
Spatially resolved and quantitatively revealed charge transfer between single atoms and catalyst supports
Bin
Di†
,
Zhantao
Peng†
,
Zhongyi
Wu
,
Xiong
Zhou
* and
Kai
Wu
 *
*
BNLMS, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: kaiwu@pku.edu.cn; xiongzhou@pku.edu.cn
First published on 3rd December 2021
Abstract
The charge state of supported single atoms is one of the most significant aspects determining the catalytic performance of single atom catalysts (SACs) which have drawn tremendous attention in recent years. In this perspective, mainly based on our previous studies and new data inputs, charge transfer between single atoms and their supports in several model systems is explored by the measurement of local work functions (LWFs). Two types of additives to tune the electronic properties of model catalysts, alkali metals and halogens, are described. The transferred charge is spatially resolved and quantitatively revealed based on LWF mapping via the Helmholtz equation. On average, Cs transfers more electrons than K does, echoing its lower first ionization energy. In contrast, Au and bromine atoms draw electrons from supports of metals like Cu and oxides like CuO. These insights into charge transfer at the atomic level are vital to understand their catalytic and promoting effects.
Introduction
Single atom catalysts (SACs), which essentially involve single-atom active sites on supports, have been extensively studied ever since the pioneering series of work by Zhang and co-workers in 2011.1–11 Single atoms, which can be envisioned as being downsized from nanoparticles (NPs), may exhibit a superb catalytic performance in various catalytic reactions due to their low coordination number, strong metal–support interaction and quantized band structure or discrete energy levels. However, the inherent ineffectiveness for reactions that require simultaneous participation of more active metal atoms frequently prevents SACs from working, in addition to ever-changing interactions between the reactants and supported single atoms,4,12–14 the available chemical environment and metal–support interactions.15–18 The single active sites make SACs conceptually resemble a homogeneous catalyst, which enables the exploration of the catalytic mechanism by excluding the ensemble effect of supported NPs with a size distribution.6 More importantly, the intrinsic properties of the single atoms are actually non-equal due to their non-uniform chemical environments on the support surfaces.6One of the crucial factors discerning the catalytic activity is the charge state of the supported metal species that originates from charge transfer between the metal species and its support,19 though a fierce debate on this has been going on. For example, both positively2,3,20–23 and negatively17,18,24,25 charged supported gold (Au) species are proposed to be responsible for the enhanced catalytic activity. Qiao et al.1 reported that positively charged Pt single atoms were responsible for the excellent catalytic activity for CO oxidation while Therrien et al.26 claimed that neutral Pt single atoms deposited on another support were also active. Therefore, a model system consisting of monodispersed single metal atoms on a well-defined oxide substrate serves as an ideal one where the charge state effect of an individual metal atom can be explored at the atomic level.
In contrast to extensively adopted approaches to anchoring single atoms, a new strategy to stabilize single atoms on supports is proposed in our previous study,8 where the oxide support is made as thin as possible so that its surface free energy can be effectively augmented by its underlying bulk metal substrate. Once the surface free energy of the ultrathin oxide support becomes comparable with that of the metal species or chemical potential of an individual metal atom, the single metal atom can then be thermally stabilized on the ultrathin oxide film without aggregation. Based on this strategy, a thermally stable single-Au-atom model catalyst was successfully prepared by Zhou et al.27via thermal deposition of Au atoms onto a monolayered copper oxide (CuO) film grown on a Cu(110) substrate, and the Au single atoms did not aggregate up to 400 K. Therrien et al.26,28 obtained stable single Pt atoms on a Cu2O(111)-like single-layer film grown on the Cu(111) substrate and achieved low-temperature CO oxidation and water activation.
Apart from active metal atoms serving as catalytic centres in heterogeneous catalysts, additives are often introduced to further promote the performance of catalysts.29–46 Among the additives, alkali metals29–39 and halogens40–46 are two types of promoters that are employed to tune the electronic properties of supported catalysts due to their capabilities47–49 of electron donation and acceptance, respectively. As such, both promoters can induce an intense interfacial charge transfer that greatly changes the electronic properties of the metal species and support in the heterogeneous catalyst.
In the past, conventional surface techniques providing statistical and averaged information have been applied to study alkali metals and halogens on various substrates.50–65 It's generally recognized that alkali metals and halogens on metal substrates result in the decrease and increase of the work functions (WFs) of metal substrates, respectively.50,55,58–67 The same scenario also holds true on oxide surfaces where the introduced alkali metals often invoke more localized charge transfer.68,69 However, such a picture for the charge transfer between these two types of promoters and their supports remains actually vague and ambiguous at the atomic level. Therefore, quantitatively unravelling the functionalizing range and extent of the impact by the charge transfer is of great significance to clarify the promoter effect of alkali metals and halogens. In our group, Peng et al.70 recently showed that potassium cations (K+) were spontaneously formed on a monolayered CuO film grown on Cu(110), leading to the decrease of the local WF (LWF) around each K+ cation, while Chen et al.71 observed that K atoms deposited on a monolayered ceria island grown on Pt(111) induced a significant decrease of the apparent LWF over the whole island.
The LWFs can be measured by scanning probe microscopy techniques with an extremely high spatial resolution, including scanning tunnelling microscopy (STM) and Kelvin probe force microscopy.72–85 These techniques have been extensively applied to acquire the information of charge transfer82–84 or redistribution.73,74,85 Moreover, the surface dipole change86–88 could be precisely extracted from the LWF variation (Δϕ). The methodology based on tunnelling current versus tip–sample distance (I–z) curves acquired by STM to deduce the LWF has been well established. Albrecht et al.89 have elucidated that it is not suitable to determine Δϕ on the atomic length scale via I–z spectroscopy, while Huang et al.81 have demonstrated that it is bias voltage dependent upon the LWF measurements of thin oxide films on metal substrates, and LWF mappings with high spatial resolution can be accurately obtained at bias voltages close to zero. In principle, the apparent barrier height (ϕa), also known as apparent WF which is approximately equal to an average WF of the tip (ϕt) and the sample (ϕs), can be quantitatively obtained by recording the tunnelling current as a function of the tip height. It can be expressed by the following equations,79,90–92
ϕa = 0.925 × (d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnI/dlnz)2 lnI/dlnz)2 |
| ϕa = (ϕt + ϕs)/2 |
Under the same tip conditions, the Δϕ between two sampling spots, denoted as A and B, on the surface can be derived as,
| Δϕ = ϕs(B) − ϕs(A) = 2 × [ϕa(B) − ϕa(A)] |
Treating the single atoms as the adsorbates, the Helmholtz equation93 can be used to describe the variation of the work function:
| Δϕ = μσ/ε0 |
| Δϕ = qlσ/ε0 = ρl/ε0 |
| Q = ∫ρdS = ∫ΔϕdS/lε0 |
In the past few decades, plenty of SACs have been prepared and reported.2,6–8,94–111 However, the insight at the atomic level of charge transfer between single atoms and their supports is still elusive. A clear picture of charge transfer can obviously help understand the extraordinary catalytic performance of SACs as well as the promoter effects of alkali metals and halogens in conventional heterogeneous catalysis.
In this perspective, we summarize some of our recent studies on and new data inputs of charge transfer by means of apparent LWF mapping or Δϕ mapping, and present an atomic view of the interaction between single atoms and substrates, demonstrating the availability and powerfulness of such a methodology. As mentioned above, three typical model systems will be described herein, namely, oxide-supported single Au atoms, oxide-supported alkali metal atoms and metal-supported halogen atoms.
Oxide-supported single Au atoms
By application of the strategy proposed by our group to mediate the surface free energy, a stable Au single-atom model catalyst on a monolayered CuO film grown on Cu(110) has been successfully prepared.27 The CuO monolayer with a unit cell of 0.36 nm × 0.51 nm, being 1 × 2 times those for the pristine Cu(110) surface, which has been well confirmed,112–116 was prepared by the exposure of the Cu(110) substrate to O2 at room temperature (RT) and subsequent annealing at 500 K. The white protrusions in the STM image of the CuO monolayer with an atomic resolution represent the Cu cations, as shown in Fig. 1a. At RT, Au atoms are deposited on the CuO monolayer and exist as single atoms (yellow protrusions in Fig. 1a). High-resolution STM imaging (inset in Fig. 1a) reveals that the Au single atom is located at the hollow site of four Cu cations, i.e., the bridge site of two O anions along the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] direction where the Au single atom is stabilized by the Au–O interaction, as confirmed by the X-ray photoelectron spectroscopy (XPS) spectra of O 1s peaks at 531.0 eV in Fig. 1f. This Au single-atom model catalyst remains unchanged up to 400 K.
0] direction where the Au single atom is stabilized by the Au–O interaction, as confirmed by the X-ray photoelectron spectroscopy (XPS) spectra of O 1s peaks at 531.0 eV in Fig. 1f. This Au single-atom model catalyst remains unchanged up to 400 K.
 | ||
| Fig. 1 Au single atoms on the CuO monolayer before and after the reaction with CO. (a) STM image of the Au single atoms on the CuO monolayer obtained by the deposition of 0.05 ML Au at RT. Inset: enlarged STM image of the Au single atoms. (b) Δϕ mapping of an individual Au atom on the CuO monolayer. The black circle indicates the location of the Au single atom and the grey grid represents the lattice of the Cu cations. (c) STM image of the Au single atoms exposed to 18 L CO at RT. (d) Enlarged STM image of the square area marked in panel d. XPS spectra of the (e) Au 4f and (f) O 1s peaks of different samples. Red: normal O anions in the CuO monolayer; green: O anions in the CuO monolayer that interact with the Au atoms; blue: chemisorbed O species. Reproduced with permission (a and c–f).27 Copyright 2018, American Chemical Society. | ||
The Δϕ mapping of the Au single atoms on the CuO monolayer is shown in Fig. 1b. An obvious increase of the LWF appears at the Au single atom site and its neighbouring area within about 0.5 nm around the Au atom, which implies the superposition of the intrinsic surface dipole by an induced dipole pointing to the vacuum. The LWF increase means negative charging of the Au single atom, which is also evidenced by the downshifted binding energy (BE) of the Au single atom with respect to a very thick neutral Au film, as shown in Fig. 1e. Therefore, the LWF increase is a consequence of electron transfer from the CuO substrate to the Au single atom. Consequently, a positively charged zone on the CuO substrate is created underneath the formed Au atom. It should be pointed out that the maximum value of Δϕ does not appear precisely at the location of the Au atom. Such a local charge transfer could alter the electronic properties of the underlying CuO support around the Au atom.
The negatively charged Au atoms are active for CO oxidation. Fig. 1c shows the STM image of the Au SAC upon its exposure to CO at RT where dark features routinely appear nearest to the Au single atoms. High-resolution STM imaging (Fig. 1d) reveals that the dark features are precisely located at the positions of the O anions nearest to the single Au atoms and hence ascribed to lattice O vacancies in the CuO monolayer which are generated by CO oxidation with adjacent lattice O anions. This is also proved by the BE change of the O 1s peak at 531.0 eV. It's noticeable that once one of the two equivalent O anions nearest to the single Au atom is depleted upon CO oxidation, the negatively charged Au atom becomes electronically neutralized and hence inactive for further CO oxidation. Such changes in the charging state of the Au atom and its reaction activity towards the CO oxidation are also confirmed by the XPS data as shown in Fig. 1e and f. However, the activity of the Au atom can be restored by fixing the O vacancies with incoming oxygen molecules at higher temperatures, which again supports that the negatively charged Au atom is active for the CO oxidation.
Obviously, the charge transfer from the monolayered CuO support to the Au single atoms plays a key role in the CO oxidation where the negatively charged Au atoms are active and their neutralized counterparts are inactive. However, the apparent charge transfer extent, i.e. the number of electrons transferred onto each Au atom from the CuO substrate, and the influencing regime of the negatively charged Au atom on the CuO substrate cannot be quantitatively disclosed by conventional surface techniques like XPS. According to Δϕ mapping, the transferred charge can be calculated as Q = ∫ΔϕdS/lε0. In addition, it's frequently noticed that the charge transfer of single atoms locally takes place around each atom within a small area. For instance, the single Au atom transfers the charge within an area of about 0.5 nm in diameter. Therefore, ∫ΔϕdS is simply estimated as average  times the influenced area. Here,
times the influenced area. Here,  l is the height of the single Au atom, about 0.2 nm. Consequently, the transferred charge onto each Au atom is about 0.03e. This quantitatively reveals how intensely the Au single atom electronically interacts with its underlying CuO substrate. It also helps rationalize the experimental observation that the incoming CO can only deplete the lattice O nearest to the negatively charged Au atom, leading to the loss of its activity towards CO oxidation upon generation of one O vacancy nearest to it. Therefore, the high-resolution LWF mapping of a modelled SAC turns out to be very powerful and can provide quantitatively revealed and spatially resolved information to help understand the catalytic performance of SACs at the sub-nanometric level.
l is the height of the single Au atom, about 0.2 nm. Consequently, the transferred charge onto each Au atom is about 0.03e. This quantitatively reveals how intensely the Au single atom electronically interacts with its underlying CuO substrate. It also helps rationalize the experimental observation that the incoming CO can only deplete the lattice O nearest to the negatively charged Au atom, leading to the loss of its activity towards CO oxidation upon generation of one O vacancy nearest to it. Therefore, the high-resolution LWF mapping of a modelled SAC turns out to be very powerful and can provide quantitatively revealed and spatially resolved information to help understand the catalytic performance of SACs at the sub-nanometric level.
Oxide-supported single alkali metal atoms
Using the same monolayered CuO film grown on Cu(110), one can also explore the charge transfer between an individual alkali metal atom and its support, as recently reported by Peng et al.70 from our group.Thermally deposited K atoms at low coverage on the CuO monolayer are spontaneously ionized at RT and become positively charged. The K cations (termed KA) are monodispersed, as shown in Fig. 2a. Its atomically resolved STM image in Fig. 2b demonstrates that KA is located on the right top of a Cu cation or at the bridge site of two O anions along the [001] direction. Unlike the situation on the hydroxylated TiO2(110) surface where the K atoms can be manipulated by the STM tip to preferentially move along the [001] direction,117 the KA cations on the CuO monolayer are extremely immobile. As KA is positively charged, it is anticipated that the LWF around KA will decrease due to the formation of an opposite surface dipole pointing from the K cation to the induced image charge underneath the surface. The Δϕ mapping around KA in Fig. 2c indeed shows a conspicuous LWF decrease within about 1.0 nm around KA, in agreement with the XPS-measured BE of the K 2p feature (Fig. 2g) that a lower BE shift is observed for the sample with single K atoms (0.5 ML) with respect to the reference sample with high coverage (4 ML) K. Similarly, the transferred charge can be estimated by LWF mapping to be about 0.22|e| per K atom. Compared to the Au anion, both the influencing area and transferred charge of KA are larger than those of the Au single atom.
 | ||
| Fig. 2 (a) Large-scale STM image of monodispersed K cations (about 0.1 ML). (b) High-resolution STM image of a K cation supported on an ultrathin CuO film, termed KA. (c) Δϕ mapping of KA. The cyan grid schematically represents the periodical Cu2+ lattice arrangement. (d) After high bias pulse treatments of all K cations in (a), most KA cations switch into KB. (e) High-resolution STM image of KB. Inset: the quartering-encircled KA highlights four possible jumping directions of the K species during the switching processes. The corresponding jumping direction for each KB is also highlighted by the straight arrowheads in (d). (f) Δϕ mapping of KB. (g) XPS measurements of the pristine ultrathin CuO film (black curve), 0.5 ML K (red curve), and 4.0 ML K (blue curve) deposited onto the ultrathin CuO film. Reproduced with permission.70 Copyright 2020, Wiley-VCH. | ||
Upon applying a sudden voltage pulse of +6 V on the KA cation, the protrusion turns into a rectangular depression (termed KB) which is randomly located at one of the four hollow sites surrounded by four Cu2+ cations nearest to the manipulated KA, as shown in Fig. 2d and e. This tip manipulation process is reversible by switching the polarity of the pulsed voltage, i.e., from +6.0 V to −6.5 V, the KB species switches back to KA again, but the restored KA may jump back on top of either of the four equivalent Cu2+ cations nearest to KB. Because of no obvious influence on the LWF of the CuO substrate around the KB species, it is deducted that KB is actually a neutral K atom, as shown in Fig. 2f.
Cesium (Cs) is bigger in size and lower in first ionization energy than K. Similarly, Cs atoms are also monodispersed on the monolayered CuO substrate upon their thermal deposition at RT. However, the monodispersed Cs atoms may sit on two different sites, either on top of the Cu cation, termed CsT, or at the hollow site surrounded by four Cu cations, termed CsH, as shown in Fig. 3a and b, respectively. To figure out the charge transfer between these two Cs species and the underlying CuO substrate, the apparent LWFs are measured and the Δϕ mappings around CsT and CsH are depicted in Fig. 3d and e, respectively. With reference to its corresponding STM image, it is clear that the apparent LWF on the Cu–O chains becomes higher than that between two Cu–O chains and the maxima of the apparent LWFs spatially appear on the Cu cations. This is counterintuitive because cations generally induce a decrease in the LWF of the underlying substrate. A possible explanation is that the LWF changes induced by the cations and anions are not precisely on the ion sites, but rather deviate from the ion locations on the atomic scale, which is similar to the observations of the Au anion and K cation described above. Furthermore, similar to the case for the KA species, the presence of CsT results in an obvious decrease in the LWF of the CuO substrate, covering an area of about 1.3 nm around CsT. Thus, the transferred charge of CsT is deduced to be about 0.73|e|. A much larger charge transfer of Cs than K actually agrees with the fact that Cs possesses a much lower first ionization energy. At a coverage of 0.8 ML Cs on the CuO substrate (here one monolayer being defined as one Cs atom per CuO unit cell), the BE of Cs 3d5/2 is measured to be 725.0 eV in XPS, as shown in Fig. 3g, which is very close to that in Cs2O, 725.1 eV, but much lower than that for metallic Cs, 726.3 eV, according to a report in the literature.118 The XPS measurements substantiate that the Cs atoms are positively charged on the CuO substrate. It seems quite reasonable, but turns out to be very complicated when the LWF measurements are carried out. Fig. 3e shows that the apparent LWF decreases in an area of about 1.1 nm around the CsH cation on the CuO substrate. The transferred charge of CsT is deduced to be 0.52e. Although both CsT and CsH are cationic according to the XPS measurements, the minimum of the apparent LWF caused by the CsH atom is lower by about 0.4 eV than that by the CsT atom, meaning that the charge transfer is site-dependent.
 | ||
| Fig. 3 High-resolution STM images of the Cs cations located on the (a) top and (b) hollow sites of the Cu cations, termed CsT and CsH, respectively. (c) Application of a +5.0 V bias pulse to switch the charge state from CsT in (a) to another one, termed CsH0. The white grids in (a)–(c) represent the lattice of the Cu cations. Image conditions: Vbias = 50 mV and It = 600 pA. Δϕ mappings of (d) CsT, (e) CsH and (f) CsH0. The black grids in (d)–(f) represent the lattice of the Cu cations. Solid circles in (d) and (e) indicate the locations of CsT and CsH, respectively. (g) XPS spectra of the Cs 3d peaks of pristine (black curve) and 0.8 ML Cs (red curve) covered ultrathin CuO films. | ||
The charge state of CsT can be manipulated by the STM tip with an application of a sudden pulsed bias voltage. In Fig. 3c, the protrusion (CsT) turns into a rectangular depression (termed CsH0), which is randomly located on one of the four hollow sites fenced by four nearest Cu2+ cations, upon applying a bias voltage pulse of +5.0 V, on CsT. Similar tip manipulation on the CsH atom, however, leads to its desorption from the CuO substrate upon applying a bias voltage pulse of +4.0 V. These experimental facts indicate that the CsT and CsH atoms interact with the underlying CuO substrate to different extents.
Fig. 3f shows the Δϕ mapping around CsH0 after the tip manipulation on CsT as shown in Fig. 3a. The LWF slightly decreases at the locations of four Cu cations, as marked by the black dashed circles in Fig. 3f. The corresponding STM topographical image is given in Fig. 3c where the four Cu cations marked by the white solid circles become a little bit higher than those of the normal Cu cations previously snugged in the Cu–O chains and the four Cu cations marked by the white dashed circles cannot be topographically imaged. Compared to CsT and CsH, the influence of CsH0 on the LWF of the monolayered CuO substrate is negligible, demonstrating that CsH0 is essentially a neutral atom. The relocation and charge neutralization of CsT may possibly alter the geometric and electronic properties of its underlying CuO substrate, as deducted from the combined measurements of the STM image and the LWF variation mapping. However, a reverse voltage pulse applied on CsH0 cannot trigger the opposite process from CsH0 to CsT, indicating that the positive-to-neutral switch of the charge state is irreversible for the CsT species, unlike the case for the KA species. The discrepancies between the charge states of individual Cs and K atoms on the monolayered CuO substrate actually reflect the difference in their intrinsic properties.
In contrast to the supported single Au atoms, experimentally both monodispersed K and Cs have no activity toward CO adsorption and oxidation. Due to their preferable donation nature of s-electrons and absence of valence d-electrons, alkali metals are usually employed as promotors rather than catalysts.
Interaction between supported Au and K atoms
Previous studies have suggested that the introduced Na or K cation can stabilize supported single Pt or Au atoms and further carry out water–gas shift reactions at low temperatures.33–36 It's also suggested that the K–(OH) species formed by deposited K on hydroxylated TiO2(110) are the active sites for Au clusters.39 Therefore, the exploration of possible charge transfer between individual Au and alkali metal (K and Cs) atoms on an ultrathin CuO substrate should be of great interest in terms of the promoter effect of alkali elements on the activities of metallic ensembles in heterogeneous catalysis. To do this, Au and K are co-deposited onto a CuO substrate at RT and the LWFs around them are mapped. It's revealed that, at low coverage, most Au and K atoms remain monodispersed on the CuO monolayer as if they were deposited separately. For example, Fig. 4a shows the co-existence of three K single atoms and one K–Au pair, the latter being marked by the yellow square and its inter-atom separation being about 0.31 nm. The precise locations of the Au and K atoms in the K–Au pair are highlighted by the small and big circles, respectively. The apparent height of the paired K–Au is higher than that of an individual Au or K atom under the same scanning conditions, which suggests that an interaction between the K and Au atoms is established. Fig. 4b shows the corresponding Δϕ mapping of the K–Au pair in Fig. 4a. The K–Au pair electronically interacts with its underlying CuO substrate within a region of about 1.9 nm in diameter, as marked by the dashed circle. To our surprise, the existing K atom (supposed to be positively charged) does not enhance the charge state of the nearby Au atom (supposed to be negatively charged), but rather they electronically cancel each other because the overall LWF of the K–Au pair in their core area in Fig. 4b approaches that of the bare CuO substrate. It looks like the positively charged K cation and negatively charged Au atom become electronically “neutralized” when they stick together. Meanwhile, the LWFs of the other three K atoms do not noticeably change with respect to that in Fig. 2c. Both experimental observations again imply that the charge transfer between the CuO substrate and K or Au is not long-range. The Au atom in the Au–K pair resembles its counterpart in a neutral state which is formed once one of its neighbouring lattice O atoms is removed by reaction with CO, as shown in Fig. 1. Therefore, the Au in the Au–K pair does indeed lose its activity towards CO oxidation, as confirmed by our experiments. When the Au stays far away from the K atoms, the LWFs around the Au and K atoms of the underlying CuO substrate do not interfere with each other, and the K atoms serve as an electronic spectator and hence do not influence the activity of the negatively charged Au atoms. It's deduced from our experiments that the interaction of Au with K in the K–Au pair weakens the Au–O interaction. This is similar to the situation where an Au single atom toward CO oxidation loses its activity once an O vacancy nearest to Au is created.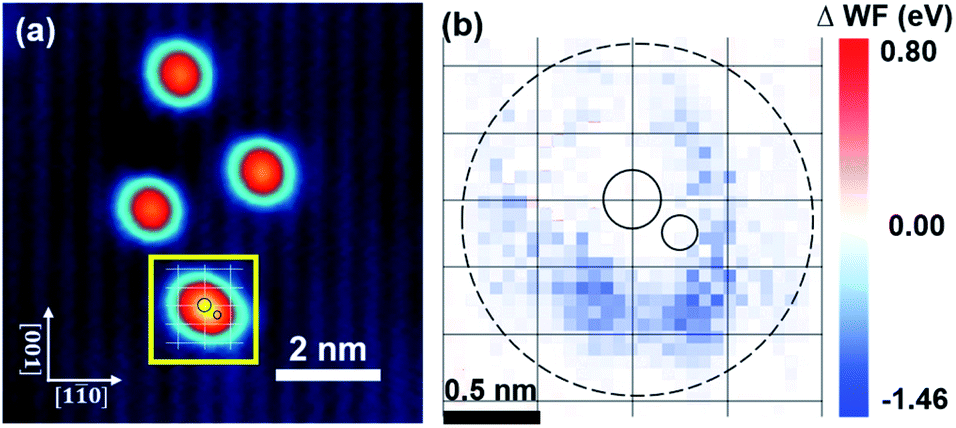 | ||
| Fig. 4 (a) STM image of co-deposited Au and K atoms on the CuO monolayer. The crossing points of the white grid represent the Cu cations in the CuO lattice. Small and big circles indicate the Au and K atoms, respectively. (b) Δϕ mapping of the region marked by the yellow square in (a). The crossing points of the black grid are the locations of the Cu cations. Small and big solid circles highlight the locations of the Au and K atoms, respectively. Image conditions: Vbias = 100 mV and It = 70 pA. | ||
Bromine single atoms on Cu(111)
In contrast to alkali metals, halogens are another class of electronic additives serving as an electron acceptor. The charge transfer between halogen atoms and their underlying metal surfaces plays a crucial role in promoting the catalytic activity. Two methods have recently been explored in our group to prepare Br atoms on a surface. One is by decomposition of bromobenzene, which was evaporated onto a Cu(111) substrate and then annealed to about 500 K. The produced biphenyl molecules via the Ullmann coupling of the bromobenzene desorbed at this temperature and the remaining Br atoms assembled into islands with regular edges. The LWF around the Br island on the Cu substrate is increased by at least 1.5 eV, indicating significant electron transfer from the Cu(111) substrate to the Br atom.120 The other is by AgBr electrolysis in an evaporator. In this method, Br atoms are directly deposited onto a substrate that is unnecessarily heated. Then, highly monodispersed Br atoms are achieved, as shown in Fig. 5a. The LWF around a single Br atom (Fig. 5b) on the Cu substrate is increased by about 0.8 eV (Fig. 5c), also confirming electron transfer from the Cu(111) substrate to the Br atom. These results are in great agreement with the conventional picture that halogen atoms typically withdraw electrons from the metal substrate, as measured by ultraviolet photoemission spectroscopy (UPS) or a Kelvin probe.55,58–63,65 The transferred charge onto a Br atom deduced from the LWF mapping is about 0.19e. This shows again that the spatially resolved charge state of an additive provides more microscopic information that is not available from conventional surface techniques.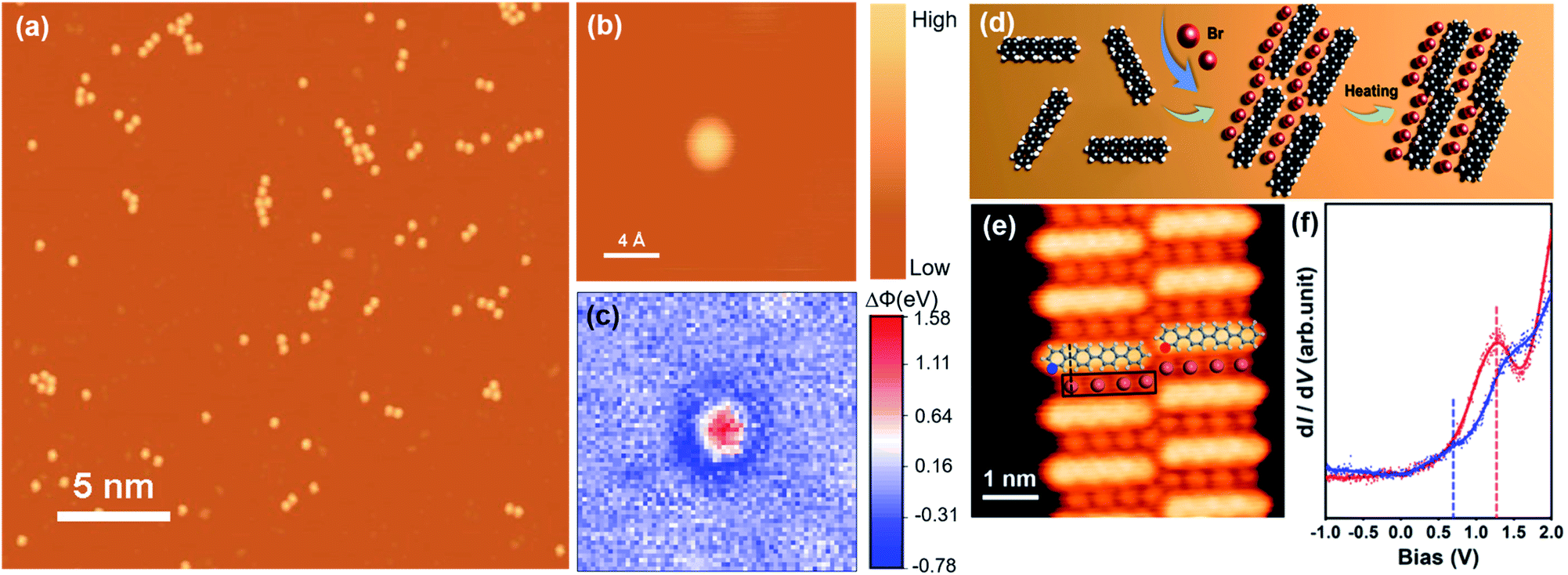 | ||
| Fig. 5 (a) STM image of highly monodispersed Br atoms on Cu(111). (b) STM image of a single Br atom and (c) its corresponding Δϕ mapping. (d) A schematic process of the coupling reaction mediated by the Br atoms. (e) High-resolution STM image of co-assembled Br atoms and Ph4. (f) dI/dV spectra sampled at the sites highlighted by the coloured dots in (e). Reproduced with permission (d–f).119 Copyright 2020, American Chemical Society. | ||
In fact, the microscopic picture of the charge transfer between the metal substrate and edge/internal Br atoms of the island is quite counterintuitive to the traditional one that the Br atoms at edges usually experience a more serious corrugation of electron density and hence cause a higher WF variation.120 However, the traditional picture lacks the information that the electron density of the edge Br atoms can be smeared out by their contact with more neutral metal atoms in the substrate and the Smoluchowski smoothening effect.121 This discrepancy between the macroscopic and microscopic pictures reminds that the charge transfer at a microscopic interface strongly depends on its locality, and hence the promoter effect on the catalytic performance in heterogeneous catalysis should be cautiously interpreted.
Lin et al.119 reported that the introduced Br atoms on Cu(111) promoted the selectivity toward the coupling reaction of p-quaterphenyl (Ph4) and p-quinquephenyl (Ph5). The co-assembly of the Br atoms and Ph4/Ph5 with a specific ratio can efficiently increase their collision probability between two neighbouring Ph4 or Ph5 molecules (Fig. 5d). It is proven that the electronic structure of the reacting molecules can also be modified by the Br atoms nearby (Fig. 5e and f). Moreover, Liu et al.122 have also proven that the Br atoms can promote the C–H bond activation of terminal alkynes on Ag(111) at room temperature.
Summary and prospects
Based on our recent studies and new data inputs, typical cases of charge transfer between single atoms and their supports on the atomic scale have been gathered in this perspective by stressing that a specific microscopic picture has to be established to understand the principles that govern the superb performance of SACs in various situations. It's widely appreciated that SACs possess the highest dispersion of and hence consume the least amount of precious metals. The exposed single metal atoms in SACs might be the most active due to their lowest coordination number on supports. Unfortunately, SACs are also thermally vulnerable because their single metal atoms can feasibly aggregate and cluster at elevated temperatures. Moreover, a single metal atom can form a strong interaction with its support so that the charge state and corresponding catalytic performance of the same single metal atom remarkably vary at different locations on the support. This is of particular importance in a nanosized SAC system where a single metal atom may be anchored to the corners, edges, kinks, steps, terraces and defects of the support. This may explain why many reported results of similar SACs are contradictory and even hard to reproduce in the literature.Previously, people can only rely on density functional theory and other theories to estimate the charge state and electron transfer between a single atom and its support. But sophisticated theories have yet to be fully developed to treat locally charged systems, ionic oxides, non-conducting supports and ensembles of strong electronic interactions. On the other hand, conventional techniques in catalysis and surface science cannot experimentally provide microscopic information of the charge state and electron transfer in SACs. It becomes even more problematic in the cases where the metal atoms in later d and f blocks in the periodic table of elements may bilaterally donate and withdraw electrons from their supports.
To date, the only available techniques that can probe the local charge state and electron transfer in SACs are high-resolution spectroscopy techniques like STM and AFM. Only a limited number of model systems have been explored so far, and therefore a comprehensive database is still far from completion and people do not have a finger-pointing reference to understand the electronic properties of SACs. Nonetheless, scanning probe microscopy techniques turn out to be quite promising in principle to acquire local electronic information which is one of the most important aspects that dictate the catalytic performance of SACs.
Even with the limited amount of spatially resolved and quantitatively revealed information on the charge state and electron transfer of single atoms including Au, K and Cs on monolayered CuO as well as Br on Cu(111), one can immediately find that the situation becomes more complicated at the atomic level. The spatially resolved LWF variation caused by single atoms shows an obvious symmetry break which does not seem to be originating from the asymmetry of the STM tip, but instead from very local geometric and electronic corrugations that await further experimental and theoretical explorations. In addition, single-atom-induced variations of the LWFs at the atomic level are not fully consistent with the simple picture, based on the information acquired by conventional XPS and UPS techniques, that the generated anions and cations increase and decrease the apparent WFs of the substrates, respectively. Yet, semi-quantitative or quantitative estimations of the charge transfer between single atoms and their underlying substrates could be deducted from the variations of the apparent LWFs.
In terms of the conventional promoter effect of alkali or halogen elements on the catalytic performance in heterogeneous catalysis, the described charge transfer between single Au and K atoms at a short distance on the CuO monolayer is so localized that they electronically cancel each other. This means that the charge transfer might take place directly in-between them or indirectly via the CuO substrate. Such a microscopic discovery suggests that the traditional explanation of alkali and halogen elements by electronic effects may fail on strongly ionic oxide or non-conducting supports in heterogeneous catalysis. Even on metallic substrates, the variations of the LWFs around the Br atoms in various spots within the Br islands are indicative that the extent of the charge transfer is very sensitive to the microscopic environment surrounding the concerned Br atom.
In summary, spatially resolved LWF measurements and quantitatively revealed electron transfer between single atoms and their underlying substrates do help us understand SACs at the atomic level. Single atoms may form strong interactions with the supports such that their catalytic performance becomes very sensitive to their specific locations on the supports. Therefore, a simple and general claim of the high efficiency of SACs could be misleading. This also applies to our understanding of the promoter effects of alkali and halogen elements in heterogeneous catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was jointly supported by the NSFC (21821004, 21932001 and 21902174) and MOST (2017YFA0204702), China.Notes and references
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- B. Qiao, J.-X. Liang, A. Wang, C.-Q. Xu, J. Li, T. Zhang and J. J. Liu, Nano Res., 2015, 8, 2913–2924 CrossRef CAS.
- J. Lin, Y. Chen, Y. Zhou, L. Li, B. Qiao, A. Wang, J. Liu, X. Wang and T. Zhang, AIChE J., 2017, 63, 4003–4012 CrossRef CAS.
- R. Lang, W. Xi, J. C. Liu, Y. T. Cui, T. Li, A. F. Lee, F. Chen, Y. Chen, L. Li, L. Li, J. Lin, S. Miao, X. Liu, A. Q. Wang, X. Wang, J. Luo, B. Qiao, J. Li and T. Zhang, Nat. Commun., 2019, 10, 234 CrossRef PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- X. Li, X. Yang, Y. Huang, T. Zhang and B. Liu, Adv. Mater., 2019, 31, e1902031 CrossRef PubMed.
- J. Zhou, Z. Xu, M. Xu, X. Zhou and K. Wu, Nanoscale Adv., 2020, 2, 3624–3631 RSC.
- Z. Li, Y. Chen, S. Ji, Y. Tang, W. Chen, A. Li, J. Zhao, Y. Xiong, Y. Wu, Y. Gong, T. Yao, W. Liu, L. Zheng, J. Dong, Y. Wang, Z. Zhuang, W. Xing, C. T. He, C. Peng, W. C. Cheong, Q. Li, M. Zhang, Z. Chen, N. Fu, X. Gao, W. Zhu, J. Wan, J. Zhang, L. Gu, S. Wei, P. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. Duan, Y. Huang, X. M. Chen, D. Wang and Y. Li, Nat. Chem., 2020, 12, 764–772 CrossRef CAS PubMed.
- C. Chu, D. Huang, S. Gupta, S. Weon, J. Niu, E. Stavitski, C. Muhich and J. H. Kim, Nat. Commun., 2021, 12, 5179 CrossRef CAS PubMed.
- Z. Zhang, J. Liu, J. Wang, Q. Wang, Y. Wang, K. Wang, Z. Wang, M. Gu, Z. Tang, J. Lim, T. Zhao and F. Ciucci, Nat. Commun., 2021, 12, 5235 CrossRef CAS PubMed.
- X. Zhou, W. Yang, Q. Chen, Z. Geng, X. Shao, J. Li, Y. Wang, D. Dai, W. Chen, G. Xu, X. Yang and K. Wu, J. Phys. Chem. C, 2016, 120, 1709–1715 CrossRef CAS.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- Z. Zuo, S. Liu, Z. Wang, C. Liu, W. Huang, J. Huang and P. Liu, ACS Catal., 2018, 8, 9821–9835 CrossRef CAS.
- J. Wang, H. Tan, S. Yu and K. Zhou, ACS Catal., 2015, 5, 2873–2881 CrossRef CAS.
- A. Sanchez, S. Abbet, U. Heiz, W.-D. Schneider, H. Häkkinen, R. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS PubMed.
- H. Tang, Y. Su, B. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. Ren, J. Huang, M. Haruta, B. Qiao, X. Liu, C. Jin, D. Su, J. Wang and T. Zhang, Sci. Adv., 2017, 3, e1700231 CrossRef PubMed.
- W. D. Schneider, M. Heyde and H. J. Freund, Chemistry, 2018, 24, 2317–2327 CrossRef CAS PubMed.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS PubMed.
- X. Zhang, H. Shi and B. Q. Xu, Angew. Chem., Int. Ed., 2005, 44, 7132–7135 CrossRef CAS PubMed.
- G. J. Hutchings, M. S. Hall, A. F. Carley, P. Landon, B. E. Solsona, C. J. Kiely, A. Herzing, M. Makkee, J. A. Moulijn, A. Overweg, J. C. Fierro-Gonzalez, J. Guzman and B. C. Gates, J. Catal., 2006, 242, 71–81 CrossRef CAS.
- M. F. Camellone and S. Fabris, J. Am. Chem. Soc., 2009, 131, 10473–10483 CrossRef PubMed.
- D. Stolcic, M. Fischer, G. Gantefor, Y. D. Kim, Q. Sun and P. Jena, J. Am. Chem. Soc., 2003, 125, 2848–2849 CrossRef CAS PubMed.
- L. D. Socaciu, J. Hagen, T. M. Bernhardt, L. Woste, U. Heiz, H. Hakkinen and U. Landman, J. Am. Chem. Soc., 2003, 125, 10437–10445 CrossRef CAS PubMed.
- A. J. Therrien, A. J. R. Hensley, M. D. Marcinkowski, R. Zhang, F. R. Lucci, B. Coughlin, A. C. Schilling, J.-S. McEwen and E. C. H. Sykes, Nat. Catal., 2018, 1, 192–198 CrossRef CAS.
- X. Zhou, Q. Shen, K. Yuan, W. Yang, Q. Chen, Z. Geng, J. Zhang, X. Shao, W. Chen, G. Xu, X. Yang and K. Wu, J. Am. Chem. Soc., 2018, 140, 554–557 CrossRef CAS PubMed.
- A. J. Therrien, K. Groden, A. J. R. Hensley, A. C. Schilling, R. T. Hannagan, M. D. Marcinkowski, A. Pronschinske, F. R. Lucci, E. C. H. Sykes and J.-S. McEwen, J. Catal., 2018, 364, 166–173 CrossRef CAS.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- O. Hinrichsen, F. Rosowski, M. Muhler and G. Ertl, Chem. Eng. Sci., 1996, 51, 1683–1690 CrossRef CAS.
- M. Muhler, F. Rosowski, O. Hinrichsen, A. Hornung and G. Ertl, in Stud. Surf. Sci. Catal., Elsevier, 1996, vol. 101, pp. 317–326 Search PubMed.
- C. Zhang, F. Liu, Y. Zhai, H. Ariga, N. Yi, Y. Liu, K. Asakura, M. Flytzani-Stephanopoulos and H. He, Angew. Chem., Int. Ed., 2012, 51, 9628–9632 CrossRef CAS PubMed.
- Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, A. U. Nilekar, G. Peng, J. A. Herron, D. C. Bell, H. Saltsburg, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2010, 329, 1633–1636 CrossRef CAS PubMed.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498–1501 CrossRef CAS PubMed.
- B. Zugic, S. Zhang, D. C. Bell, F. F. Tao and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2014, 136, 3238–3245 CrossRef CAS PubMed.
- M. Yang, J. Liu, S. Lee, B. Zugic, J. Huang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2015, 137, 3470–3473 CrossRef CAS PubMed.
- W. Ngantsoue-Hoc, Y. Zhang, R. J. O'Brien, M. Luo and B. H. Davis, Appl. Catal., A, 2002, 236, 77–89 CrossRef CAS.
- A. B. Dongil, B. Bachiller-Baeza, E. Castillejos, N. Escalona, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Catal. Sci. Technol., 2016, 6, 6118–6127 RSC.
- D. C. Grinter, E. R. Remesal, S. Luo, J. Evans, S. D. Senanayake, D. J. Stacchiola, J. Graciani, J. F. Sanz and J. A. Rodriguez, J. Phys. Chem. Lett., 2016, 7, 3866–3872 CrossRef CAS PubMed.
- C. Park and M. A. Keane, ChemPhysChem, 2001, 2, 733–741 CrossRef CAS PubMed.
- M. Vadekar and I. Pasternak, Can. J. Chem. Eng., 1970, 48, 216–218 CrossRef CAS.
- Y. Kiso, M. Tanaka, H. Nakamura, T. Yamasaki and K. Saeki, J. Organomet. Chem., 1986, 312, 357–364 CrossRef CAS.
- V. Choudhary and P. Jana, J. Catal., 2007, 246, 434–439 CrossRef CAS.
- S. T. Marshall and J. W. Medlin, Surf. Sci. Rep., 2011, 66, 173–184 CrossRef CAS.
- R. M. Lambert, R. L. Cropley, A. Husain and M. S. Tikhov, Chem. Commun., 2003, 1184–1185, 10.1039/b302620e.
- C. T. Campbell, J. Catal., 1986, 99, 28–38 CrossRef CAS.
- S. Y. Davydov and G. I. Sabirova, Tech. Phys. Lett., 2011, 37, 515–518 CrossRef CAS.
- S. Ahmad, P. Miró, M. Audiffred and T. Heine, Solid State Commun., 2018, 272, 22–27 CrossRef CAS.
- Z.-Y. Wang and D.-J. Shu, J. Phys. Chem. C, 2021, 125, 19259–19267 CrossRef.
- T. Aruga and Y. Murata, Prog. Surf. Sci., 1989, 31, 61–130 CrossRef CAS.
- H. P. Bonzel, Surf. Sci. Rep., 1988, 8, 43–125 CrossRef.
- R. D. Diehl and R. McGrath, Surf. Sci. Rep., 1996, 23, 43–171 CrossRef CAS.
- H. Tochihara and S. Mizuno, Prog. Surf. Sci., 1998, 58, 1–74 CrossRef CAS.
- R. G. Jones, Prog. Surf. Sci., 1988, 27, 25–160 CrossRef CAS.
- W. Kai, W. Dezheng, D. Junzhuo, W. Xuming, C. Yuming, Z. Maosheng, Z. Runsheng and G. Xiexian, Surf. Sci., 1992, 264, 249–259 CrossRef.
- K. Wu, D. Wang, X. Wei, Y. Cao and X. Guo, J. Catal., 1993, 140, 370–383 CrossRef CAS.
- K. Wu, D. Wang, X. Wei, Y. Cao and X. Guo, Surf. Sci. Lett., 1993, 285, L522–L524 CrossRef CAS.
- R. G. Jones and M. Kadodwala, Surf. Sci., 1997, 370, L219–L225 CrossRef CAS.
- P. Dowben and R. Jones, Surf. Sci., 1979, 84, 449–461 CrossRef CAS.
- P. Dowben and R. Jones, Surf. Sci., 1979, 88, 348–366 CrossRef CAS.
- E. Bertel, K. Schwaha and F. Netzer, Surf. Sci., 1979, 83, 439–452 CrossRef CAS.
- E. Bertel and F. Netzer, Surf. Sci., 1980, 97, 409–424 CrossRef CAS.
- S. K. Jo and J. White, Surf. Sci., 1992, 261, 111–117 CrossRef CAS.
- C. Jowett and B. Hopkins, Surf. Sci., 1970, 22, 392–410 CrossRef CAS.
- C. Benndorf and B. Krüger, Surf. Sci., 1985, 151, 271–288 CrossRef CAS.
- N. D. Lang, Phys. Rev. B: Solid State, 1971, 4, 4234–4244 CrossRef.
- Q. Zhu and S.-q. Wang, J. Electrochem. Soc., 2016, 163, H796–H808 CrossRef CAS.
- R. Lindsay, E. Michelangeli, B. G. Daniels, M. Polcik, A. Verdini, L. Floreano, A. Morgante, J. Muscat, N. M. Harrison and G. Thornton, Surf. Sci., 2003, 547, L859–L864 CrossRef CAS.
- U. Martinez, L. Giordano and G. Pacchioni, J. Chem. Phys., 2008, 128, 164707 CrossRef PubMed.
- Z. Peng, B. Di, W. Li, D. Liu, X. Wen, H. Zhu, H. Song, Y. Zhang, C. Yin, X. Zhou and K. Wu, Angew. Chem., Int. Ed., 2020, 59, 14321–14325 CrossRef CAS PubMed.
- H. Chen, W. Rong, Z. Huang, Z. Peng, Z. Xu, J. Zhou, B. Di, X. Zhou and K. Wu, J. Chem. Phys., 2019, 151, 184703 CrossRef PubMed.
- L. Olesen, M. Brandbyge, M. R. Sørensen, K. W. Jacobsen, E. Lægsgaard, I. Stensgaard and F. Besenbacher, Phys. Rev. Lett., 1996, 76, 1485 CrossRef CAS PubMed.
- L. Vitali, G. Levita, R. Ohmann, A. Comisso, A. De Vita and K. Kern, Nat. Mater., 2010, 9, 320–323 CrossRef CAS PubMed.
- F. Mohn, L. Gross, N. Moll and G. Meyer, Nat. Nanotechnol., 2012, 7, 227–231 CrossRef CAS PubMed.
- X. Wen, Y. Lin, Z. Huang, M. Diao, W. Zhao, J. Dai, L. Xing, H. Zhu, Z. Peng, D. Liu and K. Wu, J. Phys. Chem. Lett., 2019, 10, 6800–6806 CrossRef CAS PubMed.
- T. Konig, G. H. Simon, H.-P. Rust, G. Pacchioni, M. Heyde and H.-J. Freund, J. Am. Chem. Soc., 2009, 131, 17544–17545 CrossRef CAS PubMed.
- S. Schintke, S. Messerli, M. Pivetta, F. Patthey, L. Libioulle, M. Stengel, A. De Vita and W. D. Schneider, Phys. Rev. Lett., 2001, 87, 276801 CrossRef CAS PubMed.
- T. Yamada, J. Fujii and T. Mizoguchi, Surf. Sci., 2001, 479, 33–42 CrossRef CAS.
- J. Jia, K. Inoue, Y. Hasegawa, W. Yang and T. Sakurai, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 1193 CrossRef CAS.
- T. Konig, G. H. Simon, H.-P. Rust and M. Heyde, J. Phys. Chem. C, 2009, 113, 11301–11305 CrossRef CAS.
- Z. Huang, Z. Xu, J. Zhou, H. Chen, W. Rong, Y. Lin, X. Wen, H. Zhu and K. Wu, J. Phys. Chem. C, 2019, 123, 17823–17828 CrossRef CAS.
- A. Sasahara, C. L. Pang and H. Onishi, J. Phys. Chem. B, 2006, 110, 17584–17588 CrossRef CAS PubMed.
- L. Gross, F. Mohn, P. Liljeroth, J. Repp, F. J. Giessibl and G. Meyer, Science, 2009, 324, 1428–1431 CrossRef CAS PubMed.
- Y. Zhang, O. Pluchery, L. Caillard, A. F. Lamic-Humblot, S. Casale, Y. J. Chabal and M. Salmeron, Nano Lett., 2015, 15, 51–55 CrossRef CAS PubMed.
- B. Mallada, A. Gallardo, M. Lamanec, B. de la Torre, V. Spirko, P. Hobza and P. Jelinek, Science, 2021, 374, 863–867 CrossRef CAS PubMed.
- P. S. Bagus, V. Staemmler and C. Woll, Phys. Rev. Lett., 2002, 89, 096104 CrossRef PubMed.
- A. Michaelides, P. Hu, M. H. Lee, A. Alavi and D. A. King, Phys. Rev. Lett., 2003, 90, 246103 CrossRef CAS PubMed.
- T. C. Leung, C. L. Kao, W. S. Su, Y. J. Feng and C. T. Chan, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 195408 CrossRef.
- F. Albrecht, M. Fleischmann, M. Scheer, L. Gross and J. Repp, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 235443 CrossRef.
- N. D. Lang, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 10395–10398 CrossRef PubMed.
- T. Aoki and T. Yokoyama, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 155423 CrossRef.
- M. Becker and R. Berndt, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 035426 CrossRef.
- H. Geistlinger, Sens. Actuators, B, 1993, 17, 47–60 CrossRef CAS.
- H. Schwarz, Catal. Sci. Technol., 2017, 7, 4302–4314 RSC.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Acc. Chem. Res., 2018, 52, 237–247 CrossRef PubMed.
- M. T. Darby, M. Stamatakis, A. Michaelides and E. C. H. Sykes, J. Phys. Chem. Lett., 2018, 9, 5636–5646 CrossRef CAS PubMed.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- S. Liang, C. Hao and Y. Shi, ChemCatChem, 2015, 7, 2559–2567 CrossRef CAS.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- Z. Li, D. Wang, Y. Wu and Y. Li, Natl. Sci. Rev., 2018, 5, 673–689 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- M. J. Hülsey, J. Zhang and N. Yan, Adv. Mater., 2018, 30, 1802304 CrossRef PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2018, 119, 1806–1854 CrossRef PubMed.
- Y. Guo, R. Lang and B. Qiao, Catalysts, 2019, 9, 135 CrossRef.
- G. S. Parkinson, Catal. Lett., 2019, 149, 1137–1146 CrossRef CAS PubMed.
- Z. Chen, L. Chen, C. Yang and Q. Jiang, J. Mater. Chem. A, 2019, 7, 3492–3515 RSC.
- D. Liu, Q. He, S. Ding and L. Song, Adv. Energy Mater., 2020, 10, 2001482 CrossRef CAS.
- J. C. Liu, Y. G. Wang and J. Li, J. Am. Chem. Soc., 2017, 139, 6190–6199 CrossRef CAS PubMed.
- D. J. Coulman, J. Wintterlin, R. J. Behm and G. Ertl, Phys. Rev. Lett., 1990, 64, 1761–1764 CrossRef CAS PubMed.
- F. Jensen, F. Besenbacher, E. Laegsgaard and I. Stensgaard, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 10233–10236 CrossRef CAS PubMed.
- K. Kern, H. Niehus, A. Schatz, P. Zeppenfeld, J. Goerge and G. Comsa, Phys. Rev. Lett., 1991, 67, 855–858 CrossRef CAS PubMed.
- V. Pouthier, C. Ramseyer, C. Girardet, P. Zeppenfeld, V. Diercks and R. Halmer, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 9998 CrossRef CAS.
- L. Ruan, F. Besenbacher, I. I. Stensgaard and E. Laegsgaard, Phys. Rev. Lett., 1993, 70, 4079–4082 CrossRef CAS PubMed.
- A. Yurtsever, Y. Sugimoto, M. Abe, K. Matsunaga, I. Tanaka and S. Morita, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 085413 CrossRef.
- G. Ebbinghaus and A. Simon, Chem. Phys., 1979, 43, 117–133 CrossRef CAS.
- Y. Lin, Z. Huang, X. Wen, W. Rong, Z. Peng, M. Diao, L. Xing, J. Dai, X. Zhou and K. Wu, ACS Nano, 2020, 14, 17134–17141 CrossRef CAS PubMed.
- Z. Huang, Y. Lin, C. Han, Y.-Y. Sun, K. Wu and W. Chen, J. Phys. Chem. C, 2021, 125, 7944–7949 CrossRef CAS.
- R. Smoluchowski, Phys. Rev., 1941, 60, 661–674 CrossRef CAS.
- J. Liu, Q. Chen, Q. He, Y. Zhang, X. Fu, Y. Wang, D. Zhao, W. Chen, G. Q. Xu and K. Wu, Phys. Chem. Chem. Phys., 2018, 20, 11081–11088 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |