
Chemical etching induced microporous nickel backbones decorated with metallic Fe@hydroxide nanocatalysts: an efficient and sustainable OER anode toward industrial alkaline water-splitting†
Nabeen K.
Shrestha
 *a,
Supriya A.
Patil
b,
Jonghoon
Han
a,
Sangeun
Cho
a,
Akbar I.
Inamdar
a,
Hyungsang
Kim
*a and
Hyunsik
Im
*a
*a,
Supriya A.
Patil
b,
Jonghoon
Han
a,
Sangeun
Cho
a,
Akbar I.
Inamdar
a,
Hyungsang
Kim
*a and
Hyunsik
Im
*a
aDivision of Physics and Semiconductor Science, Dongguk University, Seoul 04620, South Korea. E-mail: nabeenkshrestha@hotmail.com; hskim@dongguk.edu; hyunsik7@dongguk.edu
bDepartment Nanotechnology & Advanced Materials Engineering, Sejong University, Seoul-05006, South Korea
First published on 21st March 2022
Abstract
Development of cost-effective and highly efficient electrocatalysts for water splitting is crucial to produce affordable and sustainable green-hydrogen energy that can alleviate the current overreliance on fossil fuels. This work demonstrates the simple immersion-based chemical etching of nickel foam (NF) in an ethanolic FeCl3 solution to generate microporous nickel (Ni) backbones decorated with hierarchically structured metallic Fe doped Ni–Fe-hydroxide nanoparticles serving as a highly promising oxygen evolution reaction (OER) electrode in alkaline water. The optimally etched NF-based OER electrode exhibits a low Tafel slope of 47.3 mV dec−1 and a low overpotential of 220, 270, and 310 mV at 10, 100, and 500 mA cm−2, respectively. Intriguingly, this electrode also exhibits a perfectly reversible OER and HER performance between +400 and −40 mA cm−2 with no evidence of electrode potential decay for 80 h. Importantly, when used with an industrial-type 30 wt% KOH aqueous electrolyte and compared to a benchmark Pt/C(20wt%)‖IrO2-based cell, the electrolyzer exhibits a lower cell voltage of 1.52 (vs. 1.56 V of Pt/C(20wt%)‖IrO2-cell), 1.62 (vs. 1.79), 1.69 (vs. 1.92) and 1.79 (vs. 2.08) V at 10, 50, 100, and 240 mA cm−2, respectively, with the cell voltage maintained for ∼100 h.
1 Introduction
Hydrogen economy is likely to be one of the most promising and competitive emission–reduction pathways for long-term global decarbonization.1–3 Water is the most abundantly available inexhaustible and fossil-free source of hydrogen from which green hydrogen fuel can be produced via electrochemical water-splitting technology, provided that the electrolyzer is powered by green-electricity obtained from renewable sources. However, water electrolysis is technically hindered primarily by the sluggish oxygen evolution reaction (OER) at the anode.4–7 In addition, noble-metal-based state-of-the-art catalysts such as Pt, RuO2, or IrO2 have been employed to allow water electrolysis at a lower overpotential, but these increase the costs associated with the large-scale production of hydrogen.8In order to achieve sustainable water-splitting, various transition metals, metal alloys, and associated compounds have been extensively investigated as alternatives to conventional noble-metal-based catalysts.9–20 Recently, Liu and coworkers have reported various Ni–Fe-based electrocatalysts, such as NiFe LDH/MoS2, NiFeP/MXen, N-CNTs@NiS2/Fe7S8, and Ni1−xFex-P/PO3 heterostructures, that have exhibited excellent OER performance and durability.21–24 As an alternative to 2D flat substrates, electrically conductive foam substrates, particularly nickel foam (NF), have been widely employed as 3D porous diffusion substrates to accommodate catalysts and achieve high mass and electron transportation.25–29 An uncontrolled high catalyst loading on NF usually obtained via the common deposition techniques such as drop-casting, hydrothermal, and chemical bath deposition routes, however, often leads to the deposits being stacked unevenly on top of each other. As a result, despite the high catalyst loading, stack deposition not only limits the access of the electrolyte to catalytic sites but also hinders the facile release of gas bubbles, plaguing the catalytic activity enhancement.
Herein, in contrast to the commonly used direct deposition technique for the loading of catalysts and electrochemical anodization route used to produce a porous electrode, the present study is based on the simple immersion-based chemical etching of NF in an ethanolic FeCl3 solution, which produces micro- and nano-pores in the NF backbones, and the in situ growth of a thin film of metallic Fe-doped hierarchically structured Ni–Fe-hydroxide (hereinafter referred as Fe@Ni–Fe-hydroxide) nanoparticles with a relatively thinner than those deposited via traditional deposition routes. The proposed chemical etching technique enhances active sites by preventing the over-loading of the catalyst nanoparticles, strongly binds them to the substrate, and maintains the 3D hierarchical porous network morphology of the NF backbones. This enhances mass transport, facilitating the release of O2 bubbles from the surface, and promoting the diffusion of the electrolyte to the catalytic sites. In addition, the electron transport characteristics of the anode are strengthened, thus minimizing the issues associated with severe O2 bubble dissipation and the low utilization of catalytic active sites due to the overgrowth of the catalyst. The aggressive evolution and trapping of gas bubbles at high current densities, in fact, block the active sites, thus hindering the access of the electrolyte and limiting the available active sites. For this reason, the effective detachment of these gas bubbles is essential for an efficient reaction, particularly at high currents or potentials.30,31
Most importantly, in contrast to pristine NF, optimally etched NF in an ethanolic FeCl3 solution for 18 h (Fe-18h/NF) exhibits remarkably higher electrocatalytic activity, particularly with regard to the OER, with a low Tafel slope of 45.2 mV dec−1 and a reasonably low overpotential of 220, 270, and 310 mV at 10, 100, and 500 mA cm−2, respectively, in a 1 M KOH electrolyte. In addition, the long-term electrochemical durability of the electrode for water-splitting is demonstrated at a current bias of 240 mA cm−2 for about 100 h using both a laboratory-scale prototype aqueous electrolyte (1 M KOH) and an industrial-type 30 wt% KOH electrolyte. It is worth noting that the OER performance of the chemically etched NF is superior to that of a state-of-the-art IrO2/NF catalyst and is competitive with Ni–Fe and other transition-metal-based electrocatalysts deposited on NF (Table S1†). The proposed immersion-based chemical etching strategy for the fabrication of a high-performance NF-based OER electrode is also highly scalable and thus shows great promise for use in commercial water-splitting applications.
2 Results and discussion
2.1 Physical and chemical characterization of the etched NF
Fig. S1† displays images of the pristine NF before and after itching for various durations of 6, 12, 18 and 24 h in 200 μmol mL−1 ethanolic FeCl3 at 50 °C. As the etching time increased, the NF became thinner and more fragile. Etching was more pronounced on the top tip when the NF was placed vertically in the etching bath. Thus, 1 cm−2 of the etched NF from the bottom part was selectively employed as the active area of the electrode for electrocatalysis. Microscopic examination of the foam was conducted via electron microscopy. Fig. S2a† displays an SEM image of pristine NF, showing a highly porous 3D network of Ni backbones. A magnified view of the backbones (inset of Fig. S2a†) shows Ni crystals with crystal boundaries, while a highly magnified view (Fig. S2b†) shows the surface covered with very fine grains. When the NF was immersed in 200 μmol mL−1 of ethanolic FeCl3 as an etchant at 50 °C, pitting corrosion was observed in the early stages, as illustrated by the presence of pin-holes on the NF surface after etching for 6 h (Fe-6h/NF; Fig. S2c†). As the immersion continued, new pits emerged and grew, forming micropores on the Ni backbones (Fig. S2e, g, and i).† NF with a uniform distribution of pores of about 20 μm was observed following the immersion performed for 18 h (Fe-18h/NF; Fig. 1a and S2g†). These pores act as diffusion paths for the electrolyte to reach active sites and for the rapid dissipation of gas from the active sites, leading to a facile mass and charge transport during water electrolysis. In addition to pore formation, the immersion of the NF in the etchant led to the in situ growth of dense spherical nanoparticles and their uniform distribution on the Ni backbones (Fig. 1b). The observed pitting of the Ni backbones (Fig. S2c†) was due to the strong corrosive action of the FeCl3 etchant, while the chemical interactions between Fe3+ ions from the etchant, Ni2+ ions generated during the etching of the NF, and the ethanol solvent with some dissolved moistures were the key driving source responsible for the in situ deposition of hydroxide nanoparticles. The pore size, pore density, and size of the nanoparticles were found to vary progressively with the etching time (Fig. S2†). In particular, a longer etching duration led to a higher pore density. However, prolonged etching (e.g., the Fe-24h/NF sample) led to a partial collapse of the pore structure (Fig. S2i†). On the other hand, when the etching was conducted at a lower temperature or in an etchant with a lower concentration of FeCl3, no pore formation was obtained. Meanwhile, the concentration of etchant could not be increased beyond 200 μmol mL−1 due to limitation of solubility of FeCl3 (see the Experimental section). Considering the energy costs and complexity of maintaining a symmetrical electrical field around the entire 3D NF network required in anodization route for pore formation,32,33 the simple immersion-based chemical etching strategy proposed in the present study represents a highly promising, cost-effective approach to the mass production of microporous Ni backbones from NF.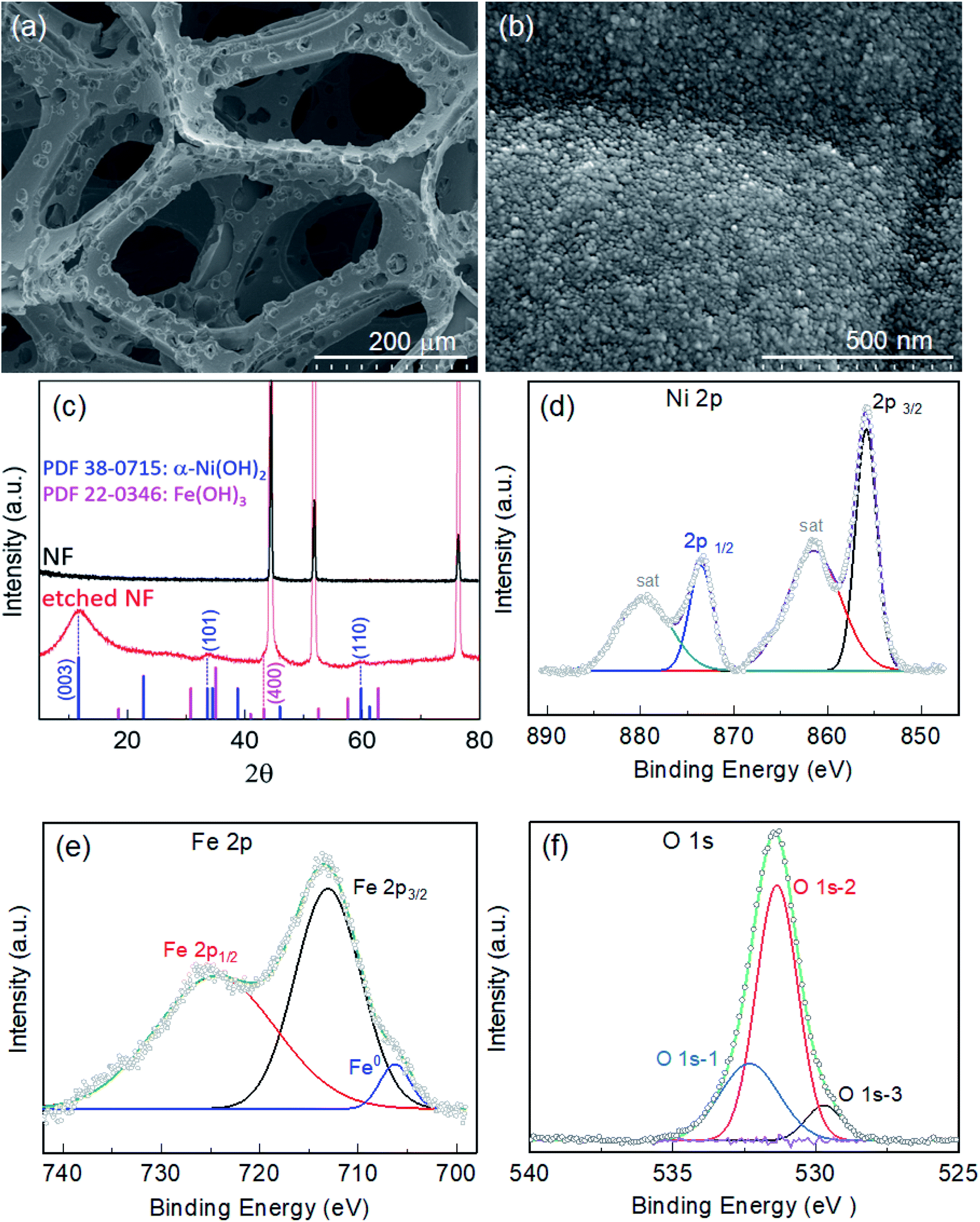 | ||
| Fig. 1 SEM images of nickel foam (NF) etched for 18 h (Fe-18h/NF) in an ethanolic FeCl3 solution at 50 °C: (a) surface view showing microporous Ni backbones at low magnification, and (b) a high-magnification view showing hierarchically-structured metallic Fe@Ni–Fe-hydroxide nanoparticles with a diameter of ∼10 nm. (c) XRD patterns of pristine NF and NF etched for 18 h. XPS spectra of (d) Ni 2p, (e) Fe 2p, and (f) O 1s for the Fe-18h/NF sample. | ||
The crystal structure of the etched NF was investigated using XRD. Fig. 1c depicts the XRD patterns obtained from pristine and etched NF samples. Compared with the pristine NF, the Fe-18h/NF sample had additional peaks other than those representing the cubic phase of the Ni substrate (JCPDS 04-0850) at about 2θ = 11.1°, 34.5°, and 60.7°. The positions of these peaks corresponded to JCPDS 38-0715, indicating the formation of α-Ni(OH)2.34–36 In addition, a small broad peak was also observed at about 43.04°, which is closely corresponded to diffraction from the (400) plane of the Fe(OH)3 cubic phase (JCPDS 022-0346). To determine the chemical composition and elemental binding states of the samples, XPS analysis of the samples was conducted. Fig. S3a† displays the elemental XPS survey spectrum, revealing that the surface of the etched NF was covered with a film composed of Cl, O, Ni, and Fe. The binding states of Ni and Fe were further investigated based on their high-resolution 2p XPS spectra. In addition to the two satellite peaks, the Ni 2p spectrum (Fig. 1d) exhibited a doublet at 873.38 eV (Ni 2p1/2) and 855.81 eV (Ni 2p3/2), which was in accordance with Ni2+ in Ni(OH)2.37,38 On the other hand, the doublet observed in the Fe 2p spectrum (Fig. 1e) at 724.55 eV and 712.86 eV was ascribed to Fe3+ in Fe(OH)3.38 In addition to the Fe3+ peaks, the deconvoluted Fe 2p spectrum also revealed a small metallic Fe0 peak at about 706.34 eV. The in situ growth of hydroxides on the etched NF was confirmed by the O 1s spectrum (Fig. 1f), which was deconvoluted into three binding peaks at 532.3, 531.3, and 529.72 eV. The first peak at the highest binding energy (O 1s-1) corresponded to surface-absorbed water molecules, while the middle peak (O 1s-2) corresponded to lattice M–OH (M = Ni, Fe) chemisorbed hydroxyl species, and the third peak (O 1s-3) at the lowest binding energy corresponded to oxygen atoms bound to metals.38,39 Here, the high O 1s-2 binding energy signal at 531.3 eV was found to account for 64.53% of the total O 1s area, thus it could be clearly assigned to the presence of hydroxide species.
Overall, the XRD and XPS findings suggest that the nanoparticles were composed of metallic Fe-composited nanocrystalline Ni(OH)2 and Fe(OH)3. Interestingly, it is worthy to be noted that, without the addition of a reducing agent to the etchant, the deposition of metallic Fe nanoparticles was observed. Based on the standard redox potential of 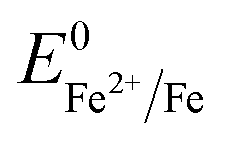 (−0.44 V) and
(−0.44 V) and 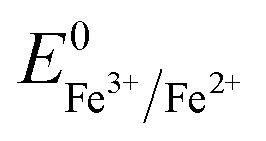 (+0.77 V), we estimated the standard reduction potential of the Fe3+ to Fe0 redox system to be
(+0.77 V), we estimated the standard reduction potential of the Fe3+ to Fe0 redox system to be 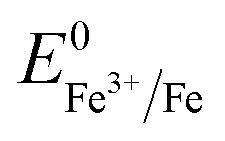 (+0.037 V). The details are presented in Section SI-1 of the ESI.† Because the standard reduction electrode potential of Ni is
(+0.037 V). The details are presented in Section SI-1 of the ESI.† Because the standard reduction electrode potential of Ni is  and the XPS spectrum does not reveal the presence of Fe2+ species in the sample (Fig. 1e), it could be assumed that there was a direct reduction of Fe3+ to Fe0. However, based on the redox potential of
and the XPS spectrum does not reveal the presence of Fe2+ species in the sample (Fig. 1e), it could be assumed that there was a direct reduction of Fe3+ to Fe0. However, based on the redox potential of 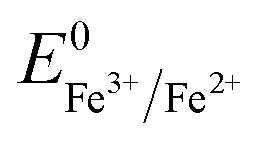 (+0.77 V) and
(+0.77 V) and 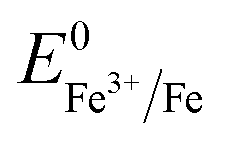 (+0.037 V), the reduction of Fe3+ to Fe2+ is more thermodynamically favorable than the reduction of Fe3+ to Fe0. Hence, the direct reduction of Fe3+ to Fe0 appears unlikely. Another possibility is that the –OH moiety of the alcohol has reducing properties and can act as a reducing agent. Although the mechanism for the deposition of Fe on the NF in this work is not yet clearly understood, the reduction of various Fe oxides to metallic Fe by ethanol at high temperatures has been reported previously.40 The introduction of the metallic Fe phase can contribute to an increase in the electrical conductivity of the in situ grown hydroxide film, thus facilitating electron and charge transport across the catalytic site/NF electrode and the catalytic site/electrolyte interfaces, respectively.
(+0.037 V), the reduction of Fe3+ to Fe2+ is more thermodynamically favorable than the reduction of Fe3+ to Fe0. Hence, the direct reduction of Fe3+ to Fe0 appears unlikely. Another possibility is that the –OH moiety of the alcohol has reducing properties and can act as a reducing agent. Although the mechanism for the deposition of Fe on the NF in this work is not yet clearly understood, the reduction of various Fe oxides to metallic Fe by ethanol at high temperatures has been reported previously.40 The introduction of the metallic Fe phase can contribute to an increase in the electrical conductivity of the in situ grown hydroxide film, thus facilitating electron and charge transport across the catalytic site/NF electrode and the catalytic site/electrolyte interfaces, respectively.
To acquire additional structural information, TEM analysis of the samples was conducted. In line with the SEM results shown in Fig. 1b, a TEM image of the Fe-18h/NF sample also revealed that the hydroxide film grown on the NF was composed of nearly spherical nanoparticles with a diameter of approximately 10 nm (Fig. 2a). HR-TEM images of these nanoparticles (Fig. 2b) revealed a lattice spacing of about 2.67 Å, 1.54 Å, and 1.51 Å corresponding to the (101), (110), and (113) planes, respectively, of α-Ni(OH)2. Similarly, a lattice spacing of about 2.10 Å corresponding to the (400) planes of the cubic Fe(OH)3 phase was also detected. This observation was also supported by the small broad XRD peak located at a 2θ of 43.04° (Fig. 1c) corresponding to the (400) plane of Fe(OH)3. In addition, the XPS Fe 2p spectrum (Fig. 1e) and O 1s spectrum (Fig. 1f), particularly the O 1s-2 peak corresponding to chemisorbed hydroxyl species (M–OH, where M = Ni, Fe), also indicated the presence of Fe(OH)3. However, Fe0 phase was also not resolved in the HR-TEM image, possibly due to the amorphous state. These findings were also supported by the diffused SAED ring and dot patterns observed in Fig. 2c, indicating the coexistence of polycrystalline and amorphous phases on the etched NF surface. The amorphous phases or those with a low degree of crystallinity suggest the presence of defects in the nanoparticles, leading to a higher surface density of unsaturated sites that act as active catalytic centers, thus having a significant impact on the catalytic performance in favor of water-splitting.41,42 It is worth noting that one such lattice defect (localized amorphization) was detected on the (110) plane, as shown in the HR-TEM image in Fig. 2b.
 | ||
| Fig. 2 TEM analysis of a Fe-18h/NF electrode: (a) TEM image showing ∼10 nm diameter particles, and (b) HR-TEM image showing the d-spacings with lattice defects on the (110) plane in the inset image. (c) SAED image showing diffused rings and dot patterns, indicating the coexistence of polycrystalline and amorphous phases. (d) HAADF mapping images showing the uniform distribution of the constituent elements. | ||
Furthermore, high-angle annular dark-field (HAADF) images obtained from STEM clearly displayed the uniform distribution of Ni, Fe, and O (Fig. 2d). Despite thoroughly cleaning the etched NF substrates, in accordance with the XPS survey result, the presence of Cl was also observed in the HAADF-STEM images. While examining the Cl 2p XPS spectrum of the Fe-18h/NF sample, a Cl 2p3/2 peak centered at 198.3 eV was observed, as shown in Fig. S3b.† This peak position is closer to that of NiCl2 while that of the FeCl3 from the etchant if presents should be located at about 199.7 eV.43,44 This finding indicates that the hydroxide film was doped with Cl. A similar result of F-incorporation from the fluoride etchant and Cl-incorporation from the metal chlorides has been reported previously.33,38
2.2 Electrocatalytic performance
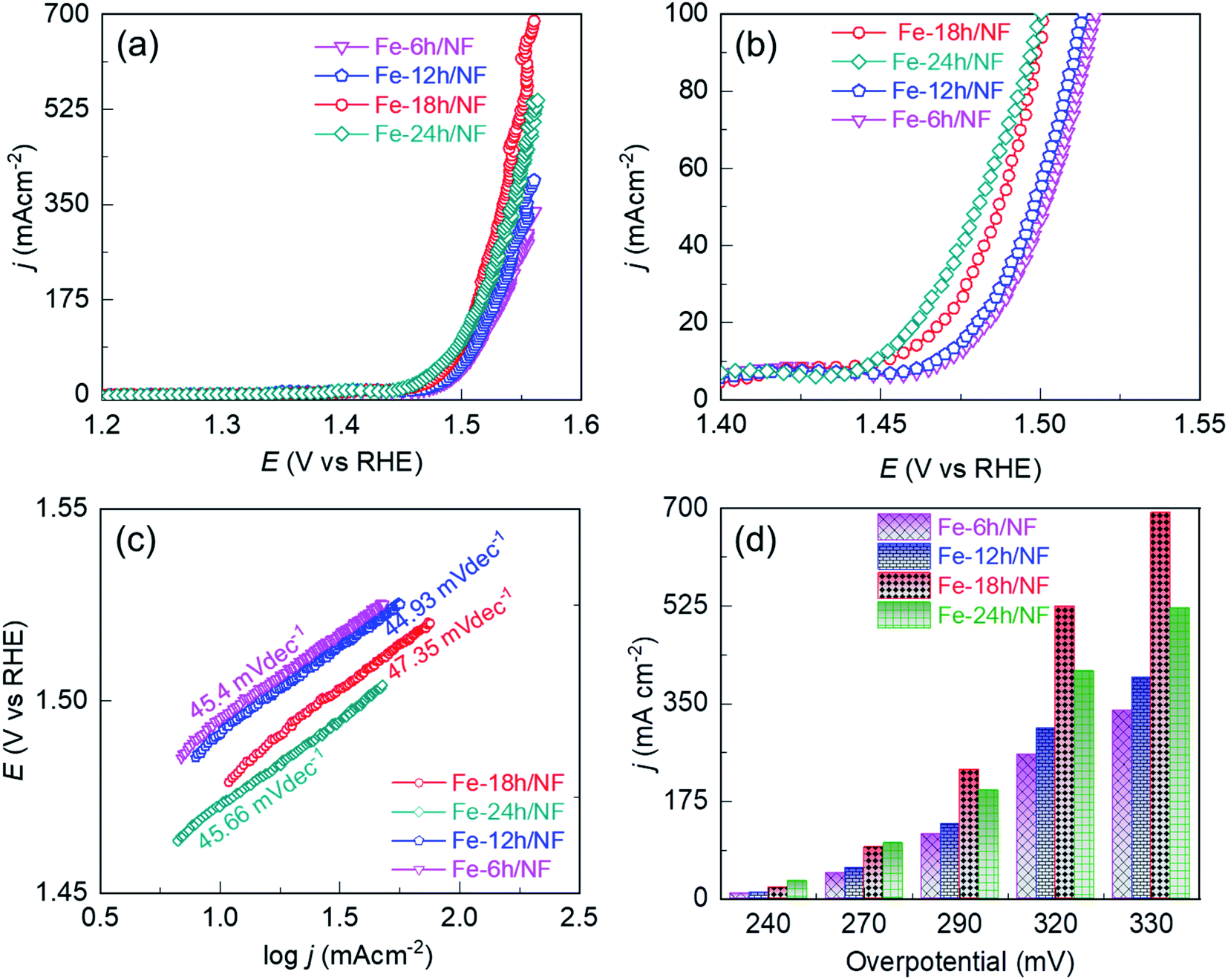 | ||
| Fig. 3 Comparison of the electrocatalytic performance of the etched NF in a 1 M KOH solution: iR compensated anodic linear sweep voltammograms (LSVs) of the (a) NFs etched for 6, 12, 18, and or 24 h, (b) magnified view of (a) below 100 mA cm−2, (c) corresponding Tafel slopes extracted from the LSVs, and (d) current density profiles with respect to the OER overpotential. | ||
To access the electrocatalytic activity on OER at a low current density, polarization was conducted at a slow scan rate of 1 mV s−1. The slow scan rate was essential particularly to suppress the oxidation peak observed at about 1.4 V vs. RHE, as shown in Fig. S4† and accessing the OER overpotential at a low current density of 10 mA cm−2. This oxidation peak is attributed to the electrochemical oxidation of (Fe–Ni)–OH phase to the catalytically favorable OER active (Fe/Ni)–OOH phase. It is worth noting that two distinct potential bias regimes were identified in the LSV curves (Fig. 3a). In the lower bias regime below 100 mA cm−2 (Fig. 3b), the OER was kinetically controlled, with each electrode exhibiting a similar Tafel slope (Fig. 3c). The OER performance in this regime is governed by the physical, chemical, and electronic properties of the catalytic sites and their interaction with the current collector. In particular, compared with Fe-24h/NF, the Fe-18h/NF electrode exhibited a lower overpotential, indicating a better OER performance for the electrode (Fig. 3d). In contrast, the polarization curves for the Fe-18h/NF electrode in the higher bias regime exhibited a significant increase in OER activity, outperforming the Fe-24h-/NF electrode. The enhanced OER activity of the Fe-18h/NF electrode in this regime can be attributed to high mass and electron transport, which facilitated the detachment of O2 bubbles and the diffusion of the electrolyte towards the catalytic sites. These mass and electron transport characteristics of the Fe-18h/NF electrode are likely to be the result of its well-defined porous structure and uniform distribution of catalyst particles. In contrast, the excess corrosion of the NF leading to the partial collapse of the pore structure and the over-growth of nanoparticles (ESI Fig. S2i†) is likely to be the key factor that hindered mass and electron transport indisputably in the Fe-24h/NF electrode, thus decreasing its OER activity. As evidence to support the enhanced mass transfer of the Fe-18h/NF electrode, chronopotentiometric curves for the closely competing Fe-18h/NF- and Fe-24h/NF-based OER electrodes were obtained and the impact of the gas bubble build-up on the surface of the electrode was evaluated at a current density of 350 mA cm−2. Fig. S5† presents the large fluctuation in the OER potential for the Fe-24h/NF electrode, which was caused by the slower growth and release of the O2 bubbles. This clearly indicates that the detachment of the O2 bubbles from the Fe-18h/NF electrode was much easier than that from the Fe-24h/NF electrode, highlighting the enhanced mass transfer of the Fe-18h/NF electrode. Regarding the Fe-12h/NF and the Fe-6h/NF electrodes, they had a similar overpotential and Tafel slope in the kinetically controlled OER regime, while the Fe-12h/NF electrode exhibited greater OER performance in the higher bias regime.
The significant enhancement of the OER activity of the etched NF depending on the pore density and uniformity of the catalyst loading in the high potential bias regime was apparent at an overpotential (η) of 290, 320, and 330 mV (Fig. 3a and d), with the Fe-18h/NF electrode, which had the highest pore density and a uniform catalyst loading (ESI Fig. S2†), exhibiting the highest OER current density of 692.09 mA cm−2. This is because higher pore density and ordered structure shortens the dwell time of gas-bubbles in the internal space of catalyst layer. Moreover, the larger 20 μm pores on the 3D-nework Ni-backbones could facilitate for the rapid dissipation of the bubbles, thereby overcoming the problem of severe coalescence of bubbles often encountered in the case of smaller sized pores.26 In contrast to the Fe-18h/NF electrode, the NF electrodes etched for 6, 12, and 24 h had lesser number of pore density and lesser amount of catalyst particle loading for the first two samples while the agglomerated catalyst particles were obtained for the NF electrodes etched for 24 h. In addition, these samples also showed smaller electrochemically active surface area (ECSA) in Fig. S6,† indicating the lesser concentration of catalytically active sites.45 Consequently, a significantly lower OER current density at the same η of 330 mV (337.08, 396.27, and 521.33 mA cm−2, respectively) was exhibited by these 3 samples. Thus, based on the pore-density (Fig. S2†) and ECSA (Fig. S6†), the NF electrodes etched for 6, 12, 18, and 24 h exhibited the OER performance in the following order: Fe-18h/NF > Fe-24h/NF > Fe-12h/NF > Fe-6h/NF (Fig. 3).
The geometrical area-based current density (Fig. 3) is related to the ECSA of the electrodes. Hence, to investigate the influence of electrode morphology on the intrinsic catalytic activity, we attempted to eliminate the influence of the ECSA by normalizing the geometrical-area-based current density to the ECSA of each sample. To achieve this, the double-layer capacitance (Cdl) was first determined from a cyclic voltammogram cascade measured in a non-faradaic potential window (Fig. S6a–e†). The ECSA was then determined according to ECSA = Cdl/Cs, where Cs is the specific capacitance of the electrode (Fig. S6g†).45 The resulting ECSA-independent current density (jECSA) at a given overpotential is presented in Fig. S6f and S7.† Based on jECSA, it is evident that the electrode with the most uniform pore and catalyst nanoparticle distribution on the Ni backbones had the highest catalytic activity, and vice versa.
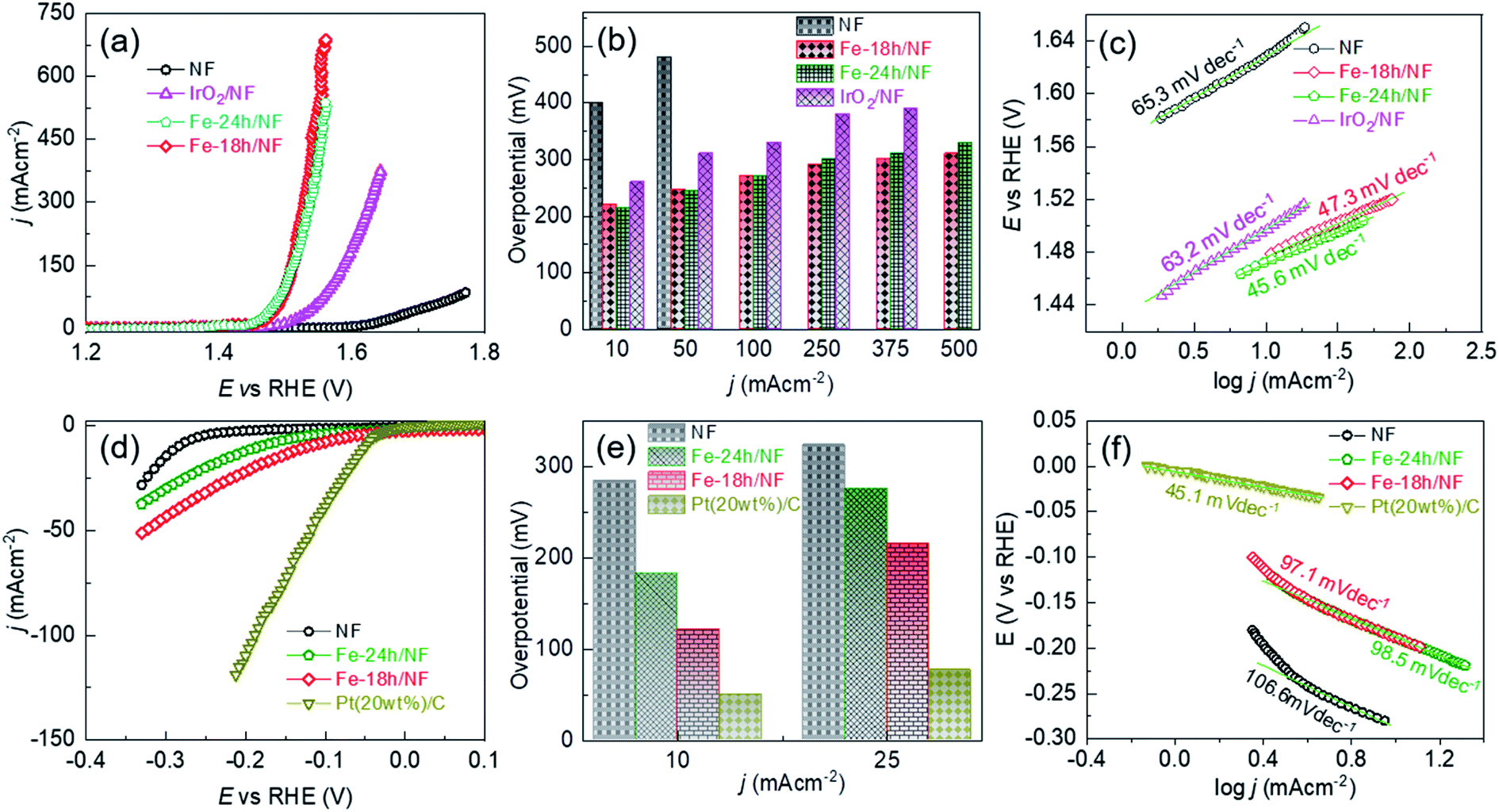 | ||
| Fig. 4 Comparison of the electrocatalytic performance of the etched NFs and a benchmark catalyst in a 1 M KOH solution: (a) iR compensated anodic LSVs for the pristine NF, etched NF, and benchmark IrO2/NF electrodes, (b) overpotential profiles with respect to the OER current density, and (c) corresponding Tafel slopes extracted from the LSVs. (d) iR compensated cathodic LSVs of the pristine NF, etched NFs, and benchmark Pt(20wt%)-C/NF electrodes, (e) overpotential profiles with respect to the HER current density, and (f) corresponding Tafel slopes extracted from the LSVs. | ||
The OER performance of the Fe-18h/NF electrode was also remarkably superior to that of a state-of-the-art IrO2/NF reference electrode, which had a higher η value of 260, 310, 330, 380, and 390 mV for OER current densities of 10, 50, 100, 250, and 375 mA cm−2, respectively. The OER performance of the Fe-18h/NF electrode was also highly competitive with those reported for benchmarked Ni-, Fe-, and Ni–Fe-based OER electrocatalysts (details are provided in ESI Table S1†). Most importantly, in contrast to the stringent electrochemical anodization process for obtaining porous Ni-backbone on NFs,32,33 the simple chemical etching process in the present study not only produced a uniform distribution of micropores on the Ni backbones, but led to an outstanding OER performance, achieving a current density of 500 mA cm−2 at η = 310 mV, thus out-performing electrochemically etched NF-based electrodes. In particular, electrochemically etched NF electrodes reported an OER current density of 500 mA cm−2 at higher η of 334 (ref. 32) and 350 mV.33 The OER performance of the Fe-18h/NF electrode was also even better than that of a porous Ni electrode (10 mA cm−2 @ η = 300 mV) obtained via phase inversion,46 and a spiky Ni electrode (100 mA cm−2 @ η = 300 mV in a 25 wt% KOH electrolyte) obtained via femtosecond laser structuring.47 The obtained superior OER performance of the etched NF electrode of this work over the already reported nanostructured Ni electrodes can be attributed to the synergistic interplay among the uniformly distributed metallic Fe@Ni–Fe-hydroxide nanocrystals, lattice defects and gas/electrolyte diffusible microporous Ni-backbones.
ESI Fig. S8† shows that the Fe-18h/NF electrode had the highest ECSA (719.09 cm−2) followed by Fe-24h/NF (695.90 cm−2), IrO2/NF (402.27 cm−2), and pristine NF (387.73 cm−2). Thus, in addition to having a uniform distribution of micropores and catalyst nanoparticles on the Ni backbones, the superior OER performance of the etched NF electrodes can also be attributed to the abundantly available electrochemical active sites. ESI Fig. S9† presents Nyquist plots and corresponding fitted parameters extracted from electrochemical impedance spectra measured at a bias of 1.5 V vs. RHE in a 1 M KOH electrolyte. The Fe-18h/NF and the Fe-24h/NF electrodes exhibited a significantly lower charge transfer resistance than did the IrO2/NF and pristine NF electrodes, leading to the faster charge transfer between the electrochemically active catalytic sites and the electrolyte during the OER. The accelerated faradaic response and the consequently superior OER kinetics for the etched NF electrodes can be attributed to the synergic effect between the metallic Fe and the hydroxides in establishing a good electrical contact across the interface between the parent hydroxides ((Fe/Ni)–OH) and the corresponding oxyhydroxides ((Fe/Ni)–OOH) formed during the application of the anodic bias, thus decreasing the Schottky barriers.48–50
To provide further insight into the electrode kinetics during the OER process, the Tafel slopes derived from the corresponding anodic polarization curves (Fig. 4a) were estimated. As shown in Fig. 4c, both etched NF electrodes exhibited similar Tafel slopes (47.3 and 45.6 mV dec−1). However, these slopes were much lower than those of the pristine NF (65.3 mV dec−1) and the IrO2/NF (63.2 mV dec−1) electrodes. These exceptionally low Tafel slopes are probably due to the coexistence of metallic Fe atoms and Ni–Fe-hydroxide nanocrystalline-clusters, which promotes faster and highly efficient OER kinetics. The HER performance of the electrodes was also assessed, as shown in Fig. 4d–f. The state-of-the-art Pt(20wt%)-C/NF-based HER electrode produced the best performance with a low HER overpotential and a low Tafel slope. Nevertheless, the Fe-18h/NF electrode also exhibited a significantly enhanced HER performance in comparison to the Fe-24h/NF and pristine NF electrodes, with a lower HER overpotential and Tafel slope.
 | ||
| Fig. 5 Electrochemical reversibility and stability in a 1 M KOH solution: (a) chronopotentiometric response when periodically polarizing the Fe-18h/NF electrode between +400 and −40 mA cm−2 for 80 h, showing perfect electrochemical reversibility between the OER and HER. (b) Cell voltages (without iR compensation) between two identical Fe-18h/NF electrodes at a stress bias of 10, 50, 100, 200, or 240 mA cm−2 for 120 h, showing high long-term electrochemical stability. Without iR compensated LSVs of the cell before and after the 120 h stability test: (c) anodic LSVs for the Fe-18h/NF anode with respect to the Fe-18h/NF cathode, and (d) cathodic LSVs for the Fe-18h/NF cathode with respect to the Fe-18h/NF anode. Inset image in (d) shows the cell voltage profile with respect to the current density, extracted from the LSVs of the anode and cathode after the stability test shown in (c) and (d). | ||
The electrochemical durability of the Fe-18h/NF electrode was investigated further under a stress bias of 10, 50, 100, 200, and 240 mA cm−2 between two Fe-18h/NF electrodes working as the cathode and anode of an electrolyzer for 24 h (total 120 h) in a 1 M KOH aqueous electrolyte. The obtained chronopotentiometric traces revealed no apparent change in the cell voltage (Vcell) for each bias, representing a high electrochemical durability for up to 120 h (Fig. 5b). This finding was supported by the similar anodic and cathodic LSVs for the anode and cathode obtained before and after the 120 h durability test (Fig. 5c and d). The slightly higher current density for the electrolyzer after the durability test could be due to the increase in the number of active sites during the long-term in situ activation of the electrodes in the stability test. Note that the cell voltage (Vcell) is the potential difference measured between two electrodes of the electrolyzer, which has real physical meaning in practical applications. In this case, iR losses are usually not accounted for. Meanwhile, the working electrode potential, E (V vs. RHE) in a half-cell configuration was compensated for iR loss to precisely determine the overpotential (i.e., the extra potential required by the electrode over the thermodynamic potential) for the OER and HER using the following relations: ηOER = ERHE − 1.23 V for OER and ηHER = ERHE − 0.0 V for HER, where ERHE is the electrode potential measured in the scale of RHE, and 1.23 V and 0.0 V are the thermodynamic potentials for OER and HER, respectively. Consequently, some small overpotential differences can be observed in between E (V vs. RHE) and Vcell in Fig. 4a, d and 5c, d.
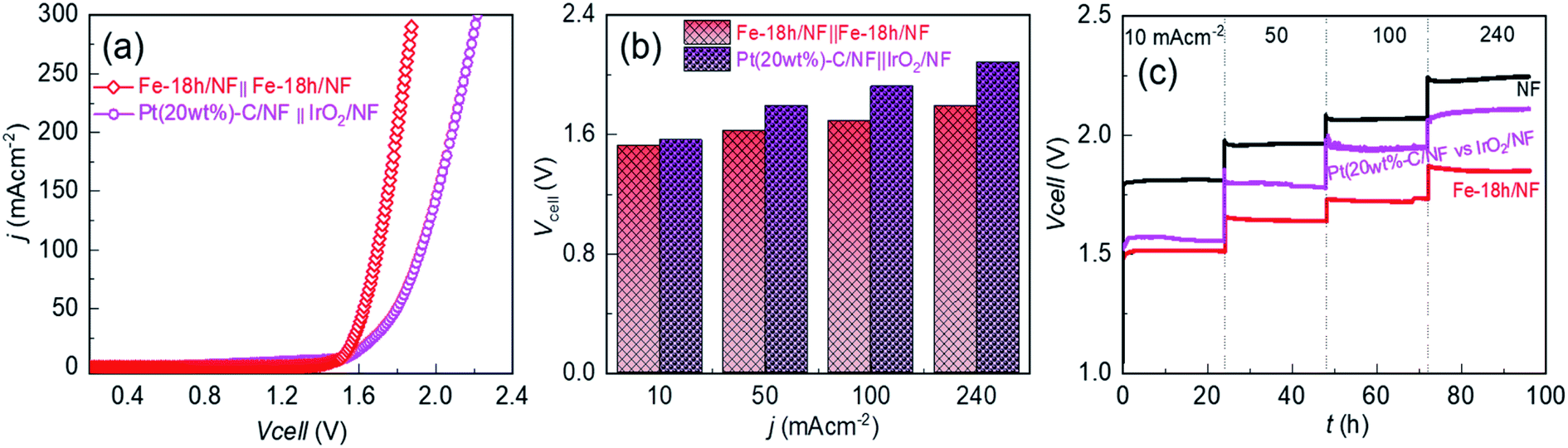 | ||
| Fig. 6 Electrochemical activity and durability in an industrial-type 30 wt% KOH solution: (a) without iR compensated anodic LSV for the Fe-18h/NF anode with respect to the Fe-18h/NF cathode, and anodic LSV for the IrO2/NF anode with respect to the Pt(20wt%-C)/NF cathode. (b) Cell voltage profiles with respect to the current density, extracted from the corresponding LSVs. (c) Cell voltages (without iR compensation) between two identical electrodes at a stress bias of 10, 50, 100, or 240 mA cm−2 for 96 h, showing the outstanding long-term electrochemical durability of the Fe-18h/NF electrode. | ||
To provide further insight into the material durability of the electrode after long-term electrochemical stability testing for 96 h, the Fe-18h/NF anode was examined using XPS, XRD, SEM, and STEM. Compared to Fig. 1d, e, ESI Fig. S12† shows that the 2p peaks of the Ni 2p and Fe 2p XPS spectra of the electrode after the stability test shifted slightly toward a higher binding energy by 0.18 and 0.20 eV, respectively. This could be attributed to the oxidation of the hydroxides of the electrode into the corresponding (Fe–Ni)–OOH phase. This interpretation was supported by the Raman shifts (Fig. S13†), which clearly showed a pair of peaks corresponding to the M–OOH phase (M = Ni, Fe).54,55 In addition, the area under the Fe0 peak decreased while that of the O 1s peaks increased, supporting the likely oxidation of M–OH into M–OOH. The XRD analysis, however, did not reveal peaks from the M–OOH phase. This could be due to the very thin film of M–OOH that formed on the surface of the hydroxide particles or due to the formation of an amorphous M–OOH phase. On the other hand, the XRD patterns indicated the presence of the initially existing prominent diffraction peaks for the (003) and (101) planes from α-Ni(OH)2 even after the durability test (ESI Fig. S14a†). In addition, an additional diffraction peak corresponding to the (006) plane of α-Ni(OH)2 also appeared after the durability test. This crystallographic planes could be equally active for water-splitting as demonstrated by the initially existing (003) and (101) planes of the α-Ni(OH)2. As such, in situ catalytic site enhancement can be realized when the electrodes are employed for long-term water electrolysis. This could be the reason for the slight improvement in the current density of the electrolyzer after the long-term durability test (Fig. 5c and d). Most importantly, the peaks after the durability test were sharper than the initial peaks (compare ESI Fig. S14a† and 1c), indicating the improved crystallization of the in situ grown α-Ni(OH)2 nanoparticles. This finding was also supported by an SEM surface view of the electrode, which showed an increase in the particle size from ∼10 nm to ∼20 nm (compare ESI Fig. S14b† and 1b). However, the initial spherical morphology of the nanoparticles remained intact without deformation. Similarly, HAADF images (ESI Fig. S15†) obtained from STEM revealed the uniform distribution of Ni, Fe, O and Cl within the electrode, as was observed before the durability test. These findings confirm the long-term electrochemical durability of the Fe-18h/NF electrode, demonstrating that it is a promising candidate as an efficient and sustainable anode material for use in industrial alkaline water-splitting applications.
3 Conclusions
This work presents a cost-effective design for the scalable production of an efficient and sustainable alkaline OER electrode. The key interest of this work lies on the morphological engineering strategy of 3D-NF via chemical etching for obtaining porous electrode to enhance the number of active sites while minimizing the detrimental electron and mass transport (i.e., inward diffusion of OH− and outward diffusion of oxygen bubbles) limitations. A readily scalable approach to the fabrication of microporous Ni backbones decorated with metallic Fe composited Ni- and Fe-hydroxide nanoparticles on NF was demonstrated using the immersion-based chemical etching of NF in an ethanolic FeCl3 solution at 50 °C. The pores and nanoparticles grew progressively and uniformly with an increase in the etching time. Compared to advanced non-noble metal-based water-splitting catalysts, including electrochemically etched NF and benchmark Pt(20wt%)-C/IrO2-based HER/OER electrodes, the optimally etched NF demonstrated an outstanding catalytic performance, with a low OER overpotential and cell voltage producing a high current density and strong long-term electrochemical durability (∼100 h) in both 1 M KOH and industrial-type 30 wt% KOH aqueous electrolytes. The cooperative interplay between the uniformly distributed micropores and metallic Fe doped hydroxide nanocatalysts with the localized amorphization in the crystal lattice facilitating mass and charge transportation is likely to be the key for promoting the access of the electrolyte to all available catalytic sites and leading to the release of O2 bubbles from these sites, thus demonstrating an outstanding electrocatalytic performance for potential water-splitting applications. In addition, the chemically etched NF-electrode showed a high robustness upon power interruptions, which might be promising for intermittent energy resources powered water-electrolysis.4 Experimental section
4.1 Reagents and materials
Iron(III) chloride (FeCl3, ≥99.99%), potassium hydroxide (≥85%), hydrochloric acid (37%), hydrochloric acid (37%), ethanol (≥99.5%), and acetone (≥99.9%) were purchased from Sigma-Aldrich. Nickel foams (NF) with a thickness of 1.6 mm and a cell size of 450 μm was purchased from Alantum Corporation (South Korea). As the substrate, the NF was cut into 1 × 5 cm2 pieces and washed sequentially for 10 minutes in 1 M HCl, deionized water, ethanol, and acetone with an ultrasonicator. Finally, the washed NF was dried at room temperature for 24 h.4.2 Chemical etching of the NF
A freshly prepared ethanolic solution of FeCl3 (200 μM mL−1) was stored overnight for settlement of insoluble excess FeCl3. The clear solution was pipetted into a 15 mL glass reagent bottle and the washed NF substrates (1 × 5 cm2) were completely immersed in the solution for etching times of 6, 12, 18, or 24 h at 50 °C with the lid of the reagent bottles closed. After the completion of etching, the immersed NF was washed thoroughly with ethanol. Finally, the sample was dried under a stream of N2 gas.4.3 Characterization of the etched NF
The morphology of the etched NF samples was investigated using a field-emission scanning electron microscope (FE-SEM, Hitachi S-4800) and a scanning transmission electron microscope (STEM, Talos™ F200X). The elemental composition was determined using an energy dispersive X-ray (EDX) spectrometer (Oxford 6587). The distribution of the elements was analyzed by generating elemental maps using the STEM. X-ray photoelectron spectroscopy (XPS) was conducted to determine the binding states of the elements present in the samples using an ESCALAB 250Xi spectrometer (ThermoFisher) and monochromatized Al Kα X-rays for excitation. X-ray diffraction (XRD) analysis was conducted using an X'Pert diffractometer (Malvern PANalytical) equipped with a Cu Kα radiation source (λ = 0.15418 nm) in a range from (2θ) 5° to 60° with a scanning step size of 0.01°.4.4 Electrochemical measurements
Electrochemical experiments were performed in a three-electrode system configured with an NF-based electrode that served as the working electrode. A graphite rod and a saturated calomel electrode (SCE) served as the counter and reference electrodes, respectively. All voltammetry experiments were conducted using a VersaSTA-3 electrochemical workstation (Princeton Applied Research), and the recorded potential was converted to the reversible hydrogen electrode (RHE) scale according to eqn (1). | (1) |
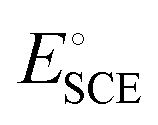 is the standard potential of an SCE electrode (0.244 V), and pH is the measured pH of the aqueous 1 M KOH electrolyte solution. Prior to measurement, the electrodes were electrochemically activated via cyclic voltammetry at a scan rate of 100 mV s−1 until stable voltammograms were obtained. To evaluate the OER and HER overpotential, linear sweep voltammograms (LSVs) were produced at a scan rate of 1 mV s−1. Electrochemical impedance spectroscopy (EIS) was conducted using the VersaSTA-3 electrochemical workstation. Unless otherwise stated, all LSVs were reported with iR drop compensation. The electrochemical stability of the electrode was measured chronopotentiometrically using a BioLogic Science Instruments electrochemical workstation, which can measure a maximum current of 240 mA.
is the standard potential of an SCE electrode (0.244 V), and pH is the measured pH of the aqueous 1 M KOH electrolyte solution. Prior to measurement, the electrodes were electrochemically activated via cyclic voltammetry at a scan rate of 100 mV s−1 until stable voltammograms were obtained. To evaluate the OER and HER overpotential, linear sweep voltammograms (LSVs) were produced at a scan rate of 1 mV s−1. Electrochemical impedance spectroscopy (EIS) was conducted using the VersaSTA-3 electrochemical workstation. Unless otherwise stated, all LSVs were reported with iR drop compensation. The electrochemical stability of the electrode was measured chronopotentiometrically using a BioLogic Science Instruments electrochemical workstation, which can measure a maximum current of 240 mA.
Author contributions
Nabeen K. Shrestha: conceptualization, investigation, experimental analysis, methodology, and writing original draft. Supriya A. Patil: methodology, experimental analysis, data curation, and writing. Jonghoon Han: methodology, and experimental analysis. Sangeun Cho: methodology, and writing & editing. Akbar I. Inamdar: experimental analysis, writing and revisions. Hyungsang Kim: conceptualization, and funding. Hyunsik Im: conceptualization, funding, and review & editing.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
The authors acknowledge the financial support from the National Research Foundation (NRF) of Korea (grant no. 2018R1D1A1B07049046, 2021R1A2B5B01001796, and 2016R1A6A1A03012877).References
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463 RSC
.
- E. S. Hanley, J. P. Deane and B. P. Ó. Gallachóir, Renewable Sustainable Energy Rev., 2018, 82, 3027 CrossRef
- M. Noussan, P. P. Raimondi, R. Scita and M. Hafner, Sustainability, 2021, 13, 298 CrossRef CAS
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 146 Search PubMed
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571 CrossRef CAS PubMed
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148 RSC
- L. Gao, C. Tang, J. Liu, L. He, H. Wang, Z. Ke, W. Li, C. Jiang, D. He, L. Cheng and X. Xiao, Energy & Environmental Materials, 2021, 4, 392 Search PubMed
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060 RSC
- F. Lyu, Q. Wang, S. M. Choi and Y. Yin, Small, 2019, 15, 1804201 CrossRef
- Y. Chen, K. Rui, J. Zhu, S. X. Dou and W. Sun, Chem.–Eur. J., 2019, 25, 703 CrossRef CAS PubMed
- A. T. A. Ahmed, S. M. Pawar, A. I. Inamdar, H. Im and H. Kim, Int. J. Energy Res., 2020, 44, 1798 CrossRef CAS
- D. V. Shinde, S. A. Patil, K. Cho, D. Y. Ahn, N. K. Shrestha, R. S. Mane, J. K. Lee and S. H. Han, Adv. Funct. Mater., 2015, 25, 5739 CrossRef CAS
- H. T. Bui, D. Y. Ahn, N. K. Shrestha, M. M. Sung, J. K. Lee and S. H. Han, J. Mater. Chem. A, 2016, 4, 9781 RSC
- S. A. Patil, S. Cho, Y. Jo, N. K. Shrestha, H. Kim and H. Im, Chem. Eng. J., 2021, 426, 130773 CrossRef CAS
- D. S. Raja, C.-L. Huang, Y.-A. Chen, Y. Choi and S.-Y. Lu, Appl. Catal., B, 2020, 279, 119375 CrossRef
- N. K. Shrestha, S. A. Patil, S. Cho, Y. Jo, H. Kim and H. Im, J. Mater. Chem. A, 2020, 8, 24408 RSC
- A. I. Inamdar, H. S. Chavan, B. Hou, C. H. Lee, S. U. Lee, S. N. Cha, H. Kim and H. Im, Small, 2020, 16, 1905884 CrossRef CAS
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. de Araujo, M. Gliech, D. Teschner, J. Zhu, W. X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed
- A. I. Inamdar, H.
S. Chavan, S. M. Pawar, H. Kim and H. Im, Int. J. Energy Res., 2020, 44, 1789 CrossRef CAS
- N. Yu, W. Cao, M. Huttula, Y. Kayser, P. Hoenicke, B. Beckhof, F. Lai, R. Dong, H. Sun and B. Geng, Appl. Catal., B, 2020, 261, 118193 CrossRef CAS
- X.-P. Li, L.-R. Zheng, S.-J. Liu, T. Ouyang, S. Ye and Z.-Q. Liu, Chin. Chem. Lett., 2022 DOI:10.1016/j.cclet.2021.12.095
- J. Chen, Q. Long, K. Xiao, T. Ouyang, N. Li, S. Ye and Z.-Q. Liu, Sci. Bull., 2021, 66, 1063 CrossRef CAS PubMed
- J.-Y. Wang, W.-T. Liu, X.-P. Li, T. Ouyang and Z.-Q. Liu, Chem. Commun., 2020, 56, 1489 RSC
- C. Huang, Y. Zou, Y.-Q. Ye, T. Ouyang, K. Xiao and Z.-Q. Liu, Chem. Commun., 2019, 55, 7687 RSC
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593 CrossRef CAS PubMed
- Y. J. Kim, A. Lim, J. M. Kim, D. Lim, K. H. Chae, E. N. Cho, H. J. Han, K. U. Jeon, M. Kim, G. H. Lee, G. R. Lee, H. S. Ahn, H. S. Park, H. Kim, J. Y. Kim and Y. S. Jung, Nat. Commun., 2020, 11, 4921 CrossRef CAS
- Y. Jo, S. Cho, J. Seo, A. T. A. Ahmed, C. H. Lee, J. H. Seok, B. Hou, S. A. Patil, Y. Park, N. K. Shrestha, S. U. Lee, H. Kim and H. Im, ACS Appl. Mater. Interfaces, 2021, 13, 53725 CrossRef CAS
- A. T. A. Ahmed, A. S. Ansari, S. M. Pawar, B. Shong, H. Kim and H. Im, Appl. Surf. Sci., 2021, 539, 148229 CrossRef CAS
- T. Kou, S. Wang, R. Shi, T. Zhang, S. Chiovoloni, J. Q. Lu, W. Chen, M. A. Worsley, B. C. Wood, S. E. Baker, E. B. Duoss, R. Wu, C. Zhu and Y. Li, Adv. Energy Mater., 2020, 10, 2002955 CrossRef CAS
- Z. Zhang, B. He, L. Chen, H. Wang, R. Wang, L. Zhao and Y. Gong, ACS Appl. Mater. Interfaces, 2018, 10, 38032 CrossRef CAS
- Y. Zhou, Y. Yang, X. Zhu, D. Ding Ye, R. Chen and Q. Liao, J. Power Sources, 2021, 507, 230260 CrossRef CAS
- X. F. Chuah, C. T. Hsieh, C. L. Huang, D. Senthil Raja, H. W. Lin and S. Y. Lu, ACS Appl. Energy Mater., 2019, 2, 743 CrossRef CAS
- Y. J. Son, K. Kawashima, B. R. Wygant, C. H. Lam, J. N. Burrow, H. Celio, A. Dolocan, J. G. Ekerdt and C. B. Mullinsn, ACS Nano, 2021, 15, 3468 CrossRef CAS PubMed
- J. Yu, S. Pan, Y. Zhang, Q. Liu and B. Li, Front. Mater., 2019, 6, 124 CrossRef
- M. Aghazadeh, M. Ghaemi, B. Sabour and S. Dalvand, J. Solid State Electrochem., 2014, 18, 1569 CrossRef CAS
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef
- X. Chen, C. Long, C. Lin, T. Wei, J. Yan, L. Jiang and Z. Fan, Electrochim. Acta, 2014, 137, 352 CrossRef CAS
- Y. Wei, C. H. Shin, E. B. Tetteh, B. J. Lee and J. S. Yu, ACS Appl. Energy Mater., 2020, 3, 822 CrossRef CAS
- Y. J. Ye, N. Zhang and X. X. Liu, J. Mater. Chem. A, 2017, 5, 24208 RSC
- M. G. Rosmaninho, F. C. C. Moura, L. R. Souza, R. K. Nogueira, G. M. Gomes, J. S. Nascimento, M. C. Pereira, J. D. Fabris, J. D. Ardisson, M. S. Nazzarro, K. Sapag, M. H. Araújo and R. M. Lago, Appl. Catal., B, 2012, 115–116, 45 CrossRef CAS
- S. Anantharaj and S. Noda, Small, 2020, 16, 1905779 CrossRef CAS PubMed
- D. Zhang, J. Z. Soo, H. H. Tan, C. Jagadish, K. Catchpole and S. K. Karuturi, Adv. Energy Mater., 2020, 2, 2000071 Search PubMed
- L. Liu, F. Tian, X. Wang, Z. Yang and X. Wang, Ionics, 2013, 19, 9 CrossRef CAS
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564 CrossRef CAS
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed
- R. Ding, S. Cui, J. Lin, Z. Sun, P. Du and C. Chen, Catal. Sci. Technol., 2017, 7, 3056 RSC
- F. Rieck Genannt Best, J. Koch, G. Lilienkamp, F. Körkemeyer, H. J. Maier, J. Caro and K. Lange, Int. J. Electrochem., 2018, 12, 1–12 Search PubMed
- H. Xu, Z. X. Shi, Y. X. Tong and G. R. Li, Adv. Mater., 2018, 30, 1705442 CrossRef
- A. Aijaz, J. Masa, C. Rösler, W. Xia, P. Weide, A. J. R. Botz, R. A. Fischer, W. Schuhmann and M. Muhler, Angew. Chem., Int. Ed., 2016, 55, 4087 CrossRef CAS
- S. Niu, Y. Sun, G. Sun, D. Rakov, Y. Li, Y. Ma, J. Chu and P. Xu, ACS Appl. Energy Mater., 2019, 2, 3927 CrossRef CAS
- N. K. Shrestha, M. Yang and P. Schmuki, Electrochem. Solid-State Lett., 2010, 13, 6 CrossRef
- J. Divisek, J. Mergel and H. Schmitz, Int. J. Hydrogen Energy, 1990, 15, 105 CrossRef CAS
- B. C. M. Martindale and E. Reisner, Adv. Energy Mater., 2016, 6, 1502095 CrossRef
- L. Bai, S. Lee and X. Hu, Angew. Chem., 2021, 60, 3095 CrossRef CAS PubMed
- H. S. Chavan, C. H. Lee, A. I. Inamdar, J. Han, S. Park, S. Cho, N. K. Shreshta, S. U. Lee, B. Hou, H. Im and H. Kim, ACS Catal., 2022, 12, 3821 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta10103j |
| This journal is © The Royal Society of Chemistry 2022 |