
A high-energy conversion-type cathode activated by amorpholization for Li rechargeable batteries†
Yongseok
Lee
ab,
Jungmin
Kang
ab,
Jinho
Ahn
ab,
Wonseok
Ko
ab,
Hyunyoung
Park
ab,
Seokjin
Lee
ab,
Sangyeop
Lee
ab,
Jung-Keun
Yoo
 *cd and
Jongsoon
Kim
*ab
*cd and
Jongsoon
Kim
*ab
aDepartment of Energy Science, Sungkyunkwan University, Suwon, 16419, Republic of Korea. E-mail: jongsoonkim@skku.edu
bSKKU Institute of Energy Science and Technology (SIEST), Sungkyunkwan University, Suwon 16419, Republic of Korea
cCarbon Composites Department, Composites Research Division, Korea Institute of Materials Science (KIMS), 797 Changwondaero, Changwon, Republic of Korea. E-mail: yoojk@kims.re.kr
dAdvanced Materials Engineering Division, University of Science and Technology (UST), Daejeon, 34113, Republic of Korea
First published on 29th April 2022
Abstract
Although conversion-type electrodes deliver larger theoretical capacities than intercalation-type electrodes, their application as practical cathodes for Li rechargeable batteries is hindered by their intrinsically sluggish kinetics and low operating voltage. In this study, we demonstrate that the conversion-type electrochemical behaviors of Cu(PO3)2 in a Li-cell system are highly enhanced by amorpholization and carbon-mixing. In particular, the presence of the (PO3)− polyanion in the structure enables a much higher operation voltage of Cu(PO3)2 relative to that of other conversion-type metal-oxide electrodes, resulting from the inductive effect by phosphorus with high electronegativity. As a result, the amorphorized Cu(PO3)2/C composite delivers not only a large reversible capacity of ∼240 mA h g−1 at 12 mA g−1 but also an average operation voltage of ∼2.8 V (vs. Li+/Li). Even at a high current density of 1200 mA g−1, up to ∼60% of the specific capacity at 12 mA g−1 is retained. Furthermore, the capacity retention after 300 cycles at 480 mA g−1 is ∼77% of the initial capacity. This outstanding power capability and cyclability of the amorphorized Cu(PO3)2/C composite differ markedly from the poor electrochemical properties of the well-crystallized Cu(PO3)2/C composites, indicating the enhanced kinetics of the conversion reaction in Cu(PO3)2 by amorpholization. In addition, the reversible conversion-reaction mechanism of Cu(PO3)2 in a Li-cell system is demonstrated through various experimental measurements.
 Jongsoon Kim | Jongsoon Kim is an assistant professor of the Department of Energy Science in Sungkyunkwan University. He obtained his BSc and MSc at the Korea Advanced Institute of Science and Technology (KAIST). Then, he completed his PhD (2014) at Seoul National University. His research laboratory focuses on development of new materials for Li-ion and Na-ion batteries using ab initio calculations and X-ray/neutron-based analyses. |
1. Introduction
Because of various environmental issues and their efficient energy storage, lithium rechargeable batteries (LRBs) with stable cycle performance and high energy densities have received great attention as promising energy storage systems (ESSs) worldwide.1–3 Today, the application of LRBs has been expanded from small electronic devices to large-scale ESSs such as electric vehicles (EVs).4–6 Because increasing the practical capacity and energy density is one of the most important issues for LRBs, there is an urgent need for the development of novel cathode materials with a high reversible capacity and average operation voltage. However, intercalation-based cathode materials only deliver limited reversible capacity not exceeding ∼220 mA h g−1, despite high redox potentials during charge/discharge.7,8 With the demands for the performance of batteries becoming increasingly stricter, a fundamental breakthrough in cathode material design is needed to upgrade LRBs to the next level.For improved Li storage in cathode materials, we focused on the application of the conversion reaction. While the storage of less than 1 mol Li+ per 1 mol [TM] cations is allowed in conventional intercalation-type electrodes, Li[TM]O2 (TM: transition metal),9,10 conversion-type electrodes can typically store 2 or 3 mol Li+ per 1 mol [TM].11–13 This finding indicates that conversion-type cathodes can achieve a much larger theoretical capacity than intercalation-type cathodes. However, the application of these materials as cathodes for LRBs requires a high average operation voltage during charge/discharge. Because the conversion reactions of general metal oxides typically occur in the low voltage range (<1 V vs. Li+/Li), conversion-type electrodes are considered suitable for anodes rather than cathodes.14–18 Moreover, the conversion reactions are accompanied by sluggish kinetics, leading to poor electrochemical behavior during charge/discharge.19–21 Thus, it is essential to address these problems for the practical application of conversion-type electrodes as cathodes for LRBs.
To increase the operation voltage of conversion-type electrodes, we attempted to maximize the inductive effect by applying polyanion-containing phosphorus with high electronegativity, inspired by the increased redox potential of Fe2+/Fe3+ in olivine LiFePO4.22 Furthermore, it has been reported that Cu-based compounds deliver a high redox potential because of the low negative standard formation enthalpy of the Cu element compared with those of other transition metals.23 Thus, we speculated that the conversion reaction of Cu(PO3)2 in the Li-cell system could result in a high operation voltage unlike those of simple metal oxides. In addition, to enhance the kinetics of the conversion reaction on Cu(PO3)2, we prepared an amorphorized Cu(PO3)2–carbon (A-CPO/C) composite. Amorphous materials composed of randomly linked clusters have been reported to possess the following attractive merits, enabling a facile conversion reaction in the Li-cell system compared with highly crystalline materials: (i) shortened diffusion paths,24 (ii) enhanced Li+ diffusion,25 and (iii) improved reaction activity.26,27 Moreover, the presence of conductive carbon in the composite provides not only high enhancement of the electrical conductivity but also suppression of morphological and structural changes.28
In this study, we demonstrated that the A-CPO/C composite based on the conversion reaction among Li, Cu, and (PO3) showed excellent electrochemical performance as a promising cathode for LRBs. At 12 mA g−1, the specific capacity of the A-CPO/C composite was ∼240 mA h g−1, corresponding to 2 mol Li storage per formula unit of Cu(PO3)2. In addition, the average operation voltage was close to ∼2.8 V (vs. Li+/Li), which is much higher than that of other conversion-type electrode materials.29,30 Even at a high current density of 1200 mA g−1, the A-CPO/C composite delivered a capacity of ∼146 mA h g−1, which is much larger than that of the low-crystalline Cu(PO3)2–carbon (LC-CPO/C) composite under the same conditions. Moreover, for 300 cycles at 480 mA g−1, the capacity retention of A-CPO/C and LC-CPO/C composites was ∼77% and ∼47%, respectively. These electrochemical data indicate that the conversion reaction of Cu(PO3)2 in the Li-cell system was highly enhanced by the synergetic effect of amorpholization and carbon coating. In addition, we confirmed that the reversible conversion-reaction mechanism Cu(PO3)2 + 2Li ↔ Cu + 2Li(PO3) occurred in the A-CPO/C composite during charge/discharge using various experimental techniques including ex situ X-ray diffraction (XRD), ex situ X-ray absorption near edge structure (XANES) analysis, ex situ extended X-ray absorption fine structure (EXAFS) analysis, X-ray photoelectron spectroscopy (XPS), and high-resolution transmission electron microscopy (HRTEM).
2. Experimental
2.1 Preparation of A-CPO/C
A-CPO/C (amorphous Cu(PO3)2/carbon composite) was prepared using CuO (Sigma Aldrich, 99%) and 2(NH4)2HPO4 (Sigma Aldrich, 98%) as precursors. Stoichiometric amounts of precursors were gathered in a Nalgene bottle with acetone and Zr balls and mixed by wet ball milling at 250 rpm for 12 h. The mixed powder in acetone was dried at 60 °C for 12 h with stirring on a hot plate, and the dried powder was pelletized and heated to 850 °C for 12 h in air. The obtained Cu(PO3)2 was placed in a silicon nitride planetary bowl (250 ml) with 30 nitride balls with a diameter of 10 π and primary ball-milled at 500 rpm for 12 h with conductive carbon (LC-CPO/C was obtained). Then the collected LC-CPO/C powders were placed in a silicon nitride planetary bowl (45 ml) with 10 nitride balls with a diameter of 3 π and secondary ball-milled at 500 rpm for 12 h (A-CPO/C was obtained). The ratios of A-CPO/C were 80 wt% Cu(PO3)2, 19 wt% Super P carbon black and 1 wt% carbon nanotubes (CNTs).2.2 Materials characterization
Bare Cu(PO3)2 and LC-CPO/C and A-CPO/C powders were characterized using XRD (PANalytical) with Cu Kα radiation (wavelength = 1.54178 Å). The 2θ range was 10–60° with a time per step of 0.13. The FullProf Rietveld program was used to analyze the XRD data. The morphology of the materials was examined using SEM (SU-8010). HR-TEM particle images and SAED patterns were obtained and EDS elemental mapping was performed using high-resolution transmission electron microscopy (HR-TEM; JEM-3010) at the National Center for Inter-university Research Facilities (NCIRF). The Cu(PO3)2 powder was analyzed using an inductively coupled plasma (ICP)-atomic emission spectrometer. The ac perturbation signal was ±5 mV and the frequency range was from 10−2 to 106 Hz. X-ray photon spectroscopy (XPS) characterization was performed using a K-alpha (Thermo Scientific Inc., U.K) model with Al Kα radiation (hν =1486.6 eV). All the XPS spectra were referenced to the C1s at a binding energy (BE) of 284.8 eV. The overall conversion reaction was revealed using XANES and EXASF spectroscopy that were performed at beamline 6D at the 3.0 GeV Pohang Light Source. The impedance of each sample was analyzed from 5 MHz to 500 mHz using a multichannel impedance analyzer system (VSP-300, Bio-Logic, Grenoble, France).2.3 Electrochemical characterization
The electrodes were fabricated from a slurry of 87.5 wt% active material (A-CPO/C or LC-CPO/C), 2.5 wt% Super P carbon, and 10 wt% polyvinylidene fluoride (PVDF) binder in N-methyl-2-pyrrolidone (NMP). The ratios of each material on electrodes were 70 wt% Cu(PO3)2 as the active material, 22 wt% conductive carbon, and 10 wt% PVDF binder. The slurry was applied on Al foil using a doctor blade and dried in an oven at 110 °C for 12 h. R2032 cells were assembled in an Ar-filled glove box using the A-CPO/C or LC-CPO/C composite electrode, a Li counter electrode, a separator (Celgard 2400), and 1.2 M LiPF6 in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC = 3:7(v/v) as the electrolyte. The electrochemical performances of the cells were evaluated by charging and discharging in the voltage range of 2.0–4.3 V at 30 °C using an automatic battery charge/discharge test system (WBCS 3000, WonATech). For the full-cell test, we used a lithiated graphite electrode as an anode. The graphite electrode was fabricated using the same procedure and ratio as the A-CPO/C electrode with the use of Cu foil. For preparation of lithiated graphite with minimization of irreversibility at the initial cycle, the graphite anode electrode was discharged to 0.001 V (vs. Li+/Li). And then, we fabricated the full-cell using the A-CPO/C electrode as the cathode and lithiated graphite as the anode. To minimize the irreversibility of graphite, the graphite electrode was pre-cycled by direct contact with Li metal in 1.2 M LiPF6 in EC:DMC = 3:7(v/v) electrolyte. Finally, R2032 coin-type full-cells were assembled with the A-CPO/C cathode and pre-cycled graphite carbon anode (capacity ratio of negative and positive electrodes of ∼1.2) in an Ar-filled glove box. The calculation of the specific capacity and energy density of the full-cell was based on the weight of the cathode material. For achieving full-cell balance, the capacity ratio of the anode (graphite) to the cathode (A-CPO/C) (N/P ratio) was retained at ∼1.2:1.
:DMC = 3:7(v/v) as the electrolyte. The electrochemical performances of the cells were evaluated by charging and discharging in the voltage range of 2.0–4.3 V at 30 °C using an automatic battery charge/discharge test system (WBCS 3000, WonATech). For the full-cell test, we used a lithiated graphite electrode as an anode. The graphite electrode was fabricated using the same procedure and ratio as the A-CPO/C electrode with the use of Cu foil. For preparation of lithiated graphite with minimization of irreversibility at the initial cycle, the graphite anode electrode was discharged to 0.001 V (vs. Li+/Li). And then, we fabricated the full-cell using the A-CPO/C electrode as the cathode and lithiated graphite as the anode. To minimize the irreversibility of graphite, the graphite electrode was pre-cycled by direct contact with Li metal in 1.2 M LiPF6 in EC:DMC = 3:7(v/v) electrolyte. Finally, R2032 coin-type full-cells were assembled with the A-CPO/C cathode and pre-cycled graphite carbon anode (capacity ratio of negative and positive electrodes of ∼1.2) in an Ar-filled glove box. The calculation of the specific capacity and energy density of the full-cell was based on the weight of the cathode material. For achieving full-cell balance, the capacity ratio of the anode (graphite) to the cathode (A-CPO/C) (N/P ratio) was retained at ∼1.2:1.
2.4 Computational details
DFT calculations31 were performed using the Vienna Ab Initio Simulation Package (VASP). We used projector-augmented wave (PAW) pseudopotentials32 with a plane-wave basis set, as implemented in VASP. Perdew–Burke–Ernzerhof (PBE) parametrization of the generalized gradient approximation (GGA)33,34 was used for the exchange-correlation functional. All the calculations were performed with an energy cutoff of 520 eV until the remaining force in the system converged to less than 0.02 eV Å−1 per unit cell. The parameters reported in the Materials Project database35 such as the U values and energy cutoff values, were used for the DFT calculations.3. Results and discussion
3.1 Material investigation for A-CPO/C
Fig. 1 shows the preparation process of the A-CPO/C composite and the differences among bare Cu(PO3)2, LC-CPO/C, and A-CPO/C composites. To achieve simultaneous amorpholization and carbon coating, we performed a high-energy ball-milling process. The amorpholization degree of the Cu(PO3)2 phase was controlled by adjusting the rotation speed of the ball-mill. The detailed preparation processes are explained in the Experimental section. The amorpholization of the Cu(PO3)2 phase provides inherent advantages for the conversion reaction in the Li-cell system, such as improved kinetics for facile Li+ diffusion and a large surface area and free volume.36 Moreover, the carbon-coating process is a well-known effective method to enhance electrical conductivity, enabling facile conversion chemistry of the electrode materials.37 Thus, we expected the A-CPO/C composite to deliver enhanced electrochemical performance as a novel conversion-type cathode for LRBs compared with that of bare Cu(PO3)2 and the LC-CPO/C composite.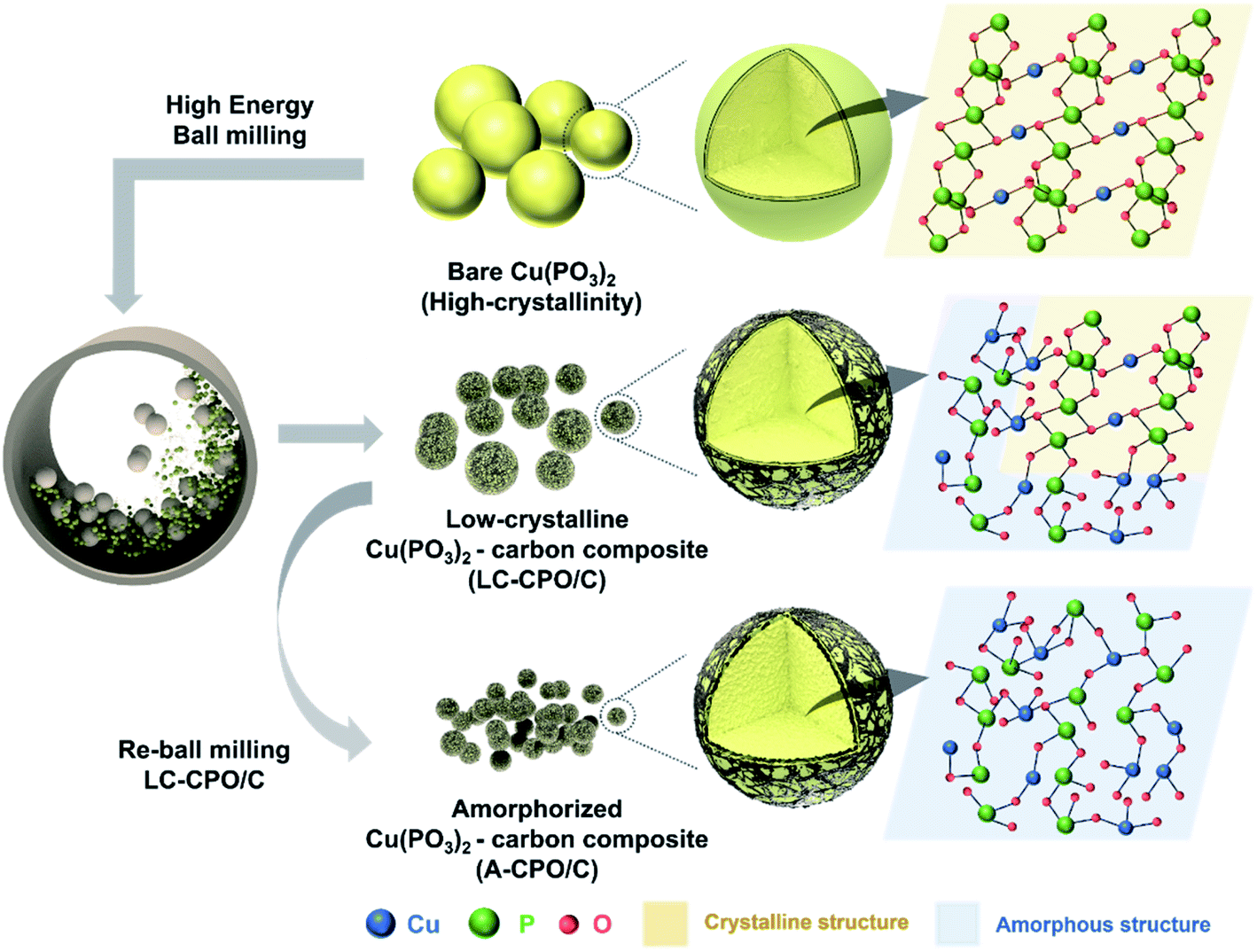 | ||
| Fig. 1 Scheme of the fabrication process from bare Cu(PO3)2 to LC-CPO/C and A-CPO/C with approximate material structure images. | ||
As shown in Fig. 2a, the XRD pattern of bare Cu(PO3)2 indicated the presence of high crystallinity and the C2/c space group. During the ball-milling process, a substantial amount of energy is applied on the particles, and thus, and the material undergoes disordering in crystallinity. The XRD pattern of A-CPO/C showed lower intensity than those of bare Cu(PO3)2 and LC-CPO/C, which is attributed to lower crystallinity of A-CPO/C than those of bare Cu(PO3)2 and LC-CPO/C. For the LC-CPO/C composite, the XRD peaks had lower intensities than those of bare Cu(PO3)2 and their shapes were broader, implying a lowered crystallinity of the LC-CPO/C composite compared with that of bare Cu(PO3)2. The reduced crystallinity of the Cu(PO3)2 phase was more prominent in the XRD pattern of the A-CPO/C composite. While the maximum XRD peak intensities of bare Cu(PO3)2 and LC-CPO/C were ∼21500 and 5600, respectively, that of A-CPO/C was only ∼1600. Moreover, only very broad and unobvious peaks appeared in the XRD pattern of A-CPO/C, indicating the formation of an amorphorized Cu(PO3)2 phase in the A-CPO/C composite. Transmission electron microscopy (TEM) analyses also revealed the difference in crystallinity between the LC-CPO/C and A-CPO/C composites (Fig. 2b and c). The selected area diffraction (SAED) pattern of the LC-CPO/C composite was composed of clear reflection spots representing the crystalline phase of Cu(PO3)2. However, there were no spots in the SAED pattern of the A-CPO/C composite, implying the presence of the amorphorized Cu(PO3)2 phase. The atomic ratio of Cu, P, and O elements in the A-CPO/C composite was investigated through elemental mappings using TEM-based energy dispersive X-ray spectroscopy (EDS). As shown in Fig. 2d, the Cu, P, and O elements were homogenously distributed in the A-CPO/C composite. In addition, the atomic ratio of Cu:P:O was confirmed to be 1:1.99:6.01, which is consistent with the inductively coupled plasma-atomic emission spectroscopy (ICP-OES) results (Table S1 (ESI†)). In addition, thermogravimetric analysis (TGA) revealed that the total carbon content in the A-CPO/C composite was ∼20 wt% (Fig. S1 (ESI†)). The overall morphologies of bare Cu(PO3)2, LC-CPO/C and A-CPO/C were compared through scanning electron microscopy (SEM) analysis (Fig. S2 (ESI†)), which indicates that A-CPO/C has a smaller average particle size than bare Cu(PO3)2 and LC-CPO/C. The average size of A-CPO/C (dA = 384.84 nm) was confirmed to be smaller than that of bare Cu(PO3)2 (dA = 3.59 μm) or LC-CPO/C (dA = 1.04 μm). The smaller particles have shorter Li+ diffusion pathways resulting in faster reaction and enlarged surface areas allowing a large contact area between the electrolyte and electrode. Therefore, the smaller particle size of A-CPO/C would help in improving its electrochemical performances compared to LC-CPO/C with a bigger particle size. Moreover, the additional ball-milling process can provide better mixing with carbon and active materials, which results in enhanced electrochemical behaviors of A-CPO/C.
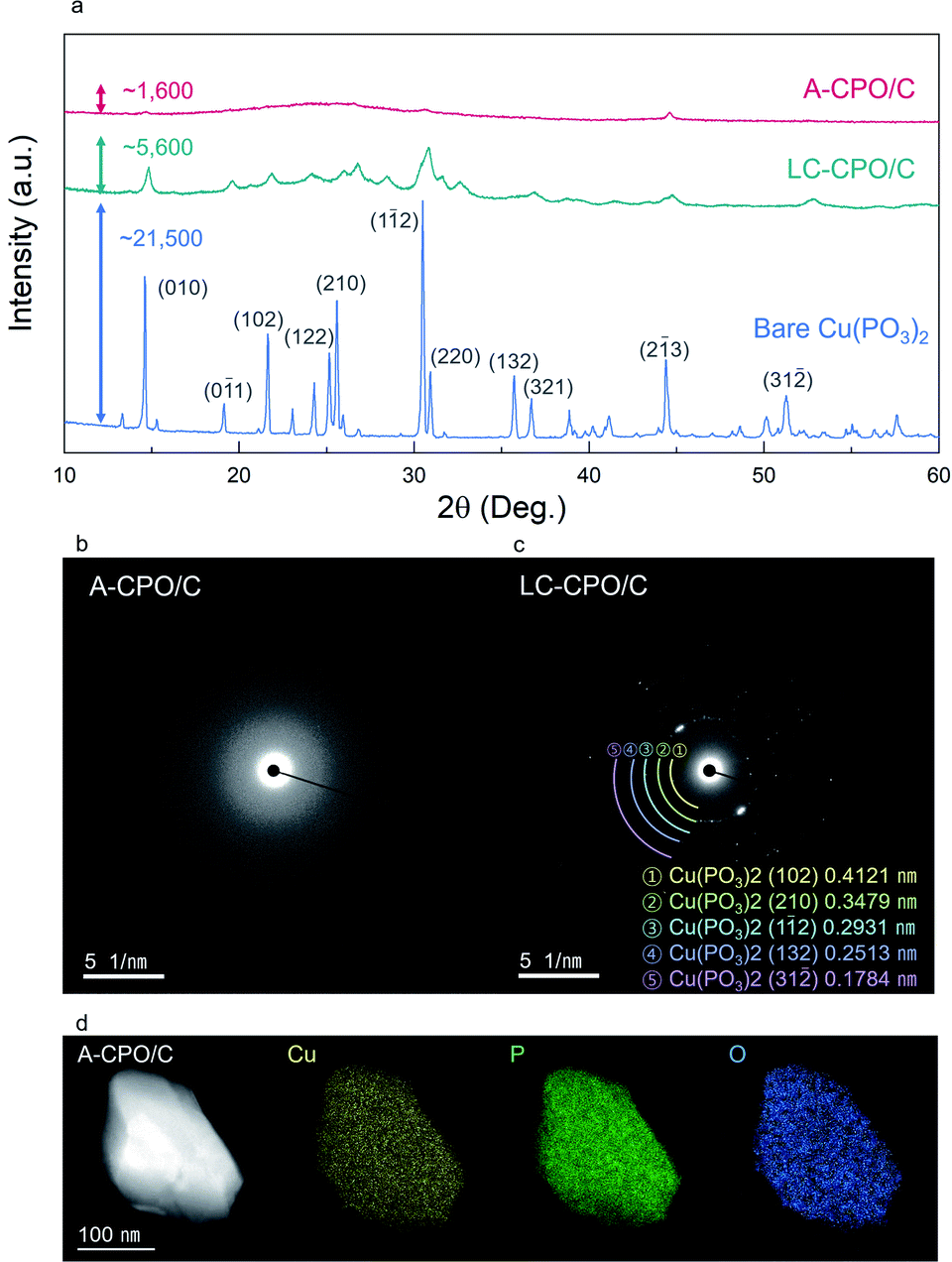 | ||
| Fig. 2 (a) Comparing XRD patterns and the maximum intensities of bare Cu(PO3)2, LC-CPO/C and A-CPO/C. TEM SAED patterns of (b) A-CPO/C, (c) LC-CPO/C, (d) EDS elemental mapping (Cu: yellow, P: green, O: blue) of A-CPO/C. | ||
Moreover, X-ray photoelectron spectroscopy (XPS) analysis was employed to confirm the valence states of the ions in the A-CPO/C composite, as the vague XRD patterns of A-CPO/C provided insufficient information for material identification. In the Cu 2p XPS spectra (Fig. 3b), a strong peak appeared at ∼934.2 eV, indicating the presence of Cu2+ ions. The satellite peak of Cu2+ was also detected at ∼943.6 eV.38,39 The O 1s XPS spectra showed two peaks at ∼531.1 and ∼532.9 eV, which are assigned to Cu–O bonds (Cu2+) and P–O bonds ((PO3)−), respectively (Fig. 3c).40,41 In terms of the P 2p XPS spectra (Fig. 3d), only one peak was observed at ∼134.6 eV, corresponding to the P–O bonds (PO3−).41 These XPS results indicate the successful preparation of the A-CPO/C composite.
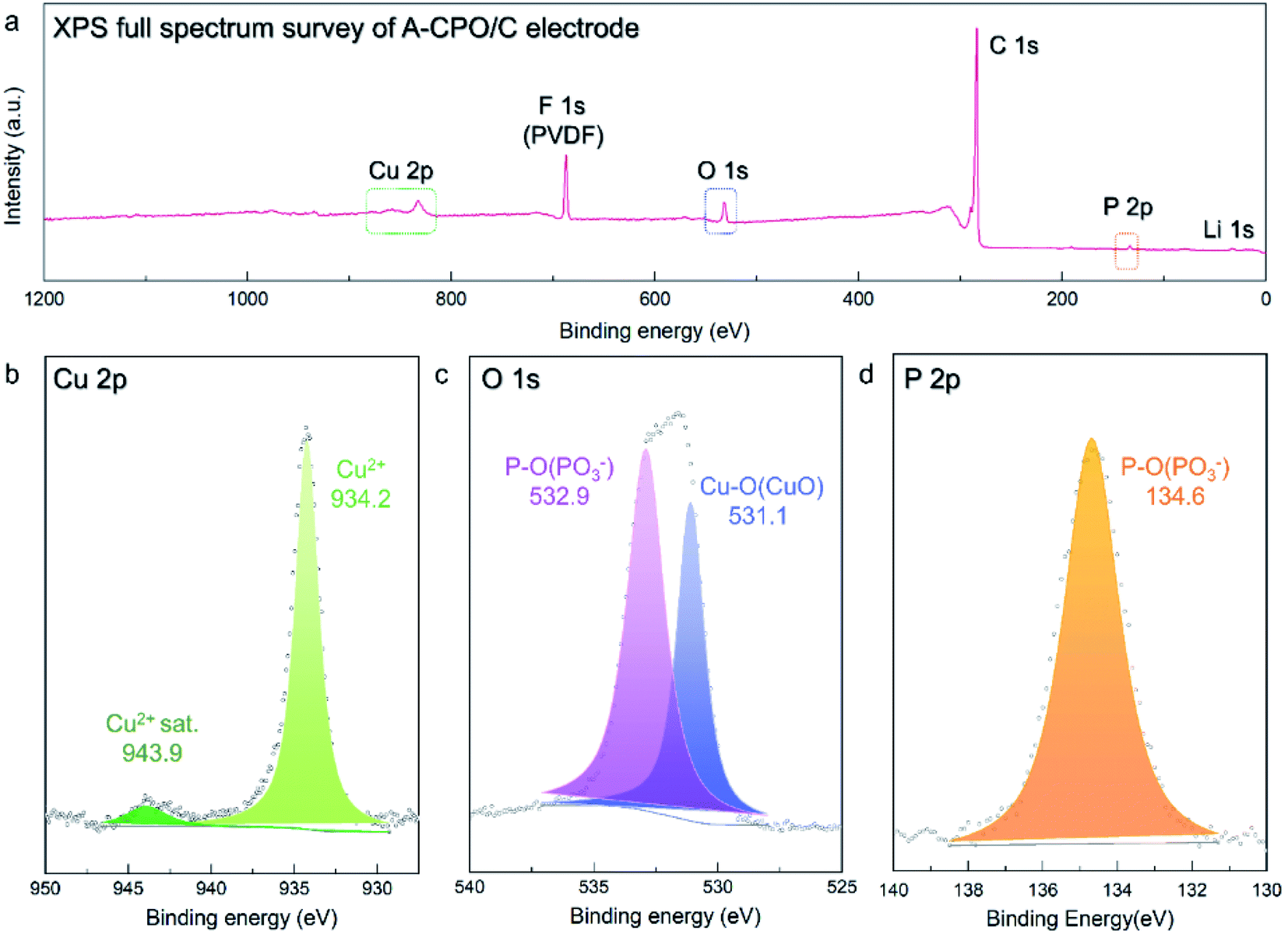 | ||
| Fig. 3 XPS spectra of A-CPO/C: (a) XPS full spectrum survey, (b) Cu 2p, (c) O 1s, and (d) P 2p. | ||
3.2 Electrochemical properties of A-CPO/C in an LRB system
In addition, to confirm the effect of the (PO3) polyanion on increasing the operation voltage for the conversion reaction, we performed first principles calculations. In the Li-cell system, Cu(PO3)2 and CuO undergo the following conversion reactions during the charge/discharge process:| Cu(PO3)2 + 2Li+ + 2e− ↔ Cu + 2LiPO3 | (i) |
| CuO + 2Li+ + 2e− ↔ Cu + Li2O. | (ii) |
The formation energies of Cu(PO3)2, CuO, Li2O and LiPO3 used for the calculation and those of theoretical redox potentials are tabulated in Table S2 (ESI†). The theoretical redox potentials of Cu(PO3)2 and CuO during each conversion reaction in the Li-cell system were confirmed to be ∼3.4 and ∼1.8 V (vs. Li+/Li), respectively. In the crystal structure of Cu(PO3)2, the oxygen (O) ions are connected with not only copper (Cu) ions but also a phosphorus (P) ion with high electronegativity, resulting in the weakened bond interaction between O and Cu ions. Thus, the inductive effect of the (PO3) polyanion enables a higher operation voltage of Cu(PO3)2 in the Li-cell system than that of CuO.
Fig. 4a–d show the electrochemical performance of the A-CPO/C and LC-CPO/C composites at various current densities in the Li-cell system in the voltage range of 2.0–4.3 V (vs. Li+/Li) after pre-cycling at 12 mA g−1. The charge/discharge curves of each composite upon pre-cycling are presented in Fig. S3 (ESI†). Compared to the LC-CPO/C electrode, the A-CPO–C electrode exhibits enhanced kinetics of the conversion reaction by amorpholization. Thus, the conversion reaction of Cu(PO3)2 + 2Li ↔ Cu + 2LiPO3 was more facile and occurred smoothly at A-CPO–C rather than at LC-CPO–C, which results in more reversible electrochemical performances of the A-CPO–C electrode during charge/discharge than those of the LC-CPO electrode. The discharge cut-off voltage for electrochemical tests of the A-CPO/C and LC-CPO/C electrodes is 2.0 V (vs. Li+/Li), which indicates that an irreversible discharge capacity by formation of an SEI layer did not affect the initial Coulombic efficiencies of the A-CPO/C and LC-CPO/C electrodes. The enhanced lithium diffusion kinetics by amorpholization enables a more reversible conversion reaction of the Cu(PO3)2 phase in the LIB system. Thus, we supposed that the A-CPO/C electrode can deliver a better initial Coulombic efficiency than the LC-CPO/C electrode. The A-CPO/C composite exhibited better power capability than the LC-CPO/C composite. At 12 mA g−1, A-CPO/C delivered a large specific capacity of ∼240 mA h g−1, corresponding to ∼2 mol Li+ storage per formula unit of Cu(PO3)2, which is connected with the following conversion reaction: Cu(PO3)2 + 2Li ↔ Cu + 2LiPO3. Moreover, we prepared another amorphorized Cu(PO3)2–carbon (A-CPO/C_1) composite through different conditions from the existing A-CPO/C composite. Fig. S4 (ESI†) shows that there is no remarkable difference of electrochemical performances between the A-CPO/C and A-CPO/C_1 composites, which indicates that amorpholization highly contributes to the improved electrochemical performances of the A-CPO/C electrode. In addition, we performed the cyclic voltammetry (CV) tests of LC-CPO/C and A-CPO/C (Fig. S5 (ESI†)). It was observed that the cathodic and anodic peaks in the CV curves of the LC-CPO/C and A-CPO/C electrodes are similar to each other, which indicates that overall electrochemical performances of both LC-CPO/C and A-CPO/C electrodes are based on the conversion reaction of the Cu(PO3)2 phase in the LRB system. In particular, the specific capacity of A-CPO/C under a high current density of 1200 mA g−1 was ∼146 mA h g−1, which is ∼3.39 times larger than that of LC-CPO/C under the same conditions. Moreover, the voltage hysteresis of A-CPO/C (ΔV = ∼0.43 V) was observed to be lower than that of LC-CPO/C (ΔV = ∼0.50 V) in the charge/discharge curves at 12 mA g−1 (Fig. S6 (ESI†)). These results imply that the amorphorized phase in A-CPO/C can result in enhanced kinetics for the facile conversion reaction of CuPO3 in a Li-cell system, as a result of the highly shortened Li+ transport paths in the structure. Furthermore, we compared the charge-transfer resistances of bare Cu(PO3)2 and the LC-CPO/C and A-CPO/C composites using electrochemical impedance spectroscopy (EIS) (Fig. S7 (ESI†)). The semicircles of each sample, which correspond to the charge transfer resistance for Li+ diffusion, indicated that A-CPO/C (42.48 Ω) delivered a lower charge-transfer resistance than not only bare Cu(PO3)2 (340.34 Ω) but also the LC-CPO/C (180.62 Ω) composite, indicating the enhanced power capability and diffusion kinetics of the A-CPO/C composite. A-CPO/C clearly exhibited a much lower charge-transfer resistance than not only bare Cu(PO3)2 but also the LC-CPO/C composite, supporting the enhanced power capability and diffusion kinetics of the A-CPO/C composite. Fig. S8 (ESI†) shows the electrochemical performances of bare Cu(PO3)2 in the LRB system.
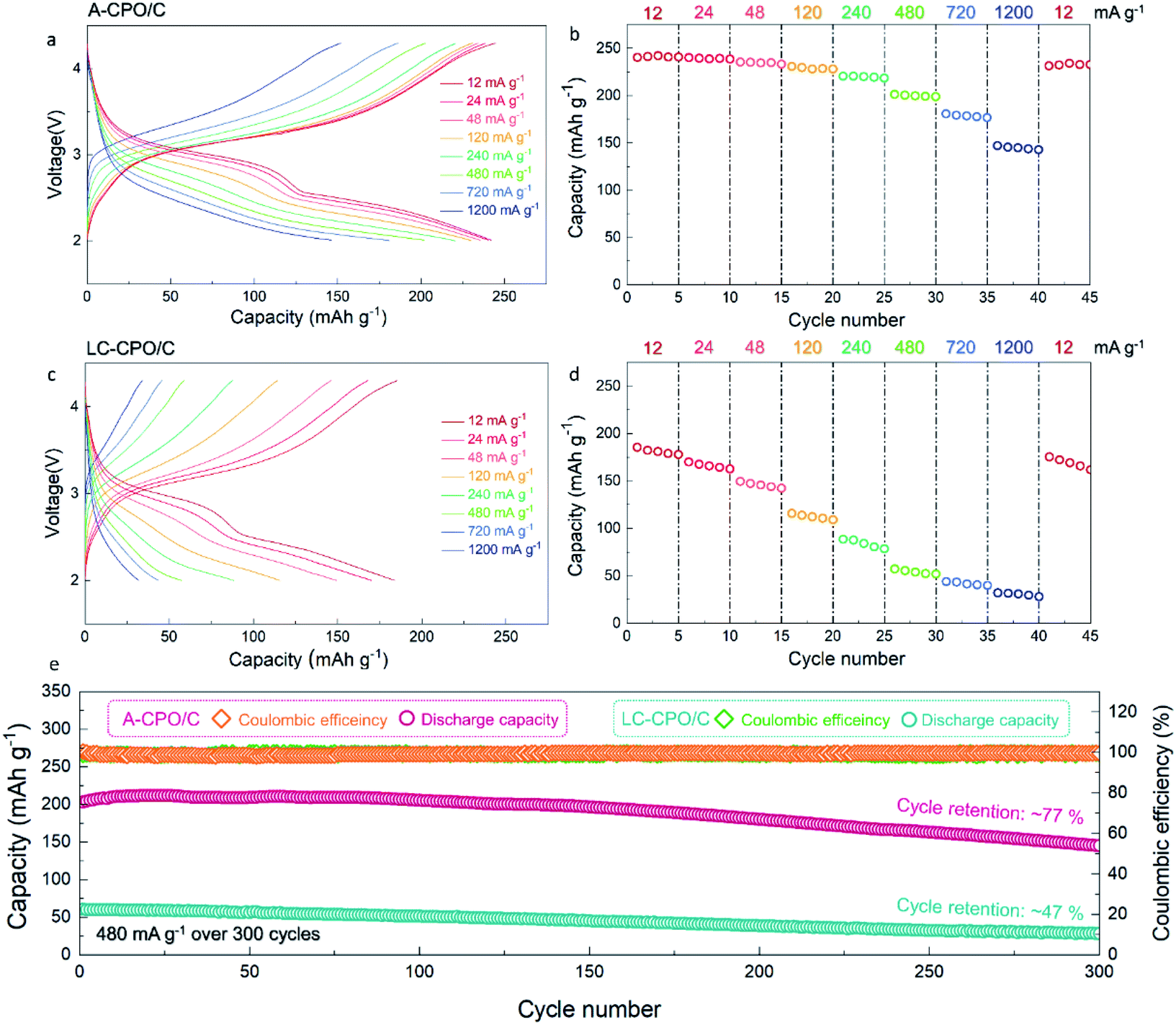 | ||
| Fig. 4 Power capability of (a) A-CPO/C and (c) LC-CPO/C at various current densities in the voltage range of 2.0–4.3 V. Rate capability of (b) A-CPO/C and (d) LC-CPO/C. (e) Cycle performances of A-CPO/C and LC-CPO/C for 300 cycles at a current density of 480 mA g−1 after an initial cycle at 30 mA g−1. | ||
In addition, the A-CPO/C composite exhibited a much more outstanding cycle performance than the LC-CPO/C composite. After 300 cycles at 480 mA g−1, the specific capacity of A-CPO/C was ∼77% of the initial capacity, with a high Coulombic efficiency (CE) of >99%. However, the LC-CPO/C composite only delivered a capacity retention of ∼47% compared with the initial capacity under the same conditions. Through XRD, SEM, and TEM analyses, it was demonstrated that the crystal structural and morphology of A-CPO/C were well maintained without critical damage, such as structural degradation or particle cracks, even after prolonged cycling (Fig. S9 (ESI†)). These experimental results imply that amorpholization and carbon coating can successfully suppress the large structural and morphological deformation that occur during the conversion reaction of Cu(PO3)2 phases. In addition, we performed the full-cell test using the A-CPO/C cathode and the lithiated graphite anode at the voltage range of 1.9–4.2 V. In terms of the graphite anode, it was known that a pre-lithiation process should be required to minimize the problem based on irreversible initial discharge capacity. Thus, we supposed that the fully lithiated graphite prepared by the pre-lithiation process can be applied as an anode for an A-CPO/C-based full-cell, because it not only provides the Li sources but also solves the irreversible capacity problem during the initial discharge. As shown in Fig. S10a (ESI†), it was verified that the specific capacity and the energy density of the full-cell at 12 mA g−1 were ∼232 mA h g−1 and ∼580 W h kg−1, respectively. In particular, the full-cell delivered an outstanding capacity retention of ∼83% after 300 cycles at 480 mA g−1 (Fig. S10b (ESI†)), which indicates that the A-CPO/C composite is a promising conversion-type cathode for high-energy LRBs.
3.3 Demonstration for the conversion reaction of Cu(PO3)2
The overall conversion reaction of Cu(PO3)2 in the Li-cell system was investigated using various ex situ analyses. As shown in Fig. 5a and b, the (111) plane of metallic Cu0 at 43° (2θ) gradually appeared during discharging to 2.0 V, implying the conversion reaction from the amorphous Cu(PO3)2 phase to the metallic Cu0 phase. After charging to 4.3 V, the metallic Cu0 phase disappeared, implying the reversibly restored amorphous Cu(PO3)2 phase. In addition, to verify the structural difference of the Cu(PO3)2 phase before and after discharging to 2.6 V, we compared the XRD patterns of the A-CPO/C electrodes discharged to 2.8, 2.6 and 2.4 V (Fig. S11 (ESI†)). During discharging to 2.8 V, the metallic Cu0 phase was not clearly detected in the XRD pattern, which implies that the mixture of Cu-(PO3)–Li exists as a solid solution. After discharging to 2.6 V, however, the signal of the metallic Cu0 phase was clearly detected in the XRD pattern. And the intensity of the XRD peak of the metallic Cu0 phase was increased even more during discharging to 2.4 V. These results indicate the existence of two plateaus in the discharge curve which is attributed to the distinct phase separation of metallic Cu0 and LiPO3. Moreover, the reversible conversion reaction of Cu(PO3)2 was confirmed through ex situ TEM and SAED analyses. Fig. 5c shows that the as-prepared A-CPO/C composite was composed of the amorphorized Cu(PO3)2 phase. After discharging, the appearance of the metallic Cu0 and LiPO3 phases was clear, indicating the occurrence of the conversion reaction of Cu(PO3)2 + 2Li ↔ Cu0 + 2LiPO3 (Fig. 5d). In the charged sample, we verified the presence of an amorphous phase with no crystallinity except a negligible faint trace of the Cu phase (Fig. 5e). These results were consistent with the ex situ XRD data.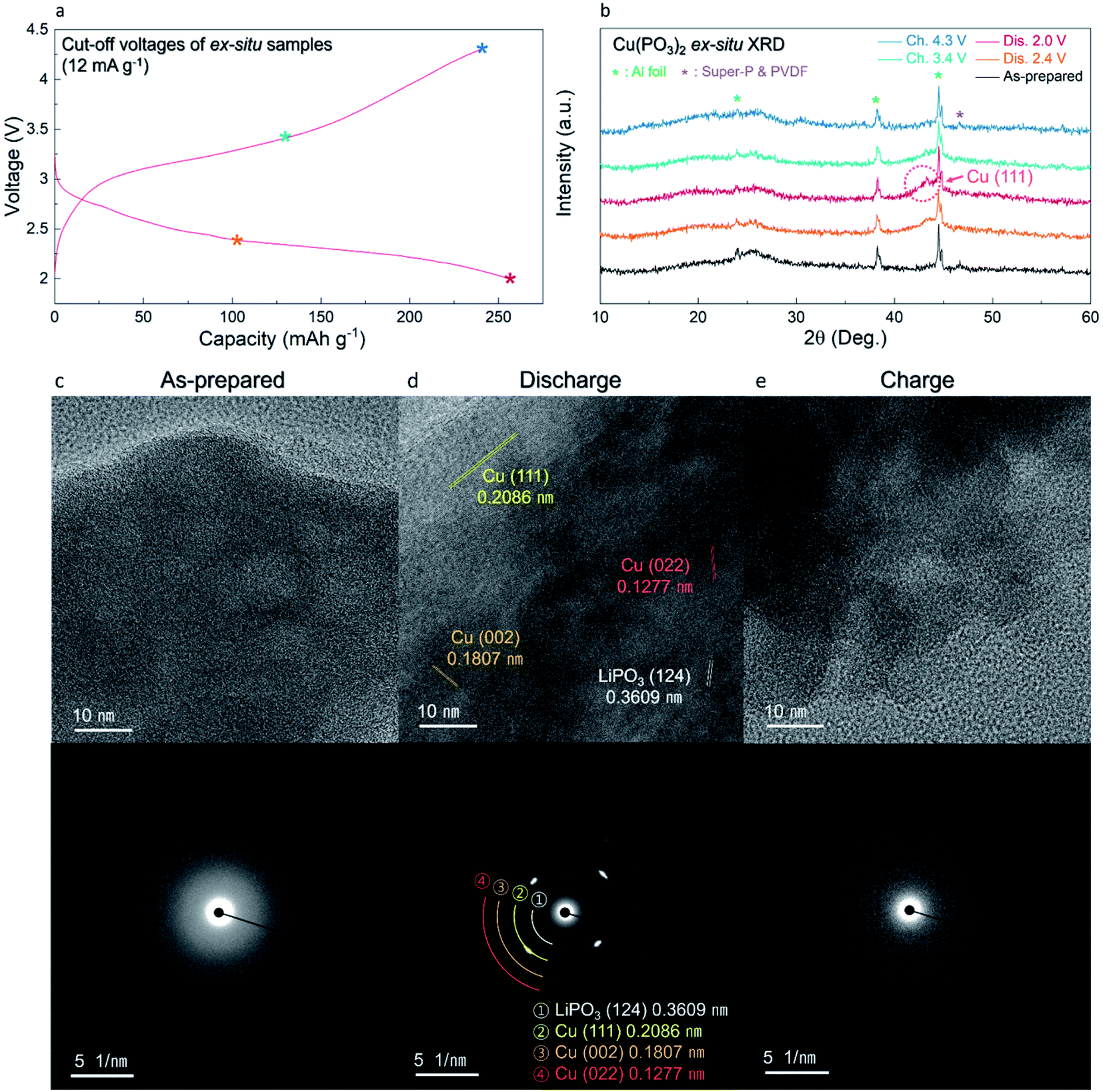 | ||
| Fig. 5 (a) Cut-off voltage points of the ex situ samples on the charge/discharge curve of A-CPO/C. (b) Ex situ XRD patterns of A-CPO/C. HRTEM images with SAED patterns of (c) as-prepared, (d) discharged, and (e) charged A-CPO/C. | ||
In addition, we demonstrated the change of the average oxidation states and local environments during charge/discharge using synchrotron-based X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses. Upon discharge, the Cu K-edge located at the energy level of Cu2+ was shifted toward low energy, corresponding to metallic Cu0 (Fig. 6a). During the charging process, the Cu K-edge returned to the original energy level. Moreover, the Fourier transform (FT) results of the EXAFS spectra indicate that the intensity of the Cu–O bond decreased during discharge whereas that of the Cu–Cu bond substantially grew because of the formation of metallic Cu0 (Fig. 6b). As the charging progressed, the intensity of the Cu–O bond grew and that of the Cu–Cu bond diminished. These results indicate that the reversible conversion reaction of Cu(PO3)2 in the Li-cell system was accomplished during charge/discharge. The conversion reaction of Cu(PO3)2 in the Li-cell system was also confirmed through ex situ XPS analyses (Fig. 6c and d). In the Cu 2p XPS spectra, it was verified that the Cu2+ peak (934.2 eV) and the satellite peak of Cu2+ (943.9 eV) disappeared and the metallic Cu0 peak (932.6 eV) appeared during discharge, whereas the Cu2+ peak and the satellite peak of Cu2+ were detected after charge. Through the O 1s XPS spectra, we confirmed that the Cu–O bonds reversibly appeared/disappeared during charge/discharge, whereas the P–O bond in the (PO3)− polyanion was clearly detected in all the samples, implying that the (PO3)− polyanion was well retained without decomposition during charge/discharge and that the bonding interaction of the (PO3)− polyanion changed reversibly between Cu2+ and Li+ (Fig. S12 (ESI†)). These results indicate the formation of LiPO3 and metallic Cu0 during the conversion reaction of Cu(PO3)2 in an LRB system.
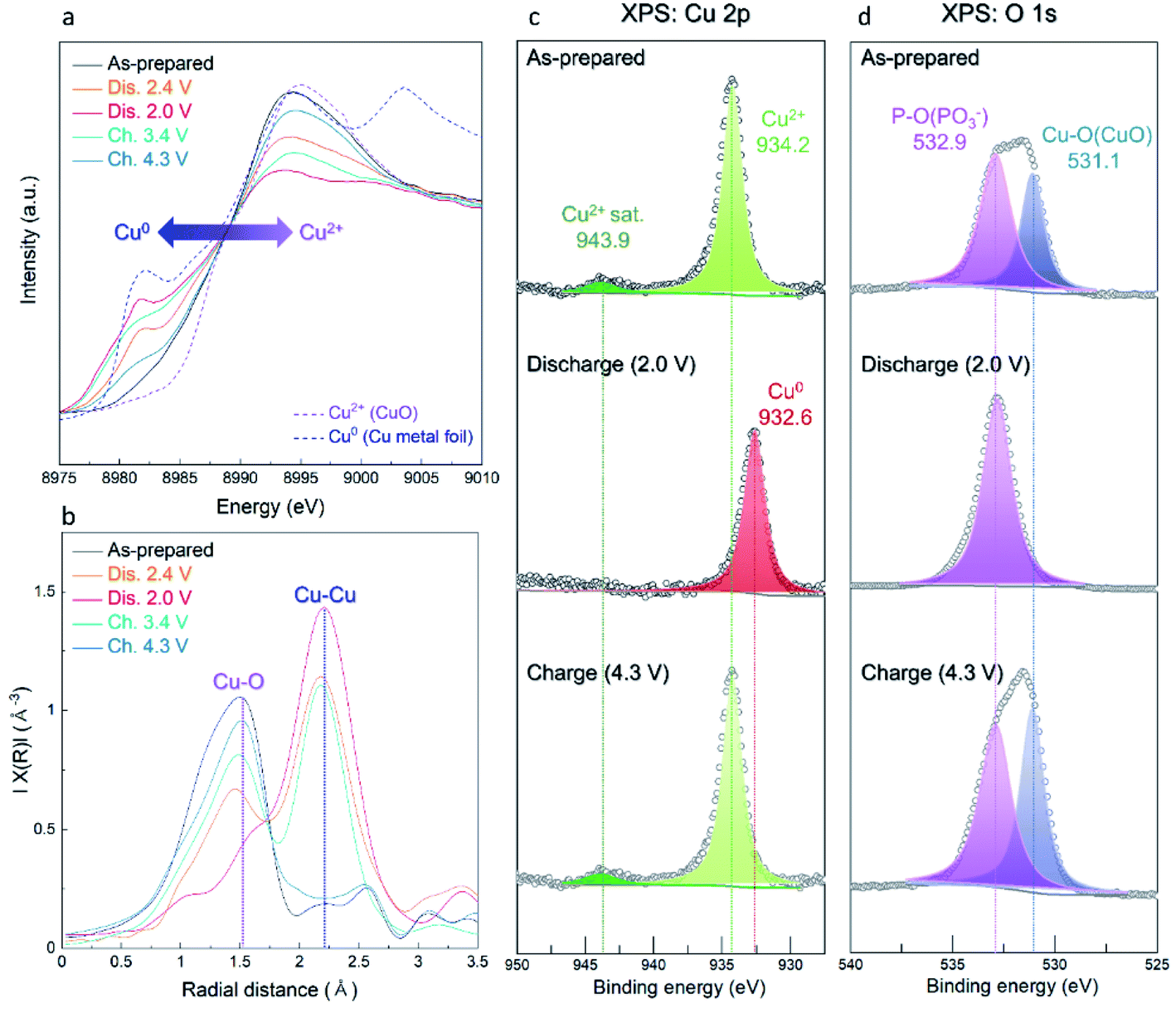 | ||
| Fig. 6 Ex situ measurements of A-CPO/C electrodes for (a) XANES spectra, (b) EXAFS spectra, (c) XPS Cu 2p spectra, and (d) XPS O 1s spectra. | ||
4. Conclusions
In summary, we demonstrated that the amorphorized Cu(PO3)2–carbon composite, A-CPO/C, can exhibit outstanding electrochemical performance such as large reversible capacity, outstanding power capability, and stable cycle performance as a promising cathode for LRBs. The reversible conversion reaction of Cu(PO3)2 + 2Li ↔ Cu0 +2LiPO3 in the Li-cell system enables not only a large capacity of ∼240 mA h g−1 at 12 mA g−1 but also an average operation voltage of ∼2.8 V (vs. Li+/Li) via the inductive effect of phosphorus with high electronegativity. First principles calculation results also clearly showed that the theoretical redox potential for the conversion reaction of Cu(PO3)2 in the Li-cell system was much higher than that for the conversion reaction of simple metal oxide CuO. Moreover, the amorpholization and homogeneous carbon coating provide highly enhanced electrical conductivity and kinetics for the facile conversion reaction of Cu(PO3)2 during charge/discharge. Thus, at a high current density of 1200 mA g−1, A-CPO/C exhibited a specific capacity of ∼146 mA h g−1, which is ∼3.39 times larger than that of LC-CPO/C under the same conditions. Furthermore, up to ∼77% of the initial capacity of A-CPO/C was retained after 300 cycles at 480 mA g−1, which differs from the poor cyclability of LC-CPO/C with a capacity retention of only ∼47% under the same conditions. Various ex situ techniques including XRD, XPS, TEM, and XANES and EXAFS analyses further supported the occurrence of the reversible conversion reaction of Cu(PO3)2 during charge/discharge. Although the energy density of the A-CPO/C-based full-cell was not highly attractive at present compared to that of the commercial LRBs based on well-established industrial systems (Fig. S13 (ESI†)),42–46 we believe that the development of novel conversion-based cathode materials will help form an effective strategy to design high-energy rechargeable batteries beyond commercial LRBs. We believe that our findings will help form an effective strategy to design a novel conversion-type cathode for high-energy rechargeable batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2019M3D1A2104105, 2020M2D8A2070870, 2021R1A2C1014280 and 2021M3H4A6A01041234). This work was also supported by the Korea Institute of Materials Science (KIMS) of the Republic of Korea (PNK 8060).References
- G. G. Eshetu, H. Zhang, X. Judez, H. Adenusi, M. Armand, S. Passerini and E. Figgemeier, Nat. Commun., 2021, 12, 5459 CrossRef CAS.
- J. Xie and Y. C. Lu, Nat. Commun., 2020, 11, 2499 CrossRef CAS.
- D. Deng, Energy Sci. Eng., 2015, 3, 385–418 CrossRef.
- J. Darga, J. Lamb and A. Manthiram, Energy Tech, 2020, 8, 200723 CrossRef.
- L. L. Wang, B. B. Chen, J. Ma, G. L. Cui and L. Q. Chen, Chem. Soc. Rev., 2018, 47, 6505–6602 RSC.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- R. J. Chen, T. L. Zhao, X. X. Zhang, L. Li and F. Wu, Nanoscale Horiz., 2016, 1, 423–444 RSC.
- N. Nitta, F. X. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- H. H. Sun, U. H. Kim, J. H. Park, S. W. Park, D. H. Seo, A. Heller, C. B. Mullins, C. S. Yoon and Y. K. Sun, Nat. Commun., 2021, 12, 6552 CrossRef CAS PubMed.
- N. Mohamed and N. K. Allam, RSC Adv., 2020, 10, 21662–21685 RSC.
- J. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702646 CrossRef.
- K. Jiang, Z. Chen and X. Meng, ChemElectroChem, 2019, 6, 2825–2840 CrossRef CAS.
- X. Zhou, H. Sun, H. Zhou, J. Ding, Z. Xu, W. Bin, J. Tang and J. Yang, J. Electro. Chem., 2018, 810, 41–47 CrossRef CAS.
- A. Douglas, R. Carter, L. Oakes, K. Share, A. P. Cohn and C. L. Pint, ACS Nano, 2015, 9, 11156–11165 CrossRef CAS.
- Z. Li, L. X. Yuan, Z. Q. Yil, Y. Liu and Y. H. Huang, Nano Energy, 2014, 9, 229–236 CrossRef CAS.
- Y. Yang, G. Y. Zheng, S. Misra, J. Nelson, M. F. Toney and Y. Cui, J. Am. Chem. Soc., 2012, 134, 15387–15394 CrossRef CAS.
- H. Kim, D. I. Kim and W.-S. Yoon, J. Electrochem. Sci. Tech., 2022, 13, 32–53 CrossRef CAS.
- M. J. Seong and T. Yim, J. Electrochem. Sci. Tech., 2021, 12, 279–284 CrossRef CAS.
- J. Jin, X. C. Tian, N. Srikanth, L. B. Kong and K. Zhou, J. Mater. Chem. A, 2017, 5, 10110–10126 RSC.
- X. R. Wang, W. T. Gu, J. T. Lee, N. Nitta, J. Benson, A. Magasinski, M. W. Schauer and G. Yushin, Small, 2015, 11, 5164–5173 CrossRef CAS PubMed.
- X. Y. Qiu, Q. S. Hua, L. L. Zheng and Z. Q. Dai, RSC Adv., 2020, 10, 5283–5293 RSC.
- J. Kim, H. Kim, I. Park, Y. U. Park, J. K. Yoo, K. Y. Park, S. Lee and K. Kang, Energy Environ. Sci., 2013, 6, 830–834 RSC.
- Y. Lee, C. H. Jo, J. K. Yoo, J. U. Choi, W. Ko, H. Park, J. H. Jo, D. O. Shin, S. T. Myung and J. Kim, Energy Storage Materials, 2020, 24, 458–466 CrossRef.
- J. H. Ku, J. H. Ryu, S. H. Kim, O. H. Han and S. M. Oh, Adv. Funct. Mater., 2012, 22, 3658–3664 CrossRef CAS.
- F. Kong, D. Sun, Y. Rao, R. Zhang, Z. Chen, D. Wang, X. Yu, H. Jiang and C. Li, Appl. Surf. Sci., 2022, 573, 151490 CrossRef CAS.
- K. Q. Xu, Y. S. Li, J. W. Xiong, X. Ou, W. Su, G. B. Zhong and C. H. Yang, Front. Chem., 2018, 6, 366 CrossRef.
- Z. Wei, D. Wang, X. Yang, C. Wang, G. Chen and F. Du, Adv. Mater. Inter., 2018, 5, 1800639 CrossRef.
- H. Q. Li and H. S. Zhou, Chem. Commun., 2012, 48, 1201–1217 RSC.
- S. Y. Lang, X. R. Feng, J. Seok, Y. Yang, M. R. Krumov, A. M. Villarino, M. A. Lowe, S. H. Yu and H. D. Abruna, Curr. Opin. Electrochem., 2021, 25, 100652 CrossRef CAS.
- S. H. Yu, X. R. Feng, N. Zhang, J. Seok and H. D. Abruna, Acc. Chem. Res., 2018, 51, 273–281 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- V. I. Anisimov, F. Aryasetiawan and A. I. Lichtenstein, J. Phy. Condes. Matter, 1997, 9, 767–808 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, Apl Mater, 2013, 1, 011002 CrossRef.
- F. Xiong, H. Tao and Y. Yue, Front. Mater., 2020, 6, 328 CrossRef.
- S. J. Sim, S. H. Lee, B. S. Jin and H. S. Kim, Sci. Rep., 2020, 10, 11114 CrossRef CAS PubMed.
- L. Xu, J. Li, H. Sun, X. Guo, J. Xu, H. Zhang and X. Zhang, Front. Chem., 2019, 7, 420 CrossRef CAS PubMed.
- V. Krylova and M. Andrulevičius, Inter. J. Photo., 2009, 2009, 304308 Search PubMed.
- Y. Gao, F. Yang, Q. Yu, R. Fan, M. Yang, S. Rao, Q. Lan, Z. Yang and Z. Yang, Mikrochim. Acta, 2019, 186, 192 CrossRef PubMed.
- S. Wang, G. Nam, P. Li, H. Jang, J. Wang, M. G. Kim, Z. Wu, X. Liu and J. Cho, Nanoscale, 2018, 10, 15834–15841 RSC.
- F. Bian, Z. Zhang and Y. Yang, J. Energy Chem., 2014, 23, 383–390 CrossRef CAS.
- Y. Zhan, H. Yu, L. Ben, Y. Chen and X. Huang, Electrochim. Acta, 2017, 255, 212–219 CrossRef CAS.
- J. Zhang, Q. Kong, Z. Liu, S. Pang, L. Yue, J. Yao, X. Wang and G. Cui, Solid State Ionics, 2013, 245–246, 49–55 CrossRef CAS.
- C. Xu, W. Xiang, Z. Wu, Y. Xu, Y. Li, Y. Wang, Y. Xiao, X. Guo and B. Zhong, ACS Appl. Mater. Interfaces, 2019, 11, 16629–16638 CrossRef CAS.
- J. Vidal Laveda, J. E. Low, F. Pagani, E. Stilp, S. Dilger, V. Baran, M. Heere and C. Battaglia, ACS Appl. Energy Mater., 2019, 2, 7036–7044 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02167f |
| This journal is © The Royal Society of Chemistry 2022 |