 M KCl) and Tris-HCl buffer solution (containing 100 mM Tris, 5 mM KCl, 1 mM MgCl2 and 100 mM NaCl) were used in this work. The screen-printed carbon electrodes (SPCE) (carbon working electrode with a radius of 2.5 mm) were purchased from Poten Industrial Technology Co., Ltd. (Qingdao, China), with Ag/AgCl as the reference electrode and carbon counter electrode. According to the literature,28 artificial saliva was prepared by dissolving 0.2 g KCl, 0.2 g NaCl, 0.01 g MgCl2, 0.01 g CaCl2, 0.2 g K2HPO4, 0.02 g KSCN, and 0.2 g methyl-4-hydroxybenzoate in 200 mL Milli-Q water.Recombinant human TNF-α was purchased from R&D Systems and TNF-α aptamer sequences were purchased from Shanghai Sangon Biotechnology Co. Ltd. (Shanghai, China). The sequence of the molecular beacon (aptamer) targeting TNF-α was 5′-MB-TGA GGG TTG TCG CC![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) -biotin-3′, where the underlined parts have affinity to TNF-α. Also, the TNF-α aptamer sequence without modified with MB was 5′-TGA GGG TTG TCG CC -biotin-3′.
-biotin-3′, where the underlined parts have affinity to TNF-α. Also, the TNF-α aptamer sequence without modified with MB was 5′-TGA GGG TTG TCG CC -biotin-3′.
Instruments
All electrochemical measurements were performed using a CHI660E system (CHI Instrument, Shanghai). Glassy carbon (GC) disk electrodes (3 mm) were purchased from Gaoss Union (China). All electrochemical measurements used a three-electrode system consisting of screen-printed carbon electrodes. A PHS-3CW pH meter (Shanghai Honggai Instrument Co., Ltd.), ultrasonic cleaner, and ALC-210.4 electronic analytical balance were also used. Electrochemical SWV measurements were performed at room temperature by scanning the potential in the range of −0.5 to 0 V for 100 mV s−1, the rest time was set to 2 s, and the frequency was set to 10 Hz. A Shimadzu UV-vis spectrophotometer model 2450 was used to measure UV-vis absorption spectra. Scanning electron microscopy (SEM) measurements were performed with a Zeiss Merlin Compact field emission scanning electron microscope. X-ray photoelectron spectroscopy (XPS) experiments were performed using a VG Multilab 2000 spectrometer coupled with a hemispherical analyzer, multichannel detector and monochromated Al Kα source (1486.6 eV).Preparation of MB intercalating molecular beacon
MB was dissolved in deionized water, diluted to 100 μM, and then 10 μL of MB aqueous solution was added to 30 μL of molecular beacon solution (10 μM) dissolved in 100 mM Tris-HCl solution (pH = 7.4). The mixture was incubated on a shaker and left to react overnight at room temperature, and then centrifuged in an ultrafiltration tube (Millipore, 30 K) for 30 min to remove the unbound molecular beacon and MB, and finally dispersed in a centrifuge tube at 4 °C for the next experiment.Fabrication of two sensing interfaces for the TNF-α detection
To prepare the nanocomposites containing streptavidin and carbonyl GO, 100 μL of 10 μg mL−1 streptavidin was dropped into 1 mL of 1 mg mL−1 prepared carbonyl GO aqueous solution and the mixture was stirred for 2 h at room temperature. Then, 10 μL of the prepared carbonyl GO/streptavidin nanocomposite was dropped onto the working electrode and dried at room temperature to obtain an SPCE-GO/streptavidin interface. Subsequently, the carbonyl GO was blocked by heterobifunctional cross-linking using EDC and NHS, which pre-activated the carboxyl groups on the interfaces to allow easy binding to the free amino group of PEG to form a PEG polymer on the sensing interfaces. The interface modified by NH2-PEG exhibited anti-fouling with low interfacial energy and steric repulsion effect.29 The clean electrode was immersed in 100 mM NHS and 400 mM EDC in 100 mM MES solution (pH = 5.5) and reacted for 1 h to activate the carboxyl group on the surface of GO. The SPCE was taken out from the above-mentioned solution, followed by washing with deionized water. Subsequently, the SPCE-GO/streptavidin interface was incubated with 10 μL of a 10 μM mixture of aptamer (MB) and 10 μM NH2-PEG, or a mixture of 10 μM aptamer-MB and 10 μM NH2-PEG solution at 4 °C overnight to obtain the SPCE-GO-apt (MB) interface and SPCE-GO-apt-MB interface, respectively. The sensing interfaces were incubated with 5 μL 100 mM Tris-HCl solution containing a certain concentration of TNF-α cytokine for 30 min, respectively, and then immersed in Tris-HCl buffer solution for SWV with electrochemical detection. The potential range was set to 0.5–0 V, the rest time was set to 2 s and the frequency was set to 10 Hz.Statistical analysis
All experiments were repeated three times, and the total samples for each condition was five. The quantitative data in the current study are presented as mean ± standard error of the mean (SEM). Statistical analyses were performed using SPSS Statistics version 26. Independent student's t-test was used for comparison of the means, where p < 0.05 was considered to be significantly different.Results and discussion
Optimization of aptamer (MB)
Firstly, the volume of MB for the preparation of the aptamer (MB) solution was optimized. Various volumes of MB solution with a concentration of 100 μM were added to a fixed volume (10 μL) and concentration (10 μM) of aptamer solution, respectively. After purification, 10 μL nanocomposite was added to the working electrode of SPCE to perform square wave voltammetry (SWV) scanning in Tris-HCl buffer solution. As shown in Fig. 1a, the current signal intensity of MB increased with an increase in the volume of MB before reaching a plateau at 10 μL of MB, indicating that the optimized volume of MB is 10 μL. To investigate the number of MB molecules intercalated in the hairpin of one aptamer molecule, the UV-vis spectra of a series of MB solutions with different concentrations were measured (Fig. 1b) and a linear relationship between the absorbance and the MB concentration was obtained (Fig. 1c), which could be used to calculate the amount of MB released from the aptamer (MB) solution after the addition of 500 pg mL−1 TNF-α. It was calculated that one aptamer could load seven MB molecules into the hairpin structure, that is MB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aptamer = 7:1.
:aptamer = 7:1.
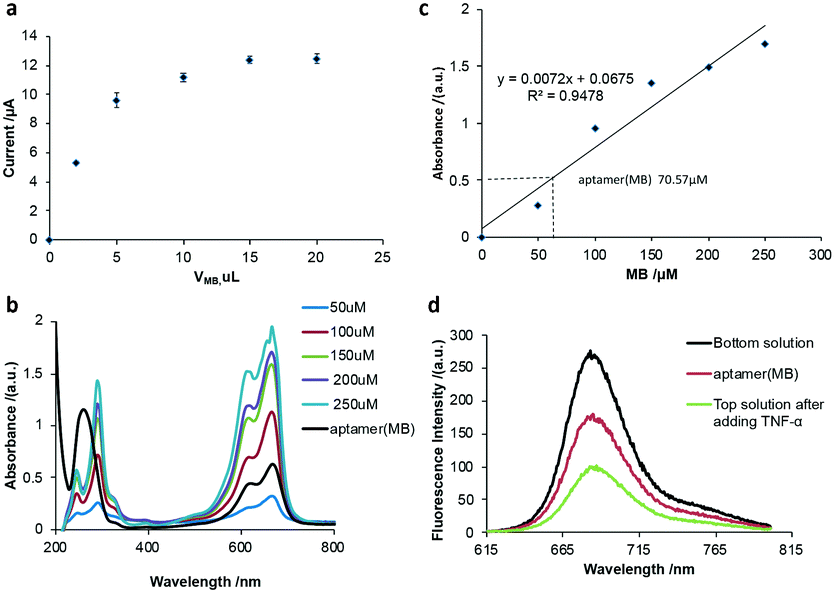 | ||
| Fig. 1 (a) SWV current of various volumes of 100 μM MB solution reacted with 10 μL of 10 μM aptamer solution in pH 7.4 Tris-HCl buffer after the addition of 100 pg mL−1 TNF-α. (b) UV-vis spectra of the prepared aptamer (MB) with different concentrations of MB. (c) Calibration curve obtained with UV-vis for different concentrations of MB solution and the prepared aptamer (MB) in (b). (d) Fluorescence spectra of aptamer (MB) before and after exposure to TNF-α with the excitation wavelength at 680 nm. | ||
To investigate if the molecular beacon would undergo a 3D configuration change in the presence of TNF-α to open the hairpin and release MB molecules, the fluorescence of MB molecules was recorded (Fig. 1d). It was observed that the fluorescence spectrum of MB showed a characteristic peak at 680 nm. After MB was intercalated in the aptamer, the aptamer (MB) solution was ultrafiltered through an ultrafiltration tube, where a fairly significant fluorescent characteristic peak appeared, corresponding to the aptamer (MB). After the addition of TNF-α and ultrafiltration through the ultrafiltration tube, a strong fluorescent characteristic peak appeared in the liquid filtered from the ultrafiltration tube (bottom solution), and a relatively weak fluorescence peak was observed in the top solution, indicating that after the addition of TNF-α, the stem loop of the aptamer opened to release MB, as shown in Fig. 1d, suggesting the successful binding of MB to the aptamer (molecular beacon).
Characterization of aptamer (MB)
As shown in Fig. 2, obvious characteristic redox peaks appeared in the MB solution at around −0.2 V in the CV diagram, which are consistent with previous report,30 while the redox peak of the aptamer (MB) solution was very weak at around −0.2 V. Because MB was intercalated in the base pair of the aptamer by π–π conjugation, the electrical communication between MB and the electrode was weakened, indicating that MB was successfully loaded in the aptamer. Besides, the two characteristic absorption peaks at 294 nm and 670 nm of MB are shown in Fig. 2b, which are attributed to the π–π* transition of the benzene ring and n–π* transition of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and consistent with the literature,31 respectively. Moreover, the aptamer has an absorption peak at 260 nm. When MB was loaded in the aptamer by π–π conjugation, the UV spectrum of the aptamer (MB) not only presented the characteristic peak of the aptamer, which was red-shifted from 260 to 272 nm, but also presented the characteristic peak of MB at 670 nm, indicating that MB was successfully bound to the aptamer. In addition, the fluorescence spectra of MB and aptamer (MB) without and with TNF-α are also shown in Fig. 1d, not only indicating the successful preparation of the aptamer (MB) but also confirming the TNF-α triggered opening of the hairpin structure of aptamer. The concentration of aptamer (MB) modified on the GO–streptavidin surface was also optimised by recording the current. It was observed that the current corresponding to MB increased with an increase in the concentration of aptamer (MB) before it reached a plateau at 10 μM. Thus, 10 μM aptamer (MB) was used as the optimised concentration for fabricating the SPCE-GO-Apt (MB)/PEG sensing interfaces.
N and consistent with the literature,31 respectively. Moreover, the aptamer has an absorption peak at 260 nm. When MB was loaded in the aptamer by π–π conjugation, the UV spectrum of the aptamer (MB) not only presented the characteristic peak of the aptamer, which was red-shifted from 260 to 272 nm, but also presented the characteristic peak of MB at 670 nm, indicating that MB was successfully bound to the aptamer. In addition, the fluorescence spectra of MB and aptamer (MB) without and with TNF-α are also shown in Fig. 1d, not only indicating the successful preparation of the aptamer (MB) but also confirming the TNF-α triggered opening of the hairpin structure of aptamer. The concentration of aptamer (MB) modified on the GO–streptavidin surface was also optimised by recording the current. It was observed that the current corresponding to MB increased with an increase in the concentration of aptamer (MB) before it reached a plateau at 10 μM. Thus, 10 μM aptamer (MB) was used as the optimised concentration for fabricating the SPCE-GO-Apt (MB)/PEG sensing interfaces.
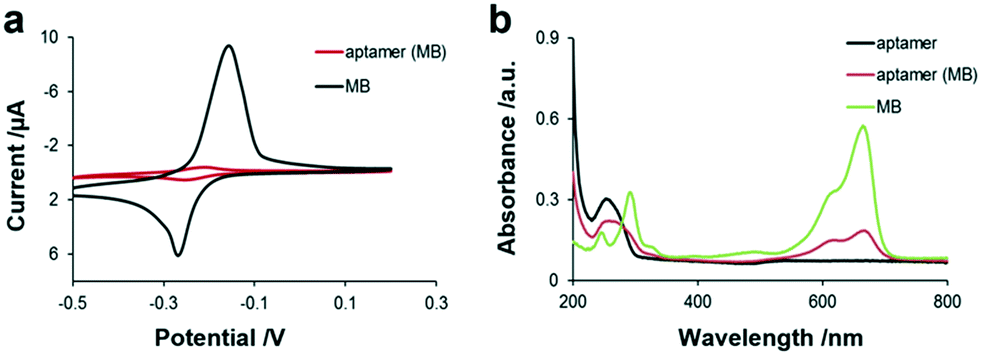 | ||
| Fig. 2 (a) Electrochemistry of aptamer (MB) probes and MB in 100 mM Tris-HCl buffer solution. (b) UV-vis of MB (5 μM), aptamer (10 μM), and aptamer (MB) in 100 mM Tris-HCl buffer solution. | ||
XPS characterization of the stepwise-fabricated sensing interface
X-ray photoelectron spectroscopy (XPS) was used to characterize the stepwise fabrication of the sensing interfaces. As shown in Fig. 3a, there are three peaks at around 284.7 eV, 286.4 eV and 288 eV in the XPS measurement of the C 1s core level spectrum for GO/streptavidin fabricated carbon surfaces. Peaks at 286.4 eV and 288 eV are assigned to the carbon atoms of –C–O–C– and –COOH– of the mixed layers.31,32 In addition, the peaks at 389.9 and 400.05 eV of N 1s correspond to –CN– and –NH2 of streptavidin,33 respectively (Fig. 3c). After SPCE was modified with GO–streptavidin, the SPCE surface was immersed in EDC/NHS solution for 1 h and incubated with the prepared aptamer (MB) terminated with biotin at the 3′ terminal. As shown in Fig. 3d, the peaks at 400.9 eV and 402.0 eV in the N 1s core spectra are assigned to the nitrogen of the NHS ester and the protonated tertiary amine of EDC, respectively.34 Also, the peak at 399.5 eV is assigned to the nitrogen of –CO–NH–. Moreover, the peak at 288 eV shifted to 287.7 eV, corresponding to the carbon of –CO–NH–, indicating the successful binding of the amine-terminated PEG (Fig. 3b). Simultaneously, the S2p peaks at 163.8 eV, 165.09 eV and 168.0 eV can be assigned to C–S–C of MB,35 C–S–C of streptavidin36 and SO32− of biotin,37 respectively (Fig. 3d). Furthermore, the peak of Cl2p at 198.0 eV corresponds to the chlorine species of MB38 (Fig. 3f), and the P2p peak at 133.20 eV is assigned to the phosphorus species in the aptamer39 (Fig. 3g). The wide spectra for the SPCE-GO/streptavidin and SPCE-GO-Apt (MB)/PEG interfaces are shown in Fig. 3h, showing the significant difference in each element after the fabrication of the aptamer (MB).
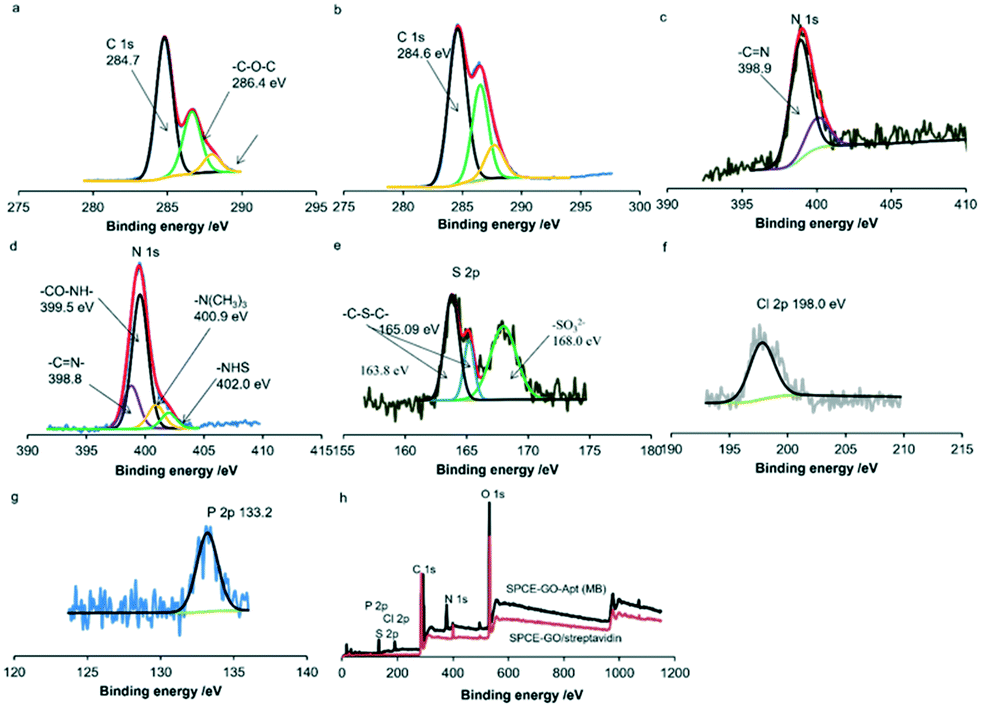 | ||
| Fig. 3 C 1s core level spectra for interfaces of (a) SPCE-GO/streptavidin and (b) SPCE-GO-Apt (MB). N 1s core level spectra for interfaces of (c) SPCE-GO/streptavidin and (d) SPCE-GO-Apt (MB)/PEG. (e) P2p, (f) S2p and (g) Cl2p core level spectra for SPCE-GO-Apt (MB). (h) Wide spectra for SPCE-GO/streptavidin and SPCE-GO-Apt (MB)/PEG interfaces. | ||
Electrochemical response of the fabricated sensing interfaces to TNF-α
As shown in Fig. 4a, before the addition of TNF-α, the sensing interface of SPCE-GO-Apt-MB presented a signal peak at around −0.3 V, which is the characteristic peak of MB. After the addition of TNF-α, the hairpin structure of the aptamer opened because of the strong affinity between TNF-α and aptamer-MB, followed by the MB molecule at the 5′ end moving away from the SPCE working electrode, resulting in a significant decline in the peak of MB. Fig. 4b shows the SWV response of the SPCE-GO-Apt (MB) sensing interface to TNF-α, which displayed a weak MB signal peak at −0.3 V in the absence of TNF-α. However, it is obvious that a very prominent MB peak with the addition of TNF-α was observed due to the structure switching to release the intercalated MB from the hairpin structure of the aptamer (MB), making it accessible to be detected by the SPCE. The SWV and calibration curves of the two sensing interfaces (SPCE-GO-Apt (MB) and SPCE-GO-Apt-MB) for the detection of TNF-α at different concentrations are presented in Fig. 4. As shown in Fig. 4c and d, with an increase in the concentration of TNF-α at the SPCE-GO-Apt (MB) sensing interface, the hairpin structure of the aptamer transformed to release the loaded MB molecules and the current signal strength of MB increased accordingly. The SPCE-GO/Apt (MB) sensing interface could detect TNF-α in the linear range of 1–400 pg mL−1, with a detection limit of 1 pg mL−1 and R2 of 0.9909. In contrast, for the SPCE-GO-Apt-MB sensing interface, the single MB molecule labeled on the aptamer was far away from the electrode surface with an increase in the concentration of TNF-α, and thus the current signal intensity of MB correspondingly decreased. The sensing interface SPCE-GO-Apt-MB responded to TNF-α in the linear range of 10–300 pg mL−1 with a detection limit of 10 pg mL−1 and R2 value of 0.9791 (Fig. 4e and f). Therefore, compared with the SPCE-GO-Apt-MB interface, the SPCE-GO-Apt (MB) interface could respond to TNF-α more sensitively, which is desirable for the detection of TNF-α in low abundance.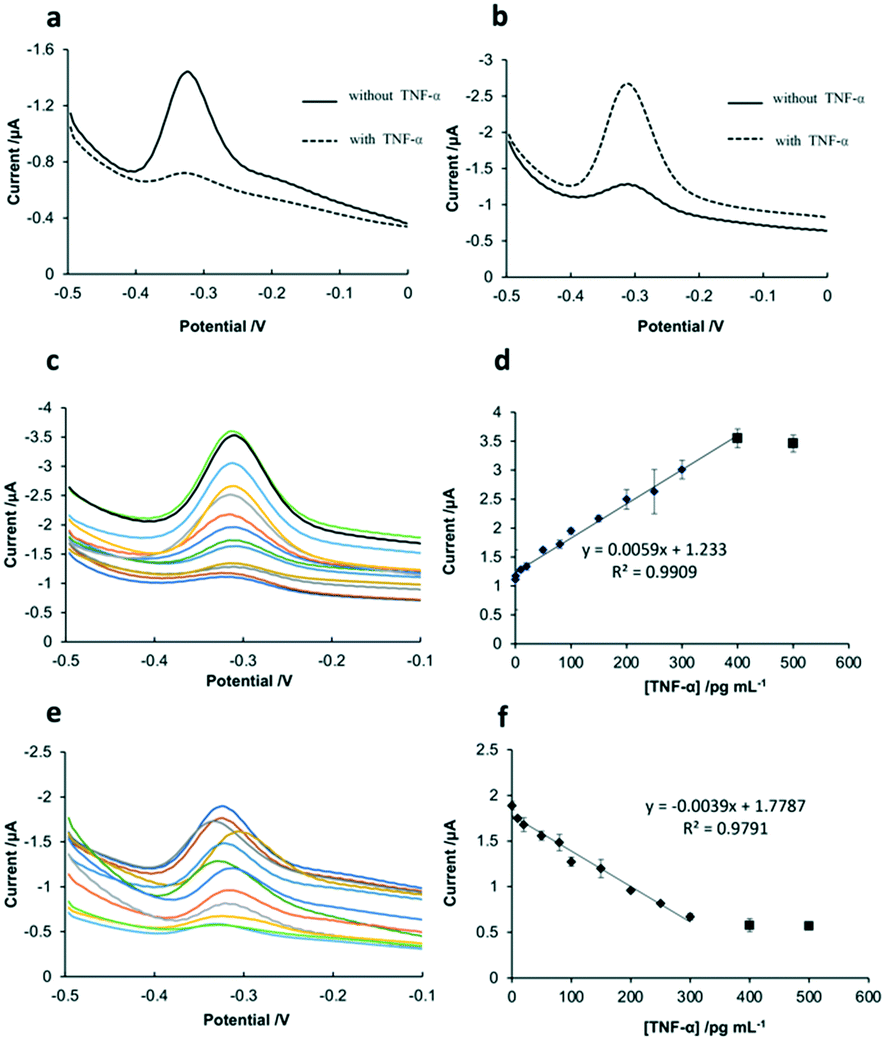 | ||
| Fig. 4 SWV response curves of (a) SPCE-GO-Apt-MB sensing interface and (b) SPCE-GO-Apt (MB) sensing interface before and after the addition of 100 pg mL−1 TNF-α. (c) SWV of SPCE-GO-Apt(MB) interface after exposure to different concentrations of TNF-α. (d) Calibration curve of SPCE-GO-Apt (MB) interface obtained by SWV in (c). (e) SWV of SPCE-GO-Apt-MB interface after exposure to different concentrations of TNF-α. (f) Calibration curve of SPCE-GO-Apt-MB interface obtained by SWV in (e). | ||
Comparison of the stability of the two sensing interfaces
To compare the stability of the SPCE-GO/Apt-MB sensing interface and the SPCE-GO/Apt (MB) sensing interface, these two interfaces were subjected to different conditions, as follows: (1) the two interfaces were placed in a Tris-HCl buffer solution and ultra-sonicated for 5 s; (2) the two interfaces were placed in 100 mM Tris-HCl buffer (pH 7.4) for cyclic voltammetry (CV) scanning for 10 cycles; (3) the two interfaces were stored at 4 °C for 7 days; and (4) the two interfaces were stored at 4 °C for 40 days. Then these sensing interfaces were applied for the detection of TNF-α (100 pg mL−1). It can be seen from the data in Table 1 that all the signals remained higher than 90% of the original signal without any treatment except for the condition for 40 days at 4 °C, which decreased to less than 90%. Therefore, these two sensing interfaces have similar stability for the detection of TNF-α.| Compared to the original signal (%) | Ultrasonicated for 5 s | CV scanning for 10 cycles | Stored for 7 days at 4 °C | Store for 40 days at 4 °C |
|---|---|---|---|---|
| SPCE-GO-Apt-MB | 92.01 | 95.18 | 90.68 | 82.56 |
| SPCE-GO-Apt (MB) | 95.16 | 96.32 | 91.33 | 87.52 |
Factors affecting the performance of SPCE-GO-Apt (MB) sensing interface
The key factors affecting the sensing performance of SPCE-GO-Apt (MB), including pH, concentration of Na+, temperature and incubation time, were systematically investigated to obtain the optimal performance (Table S1†). The optimal criteria for these parameters were determined based on the electrochemical signal of MB. The optimal factors are presented in detail in the ESI† (Fig. S1).To further explore the selectivity of the prepared SPCE-GO-Apt (MB) sensing interface, four potential interfering proteins including interleukin IL-6 (0.5 mg mL−1), prostate specific antigen PSA (1 mg mL−1), bovine serum albumin BSA (2 mg mL−1) and mouse immunoglobulin IgG (1 mg mL−1) were added to the assay buffer in the presence of 50 pg mL−1 TNF-α. Subsequently, the current signal response of this interface to each cytokine was recorded (Fig. S1e†), and the current of the interfering protein was higher than 88% of the original signal, which was with only 50 pg mL−1 TNF-α, indicating that little interference occurred in the presence of these chemicals. Thus, it can be seen that our designed SPCE-GO-Apt (MB) sensing interface has good selectivity. The sensing interface after exposure to IL-6 (0.5 mg mL−1), PSA (1 mg mL−1), BSA (2 mg mL−1) and IgG (1 mg mL−1), respectively, did not show significant changes comparing to the sensing interface after exposure to Tris buffer, suggesting that this sensor has good specificity. Fig. S1f† shows the reproducibility of the SPCE-GO-Apt (MB) sensing interface. Because the molecular beacon configuration change is reversible, after the SPCE-GO-Apt (MB) interface released all the MB molecules, the sensing interface could be regenerated by exposing the sensing interface to a high concentration of MB, and thus MB would be intercalated in the hairpin structure again. The response of the regenerated SPCE-GO-Apt (MB) interface to TNF-α was compared, and it was observed that the sensor retained above 80% of its signal response after 5-times regeneration (Fig. S1f†). Thus, the SPCE-GO-Apt (MB) interface can be regenerated.
Detection of salivary TNF-α using the SPCE-GO-Apt (MB) sensing interface
Salivary TNF-α, as a potential biomarker in oral cancer, was found to have a content of 63.94 ± 56.05 pg mL−1 in an oral squamous cell carcinoma group, while its level was 5.75 ± 3.98 in the control group.40 Although the salivary TNF-α concentration level is still controversial, most of studies proved that it is significantly elevated in patients with oral cancer.41–43 Thus, the prepared SPCE-GO-Apt (MB) sensing interface was applied to measure TNF-α in saliva. ELISA was used to evaluate its performance. According to the recovery results obtained (Table 2), the SPCE-GO-Apt (MB) sensing interface detected 5 pg mL−1, 20 pg mL−1, and 100 pg mL−1 in saliva with a recovery rate of 92.9–103.9%, suggesting the excellent reliability of the sensing interface for the determination of TNF-α in relatively complex samples (such as saliva). The performance of the sensing interface for the detection of TNF-α was comparable to that of ELISA, where a low concentration of TNF-α (5 pg mL−1) was not detected by ELISA, suggesting that the sensing interface presented herein is more sensitive than ELISA. Thus, this fabricated SPCE-GO-Apt (MB) has potential to be used as a reliable tool to detect TNF-α in saliva towards the early diagnosis of oral cancer.| Spiked concentration (pg mL−1) | Detected concentration by biosensor herein (pg mL−1) | Recovery (%) | Detected concentration by ELISA (pg mL−1) | Recovery (%) |
|---|---|---|---|---|
| Notes: a recovery = (detected concentration/spiked concentration) × 100%. | ||||
| 5.00 | 4.83 ± 6.09 | 96.6 | N/A | N/A |
| 20.00 | 20.78 ± 3.92 | 103.9 | 19.54 ± 5.67 | 97.7 |
| 100.00 | 92.91 ± 3.09 | 92.9 | 95.86 ± 4.21 | 95.9 |
Conclusion
In this study, we developed an electrochemical molecular beacon-based biosensor on an SPCE for the detection of salivary TNF-α by intercalating MB molecules in the hairpin part of a molecular beacon to achieve an SPCE-GO-Apt (MB) sensing interface. It was observed that 7 MB molecules could be loaded in one molecular beacon. Electrochemistry, XPS, UV-vis, etc. were used to characterize the sensing interface. Factors such as pH, temperature, sodium ion concentration, and incubation time were investigated to achieve a desirable sensing performance. For comparison, an SPCE-GO-Apt-MB sensing interface was prepared in which one MB was covalently labeled on one molecular beacon. Under the optimized conditions, the SPCE-GO-Apt (MB) sensing interface was capable of detecting TNF-α with a linear range of 1–400 pg mL−1 and a detection limit of 1 pg mL−1, while SPCE-GO-Apt-MB provides a linear range of 10–300 pg mL−1 and detection limit of 10 pg mL−1. Thus, SPCE-GO-Apt (MB) was more stable and sensitive to detect TNF-α than SPCE-GO-Apt-MB. The SPCE-GO-Apt (MB) sensing interface could be regenerated, and thus is suitable for the development of a recyclable device for the POC detection of cytokines in saliva. Although SPCE-GO-Apt (MB) has advantages over SPCE-GO-Apt-MB in sensitivity, releasing an external chemical (such as methylene blue) to the detection environment will cause safety concerns for in vivo applications, which makes SPCE-GO-Apt (MB) suitable for in vitro detection. With the aid of nanotechnology, SPCE-GO-Apt-MB is promising to develop an oral wearable device, which can be used for continuous monitoring of analytes in saliva towards point-of-care digital analysis of oral health conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (Grant 22174121), Shenzhen Bay Open Laboratory Fund 2021 by Shenzhen Bay Laboratory, the University Development Fund (UDF01002012) of Chinese University of Hong Kong, Shenzhen.References
- M. Kumar, R. Nanavati, T. G. Modi and C. Dobariya, J. Cancer Res. Ther., 2016, 12, 458 CrossRef CAS PubMed.
- Y.-F. Su, Y.-J. Chen, F.-T. Tsai, W.-C. Li, M.-L. Hsu, D.-H. Wang and C.-C. J. Yang, Diagnostics, 2021, 11, 1287 CrossRef CAS PubMed.
- Z. Khurshid, M. S. Zafar, R. S. Khan, S. Najeeb, P. D. Slowey and I. U. Rehman, Adv. Clin. Chem., 2018, 86, 23–70 CrossRef CAS PubMed.
- C. Liu, D. Chu, K. Kalantar-Zadeh, J. George, H. A. Young and G. Liu, Adv. Sci., 2021, 8, 2004433 CrossRef CAS PubMed.
- J.-M. Zhang and J. An, Int. Anesthesiol. Clin., 2007, 45, 27 CrossRef PubMed.
- B. J. Ferguson, S. Marler, L. L. Altstein, E. B. Lee, M. O. Mazurek, A. McLaughlin, E. A. Macklin, E. McDonnell, D. J. Davis and A. M. J. Belenchia, Brain, Behav., Immun., 2016, 58, 57–62 CrossRef CAS PubMed.
- Z. Shen, J. Huang, H. Wei, H. Niu, B. Li, R. Li and G. Liu, Bioelectrochemistry, 2020, 134, 107532 CrossRef CAS PubMed.
- G. Dranoff, Nat. Rev. Cancer, 2004, 4, 11–22 CrossRef CAS PubMed.
- T. Diesch, C. Filippi, N. Fritschi, A. Filippi and N. Ritz, Cytokine, 2021, 143, 155506 CrossRef CAS PubMed.
- M. M. A. Chiamulera, C. B. Zancan, A. P. Remor, M. F. Cordeiro, F. O. Gleber-Netto and A. R. Baptistella, BMC Cancer, 2021, 21, 1–16 CrossRef PubMed.
- N. N. Scheff, Y. Ye, A. Bhattacharya, J. MacRae, D. H. Hickman, A. K. Sharma, J. C. Dolan and B. L. Schmidt, Pain, 2017, 158, 2396 CrossRef CAS PubMed.
- J. W. Chadwick, R. Macdonald, A. A. Ali, M. Glogauer and M. A. Magalhaes, Front. Oncol., 2021, 11, 741013 CrossRef PubMed.
- N. Favre, G. Bordmann and W. Rudin, J. Immunol. Methods, 1997, 204, 57–66 CrossRef CAS PubMed.
- Y. Liu, T. Kwa and A. Revzin, Biomaterials, 2012, 33, 7347–7355 CrossRef CAS PubMed.
- Y. Lu, Q. Zhou and L. Xu, Front. Bioeng. Biotechnol., 2021, 758 Search PubMed.
- G. Liu, C. Jiang, X. Lin and Y. Yang, View, 2021, 2, 20210003 CrossRef CAS PubMed.
- M. Qi, Y. Zhang, C. Cao, M. Zhang, S. Liu and G. Liu, Anal. Chem., 2016, 88, 9614–9621 CrossRef CAS PubMed.
- F. G. Bellagambi, A. Baraket, A. Longo, M. Vatteroni, N. Zine, J. Bausells, R. Fuoco, F. Di Francesco, P. Salvo and G. S. Karanasiou, Sens. Actuators, B, 2017, 251, 1026–1033 CrossRef CAS.
- C. Cao, F. Zhang, E. M. Goldys, F. Gao and G. Liu, TrAC, Trends Anal. Chem., 2018, 102, 379–396 CrossRef CAS.
- B. P. Crulhas, D. Hadley, Y. Liu, D.-S. Shin, G. Stybayeva, M. Imanbekova, A. E. Hill, V. Pedrosa and A. Revzin, Anal. Methods, 2017, 9, 4527–4532 RSC.
- S. Zhang, X. Hu, X. Yang, Q. Sun, X. Xu, X. Liu, G. Shen, J. Lu, G. Shen and R. Yu, Biosens. Bioelectron., 2015, 66, 363–369 CrossRef CAS PubMed.
- Z. Shen, S. Ni, W. Yang, W. Sun, G. Yang and G. Liu, Sens. Actuators, B, 2021, 336, 129747 CrossRef CAS.
- G. Liu, C. Cao, S. Ni, S. Feng and H. Wei, Microsyst. Nanoeng., 2019, 5, 1–11 CrossRef PubMed.
- C. Cao, R. Jin, H. Wei, W. Yang, E. M. Goldys, M. R. Hutchinson, S. Liu, X. Chen, G. Yang and G. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 33078–33087 CrossRef CAS PubMed.
- D. Basnig, N. Vilà, G. Herzog and A. Walcarius, J. Electroanal. Chem., 2020, 872, 113993 CrossRef CAS.
- J. Pilas, T. Selmer, M. Keusgen and M. J. Schöning, Anal. Chem., 2019, 91, 15293–15299 CrossRef CAS PubMed.
- T. Zhang, Z. Xu, H. Xu, Y. Gu, Y. Xing, X. Yan, H. Liu, N. Lu, Y. Song, S. Zhang, Z. Zhang and M. Yang, Sens. Actuators, B, 2019, 288, 469–475 CrossRef CAS.
- J. Pytko-Polonczyk, A. Jakubik, A. Przeklasa-Bierowiec and B. Muszynska, J. Physiol. Pharmacol., 2017, 68, 807–813 CAS.
- C. Jiang, G. Wang, R. Hein, N. Liu, X. Luo and J. J. Davis, Chem. Rev., 2020, 120, 3852–3889 CrossRef CAS PubMed.
- G. Zaitseva, Y. Gushikem, E. S. Ribeiro and S. S. Rosatto, Electrochim. Acta, 2002, 47, 1469–1474 CrossRef CAS.
- P. Jing, W. Ji, X. Yuan, M. Ikezawa, L. Zhang, H. Li, J. Zhao and Y. Masumoto, J. Phys. Chem. Lett., 2013, 4, 2919–2925 CrossRef CAS.
- G. Liu, S. Wang, J. Liu and D. Song, Anal. Chem., 2012, 84, 3921–3928 CrossRef CAS PubMed.
- Z. Yang, Z. Xie, H. Liu, F. Yan and H. Ju, Adv. Funct. Mater., 2008, 18, 3991–3998 CrossRef CAS.
- E. Chow, E. L. S. Wong, T. Böcking, Q. T. Nguyen, D. B. Hibbert and J. J. Gooding, Sens. Actuators, B, 2005, 111–112, 540–548 CrossRef CAS.
- J. Wang, L. Kou, Z. Huang and L. Zhao, RSC Adv., 2018, 8, 21577–21584 RSC.
- C.-M. Yam, C.-M. Pradier, M. Salmain, P. Marcus and G. Jaouen, J. Colloid Interface Sci., 2001, 235, 183–189 CrossRef CAS PubMed.
- M. F. Elahi, G. Guan, L. Wang and M. W. King, Materials, 2014, 7, 2956–2977 CrossRef CAS PubMed.
- M. Cuartero, R. G. Acres, J. Bradley, Z. Jarolimova, L. Wang, E. Bakker, G. A. Crespo and R. De Marco, Electrochim. Acta, 2017, 238, 357–367 CrossRef CAS.
- S. Bizid, S. Blili, R. Mlika, A. Haj Said and H. Korri-Youssoufi, Anal. Chim. Acta, 2017, 994, 10–18 CrossRef CAS PubMed.
- G. Deepthi, S. Nandan and P. G. Kulkarni, Asian Pac. J. Cancer Prev., 2019, 20, 2087 CrossRef CAS PubMed.
- M. Ameena and R. Rathy, J. Int. Oral Health, 2019, 11, 92 CrossRef.
- M. Polz-Dacewicz, M. Strycharz-Dudziak, J. Dworzański, A. Stec and J. Kocot, Infect. Agents Cancer, 2016, 11, 1–8 CrossRef PubMed.
- R. Krishnan, D. K. Thayalan, R. Padmanaban, R. Ramadas, R. K. Annasamy and N. Anandan, Asian Pac. J. Cancer Prev., 2014, 15, 7141–7148 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sd00035k |
| This journal is © The Royal Society of Chemistry 2022 |