
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the mechanism of nitroaromatic vapour uptake in PDMS-based pre-concentrators using 4-nitrotoluene†
Beta Z.
Poliquit
,
Paul L.
Burn
 * and
Paul E.
Shaw
*
* and
Paul E.
Shaw
*
Centre for Organic Photonics & Electronics, School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: p.burn2@uq.edu.au; p.shaw3@uq.edu.au
First published on 22nd June 2022
Abstract
Pre-concentration is an important tool to improve the detection of low vapour pressure analytes. Quantification of the amount of analyte sorbed and released and how it relates to the pre-concentrator material and analyte of interest is important for analyte detection and pre-concentrator material development. In particular, establishing the diffusion mechanism enables correlations between the type of sorbing phase and the sorption mode to be identified, which may then provide strategies to improve pre-concentrator materials. We describe the development of a UV-vis absorption spectroscopy method to monitor the real time uptake of analyte vapours by pre-concentrator materials and apply it to characterising the uptake of the nitroaromatic 4-nitrotoluene (pNT) by three pre-concentrator materials under continuous flow conditions: poly(dimethylsiloxane) (PDMS), PDMS:1,4-divinylbenzene (DVB), and PDMS-co-DVB. The results show that PDMS and PDMS:DVB films up to ∼100 μm thick initially exhibit pNT uptake consistent with Case II diffusion as opposed to a Fickian process. In contrast, the PDMS-co-DVB films exhibited more complex behaviour, with mass uptake kinetics consistent with anomalous diffusion that varies with film thickness, which we attribute to the presence of different phases in the films. The pNT uptake for the thicker DVB-containing films under continuous flow was lower than that of PDMS although the rate of diffusion was faster, which was attributed to the presence of large pores in these materials. The results demonstrate that the mechanism of diffusion of analyte vapours into PDMS-based pre-concentrators is composition dependent and impacts the balance between the rate of analyte uptake and the total analyte uptake during continuous flow measurements.
1. Introduction
There are two key difficulties associated with the direct detection of trace-level quantities of chemical vapours in air. First, the vapour sample will likely be composed of a mixture of chemicals (analytes) and second, the amount of chemical vapour in the air can be lower than the sensitivity and detection limit of the analytical instrument used to determine the presence of the analyte. To address the latter limitation, a pre-concentration step is performed to increase the concentration of the desired component. In 1990, Pawliszyn et al. introduced a pre-concentration technique known as solid phase microextraction (SPME).1,2 SPME devices contain a polymeric sorbent material, which collects the dispersed analytes from the sample matrix, e.g., a sample of air. The collected analyte is then released in a concentrated burst either by heating or dissolution in a solvent for delivery to the analytical instrument.Pre-concentrators generally have poor selectivity and sorb a wide range of analytes with similar properties (for example, polarity), relying on the analytic instrument to provide differentiation of the chemicals in the sample mixture.3 Furthermore, the quantity of analyte collected is typically inferred by measuring how much is desorbed from the pre-concentrator using the analytical instrument, which requires numerous time-based sequential sampling-measurement iterations.4,5 This approach for determining how much analyte is sorbed is not ideal as only the analyte released from the SPME is detected, which may be less than the amount sorbed. Indeed, the rate and amount of analyte released can have a strong temperature dependence.4 It is therefore of interest to be able to study the analyte sorption process over time both in terms of the rate of up-take and quantitatively assessing the amount of analyte sorbed. It has been reported that the sorption of analytes into SPMEs is governed by Fick's law of diffusion, whereby diffusion is controlled by a concentration gradient.5,6 However, polymeric materials can swell upon absorption of the analyte, which is a characteristic of Case II and Super Case II diffusion.7,8 Case II and Super Case II diffusion differ in that while the analyte diffuses into the film as a distinct front, in the latter case the front accelerates during the diffusion process. These subtleties in diffusion cannot be observed by the standard sorption-release measurements. In this regard, establishing the diffusion mechanism enables correlations between the type of sorbing phase and the sorption mode to be identified, which may then provide strategies to improve SPMEs, including the choice of materials and their selectivity.
In this study, we describe the development of a novel approach, based on in situ UV-vis spectroscopic measurements, to continuously monitor the analyte sorption with respect to time in PDMS-based pre-concentrator films under a constant supply of analyte vapour. The advantage of using absorbance measurements is that it provides an unambiguous quantitative measurement of the analyte present in the film. Analyte vapour uptake measurements were performed on three different types of pre-concentrator materials – poly(dimethylsiloxane) (PDMS), PDMS:1,4-divinylbenzene (DVB) blends, and PDMS-co-DVB (copolymerised material). We demonstrate the method using p-nitrotoluene (pNT) as an exemplar for explosives detection. The observed time-dependence of the uptake is consistent with Case II diffusion, i.e., the swelling driven process that results in a defined front propagating through the film at constant velocity. From the data, we calculated the front velocity for each PDMS-based material. The analyte diffusion mechanism as well as the calculated values of the front velocity confirm the spontaneous nature of the analyte uptake process. They also provide concrete evidence of the relationship between the physical and chemical attributes of the different film compositions, such as, the presence of pores, and the crystallinity and polarity of the polymer, to the analyte uptake. This new approach clarifies how nitroaromatic vapours diffuse into PDMS-based pre-concentrator materials and provide a quantitative approach for evaluating future pre-concentrator materials.
2. Experimental
2.1 Preparation of pre-concentrator films
The details of the sol–gel preparation, film fabrication and characterisation have been previously reported.9,10 In summary, the polymer films were made by dip-coating a pre-cleaned and pre-activated fused silica disc into a precursor dichloromethane solution containing the macromonomer (hydroxy-terminated poly(dimethylsiloxane), 750 cSt HO-PDMS), crosslinker (methyltriethoxysilane; MTEOS), additive or co-monomer (1,4-divinylbenzene; DVB), end capping polymer (poly(methylhydrosiloxane);![[M with combining overline]](https://www.rsc.org/images/entities/char_004d_0305.gif) n: 1700–3200 g mol−1 PMHS), acid catalyst (trifluoroacetic acid; TFA 99%) and an initiator (benzophenone, BP). The number of dip-coating cycles was varied between 2 to 7 to control the film thickness in the range of 10–50 μm. The substrates were coated on one side by protecting the other side with polyimide (Kapton) tape during dip-coating. The tape was removed immediately after dip-coating. The films were dried overnight at 65 °C in air and were then pre-conditioned at 250 °C for 4 h under argon. The films were stored in air in a sealed plastic container at room temperature between measurements. The thickness of the films was determined using a Veeco Dektak 150 surface profiler. At least 4 measurements were taken across the center of the film to determine uniformity. The calculated standard deviation is reflected as error values of the film thickness.
n: 1700–3200 g mol−1 PMHS), acid catalyst (trifluoroacetic acid; TFA 99%) and an initiator (benzophenone, BP). The number of dip-coating cycles was varied between 2 to 7 to control the film thickness in the range of 10–50 μm. The substrates were coated on one side by protecting the other side with polyimide (Kapton) tape during dip-coating. The tape was removed immediately after dip-coating. The films were dried overnight at 65 °C in air and were then pre-conditioned at 250 °C for 4 h under argon. The films were stored in air in a sealed plastic container at room temperature between measurements. The thickness of the films was determined using a Veeco Dektak 150 surface profiler. At least 4 measurements were taken across the center of the film to determine uniformity. The calculated standard deviation is reflected as error values of the film thickness.
2.2 Instrumentation
Fig. 1 presents a schematic diagram of the custom-built set-up assembled for the dynamic analyte sorption measurements, all of which were undertaken at an ambient temperature of 23 ± 1 °C. The sample chamber (Fig. 1a) was custom-made in the Faculty of Science mechanical workshop at The University of Queensland (Australia) and designed to be mounted inside a Varian Cary 5000 UV-vis-NIR absorption spectrometer. The sealable stainless steel sample chamber had outer dimensions of 8.1 cm × 11.1 cm × 5.1 cm, an inner volume of approximately 400 mL, and inlet and outlet ports for the vapour delivery and egress. pNT was chosen as the analyte due to its relatively high vapour pressure, which facilitates reliable calibration of the vapour source, provides an unambiguous absorption signal, and ensures that equilibrium is achieved on a timescale of hours, all of which are critical for the collection of reliable data. The vapour delivery system (Fig. 1b) consisted of two mass flow controllers (MFC-1 and MFC-2) connected to a nitrogen gas supply with MFC-2 delivering nitrogen through a calibrated coiled stainless-steel tube coated on the inside with pNT. The concentration of pNT supplied by MFC-2 was determined to be 19 ± 5 ppm (ESI† Fig. S1). The pNT concentration was not diluted using MFC-1 and the flow was maintained at 1.0 L per minute to ensure that the atmosphere in the sample chamber rapidly reached equilibrium (see ESI† Fig. S2). The fused silica substrate coated with the pre-concentrator material was mounted in a holder attached to the lid of the sample chamber (see Fig. 1a). The sample holder is positioned on the lid of the sample chamber so that it is aligned with the optical windows to allow light transmitted through the pre-concentrator film to be continuously measured.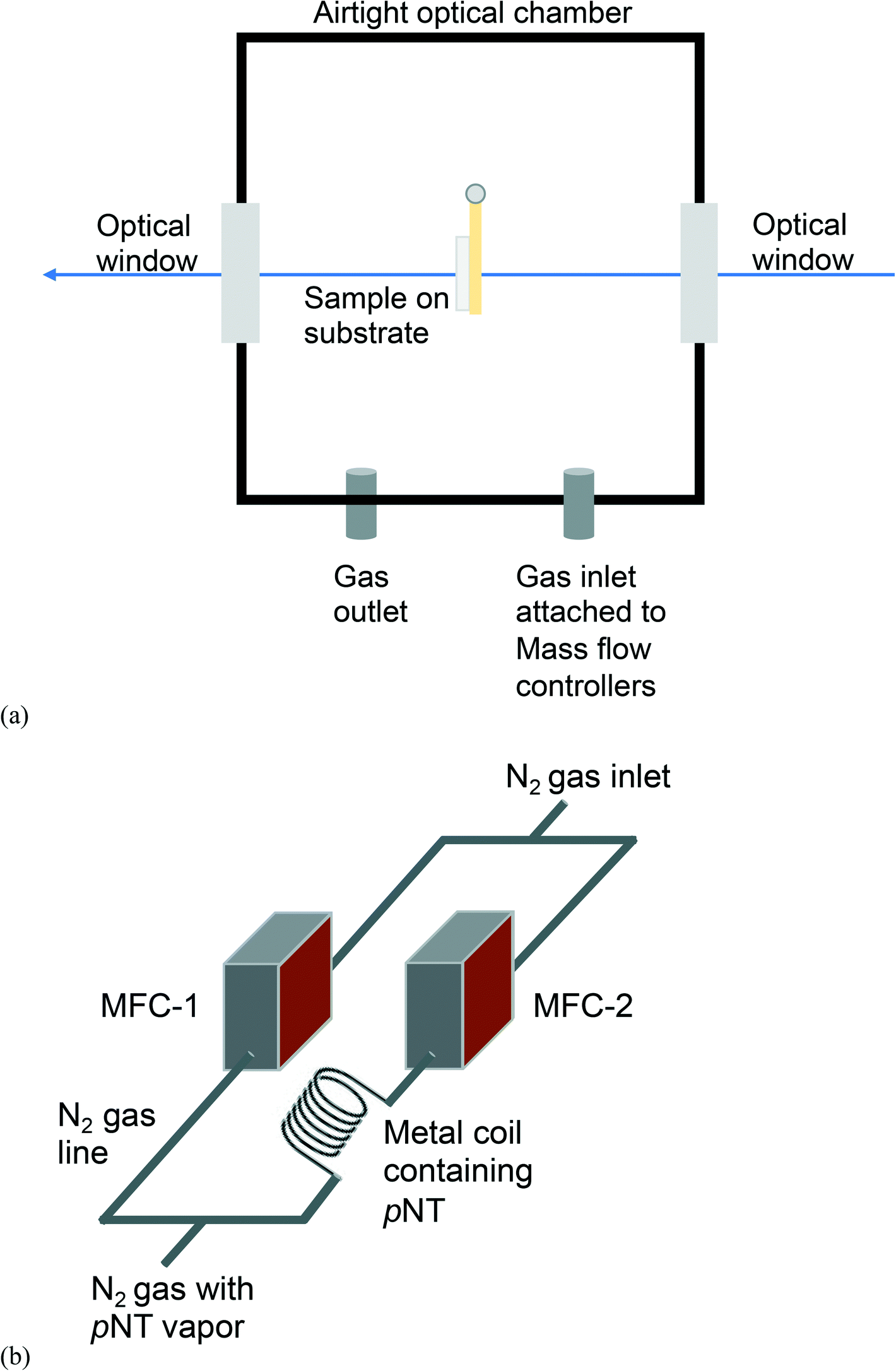 | ||
| Fig. 1 Schematic diagram of (a) the custom-built sample chamber and (b) the pNT vapour delivery system based on 2 mass flow controllers (MFC). | ||
2.3 In situ dynamic analyte sorption measurement
Prior to each test, the chamber and pre-concentrator films were pre-conditioned by venting nitrogen through the chamber while monitoring any change in the optical density of the film. Once the optical density of the sample had stabilised (changes on the order of 0.003, see ESI† Fig. S2), the analyte sorption kinetics measurements were performed. The change in the optical density of the film was measured at the λmax of the analyte (pNT = 269 nm) with 1 second intervals. The total duration of a typical measurement sequence was 7260 s where the first 60 s was nitrogen only to provide the zero reference, with the remaining 7200 s allocated for the measurement with pNT vapour. The data are presented in terms of optical density, which is directly related to the concentration of the sorbed analyte in the pre-concentrator film.3. Results and discussion
It is important to note that the pre-concentrator films had different surface topologies depending on their composition. We previously reported how PDMS films were essentially flat and uniform, the 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PDMS:DVB blend films had clearly observable holes, whilst the 3:1 PDMS-co-DVB films were composed of two phases and contained voids and spheroidal features on the surface.9 PDMS films were shown to swell upon pNT absorption leading to a change in the surface topology and a decrease in the surface roughness.10Fig. 2 shows the change in the optical density of films of (a) PDMS, (b) 3:1 PDMS:DVB blend, and (c) 3:1 PDMS-co-DVB of different film thickness upon introduction of pNT vapour into the optical chamber. The PDMS:DVB ratio 3:1 was chosen for this study based on previous results that showed it had the greatest sorption capacity and was therefore potentially the most suitable for use as a pre-concentrator.9 Note that in the plotted results the data has been offset in terms of time so that the introduction of the pNT occurs at time zero with the 60 second purging stage not shown.
:1 PDMS:DVB blend films had clearly observable holes, whilst the 3:1 PDMS-co-DVB films were composed of two phases and contained voids and spheroidal features on the surface.9 PDMS films were shown to swell upon pNT absorption leading to a change in the surface topology and a decrease in the surface roughness.10Fig. 2 shows the change in the optical density of films of (a) PDMS, (b) 3:1 PDMS:DVB blend, and (c) 3:1 PDMS-co-DVB of different film thickness upon introduction of pNT vapour into the optical chamber. The PDMS:DVB ratio 3:1 was chosen for this study based on previous results that showed it had the greatest sorption capacity and was therefore potentially the most suitable for use as a pre-concentrator.9 Note that in the plotted results the data has been offset in terms of time so that the introduction of the pNT occurs at time zero with the 60 second purging stage not shown.
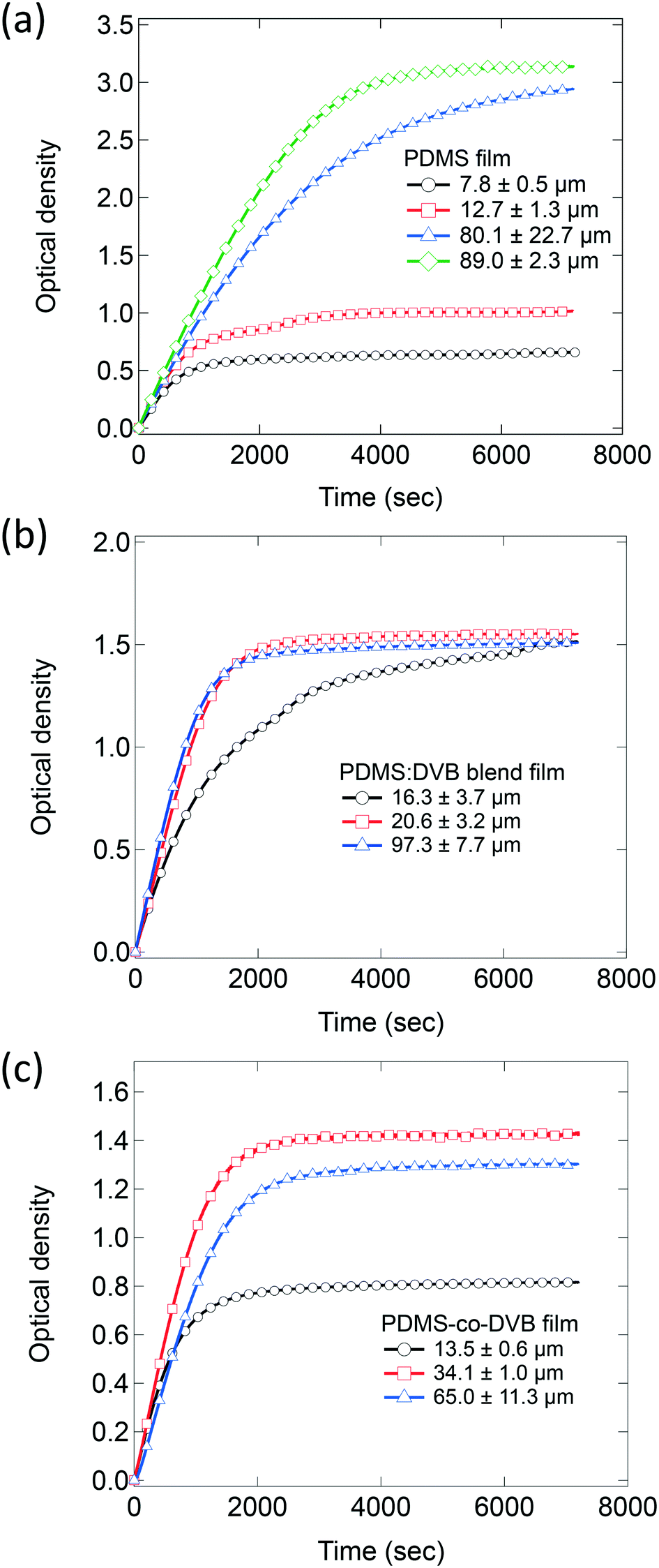 | ||
| Fig. 2 Change in the optical density at 269 nm of (a) PDMS, (b) PDMS:DVB, and (c) PDMS-co-DVB films of varying thickness upon exposure to a continuous supply of pNT vapour at 19 ± 5 ppm. The number of markers has been reduced to 1 for every 200 data points for improved clarity noting that a measurement was taken every second. | ||
Films of all three PDMS-based systems showed similar behaviour with an immediate increase in their optical densities when the flow of pNT into the chamber was initiated followed by a decrease of that rate with equilibrium reached at later times. Control measurements without a pre-concentrator film showed a much smaller change in absorbance, on the order of 0.06 after 60 s (ESI† Fig. S3), which is attributed to pNT in the atmosphere along the optical path. This confirms that sorption of the analyte into the pre-concentrator film is the primary source of the measured absorbance change. While all three systems present the same initial sorption trend with a rapid and steady uptake of analyte, they differ in terms of the time taken to achieve equilibrium and the equilibrium concentration. For example, for the PDMS films (Fig. 2a), the time taken to reach equilibrium and the equilibrium concentration both increase with film thickness. This is consistent with the analyte being sorbed into the bulk of the film rather than adsorbed onto a surface. In the latter case analyte uptake would be near-identical as the samples have the same surface area. In contrast, the films of PDMS:DVB (Fig. 2b) equilibrate at similar absorbance values (∼1.5) independent of film thickness with the thicker films equilibrating faster. This behaviour suggests that the pNT uptake might be limited to the surface of the PDMS:DVB films rather than in the bulk. Finally, for the films made of PDMS-co-DVB (Fig. 2c) the behaviour was intermediary, initially showing an increase in the amount of analyte sorbed at equilibrium with increased film thickness and then a decrease for the thickest film.
The sorption kinetics can therefore be divided into two key stages noting that as the chamber is supplied with a continuous supply of analyte vapour, the concentration of analyte in the atmosphere is constant and therefore any changes to the rate of uptake can be attributed to the sample: the first stage (stage 1) is the initial sorption phase where the film is not saturated with analyte and the analyte uptake is limited by either the rate of deposition onto the surface of the film or its diffusion into the film. Where the analyte diffuses into the bulk of the film, this process will continue until the diffusion front has reached the substrate and can no longer proceed. In this work, this process is characterised by a linear increase of the optical density over time and therefore a constant rate of mass uptake. The second key stage is the attainment of an equilibrium, which appears as a plateau in the kinetics data. That is, the rate of analyte sorption equals the rate of desorption and the amount of pNT within the film remains constant under the conditions of the measurement. There is often not a sharp transition between these two states, which can arise from, for example, the reorganisation of the polymer chains to enable the analyte to diffuse into all available sites.
The data from stage 1 in the sorption kinetics can be used to extract details of the mechanism by which the analyte diffuses into the film. Specifically, each diffusion mechanism will exhibit a distinct time-dependence of the mass uptake and can be identified from8,12
| mt = ktn, | (1) |
| At = ktn, | (2) |
Fig. 3 shows the change in the optical density of the PDMS-based pre-concentrator films over time plotted on a log–log scale. The curves show similar trends implying that the diffusion mechanism for pNT was similar regardless of the film composition or thickness, with the exception of thicker PDMS-co-DVB films. The value of n for each pre-concentrator film was taken from fitting to the data with eqn (2), where each curve was fitted and then the results averaged (see ESI† Table S1). For the PDMS and PDMS:DVB blend films the average value of n was 0.99 ± 0.01 and 0.99 ± 0.08, respectively. In other words, the initial mass uptake increased linearly with time, which is consistent with Case II diffusion. That is, a front of the analyte diffused into the films at a constant rate despite the PDMS:DVB films having voids in the surface. The value of n for the diffusion of pNT into PDMS-co-DVB was 0.92 ± 0.07, which indicates that the diffusion has “anomalous” characteristics. Anomalous diffusion of analyte is often attributed to non-uniform film morphology. For example, a rigid material with strained swelling is likely to experience non-linear analyte diffusion compared to a highly flexible material.15–17 We have previously reported that the PDMS-co-DVB films contain two phases in addition to surface voids. The spheroidal phase was assigned to poly(1,4-divinylbenzene) rich regions of the film, which would be expected to be more rigid than the PDMS containing components.9 Thus, the non-uniform film morphology is consistent with the films having anomalous diffusion characteristics.9 Furthermore, the fact that n for the PDMS:DVB film was around 1 suggests that the components within the blend were more uniformly distributed.
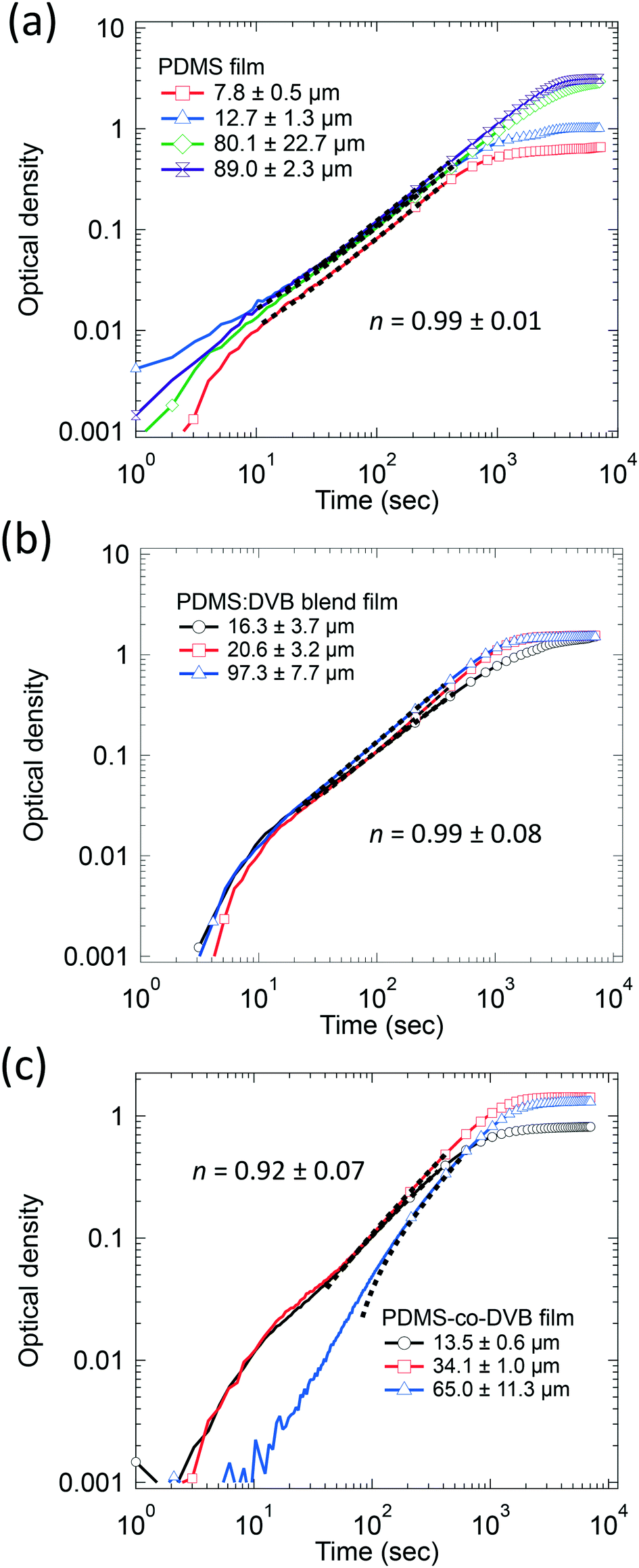 | ||
| Fig. 3 The change in the optical density over time in log–log of (a) PDMS, (b) PDMS:DVB, and (c) PDMS-co-DVB films of different thickness exposed to a continuous supply of pNT vapour at 19 ± 5 ppm. Fits to stage 1 data are represented by the black segmented line, which also represents the range of data used for the fitting (∼300–400 data points). Note that as per Fig. 2 the number of markers has been reduced to 1 for every 200 data points for improved clarity. | ||
The differences in the morphologies would be expected to impact the velocity of the diffusion front, which is a useful parameter for assessing the capabilities of the sorbing material and making comparisons. Furthermore, it also provides guidance on how long sampling needs to be undertaken before an appropriate concentration of analyte is reached for a specific analytical instrument. One simple approach to estimate the velocity of the front is based on the inflexion point of the optical density (analyte sorption) versus time plot, which for Case II and Super Case II diffusion will approximately correspond to the travel time required for the analyte front to reach the substrate. Hence, the time-averaged velocity of the diffusion front can be estimated by dividing the film thickness by the travel time. Fig. 4 shows the velocity of the diffusion front for films of each material for different thicknesses. For thin films (<20 μm), the velocity of the front is comparable across all the PDMS-based materials. However, as the film thickness was increased (>20 μm), the front velocity in the PDMS:DVB and PDMS-co-DVB films exceeded that of the PDMS films. This observation is consistent with the voids on the surface of the PDMS:DVB and PDMS-co-DVB films. That is, the voids provide pathways for the analyte to diffuse into the film structures, thus increasing the surface area for the analyte to sorb into the pre-concentrator material. It might be thought that given none of the films sorbed the analyte through a Super Case II mechanism that the front velocity would be constant independent of the film thickness. However, this was not the case with the thicker pre-concentrator films having higher front velocities. It is not entirely clear what the origin of this effect is, but it may arise from the thicker films requiring several dip-coating steps that could give rise to subtly different film morphologies. The different morphologies could arise from differences in solvent evaporation from or drying of the film with thickness, or the ageing of the sol–gel solution during the fabrication process. To probe the potential effect of ageing of the sol solution on the film morphology and velocity of the diffusional front, a series of PDMS films were made in succession from 2 dip-coating cycles using the same sol solution. The results are shown in ESI† Fig. S4 and indicate that film thickness rather than the order of film fabrication had the greatest influence on the front velocity. While the link between dipping cycles and morphology is unclear, the dip-coating cycles promote strong film surface features (e.g., surface bumps) which clearly contribute to the overall film morphology. This point is further supported by the increased light scattering with film thickness, that is, increased optical density in the transparent region of thick films compared to thin films.
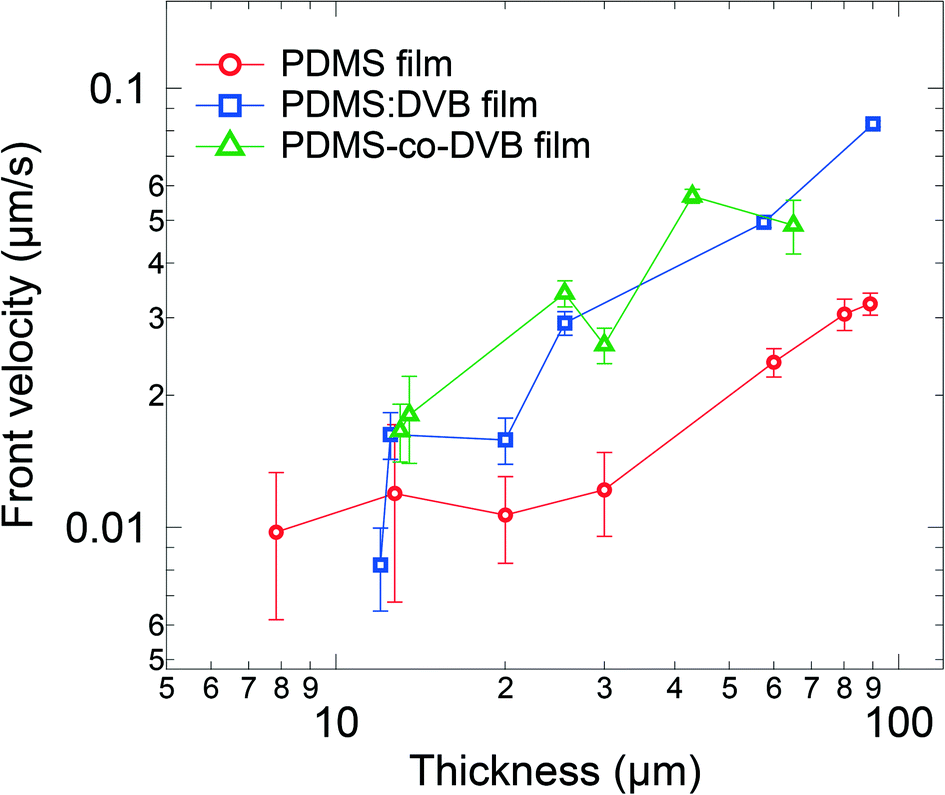 | ||
| Fig. 4 Velocity of the front in the PDMS, PDMS:DVB and PDMS-co-DVB pre-concentrator films as a function of film thickness. The velocity of the front was estimated by dividing the film thickness by the travel time. | ||
In the final part of the study, we quantitatively compared the amount of sorbed analyte at equilibrium by pre-concentrator films of each material of similar thickness (Fig. 5). Note the concentration of the pNT was 19 ± 5 ppm for these measurements. For films with a thickness of 13 μm, the PDMS:DVB films were found to sorb the greatest amount of analyte, followed by the pristine PDMS film, with the PDMS-co-DVB pre-concentrator film the least. These results are opposite to what was reported from static sorption measurements where the PDMS-co-DVB pre-concentrator films had higher analyte uptake.9 However, it is important to note at this stage that independent of the measurement type – static or dynamic – pre-concentrator films that contained a DVB or a DVB-derived component always performed better than PDMS alone. The reason why the films with DVB or a DVB-derived component perform better than PDMS arises from the fact that the π–π interactions between the analyte and pre-concentrator film plays a role in the sorption process. In the static measurements the films were placed in the headspace vapour of the analyte for defined time periods whereas in the dynamic measurements there is a continuous air flow over the films. In both cases there are both sorption and desorption processes that can occur. Given that the first step in the sorption into the film is the adsorption of the analyte onto the film surface then there might be expected to be a difference in the desorption rate from the films under the two conditions. Intuitively, one would expect that the rate of surface desorption would be higher for dynamic conditions due to the nitrogen flow. Indeed, it has been previously shown that the speed of airflow across a PDMS/DVB SPME fiber strongly affected the sorption of toluene.18 In that study, the extraction profiles for toluene were investigated using a 65 μm PDMS/DVB coating and airflow speeds ranging from 0.8 to 83.2 cm s−1 for sampling times ranging from 5 s to 1 h. It was found that the maximum adsorbed toluene was lower and occurred faster for higher airflow, which was attributed to a thinner pre-concentrator/analyte boundary layer and a more competitive adsorption process at higher flow speeds. Thus, these reported results would suggest that for the PDMS-co-DVB films the analyte is less able to sorb efficiently thoughout the film, which is consistent with the anomalous diffusion process. The results also suggest that for the PDMS-co-DVB pre-concentrator films that under dynamic conditions the analyte is preferentially sorbed in the PDMS rich regions of the film with less diffusing into the more rigid DVB-derived spheroids. In contrast, the enhanced up-take of the PDMS:DVB blend over that of the PDMS pre-concentrator film arises primarily from the π–π interactions between the analyte and DVB in the pre-concentrator film.19
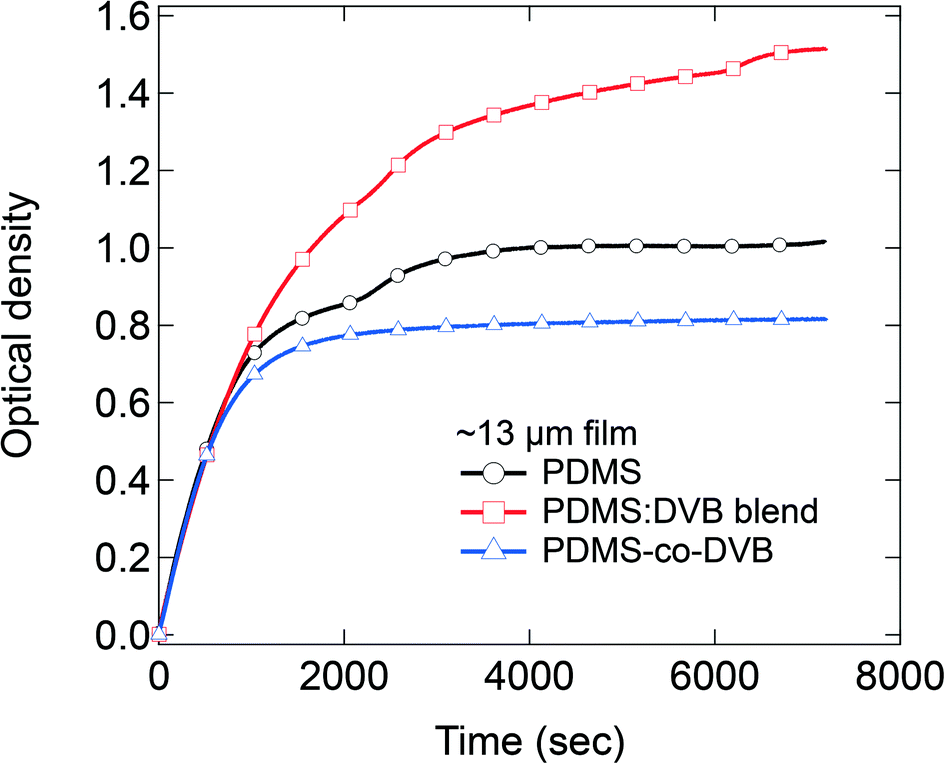 | ||
| Fig. 5 Amount of pNT sorbed in ∼13 μm films of PDMS, PDMS:DVB blend and PDMS-co-DVB. | ||
4. Conclusions
We characterised the time-resolved uptake of pNT vapour by films of PDMS, PDMS-co-DVB and a PDMS:DVB blend under a continuous flow with the aim of identifying the diffusion process. We find that both PDMS and PDMS:DVB exhibit initial mass uptake consistent with Case II diffusion for films up to ∼100 μm thick. This indicates a swelling-driven as opposed to a concentration-driven process, as has previously been assumed. In contrast, PDMS-co-DVB films feature mass uptake kinetics that are consistent with an anomalous diffusion process that varies with film thickness. One explanation for this observation is that the PDMS-co-DVB films are composed of different phases with distinct diffusion characteristics and that the amount of each phase present varies with film thickness.
We find that the relative amount of pNT sorbed by each of the PDMS-based pre-concentrator materials under a continuous flow is different from what was previously observed under static conditions. Specifically, we find that the amount of pNT vapour sorbed by PDMS was generally greater than that sorbed by PDMS-co-DVB and PDMS:DVB for the thicker films. For the thinner films the order was changed with the PDMS:DVB blend having the largest uptake at equilibrium and the PDMS-co-DVB having the least. The results show that under dynamic conditions there is a subtle trade-off between film surface area and intermolecular interactions of the analyte and the pre-concentrator material. Nevertheless, independent of the film thickness the speed of the diffusion front was greater for the films that had greater porosity, that is, the PDMS:DVB blend and PDMS-co-DVB.
The results confirm that the diffusion of analyte vapours into PDMS-based concentrators cannot be assumed to be a concentration-driven process, and that the composition of the pre-concentrator can affect the diffusion process and the balance between the rate of analyte uptake and the total analyte uptake under dynamic conditions. The development of a method for characterising analyte uptake over time therefore provides a new perspective with which to evaluate pre-concentrator materials and optimise their performance.
Conflicts of interest
There are no conflicts to declare.References
- C. L. Arthur and J. Pawliszyn, Solid Phase Microextraction with Thermal Desorption Using Fused Silica Optical Fibers, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- H. Lord and J. Pawliszyn, Evolution of solid-phase microextraction technology, J. Chromatogr. A, 2000, 885, 153–193 CrossRef CAS PubMed.
- C. Zambonin and A. Aresta, Recent applications of solid phase microextraction coupled to liquid chromatography, Separations, 2021, 8, 1–14 CrossRef.
- S. Risticevic, H. Lord, T. Górecki, C. L. Arthur and J. Pawliszyn, Protocol for solid-phase microextraction method development, Nat. Protoc., 2010, 5, 122–139 CrossRef CAS PubMed.
- J. Pawliszyn, in Applications of Solid Phase Microextraction, ed. J. Pawliszyn, The Royal Society of Chemistry, Cambridge, 1999, pp. 3–21 Search PubMed.
- S. N. Semenov, J. A. Koziel and J. Pawliszyn, Kinetics of solid-phase extraction and solid-phase microextraction in thin adsorbent layer with saturation sorption isotherm, J. Chromatogr. A, 2000, 873, 39–51 CrossRef CAS PubMed.
- M. A. Ali, Diffusion of Analyte Molecules into Fluorescent-based Explosive Sensors, The University of Queensland, 2015 Search PubMed.
- Diffusion in Polymers, ed. J. Crank and G. S. Park, Academic Press Inc., New York, US Edition, 1968 Search PubMed.
- B. Z. Poliquit, P. L. Burn and P. E. Shaw, Properties of PDMS-divinylbenzene based pre-concentrators for nitroaromatic vapors, J. Mater. Chem. C, 2020, 8, 16967–16973 RSC.
- B. Z. Poliquit, P. L. Burn and P. E. Shaw, Effect of precursor macromonomer molecular weight on poly(dimethylsiloxane) film morphology and nitroaromatic vapor sorption, Sens. Actuators, B, 2018, 270, 283–290 CrossRef CAS.
- J. Crank, The Mathematics of Diffusion, Oxford University Press, London, 2nd edn, 1979 Search PubMed.
- Diffusion in Polymers, ed. P. Neogi, Marcel Dekker, Inc., New York, 1996 Search PubMed.
- T. Owen, Principles and applications of UV-visible spectroscopy: a primer, Hewlett-Packard Company, Germany, 1996 Search PubMed.
- D. Calloway, Beer-Lambert Law, J. Chem. Educ., 1997, 74, 744 CrossRef CAS.
- M. R. Pereira and J. Yarwood, ATR-FTIR spectroscopic studies of the structure and permeability of sulfonated poly(ether sulfone) membranes. Part 2.—Water diffusion processes, J. Chem. Soc., Faraday Trans., 1996, 92, 2737–2743 RSC.
- J. Ramirez, T. J. Dursch and B. D. Olsen, A Molecular Explanation for Anomalous Diffusion in Supramolecular Polymer Networks, Macromolecules, 2018, 51, 2517–2525 CrossRef CAS.
- S. Ito, Y. Taga, K. Hiratsuka, S. Takei, D. Kitagawa, S. Kobatake and H. Miyasaka, Restricted diffusion of guest molecules in polymer thin films on solid substrates as revealed by three-dimensional single-molecule tracking, Chem. Commun., 2015, 51, 13756–13759 RSC.
- J. Koziel, M. Jia and J. Pawliszyn, Air sampling with porous solid-phase microextraction fibers, Anal. Chem., 2000, 72, 5178–5186 CrossRef CAS PubMed.
- L. Tuduri, V. Desauziers and J. L. Fanlo, Potential of solid-phase microextraction fibers for the analysis of volatile organic compounds in air, J. Chromatogr. Sci., 2001, 39, 521–529 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sd00045h |
| This journal is © The Royal Society of Chemistry 2022 |