
Thioether-based ROS responsive polymers for biomedical applications
Miryam
Criado-Gonzalez
 *a and
David
Mecerreyes
ab
*a and
David
Mecerreyes
ab
aPOLYMAT, University of the Basque Country UPV/EHU, Paseo Manuel de Lardizabal 3, 20018 Donostia-San Sebastián, Spain. E-mail: miryam.criado@ehu.eus
bIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
First published on 29th April 2022
Abstract
Reactive oxygen species (ROS) play a key role in several biological functions of living organisms such as regulation of cell signalling, production of some hormones, modulation of protein function or mediation of inflammation. In this regard, ROS responsive polymers are ideal candidates for the development of stimuli-responsive biomaterials for target therapies. Among different ROS-responsive polymers, those containing thioether groups are widely investigated in the biomedical field due to their hydrophobic to hydrophilic phase transition under oxidative conditions. This feature makes them able to self-assemble in aqueous solutions forming micellar-type nanoparticles or hydrogels to be mainly used as drug carriers for local therapies in damaged body areas characterized by high ROS production. This review article collects the main findings about the synthesis of thioether-based ROS responsive polymers and polypeptides, their self-assembly properties and ROS responsive behaviour for use as injectable nanoparticles or hydrogels. Afterward, the foremost applications of the thioether-based ROS responsive nanoparticles and hydrogels in the biomedical field, where cancer therapies are a key objective, will be discussed.
 Miryam Criado-Gonzalez | Miryam Criado Gonzalez is a postdoctoral researcher at POLYMAT-University of the Basque Country (San Sebastián, Spain). She received her PhD in 2017 from the Institute of Polymer Science and Technology-CSIC (Madrid, Spain). Then, she spent a postdoctoral period in the Institute Charles Sadron-CNRS and Institute of Health and Medical Research-INSERM in Strasbourg (France). Her research interests include the development, assembly and applications of functional polymers and peptide based materials in the biomedical field. The European Materials Research Society recognized her research on this field with a Young Scientist Award in 2021. |
 David Mecerreyes | Prof. David Mecerreyes is an Ikerbasque Research Professor at POLYMAT (https://www.polymat.eu) University of the Basque Country. His research interests are the synthesis and application of innovative polymers such as poly(ionic liquid)s, conducting polymers and redox polymers for applications in energy and health. His research on new polymers has been recognized with several awards, including an ERC Starting Grant in 2011 and Research Excellency Award in 2020 by the Spanish Royal Chemical Society (RSEQ). He is the co-author of >340 publications in peer-reviewed journals and has edited two books that have received over 20 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 citations.
000 citations.1. Introduction
The development of stimuli-responsive materials that can be employed as therapeutic agents attracts great attention in the biomedical field for drug delivery,1,2 tissue engineering,3,4 or biosensing applications.5–7 These biomaterials aim to take advantage of biological stimuli like pH,8 temperature,9 enzyme activity,10,11 or redox balance5,12 to produce specific therapeutic effects. In this regard, reactive oxygen species (ROS), which is the term given to biologically relevant oxidants present in the human body as by-products of aerobic respiration, including hydrogen peroxide (H2O2), the superoxide anion (O2˙−), hydroxyl radical (˙OH), peroxynitrite (ONOO−), single oxygen (1O2), and hypochlorite (OCl−), has gained great importance in the last decade.13 ROS can be produced endogenously, from incomplete reduction of oxygen and the enzyme NADPH oxidase in the plasma membrane, or through exogenous stimuli such as UV light, ionizing radiation, and xenobiotics.14 It is worth noting the highly concentration-dependent ROS role ranging from beneficial cell survival to non-desirable effects.15 Thus, at appropriate concentrations, ROS play a key role in several biological functions of living organisms such as regulation of cell signalling, production of some hormones, modulation of protein function, and mediation of inflammation. However, ROS overproduction results in oxidative stress causing several pathologies such as inflammatory diseases, cancer, age-related diseases, atherosclerosis, and cognitive dysfunction, among others.16,17 Therefore, the need to control ROS production and concentration paves the way for the development of tailor-made ROS sensitive biomaterials. To that aim, soft materials like polymers and peptides with sequence-defined structures and spatial-temporal self-assembly properties, which are able to mimic, replace, or be compatible with human body tissues, are excellent candidates.18–20Depending on the ROS active unit, there exist different ROS-responsive groups which have been attached to polymers, i.e., thioethers or sulfides, diselenides, thioketals, aryl boronic esters, etc.21 Among them, those bearing thioether groups have been widely investigated in the biomedical field due to their oxidative nature, which confers them the ability to be oxidized in the presence of ROS experiencing thereby a hydrophobic to hydrophilic phase transition without the need to be cleaved. This feature plays a pivotal role in the case of polypeptides containing L-methionine amino acids, which can experience a reversible redox behaviour in the presence of reductase enzymes after oxidation to methionine sulfoxide.22,23 Therefore, in this review we will focus on thioether groups including synthetic polymers and biological ROS-sensitive peptides, i.e., poly(L-methionine) and poly(L-cysteine). First, the main achievements and recent developments found in the synthesis of ROS-responsive thioether-based polymers and their self-assembly into nanoparticles and hydrogels to be employed as therapeutic agents are summarized. Apart from that, the synthesis of polypeptides, which contain polymer and peptide (L-methionine- and/or L-cysteine) chains, is also addressed. Besides, the physical–chemical and oxidative-responsive properties of the self-assembled polymers and polypeptide materials are also reported. Afterward, the employment of these thioether-based materials in the biomedical field for drug delivery, cancer, and other therapies is discussed allowing obtaining an overview of the perspectives and challenges to be tackled in this field (Scheme 1).
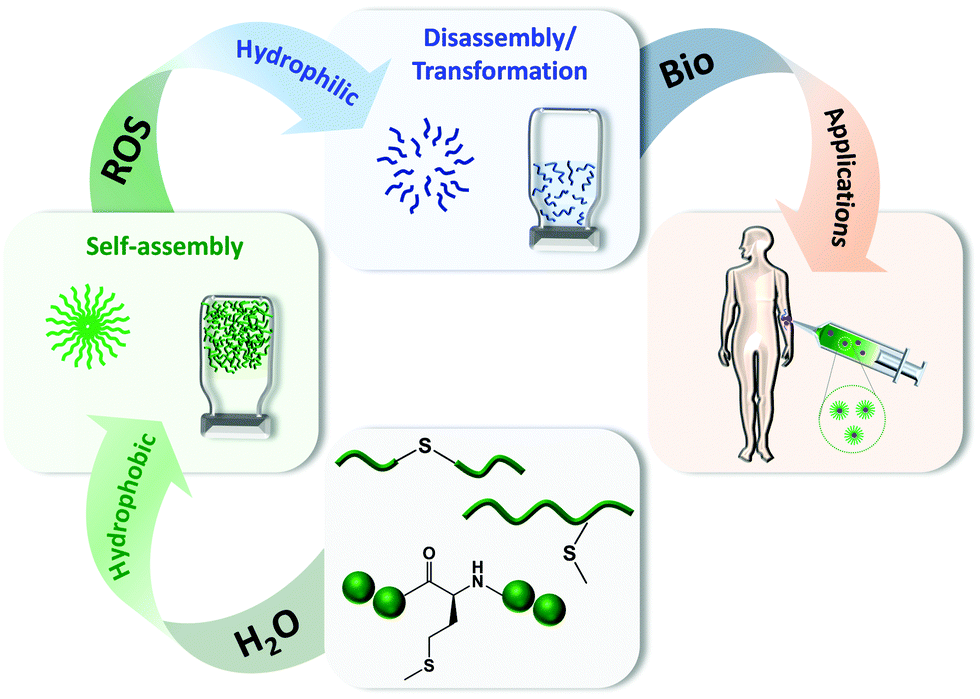 | ||
| Scheme 1 Thioether-based polymers and polypeptides that are able to self-assemble in aqueous solutions forming nanoparticles and hydrogels with the ability to be transformed or disassembled in the presence of ROS for biomedical purposes. | ||
2. Thioether ROS-responsive units
Sulfur is a non-toxic, biocompatible, and essential element in the biological system as it is found in all living cells and forms part of some essential proteins. Sulfur containing amino acids play a pivotal role in cellular maintenance and integrity due to the fact that they influence the cell redox state and its ability to detoxify reactive oxygen species, free radicals or toxic compounds.22 This is one of the reasons why thioether-containing polymers and peptides are widely used ROS-sensitive materials for biomedical applications, together with their hydrophobic–hydrophilic phase transition under oxidative conditions. Hydrophobic thioether groups present in polymers easily transform into hydrophilic sulfoxide or sulfone, under mild and strong oxidative conditions, respectively (Fig. 1A), at target human body areas with elevated ROS concentrations as a consequence of a lesion. Interestingly, the thioether group also forms part of the essential amino acid L-methionine that can also be easily oxidized into methionine sulfoxide under mild oxidation conditions, as this process is reversible by action of methionine sulfoxide reductase (MSR) A and B enzymes that reduce the methionine sulfoxide back to methionine, or by continuing the oxidation process under strong oxidant conditions to form methionine sulfone (Fig. 1B).24,25 All of these make L-methionine an interesting amino acid to form part of several peptide and polymer sequences leading to a plethora of stimuli-responsive polypeptides.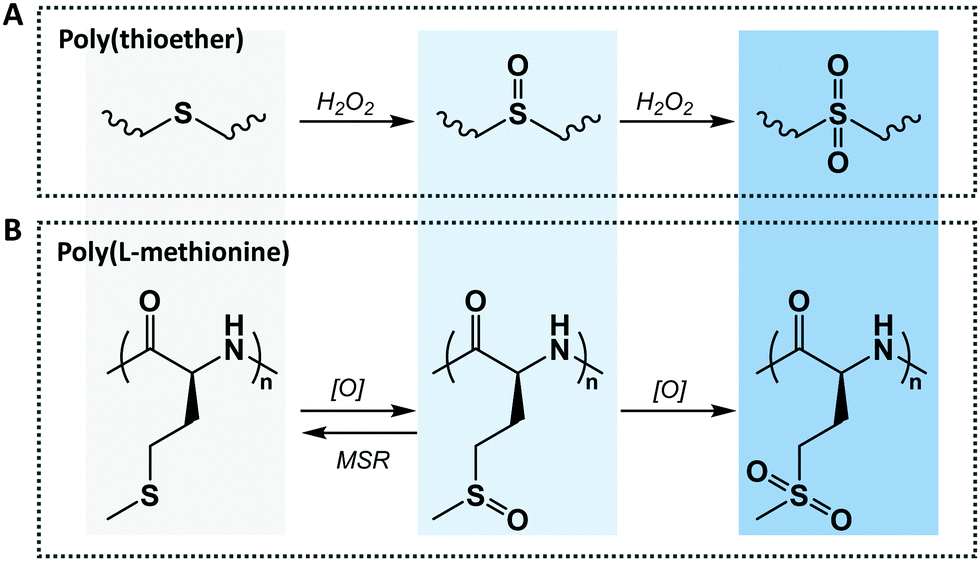 | ||
| Fig. 1 Oxidation-responsive (A) thioether and (B) L-methionine units, in polymers and peptides respectively, and their oxidation products. | ||
3. ROS-sensitive thioether-containing polymers
3.1. Synthesis and self-assembly of thioether-containing polymers
Two different strategies can be employed for the synthesis of thioether-containing polymers. The first one is based on the presence of thioether groups in the main chain, being poly(thioethers)/polysulfides, that is polymers containing C–S–C groups in their main chain, the simplest polymers with this functionality. They can be obtained by step- and chain-growth polymerization reactions. Whereas step-growth polymerizations are based on nucleophilic substitution, and nucleophilic or radical addition reactions, chain-growth reactions are mainly focused on ring-opening polymerization (ROP) strategies.26,27The hydrophobic character of thioether-containing polymers confers them the ability to self-assemble in aqueous media. A pioneering work in this field was carried out by Hubbell and co-workers, who synthesized amphiphilic triblock copolymers made of hydrophilic poly(ethylene glycol) (PEG) and hydrophobic poly(propylene sulfide) (PPS), PEG-b-PPS-b-PEG. This copolymer was able to self-assemble forming ROS-responsive polymer nanoparticles (NPs) in aqueous media through hydrophobic/hydrophilic interactions. In a second step, polymer oxidation in the presence of H2O2 resulted in hydrophilic NPs inducing their destabilization and disassembly. Besides, they also demonstrated that the hydrophobic–hydrophilic transition could be accelerated by using hypochlorite as an oxidant agent, which gave rise to the formation of more sulfone groups than in the case of H2O2 in which sulfoxide groups were the predominant ones.28
Since then, several polymers containing the thioether group in the main, side, or tail chains have been developed. ROS-responsive amphiphilic diblock copolymers made of thioether-containing polycarbonate block copolymers, mPEG-b-PS, were obtained through an enzyme-catalyzed ROP route.29 The amphiphilic mPEG-b-PS self-assembled in aqueous media into NPs that exhibited a size increase dependency with the length of the hydrophobic PS chain. Nuclear magnetic resonance (NMR) was employed to further study the oxidation behaviour in the presence of 200 mM H2O2. Results point to sulfide oxidation to sulfoxide from 12 h to 72 h, and then sulfones started to be formed, with a water contact angle decrease from 31° to 10° attributed to the enhancement in the hydrophilicity. Besides, gel permeation chromatography (GPC) proved that the polymer backbone integrity was maintained with a slight increase of the molecular weight after oxidation ascribed to the oxidation induced chain volume expansion. Biopolyesters such as poly(ε-caprolactone) (PCL), approved by the Food and Drug Administration (FDA), are extensively explored in the biomedical field.30,31 Thus, the one-step lipase-catalyzed polycondensation of hydrophilic monomethoxy poly(ethylene glycol) (mPEG) and hydrophobic poly(ε-caprolactone) (PCL) or poly(β-thioether ester) (PTE) allowed to obtain the block copolymers, mPEG-b-PCL and mPEG-b-PTE respectively, which were able to self-assemble in aqueous solution forming nanosized micelles. The critical micelle concentration (CMC) decreased with the PTE chain length due to the enhanced hydrophobicity of the micelle inner cores, and the size of the micelles increased. Moreover, the CMC value of mPEG-b-PTE was higher than that of mPEG-b-PCL because of the stronger polarity of thioether in the hydrophobic PTE segment than that of alkane in the PCL segment. These micelles were later disassembled under oxidative conditions due to the oxidising nature of thioether groups and degradable ester groups. Therefore, the combination of thioether and ester bonds in the polymer backbone makes NPs sensitive to both oxidation and hydrolysis. The results showed that mPEG-b-PTE diblock copolymers were more prone to be oxidized than mPEG-b-PCL. mPEG-b-PTE was oxidized after 2 h of contact with H2O2 (300 mM), as determined by 1H-NMR, and degraded into the low molecular weight products after 24 h of oxidation, as confirmed by GPC, whereas only a little degradation was observed for mPEG-b-PCL.32 Other examples of linear thioether containing polymers in the main chain are the thiol–ene linear step-growth photopolymerization of 2,2-(ethylenedioxy)diethanethiol (EDDT) and diallyl phthalate (DAP), diallyl adipate (DAA), or ethylene glycol dimethacrylate (EGDMA), giving rise to different poly(thioether ester)s, P(EDDT–DAP), P(EDDT–DAA),33 or P(EDDT–EGDMA)34 respectively. As previously, they were employed for the fabrication of dual oxidation- and hydrolysis-responsive NPs. P(EDDT–DAP) and P(EDDT–DAA) copolymers self-assembled in water leading to NPs with average diameters of 170 nm. P(EDDT–DAP) were fully hydrolysed after 160 h in contact with HCl solution at pH 2, whereas only 60% was oxidized in the presence of 3 M H2O2. On the contrary, P(EDDT–DAA) disassembled faster by H2O2 oxidation (20 h) than by hydrolysis (160 h). This difference can be attributed to the supplement of rigidity provided by the phenyl group in P(EDDT–DAP) forming large spherulites during crystallization, whereas P(EDDT–DAA) displays a high number of small nuclei.33 P(EDDT–EGDMA) NPs were disintegrated by ester hydrolysis (10 U esterase) and/or sulfide oxidation (300 mM H2O2) of the hydrophobic cores. Consequently, such responses can change the hydrophobic/hydrophophilic balance causing the colloids destabilization.34
The variety of thioether-containing polymers can be considerably increased by the insertion of the thioether group in the polymer side chains. Thus, the polymer main chain is not altered by the ROS action maintaining their backbone structure. In this case, thioethers can be present during the polymerization reactions of a large variety of monomers, i.e., lactones, epoxides, and acrylates, due to their low reactivity, or produced during grafting-to thiol–ene reactions or grafting-from approaches.35
One example is the synthesis of alternating copolymers by the amine–epoxy click reaction of 3-(methylthio)-propylamine (MSPA) and ethylene glycol diglycidyl ether (ED), P(MSPA-a-EG). These copolymers with hydrophobic MSPA and hydrophilic EG units were also able to self-assemble into spherical micelles with an average diameter of about 150 nm, which could be disassembled later on by the oxidation of thioether groups in MSPA units as the H2O2 concentration increased up to 166.6 mM.36 Another example is the synthesis of diblock copolymers consisting of 2-(methylthio)-ethyl glycidyl ether, and 3-(methylthio)propyl ethylene phosphate (MSPEP), mPEG-b-PMSPEP, through ring-opening polymerization of the functionalized cyclic phosphoester monomer using methoxy poly(ethylene glycol) and Sn(Oct)2 as the macroinitiator and catalyst respectively. CMC values of mPEG-b-PMSPEP decreased with the increase in the chain length of a hydrophobic PMSPEP block leading to higher thermodynamic stability. NPs with average diameters of ∼175 nm were obtained by spontaneous self-assembly in aqueous solutions. The oxidation extent was increased from 17.5% to 100% after 12 h of incubation with 1.2 and 18.8 mM H2O2, respectively.37 Besides, random and block copolymers made of PEG and 2-(methylthio)ethyl glycidyl ether (MTEGE), PEG–PMTEGE and PEG-b-PMTEGE copolymers have also been synthesized by ROP to form ∼30 nm diameter micelles. Random PEG–PMTEGE copolymers with MTEGE ratios of 5–24 mol% are water-soluble at room temperature, but turbidity is observed when they are heated exhibiting thermoresponsiveness between 88 and 28 °C respectively, which is a common feature of multifunctional PEGs bearing hydrophobic units along the polymer backbone. The incorporation of hydrophobic MTEGE moieties decreases the number of water molecules bound to PEG and their configurational entropy, resulting in an entropy-driven coil collapse upon heating, which is interesting for hypothermia diseases. In contrast, mPEG-b-PMTEGE block copolymers with MTEGE molar ratios lower than 14 mol% did not show cloud point temperatures in the range of 0–100 °C, exhibiting different aggregation behaviors in water in comparison to their random counterparts. Then, in the case of random PEG–PMTEGE copolymers, polydisperse samples with the occurrence of unimers and larger aggregates were obtained at room temperature in water, whereas for mPEG-b-PMTEGE26 with the longest hydrophobic block, micelles around 30 nm were formed. The changes in polarity from a non-polar thioether to polar sulfoxide or sulfone units strongly influences the disaggregation process. Thus, random PEG–PMTEGE copolymers in solution (5 mg mL−1) with a cloud point of 28 °C were rapidly disaggregated in 8 min by tempering them at 37 °C in the presence of 300 mM H2O2, and mPEG-b-PMTEGE26 exhibited a slower disintegration in 20 min.38 Very recently, ROS-responsive thioether-containing amphiphilic hyperbranched polymers have been synthesized from 3-(methylthio)propylamine (MTPA) and trimethylolpropane triglycidyl ether (TMPTGE), P(MTPA–TMPTGE), by amine-epoxy click reaction via an A2 + B3 one-pot approach, showing a good ability to self-assemble forming micelles with diameters around 70 nm in aqueous environments. These micelles were totally disassembled in contact with 25.7 mM H2O2 for 12 h as proven by 1H-NMR, UV-vis, and dynamic light scattering (DLS) measurements.39 Apart from that, Vasilev and co-workers designed thioether functional diblock copolymers in a two-step polymerization process. First, a well-defined AB diblock copolymer of 2-vinyl-4,4-dimethylazlactone (VDA) and N,N-dimethylacrylamide (DMA), P(VDA-b-DMA), was obtained by reversible addition–fragmentation chain transfer radical polymerization (RAFT). Then, the VDA–DMA diblock copolymer reacted with 2-(methylthio)ethylamine (MTEA) and 3-(methylthio)propylamine (MTPA) to yield two thioether functional diblock copolymers, P(MTEA-b-DMA) and P(MTPA-b-DMA) respectively. Both copolymers formed micelles in aqueous media, with CMC values of 0.1 and 0.06 mg mL−1 for P(MTEA-b-DMA) and P(MTPA-b-DMA) respectively consistent with the presence of more hydrophobic groups in the case of P(MTPA-b-DMA). These NPs, formed by a hydrophobic core containing either MTEA or MTPA units stabilized by a hydrophilic DMA corona modified with a thioether group, showed diameters in the range of 20–30 nm as confirmed by dynamic light scattering (DLS) (Fig. 2A) and transmission electron microscopy (TEM). The further oxidation process of the thioether group, with 10 mM H2O2, gave rise to a considerable decrease in the hydrodynamic diameter of the NPs from ∼24 nm to ∼9 nm due to the hydrophobic to hydrophilic phase transition and micelles disassembly.40 Biopolyesters containing side-chain thioether groups have also been synthesized exhibiting different oxidative behaviour depending on the thioether substituent. In a first step, α-substituted caprolactone-type monomers, α-ethylthio caprolactone or α-phenylthio caprolactone, were polymerized by ROP using benzyl alcohol as the initiator and stannous octoate as the catalyst at 130 °C. Then, amphiphilic block copolymers were obtained using PEG as the macroinitiator. The results showed that NPs formed with block copolymers containing ethylthio substituents could sufficiently swell and disassemble by ethyl thioether oxidation in the presence of H2O2 (5 mM) accompanied by a significant increase in polarity of the polyester block. On the other hand, NPs formed with copolymers containing phenylthio pendant groups degraded very slowly owing to the strong hydrophobic nature of the phenyl thioether, together with a reduction in the polyester block flexibility in the NPs due to the π–π stacking interaction between the phenyl thioether groups, as can be observed in the TEM images (Fig. 2B).41
 | ||
| Fig. 2 (A) Diblock copolymer MTEA–DMA with a thioether side chain employed for the synthesis of ROS-responsive micelles (left) and DLS measurements of the micelles before and after oxidation (right). Adapted and reprinted with permission from ref. 40 Copyright 2021 Elsevier Ltd. (B) ROS response of PEG–poly(α-ethylthio caprolactone) and PEG–poly(α-phenylthio caprolactone) (left) and TEM images of the NPs formed in aqueous solution before and after 24 h and 48 h oxidation (right). Scale bars = 500 nm. Adapted and reprinted with permission from ref. 41. | ||
The thioether group can also be used as the ending cap of the polymer chain. With this strategy, the backbone functions remain intact as in the case of the side-chain thioether-containing polymers. α-Methoxy,ω-vinylsulfonyl poly(ethylene glycol) (PEG–VS) was used as the ending cap of PPSES to obtain branched amphiphilic PEGylated polysulfides, PPSES–PEG. They showed improved homogeneity during the aggregation step to form spherical micelles with increased stability and higher drug loading ascribed to the branching inhibiting crystallization in the polysulfide blocks. The hydrophilization of the polysulfide cores during the oxidation with H2O2 was faster if a stoichiometric equivalence between oxidant and oxidizable groups was used.42
3.2. Multiresponsive thioether-containing polymers
Apart from single ROS-responsive polymers, several multi-responsive systems have been developed combining thioether groups with other stimuli-responsive functional groups or molecules that modulate the ROS production or confer them additional functions. The most relevant ones comprise dual effects of ROS with temperature, pH, light, or enzymes.One interesting approach is the development of dual ROS- and thermo-responsive polymers able to exhibit this synergistic effect under physiological mimicking conditions. In this regard, poly(propylene sulphide) (PPs) has been polymerized with poly(N-isopropylacrylamide), PNIPAm, using a combination of living anionic ROP and atom transfer radical polymerization (ATRP) to obtain block copolymers PPS-b-PNIPAm. These copolymers were used to form NPs in water with an average diameter of 100 nm determined by TEM. By increasing the upper temperature of the lower critical solution temperature (LCST), the micellar-type NPs experienced a contraction of the PNIPAm corona decreasing their size, at the same time that the shrunk hydrophilic corona allowed easier ROS access to the hydrophobic PPS core inducing their disassembly in contact with oxidant agents (Fig. 3A).43 Besides, thermo- and ROS-responsive triblock copolymers have been developed by thiolene polymerization of poly(ethylene glycol)diacrylate (PEGDA) and 1,2-ethanedithiol (EDT), mPEG-b-EDT-b-mPEG. The hydration of hydrophilic PEG segments made the polymer fully dissolve in water, but the temperature increase produced a collapse of the polymer as the PEG segment became dehydrated along with the increasing hydrophobic interaction between β-thioether ester segments, leading to the nanoparticles precipitation (∼110 nm diameter) in the absence of ROS. But, under oxidative conditions, NPs were dissolved by the hydrophobic–hydrophilic transition from sulphur to sulfide groups. Upon exposure to 300 mM H2O2, the EDT core was partially oxidized during the first 10 minutes leading to a slight swelling of the NPs with an increase of the hydrodynamic diameter. In the interval from 10 to 50 min, this diameter continued increasing, which indicated the concurrence of disintegration and swelling of NPs. From 60 min, the swollen NPs were totally disintegrated.44 A different approach to obtain thermo/ROS-responsive nanostructures can be the encapsulation of magnetic nanoparticles (MNPs), which experience a temperature increase in the presence of a magnetic field, into ROS-responsive polymer matrices. Thus, MNPs were successfully incorporated during PTE NP formation by miniemulsion forming MNP–PTE NPs with diameters around 150 nm and exhibiting superparamagnetic behaviour for magnetic hyperthermia therapies.45
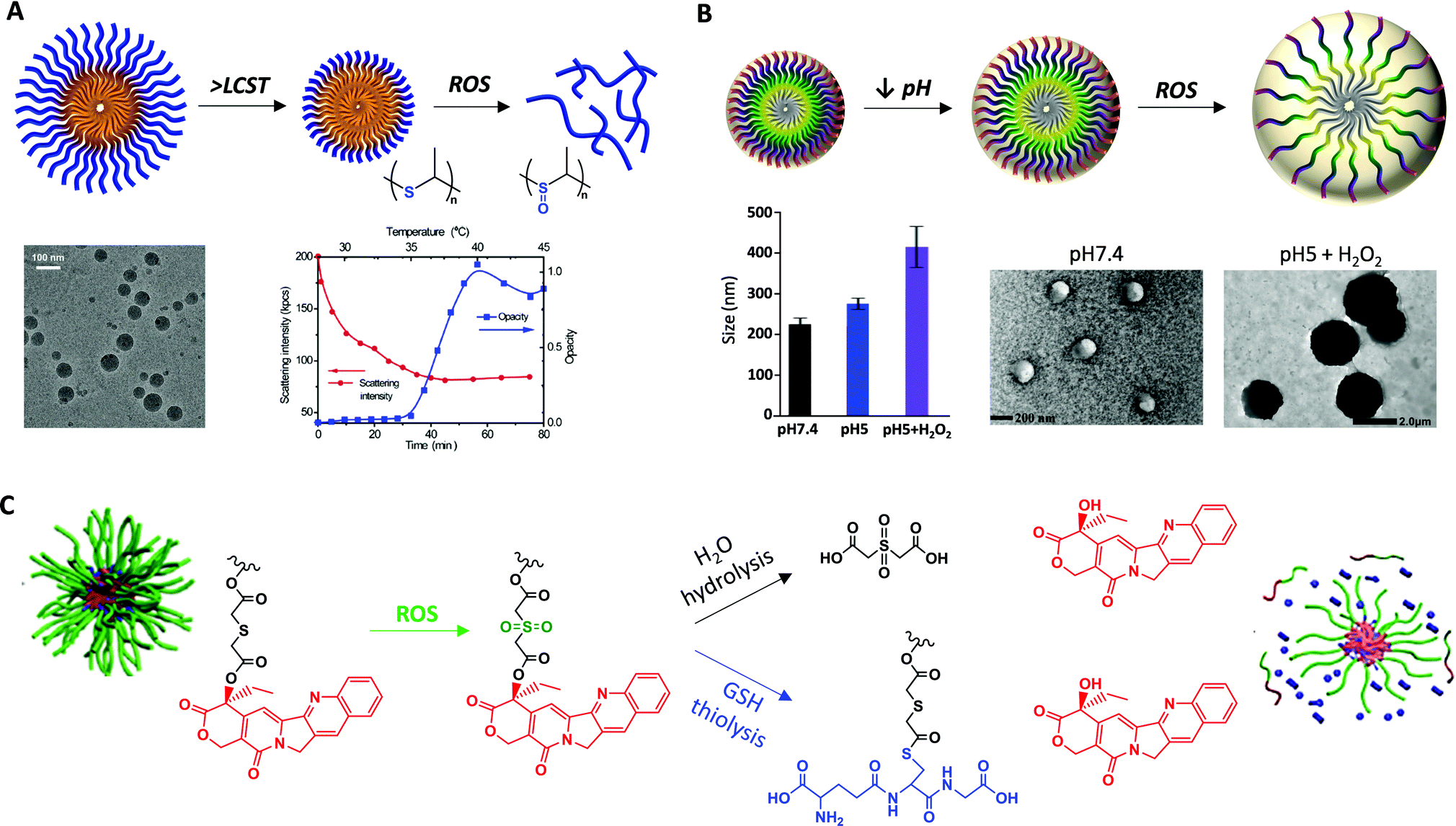 | ||
| Fig. 3 (A) Schematic illustration of the dual thermo- and ROS-responsive PPS-b-PNIPAm micelles (top). TEM images of the micelles at temperatures lower than LCST and thermal responsiveness (bottom). Adapted and reprinted with permission from ref. 43 Copyright 2017 Creative Common CC BY License. (B) Representation of the dual pH- and ROS-responsive mechanism of PEG–PPMT nanoparticles (top), and evolution of their size under different pH and oxidation conditions together with their TEM images (bottom). Adapted and reprinted with permission from ref. 47. (C) Drug release mechanism of dual-responsive amphiphilic diblock copolymer prodrug (GR-BCPs) through GSH thiolysis and ROS oxidation. Adapted and reprinted with permission from ref. 51 Copyright 2020, American Chemical Society. | ||
Taking into account that a combination of oxidative stress and reduced pH factors are common physiological stimuli used for intracellular and target drug delivery at pathological tissues, the synthesis of polymers containing thioether and amino groups led to dual ROS- and pH-responsive polymers. Methoxy-PEG-b-poly(chloromethylstyrene) block copolymers, MeO–PEG-b-PMNT, synthesized by radical polymerization were further modified with a nitroxide radical moiety, NH2–TEMPO, by an amination reaction in order to obtain dual ROS- and pH-responsive nanoparticles.46 Gong et al. synthesized another kind of block copolymer, PEG–poly(ω-pentadecalactone-b-N-methyldiethyleneamineco-3,3′-thiodipropionate) (PEG–PPMT), which is responsive to tumour-relevant acidic pH (5.0–6.5) and ROS present in tumour cells. The self-assembly of these copolymers in aqueous solutions formed nano-scaled particles that were stable under physiological pH (7.4), but swelled upon reducing the pH to 5.0, increasing their size from 215 to 416 nm, and/or the presence of ROS, reaching even 1709 nm in diameter, due to the protonation of the tertiary amines that reduced the hydrophobicity of the particle inner cores causing their swelling and oxidation of the thioether groups respectively, which also induced the hydrophilic transformation triggering the NP swelling or even the complete disassembly (Fig. 3B). When the ω-pentadecalactone (PDL) amount in the polymer chains increased, the NPs became less responsive to pH and H2O2 possibly due to the lower amine and thioether contents and higher hydrophobicity of PDL-rich nanoparticle cores. This higher hydrophobicity hindered H2O2 in the medium to reach and react with the thioether functional groups in the NPs.47
ROS formation can also be induced or enhanced by light. Aside from H2O2, photosensitizers generate other types of ROS such as singlet oxygen under irradiation at specific wavelength lights. For this purpose, photosensitizer molecules such as chlorin e6 (Ce6) have been incorporated during the polymer self-assembly process leading to the formation of dual redox- and photo-sensitive micelles with Ce6 molecules packaged into the polymer NPs. In this sense, Ce6 has been successfully encapsulated into mPEG-b-PMSPEP NPs and P(MTPA–TMPTGE) NPs, which responded to ROS generated by the photosensitizer action when they were irradiated.37,39 It is noteworthy that the development of ROS- and light-responsive NPs is also a useful approach for real-time in situ imaging of ROS. Fluorescent sulfur-tagged europium coordination polymers (CPs), selectively quenched by ROS, were prepared by simply mixing the precursors 2,2′-thiodiacetic acid (TDA) and Eu(NO3)3·6H2O in ethanol. Interestingly, the CPs underwent a morphological transformation from microcrystal to nanoparticle upon increasing the reaction temperature, while the structure and fluorescent properties were retained.48
In addition to this, a hallmark of cancer cells is the heterogeneous coexistence of overproduced ROS and intracellular glutathione (GSH) as compared with those levels in normal cells. Therefore, the design of dual redox-responsive micelles with ROS and GSH sensitive moieties plays a key role for cancer therapies. This can be achieved through the synthesis of poly(thioether ester)s, where the thioether bond is oxidized in the presence of ROS whereas the disulphide structure is cleaved in the presence of GSH.49,50 Yin et al. synthesized a dual-responsive amphiphilic diblock copolymer consisting of PEG and camptothecin (CPT)-conjugated poly(methacrylate) in the side chains via thioether bonds. The spherical micellar nanoparticles obtained in aqueous solution, with ∼50 nm diameter, showed accurate ROS- and GSH-responsive properties (Fig. 3C).51 In this line, a series of thioether phosphatidylcholines (S-PCs) and S-PC-based liposomes (S-LPs) were developed by using S-PCs with different chain lengths to modulate the phase transition temperature.52
3.3. Synthesis and self-assembly of thioether—containing polypeptides
Apart from polymers, peptides are also ideal candidates for the synthesis of stimuli-responsive biomaterials as they are formed by sequence-defined amino acid chains that endow them with specific structural, biological, and sensitive properties mimicking protein features.19,53,54 Therefore, the incorporation of thioether—containing amino acids during the polymer synthesis can tune the final properties of the developed polypeptides enlarging their range of applications. They can be incorporated as side chains, terminal groups, or linkages connected with conjugates using the amino acid L-methionine or through the etherification of L-cysteine.23,55–57 The oxidation of ROS-sensitive polypeptides also modulates the hydrophobicity and assembly properties leading to different aggregate states, secondary structures and micro/macro-structural properties of the final material. Deming and co-workers studied the self-assembly of amphiphilic peptides consisting of poly(L-methionine)65-b-poly(L-leucine0.5-stat-L-phenylalanine0.5)20, named M65(L0.5/F0.5)20, which showed α-helix conformation in this hydrophobic state. Under mild oxidation conditions and short times, the methionine unit was oxidized to L-methionine sulfoxide showing a completely disordered random coil structure. But at longer oxidation times, fully oxidized L-methionine sulfone groups were observed recovering the α-helical arrangement and hydrophobic nature due to interactions between sulfone groups and crystallization of the helices (Fig. 4).25 Keeping in mind that methionine oxidation can be reverted enzymatically by methionine sulfoxide reductases (MSR), MSR A reduces the (S)-isomer and MSR B the (R)-isomer of methionine sulfoxide,58 the authors also studied the reversibility properties from the L-methionine sulfoxide state to L-methionine in the presence of MSR enzymes.25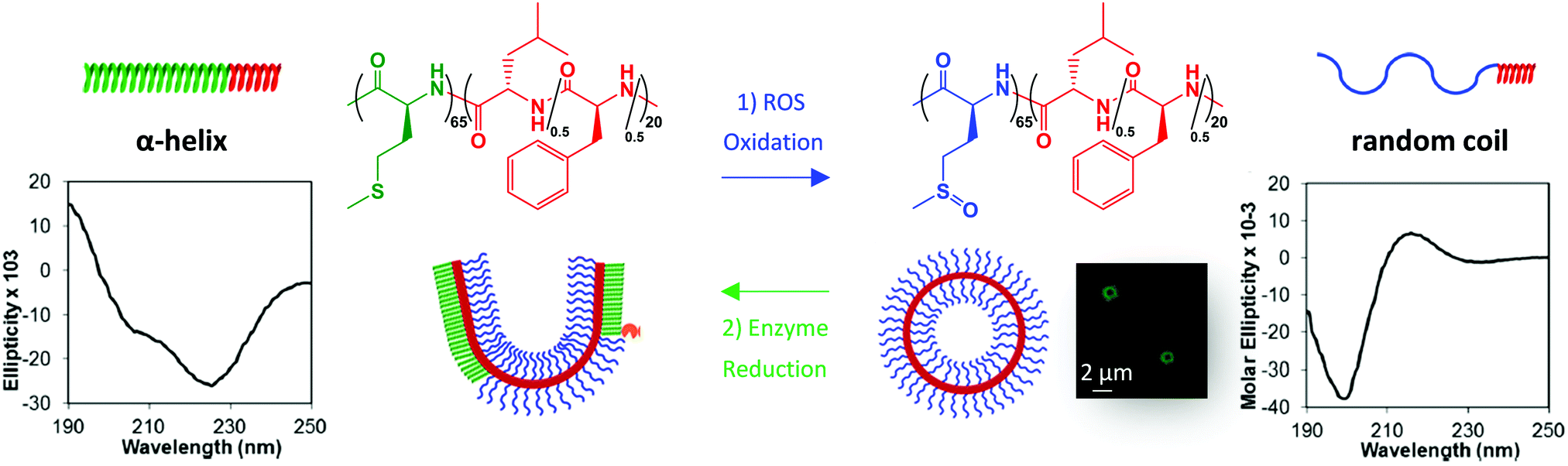 | ||
| Fig. 4 Redox properties of the M65(L0.5/F0.5)20 polypeptide containing the L-methionine ROS active unit. Circular dichroism spectra showing the structural arrangements at each state, with ellipticity measured in degrees cm2. Laser scanning confocal microscopy (LSCM) image of polypeptide self-assembly forming vesicles, and reversibility properties by MRS enzymes. Adapted and reprinted with permission from ref. 25 Copyright 2013, American Chemical Society. | ||
Therefore, the design of ROS-responsive polypeptides based on L-methionine opens a new field with broader possibilities. One simple approach is the synthesis of L-methionine poly(ester amide–PEG) (Met–PEA–PEG) that is also able to self-assemble in water forming micellar-type NPs.59 Apart from that, other L-methionine containing polypeptides have been developed for the synthesis of micellar-type nanoparticles. An example is a ROS- and protease-activated cell-penetrating peptide consisting of L-methionine as the ROS-responsive unit, a lysine chain as the cell permeable unit, and a matrix metalloproteinase (MMP)-cleavable linker, with the hydrophobic sequence PLGLAG introduced between the ML and hydrophilic PEG chain, PML–PLGLAG–PEG. The hydrophobic character of this polypeptide conferred it the ability to self-assemble in aqueous solution forming ∼100 nm micelles and showing a β-sheet conformation. The later contact of these micelles with H2O2 gave rise to a change in the secondary structure to a random coil conformation due to the oxidation of the thioether group of L-methionine amino acids with the subsequent hydrophobic to hydrophilic transition and micelle disintegration in 24 h.60 Moreover, ROS-sensitive degradable poly(amino acid)s (PAA)s, containing 6-aminohexanoic acid, L-methionine, L-hydroxyproline and L-phenylalanine, were synthesized via in situ melting polycondensation. Then, it was proven that the methionine content accelerated the poly(amino acid) degradation in oxygen-rich environments.61
Apart from the NPs, polypeptides can self-assemble forming fibers that work as precursors of hydrogel formation. That is the case of a PEG-containing peptide that conjugates the coupling of an α-acyl-brominated tetra-phenylalanine peptide (FFFF) with a disulfide-bridged polymeric PEG scaffold, MeO–FFFF–PEG–FFFF–OMe. Their further supramolecular self-assembly led to fibers formation with an average diameter of ∼10 nm, through π–π stacking of aromatic constituents forming β-sheets.62 Recently, Battaglia and co-workers carried out the one-pot synthesis of oxidation-sensitive supramolecular micelles and vesicles by polymerization-induced self-assembly (PISA) of the N-carboxyanhydride (NCA) precursor of methionine using poly(ethylene oxide) as a stabilizing and hydrophilic block in dimethyl sulfoxide (DMSO). Different morphologies ranging from spherical to wormlike micelles up to vesicles could be obtained by adjusting the hydrophobic block length and concentration. Wormlike micelles were obtained for a wide range of methionine block lengths and solid contents that could further assemble into self-standing gels, while spherical micelles were restricted to very short hydrophobic lengths. In all cases, the different constructs showed ROS induced degradation.63 To go a step further, injectable thermo-sensitive hydrogels were obtained by self-assembly of a functional triblock copolymer, which comprises a hydrophilic central PEG block flanked by two hydrophobic polypeptide blocks that contain ROS-responsive L-methionine (Me) and D-1MT, designated as P(Me–D-1MT)–PEG–P(Me–D-1MT). The hydrogels showed a hydrophobic to hydrophilic transition by oxidation in the presence of 10 mM H2O2 with a total disintegration after 3 days.64 Another example of injectable and self-healing ROS-responsive hydrogels is based on the employment of tetra-poly(ethylene glycol)-b-oligo(L-methionine) named as t-PEG56-b-OMetn. In that case, the mechanical strength of the hydrogels was markedly enhanced by increasing the length of the OMetn block in the copolymer. The copolymer assembled into an injectable and self-healing network with hydrophobic cores as cross-linkers, where the longer block length and higher contents of OMetn strengthen the hydrophobic interactions. The subsequent contact with 10 mM H2O2 gave rise to the oxidation of methionine groups and hydrophobic to hydrophilic transition inducing the gel–sol transition.65 Furthermore, dual ROS- and thermo-responsive diblock copolypeptides, composed of mPEG and poly(L-methionine), mPEG–PMet, were synthesized by ROP of L-methionine N-carboxyanhydride (Met NCA). These mPEG–PMet copolymers exhibited a thermo-induced sol–gel phase transition in aqueous environments, depending on the poly(L-methionine) block length, to form injectable hydrogels that can be disrupted in the presence of H2O2 (Fig. 5A). Hydrogels completely disappeared after 19 days contact with 1 mM H2O2, but this period could be drastically decreased to 3 days by increasing the H2O2 up to 10 mM.66 The sensitivity properties were even enlarged by transient supramolecular polymerization of β-sheet peptide monomers such as glutamic acid and L-methionine in water. They could be catalyzed by glucose oxidase (GOx) to reach glucose-fueled transient hydrogelation in response to an interplay of pH and oxidation stimuli promoted by ROS production. Interestingly, the assembly and disassembly rates of these supramolecular polymers could be modulated by the enzyme and glucose concentration tuning in turn the hydrogel stiffness. In addition to this, the incorporation of triethylene glycol chains introduced extra thermo-responsive properties to the gels. But not only that, these polypeptides exhibited reversibility properties of the methionine-based thioether side chains by enzymatic action of methionine sulfoxide reductase A (MSRA) and B2 (MSRB2).67
 | ||
| Fig. 5 (A) Oxidation of the mPEG–PMet diblock copolymer in presence of H2O2, photographs and TEM images of the gel–sol transition in presence of H2O2 (left). Circular dichroism spectra of the gels before and after oxidation (right). Adapted and reprinted with permission from ref. 66 Copyright 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (B) Schematic representation and photographs of the oxidation process of poly(L-EGxMA-C)n NPs associated with a β-sheet to random coil transformation (left). Circular dichroism spectra of the NPs before and after oxidation (right). Adapted and reprinted with permission from ref. 68 Copyright 2014, American Chemical Society. | ||
Cysteine can also be used as a thioether bond forming amino acid. OEGylated poly-L-cysteine peptides PEG45-b-P(L-EG2MA-C)22 were synthesized through ROP of the macroinitiator mPEG45-NH2 and L-EG2MA-C N-carboxyanhydride. This PEG45-b-P(L-EG2MA-C)22 could self-assemble into spherical micelles in water, exhibiting a β-sheet conformation by circular dichroism (CD), which could undergo an oxidation-triggered disassembly with a random coil transformation, due to the oxidation-responsive thioethers (Fig. 5B).68 In another work, Deming and Kramer designed glycopolypeptides by polymerization of glycosylated L-cysteine–N-carboxyanhydride (glyco-C NCA) monomers obtained by thiol–ene “click” chemistry of alkene-terminated C-linked glycosides of D-galactose or D-glucose to L-cysteine. Remarkably, these glycopolypeptides exhibited an unprecedented ability in synthetic polymers to switch chain conformation and remain water-soluble. They showed an α-helical conformation by circular dichroism (CD), but once the side-chain thioether linkages were oxidized to sulfone groups, they experienced a random-coil transformation while remaining water-soluble.69 Later on these authors also found a new class of polymers with the ability to respond either through a change in chain conformation or in water solubility. To that aim, they synthesized poly(S-alkyl-L-homocysteine)s, formed by incorporation of water solubilizing alkyl functional groups integrated with precisely positioned multiresponsive thioether linkages.70 Another example of a multiresponsive polypeptide was obtained by the reaction of mPEG–cystamine with cystamine, and bis(4-nitrophenyl) diethyl sulphide leading to mPEG-b-P(Des-a-Cys). This DMSO solubilized mPEG-b-P(Des-a-Cys) could self-assemble in water forming NPs that exhibit a dual responsive behaviour as the Des and Cys units are sensitive to ROS and GSH respectively.49 Besides, another dual responsive polypeptide, based on PEG–poly(ω-pentadecalactone-co-N-methyl-diethyleneamine sebacate-co-2,2′-thiodiethylene sebacate) (PEG–poly(PDL-co-MS-co-TS)), was synthesized via lipase-catalyzed copolymerization and named PEG–PMT. Then, it self-assembled in aqueous solutions forming micelles that showed a size increase with the length of the hydrophobic PMT segments in the block copolymers, as well as with the exposure to acidic pH and ROS. The particle size slightly increased by decreasing the pH from 7.4 to 5.0 due to the protonation of the tertiary amino groups in the polymer backbone. However, a significant increase was achieved through the simultaneous stimuli of pH 5.0 and a high ROS concentration (100 μM H2O2), due to the synergistic effect of tertiary amino group protonation and oxidation of hydrophobic thioether groups in the PMT segments to form hydrophilic sulfoxides or sulfones, and the micelle cores became more hydrophilic causing their swelling by water absorption from the medium.71
4. Biomedical applications
ROS responsive materials play a key role in the treatment of several diseases due to the abnormal levels of ROS in specific damaged areas of the human body. Normal human plasma typically contains an average of 3 μM H2O2 and respiratory lining cells are exposed to H2O2 concentrations ranging from 0.1 to 1 μM H2O2. However, the H2O2 concentration is usually elevated to 20 μM and 100 μM in the case of inflammatory lung diseases and brain ischemia respectively.66 Similar ROS (H2O2, ˙OH, 1O2, O2˙−, ROOH, ROOR’) levels of ∼100 μM are reportedly reached in some tumor cells due to the oncogene stimulation, mitochondrial mutations and chronic inflammation as compared with healthy cells.49 Furthermore, acidic pH environments are present in the endosomes (pH 5.0–5.5) and lysosomes (pH 4.5–5.0) of tumor cells.47 The tightly regulated redox balance in cells can be altered by action of the ROS produced by thioether-containing polymers and polypeptides giving rise to antagonistic, additive, or synergistic effects. Thus, the effect of the different thioether-based nanoparticles and hydrogels by themselves, as well as by action of the therapeutics loaded within them, in the final biomedical application will be discussed in this section.4.1. ROS-triggered drug release for cancer therapies
Most thioether-containing polymers and polypeptides have been used as drug carriers for biomedical applications. Among them, the encapsulation of anticancer drugs such as doxorubicin (DOX), paclitaxel (PTX), or camptothecin (CPT) within the micellar-type nanoparticles or hydrogels has been widely explored and tested for cancer therapies.Wang et al. designed a micellar-type DOX carrier from P(MSPA-a-EG) that showed ROS-triggered DOX release. By increasing the H2O2 concentration from 5 to 20 mM, the oxidation of the thioether group gave rise to a hydrophobic to hydrophilic phase transition inducing the NP disassembly, as corroborated by transmission electron microscopy (TEM), with the subsequent DOX release from 23.4% to 55.6% for 5 and 20 mM H2O2 respectively.36 In another case, PML–PLGLAG–PEG NPs loaded with DOX, named MLMP, exhibited matrix metalloproteinase (MMP)-sensitive cleavage and ROS-induced DOX release. After injection into the body, while MLMP was circulated in the blood stream reaching tumor tissues, the MMP-sensitive linkers (PLGLAG) were cleaved by excess of matrix metalloproteinase-2 (MMP-2) enzyme around the tumor tissues leading to exposure of the CPP segments and release of PEG blocks. On the other hand, poly-L-lysine chains assisted the cellular penetration by electrostatic interactions between the lipid plasma membranes. Then, the thioether groups in the methionine chains could be oxidized by the ROS excess in the cytosol of cancer cells, thereby selectively provoking the release of DOX in cancer cells and induction of the apoptotic capability. The bio-distribution study using IR-780 dye encapsulated MLMP showed superior tumor targetability with long retention (Fig. 6). The employment of DOX loaded micelles allowed a decrease in the tumor size from ∼1.86 ± 0.68 g to ∼0.13 ± 0.05 g, with greater effectiveness than free DOX that allowed a smaller reduction (∼0.66 ± 0.16 g).60 In another case, PPS–PNIPAm based NPs loaded with DOX were able to be taken by tumour MCF-7 cells and mainly reside in the cytoplasm. Interestingly, a higher quantity of DOX was released in the stimulated cells with an elevated level of ROS for more efficient cancer chemotherapies. In the absence of oxidants, the DOX release at 25 °C or 37 °C for 24 h was negligible. However, DOX release values of 31% were achieved after 10 h contact with 0.1% H2O2 at 25 °C, and significantly increased up to 51% if the treatment with H2O2 was performed at 37 °C, resulting in a synergistic release profile of the PPS–PNIPAm micelles under two stimuli. It was also proven that ROS produced by MCF-7 cells were not enough to induce the oxidation of PPS–PNIPAm micelles being necessary to stimulate them with Rosup reagent (50 μg mL−1) for 20 minutes in order to achieve cell viability reductions similar to those ones of free DOX (∼30% for 10 μg mL−1 DOX).43 Furthermore, DOX was loaded in thioether phosphatidylcholines (S-PCs) and S-PC-based liposomes (S-LPs) for chemotherapy. In vitro and in vivo tests revealed an improved drug potency of S-LPs in comparison to conventional stealth liposomes due to the ROS-triggered destruction of S-LPs after the uptake by tumor cells followed by rapid DOX release.52 On the other hand, ROS-responsive injectable and self-healing hydrogels made of t-PEG56-b-OMetn polypeptide provides the ideal environment to encapsulate DOX for controlled and site-specific drug release.65 The same approach was also employed in the case of DOX-loaded PEG45-b-P(L-EG2MA-C)22 hydrogels. Interestingly, in the presence of a catalytic amount of Fe2+ (36 μmol L−1) that exists in vivo in oxidative stressed environments (i.e., injury, cancer, and other diseases) the DOX release rate could be accelerated and increased from 43% to 66%, due to the fact that Fe2+ increases the reactivity of H2O2 (0.3% v/v).68
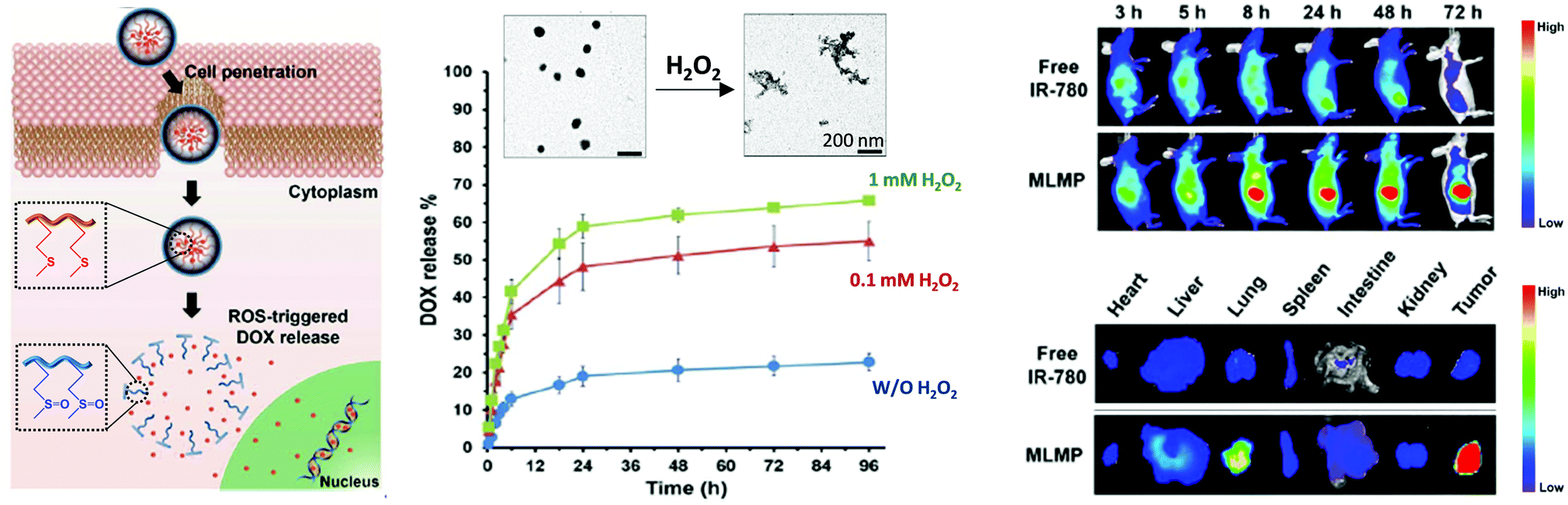 | ||
| Fig. 6 Schematic illustration of the intracellular DOX delivery from PML–PLGLAG–PEG micelles induced by ROS (left). TEM images of micelles disassembly due to oxidation of thioether groups in the presence of ROS and subsequent DOX release (centre). NIR fluorescence images at various time points for 72 h after tail vein injection of free IR-780 and MLMP encapsulated IR-780 dye, for the bio-distribution analysis using NCI-H460 tumor bearing nude mice, and ex vivo NIR images of tumors and organs at the end of the bio-distribution study (right). Adapted and reprinted with permission from ref. 60 Copyright 2017 Elsevier B.V. | ||
Apart from DOX, other drugs such as docetaxel (DTX) and gambogic acid (GA) have also been loaded in the micellar-type NPs. For example, ROS-responsive thioether-bearing polymer (TEP) NPs were used as piperlongumin (PL) drug carriers. PL-TEP NPs showed a ROS-sensitive PL release profile by the hydrophobic–hydrophilic phase transition due to the thioether oxidation in the presence of 100 mM H2O2. The in vitro tests in contact with MCF-7 cancer cells showed a more efficient cellular uptake and anticancer activity in MCF-7 cancer cells compared to free PL. What is more PL–TEP NPs showed cancer-selective cytotoxicity over normal human dermal fibroblast (HDF) cells, due to the 10% higher intracellular ROS levels of MCF-7 than that of HDF. Non-loaded NPs caused a slight ∼20% increase in intracellular ROS levels compared to untreated cells. Interestingly, PL-loaded NPs revealed a significant 95% increase in intracellular ROS levels, much higher than this one for free PL (∼59%). Therefore, the high cytotoxicity of PL and PL–TEP NPs in MCF-7 cells is closely related to the elevated intracellular ROS levels after treatment.72 Another example is DTX-loaded PEG–poly(PDL-co-MS-co-TS) micelles that were triggered synergistically by acidic pH and ROS stimuli to release ∼85% DTX and thus prohibited the growth of CT-26 tumors xenografed in vivo (70% of tumor-inhibiting efficiency), whereas they possessed minimal toxicity toward normal organs, such as liver and kidney, to be used for controlled release at intracellular tumor sites.71 Specifically, GA was encapsulated within Met–PEA–PEG NPs for treatment of prostate cancer, and the results highlighted the sensitivity of Met–PEA–PEG NPs to the high intracellular ROS level of PC3 prostate cancer cells, as well as an enhanced cytotoxicity toward PC3 and HeLa cells as compared to free GA. The GA loaded Met–PEA–PEG NPs induced a 49% apoptotic PC3 cell, while free GA led to a 36%. This could be due to the increased solubility of GA in Met–PEA–PEG NPs than free GA, and hence a higher uptake.59
4.2. Multiresponse systems for cancer therapies
The therapeutic effect of ROS-responsive polymers and polypeptides can be tuned by the action of additional stimuli such as temperature, pH, light or enzymes giving rise to multiresponse systems with enhanced functionalities.In the case of ROS- and thermo-responsive dual systems, injectable polypeptide hydrogels based on P(Me–D-1MT)–PEG–P(Me–D-1MT)) were used as carriers of immunosuppressive factor inhibitors, aPD-L1 and dextro-1-methyl tryptophan (D-1MT). These biocompatible hydrogels could not only sustain the in situ delivery of aPD-L1 and D-1MT under ROS conditions, but also decline the intratumoral ROS level. In vivo results demonstrated the ability of the aPD-L1-loaded hydrogel to naturally stimulate infiltration of immune cells and enhance the antitumor efficacy compared to free drugs (Fig. 7A).64
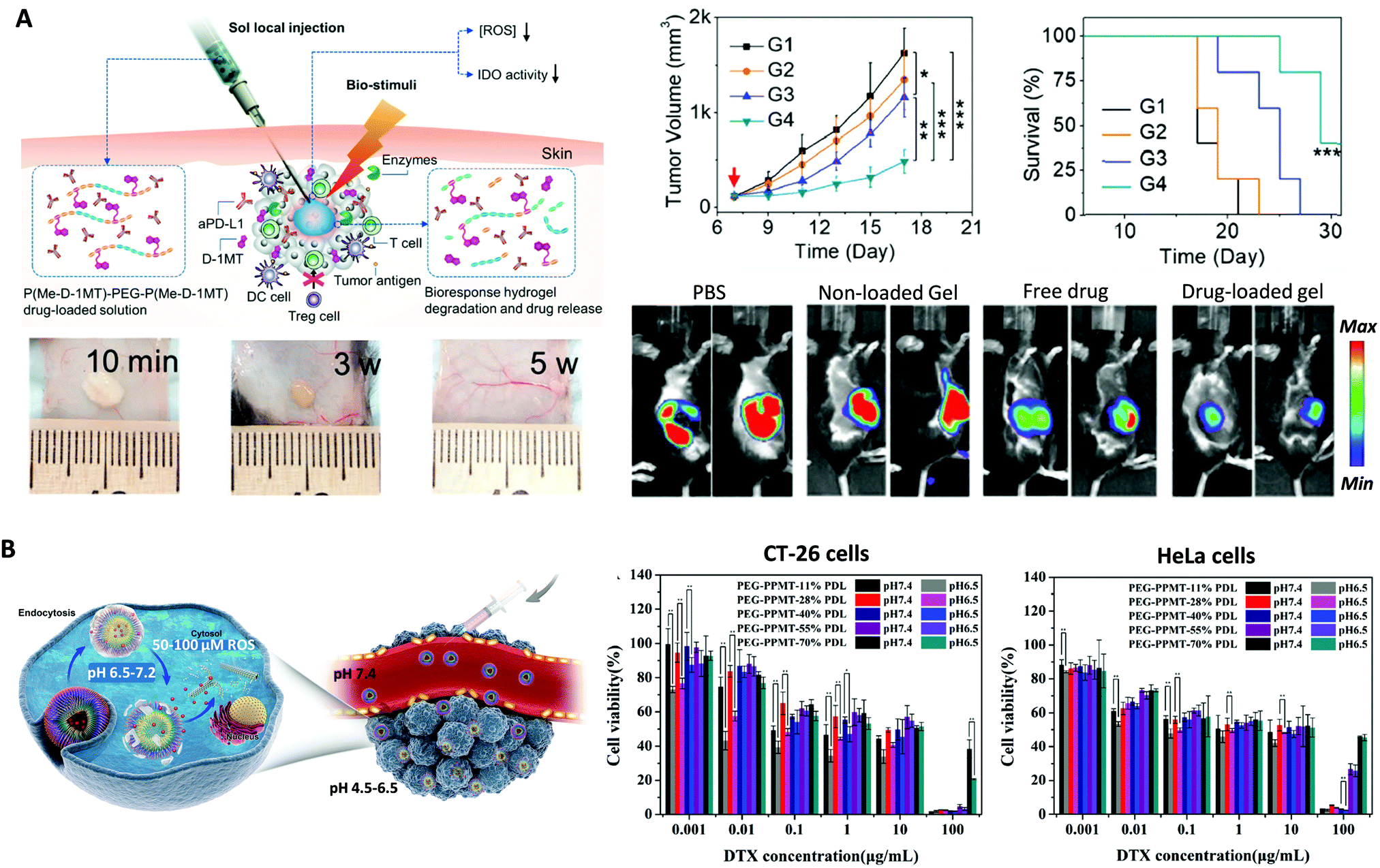 | ||
| Fig. 7 (A) Schematic illustration of localized drug loaded P(Me–D-1MT)–PEG–P(Me–D-1MT) hydrogel formation and biostimuli-triggered drug release (left-top). In vivo degradation behavior and tissue biocompatibility of the in situ-formed hydrogel (left-bottom). In vivo assays and bioluminescence imaging of the B16F10 tumors, average tumor volumes (n = 5), and average survival carves (n = 5) (right). Adapted and reprinted with permission from ref. 64 Copyright 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (B) Schematic representation of pH/ROS-triggered NPs swelling and rapid drug release upon uptake by tumor cells (left). Cytotoxicity of DTX-loaded PEG–PMMT NPs against CT-26 cells and HeLa cells (right). Data are given as mean ± SD (n = 3). Adapted and reprinted with permission from ref. 47. | ||
On the other hand, dual ROS and pH-responsive poly(ethylene glycol)-b-poly(L-lysine)-g-α-tocopheryl succinate and methionine modified with dimethylmaleic anhydride (PPT/D(DMA)) micelles loaded with DOX were fabricated. The negatively charged surface of these micelles in blood had a great ability of prolonging circulation time. They showed a charge when exposed to acidic conditions resulting in cell membrane penetration. The ROS-responsive induced disassembly gave rise to the DOX delivery to tumor cells causing their death. Moreover, α-tocopheryl succinate (TOS) segments led to an augmented intracellular ROS concentration accelerating the DOX release.73 In the case of PEG–PMMT nanoparticles, DTX was encapsulated within them to test their application in vitro with CT-26 and HeLa cells. It should be highlighted that they have excellent properties for controlled release of this anticancer drug, in a synergistic manner by acidic pH (6.5 in an extracellular tumor environment, or 5.0 in endosomes or lysosomes) and a high-ROS (100 μm H2O2) environment in tumour cells, with a 95% growth inhibition of CT-26 tumours xenografted in mice. Importantly, biosafety analyses showed minimal toxicity toward normal organs, including liver and kidneys, during in vivo antitumor treatments (Fig. 7B).47
Another strategy for the fabrication of dual responsive nanoparticles was focused on the encapsulation of a photosensitizer together with the drug leading to ROS- and light-responsive biomaterials. In this sense, nanoparticles made of ROS-responsive sulphur-containing PCLs were used as carriers of DOX and the photosensitizer Ce6. The cumulative release of DOX or Ce6 was lower than 5% in 24 h under dark conditions, whereas release values up to 40% DOX and 25% Ce6 were achieved upon red light irradiation (650 nm), with these values being higher in the case of ethylthio than phenylthio pendant groups. Thus, their activity against breast cancer cells (MCF-7) was tested in vitro showing dual photochemical induced 80% cell death upon red light irradiation, whereas normal human breast cells HBL-100 remained unaffected.41 Paclitaxel (PTX) and Ce6 have also been co-encapsulated in mPEG-b-PMSPEP NPs to achieve a photo-accelerated paclitaxel (PTX) release by the ROS generated under light irradiation. Human breast adenocarcinoma MDA-MB-231 cells were incubated with Ce6&PTX loaded NPs and then treated with or without NIR irradiation. In the absence of laser irradiation, NPs exhibited the lowest anticancer effect at each concentration, reaching a maximum of 50% cell viability reduction for 5 μg mL−1 PTX, whereas treatment with light irradiation (660 nm, 1.0 W cm−2) displayed the highest anticancer efficacy, with almost 95% cell viability reduction for 5 μg mL−1 PTX and 3 μg mL−1 Ce6. This demonstrated that the ROS-sensitive NPs could not only induce photo-triggered intracellular drug release though the hydrophobic to hydrophilic transition of the PMSPEP core but also combine chemotherapy and photodynamic therapy.37 The same pair of molecules were also incorporated within a hyperbranched polymer MTPA–TMPTGE. The results showed that HBPMT micelles loaded with Ce6 and PTX could efficiently enter MCF-7 cancer cells and display a synergic chemo-photodynamic anticancer efficacy under laser irradiation (660 nm). The results pointed to a 50% cell viability reduction for 2 μg mL−1 PTX, whereas light irradiation (660 nm) induced a 80% cell reduction showing a higher anticancer efficacy.39 Additionally, glyco-PEGylated polypeptide micelles were used as templates to generate the plasmonic composite nanoparticles and load two anticancer drugs, doxorubicin (DOX) and 6-mercaptopurine (6-MP), through physical interactions and Au–S bonds respectively, for combined chemotherapy and photo-thermal therapy. They showed near-infrared (NIR) absorption (650–1100 nm) and a temperature increase up to 30.1 °C upon continuous-wave laser irradiation (808 nm, 5 min, 2 W cm−2) for inducing the NIR-triggered cocktail drugs release. NPs were more internalized by the HepG2 than the HeLa cell line, demonstrating a LAC-targeting enhanced cytotoxicity toward HepG2, as well as the combination cocktail of chemo-photothermal therapy produced a lower half maximal inhibitory concentration than cocktail chemotherapy or photothermal therapy alone, displaying a good synergistic antitumor effect.74
The last kind of multiresponse systems are based on ROS- and GSH-responsive polymers and polypeptides. Dual-responsive amphiphilic diblock copolymer prodrug (GR-BCPs) NPs, in which the camptothecin (CPT) drug was conjugated via thioether bonds, exhibited a more efficient drug release inside tumor cells in the case of dual-responsive GR-BCPs for boosting the antitumor efficacy as compared with GSH or ROS single responsive amphiphilic diblock copolymer prodrugs (BCPs). In the presence of 10 mM GSH or 10 mM H2O2 (tumor tissues), GR-BCPs showed the fastest drug release (80% after 48 h), while negligible drug release was observed at low GSH or H2O2 concentrations (10 μm) in normal tissues. In vivo experiments were carried out in H22 mice tumor models where mouse liver cancer cells H22 could grow quickly and uniformly via subcutaneous transplantation with a 26-fold volume increase on day 18, indicating high aggressiveness of the H22 tumors. After intravenous injection of free CPT or GR-BPCs at a concentration of 20 mg kg−1, the free CPT showed very limited efficacy, with a 19.2-fold tumor volume increase, attributed to the limited tumor accumulation whereas GR-BPCs exhibited a significant low 2.9-fold tumor volume increase.51 This dual GSH and ROS responsive behaviour has also been studied in CPT-loaded mPEG-b-P(Des-a-Cys) NPs. These NPs remained stable at low levels of ROS (1 μm H2O2) and GSH (5 μm GSH) in blood circulation, with 30 wt% and 15 wt% CPT release respectively, preventing CPT toxicity in ROS/GSH balanced normal cells (e.g., fibroblast cells, etc.) or normal organs (e.g., liver, kidney, etc.), whereas it showed high redox sensitivities in cancer cells that are characterized by high levels of ROS (100 μm H2O2) or GSH (20 μm GSH) (e.g., lung, gastric, and colon cancer cells), with 97 wt% and 99 wt% CPT release respectively. The cytotoxicity of CPT loaded NPs was tested in vitro with different cancer cells, including A549 human lung adenocarcinoma epithelial cells, N87 human gastric carcinoma cells, and HCT116 human colon cancer cells. CPT loaded NPs showed a more potent toxic effect than free CPT after 48 h treatment, exhibiting IC50 values of 0.019 vs. 0.423 μg mL−1 for HCT116, 2.65 vs. 5 μg mL−1 for A549, and 0.173 vs. 5 μg mL−1 for N87 cells. In addition to this, in vivo tests were performed with mice bearing HCT116 tumors by intravenous injection of PBS, free CPT (5 mg kg−1), and CPT-loaded NPs (5 mg kg−1) with a tumor volume of ∼500 mm3. Remarkably, tumors treated with PBS and free CPT grew with time reaching a 9.3-fold increase and 4.5-fold increase after 27 days respectively. In contrast, tumor sizes injected with CPT-loaded NPs exhibited a 3.4% increase in size over 27 days and mitigated the side effects of CPT, i.e., diarrhea, nausea and vomiting.49
4.3. ROS-responsive materials for treatment of osteoporosis, diabetes, inflammatory diseases, and bio-imaging
Chen and co-workers developed mPEG–PMet hydrogels with an innate cytoprotective effect against the damage of H2O2-induced oxidative stress when they were incubated with mouse fibroblast L929 cells. The hydrogels, which showed good biocompatibility in vivo, were degraded within 6 weeks after subcutaneous injection into rats. These results suggested their potential employment as platforms for sustained drug delivery and cell-based therapies in the treatment of inflammatory diseases with local oxidative stress.66In another case, PEGylated polysulfide (PPSES–PEG) micelles loaded with antiosteoclastic rapamycin were used as synergic nanocarriers for ROS-scavenging and ROS-responsive drug release inhibiting the differentiation of inflammatory cells into osteoclasts to be employed in potential therapy against osteoporosis. It is important to highlight the key role of the ratio between oxidizable groups (amount of therapeutic/antioxidant agent) and oxidants (ROS level) at the action site to determine the efficacy of the treatment.42
ROS-responsive properties can also be affected by the encapsulation of enzymes within the ROS-responsive polymer NPs or hydrogels. To that aim, glucose oxidase (GOx) is a good candidate as it can catalyze the oxidation of glucose in the presence of oxygen producing gluconic acid and H2O2. In this sense, the hypoxia and H2O2 dual-sensitive diblock copolymer PEG–poly(Ser-S-NI) was employed to develop a glucose-responsive smart insulin patch, using a painless microneedle-array patch containing insulin-loaded vesicles (d-GRPs), which mimics pancreas activity without long-term side effects to improve diabetic patients’ health and quality of life. The glucose-responsive d-GRPs were disassociated, by H2O2 action and hypoxia generated during glucose oxidation catalyzed by GOx, releasing insulin. When d-GRPs vesicles were brought in contact with different glucose concentrations of glucose, including a typical hyperglycemic level (400 mg dL−1) and a normoglycemia level (100 mg dL−1), the results showed rapid oxygen consumption (in 20 min) at hyperglycemic levels due to the oxidation of glucose catalyzed by GOx. Nevertheless, a significantly slower oxygen consumption rate was observed in the case of normoglycemia levels. This lower oxygen level in d-GRPs vesicles was attributed to the effective H2O2 elimination ability of PEG–poly(Ser-S-NI) avoiding the GOx deactivation. Moreover, in vivo experiments proved the efficiency of this patch, loaded with 10 mg kg−1 insulin and 3 mg kg−1 GOx, to regulate the blood glucose in the chemically induced type 1 diabetic mice for 10 h (Fig. 8), whereas the healthy tissues remained unaffected.75
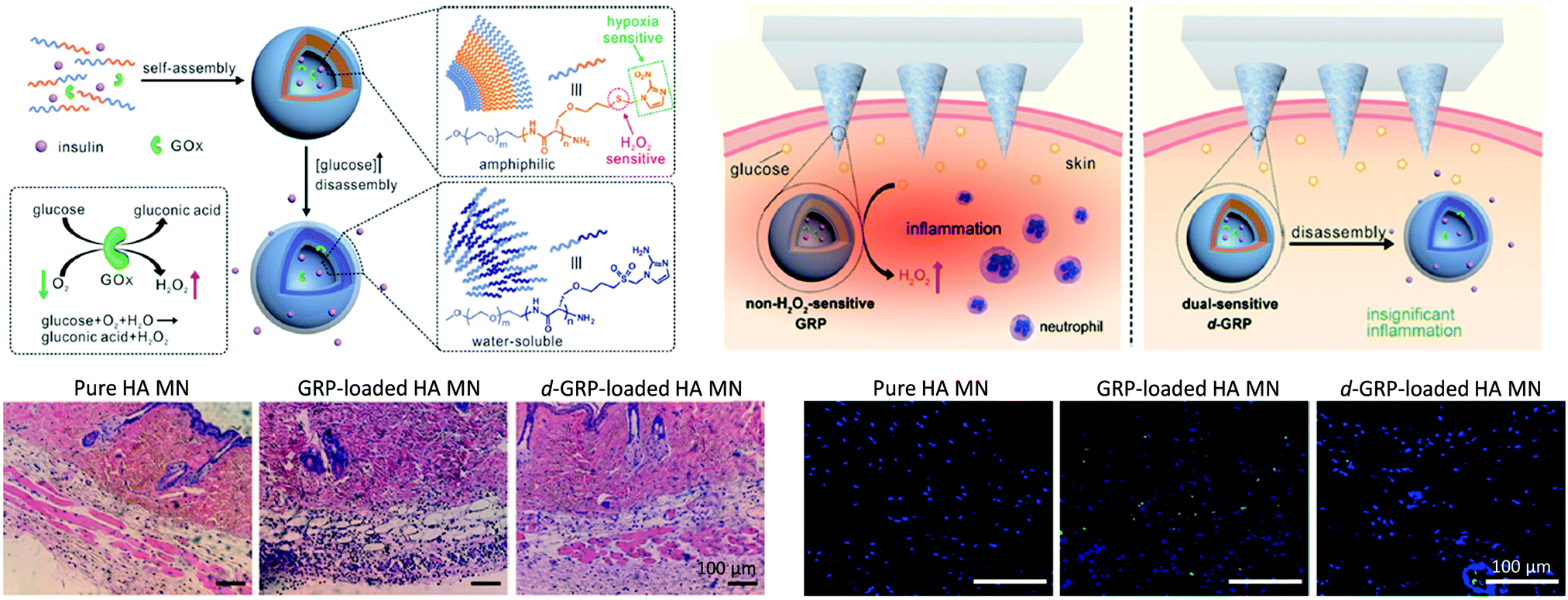 | ||
| Fig. 8 Scheme of the hypoxia and H2O2 dual-sensitive polymersome-based vesicles (d-GRPs) comprised of PEG–poly(Ser-S-NI), and d-GRP-loaded microneedle-array patch for in vivo insulin delivery triggered by a hyperglycemic (top). H&E stained images of mouse skin tissue H&E stained images and histology stain with TUNEL assay (green) and Hoechst (blue) (bottom). Adapted and reprinted with permission from ref. 75 Copyright 2017, American Chemical Society. | ||
The imaging of ROS is crucial to early detection and treatment of many life-threatening diseases. Europium(III) CPs can also act as fluorescence probes for selective monitoring of ROS in biological and environmental systems. The fluorescence of the S-tagged europium(III) CPs can be selectively quenched by ROS for sensitive and selective monitoring of ROS in live cells.48
5. Conclusions and perspectives
This article aimed to present a comprehensive overview of thioether-based polymer and polypeptide materials with the ability to respond to reactive oxygen species (ROS) present in the human body, a research topic that has attracted increasing attention in the biomedical field in the last two decades. A summary of all studies referenced here is collected in Table 1. Different amphiphilic thioether-based ROS sensitive polymers have been developed by incorporation of this functional group in the main, side or tail chains to confer tuneable self-assembling properties, from micellar-type nanoparticles (NPs) to hydrogels, and ROS sensitive features. Taking advantage of the oxidative nature of the thioether group, these materials possess the ability to be oxidized in the presence of ROS experiencing a hydrophobic to hydrophilic phase transition very interesting for the development of ROS responsive therapeutics.| Polymer/polypeptide | Drug | Self-assembly structure | Stimuli-response | Biomedical application | Ref. |
|---|---|---|---|---|---|
| mPEG-b-PS, mPEG-b-OPS | Nile red | Nanoparticles | ROS (100–400 mM H2O2) | Drug delivery | 29 |
| mPEG-b-PTE, mPEG-b-PCL | Nile red | Nanoparticles | ROS (300 mM H2O2) | Drug delivery | 32 |
| P(MTEA-b-DMA), P(MTPA-b-DMA) | Nile red | Nanoparticles | ROS (10–100 mM H2O2) | Drug delivery anti-inflammatory therapies, bacterial infections | 40 |
| PEG–PMTEGE, PEG-b-PMTEGE | Nile red | Nanoparticles | ROS (300 mM H2O2) | Drug delivery | 38 |
| P(MSPA-a-EG) | DOX | Nanoparticles | ROS (5–20 mM H2O2) | Drug delivery for cancer therapy | 36 |
| DSPE–PEG | DOX | Nanoparticles | ROS | Drug delivery | 50 |
| ML–PLGLAG–PEG | DOX | Nanoparticles | ROS (0.1–1 mM H2O2) | Drug delivery lung cancer therapy | 60 |
| DSPE–PEG | DOX | Nanoparticles, liposomes | ROS (10 mM H2O2) | Drug delivery cancer therapy | 52 |
| PEG-b-P(L-EG2MA-C) | DOX | Nanoparticles Hydrogel | ROS (5–10% H2O2, 36 μmol L−1 FeSO4) | Drug delivery cancer therapy | 68 |
| t-PEG-b-OMet | DOX | Hydrogel | ROS (10 mM H2O2) | Drug delivery cancer therapy | 65 |
| PDEGDA | PL | Nanoparticles | ROS (100 mM H2O2) | Drug delivery breast cancer therapy | 72 |
| PPSES–PEG | Rapamycin | Nanoparticles | ROS (0.05–1 mM H2O2) | Drug delivery for anti-inflammatory therapy and osteoporosis | 42 |
| Met–PEA–PEG | GA | Nanoparticles | ROS (200 mM H2O2) | Drug delivery prostate cancer therapy | 59 |
| PEG–P(Ser-S-NI) | Insulin, GOx | Nanoparticles | ROS (5 μM H2O2, 3 mg kg−1 GOx) | Drug delivery for anti-inflammatory and diabetes therapies | 75 |
| PEO–PMET | — | Nanoparticles | ROS (0.1–10 mM H2O2) | Scaffolds for tissue engineering | 63 |
| mPEG–PMet | Rhodamine 6G | Hydrogel | ROS (1–10 mM H2O2) | Drug delivery | 66 |
| PAAs | — | Hydrogel | ROS (5% H2O2) | Scaffolds for tissue engineering | 61 |
| PPS–PNIPAm | DOX | Nanoparticles | ROS (300 mM H2O2), temperature (37 °C) | Drug delivery breast cancer therapy | 43 |
| P(Me–D-1MT)-PEG–P(Me–D-1MT) | aPD-L1, D-1MT | Hydrogel | ROS (10 mM H2O2), temperature (37 °C) | Drug delivery melanoma cancer therapy | 64 |
| PPT/D(DMA) | DOX, TOS | Nanoparticles | ROS (0.1–10 mM H2O2), pH (7.4 – 5.5) | Drug delivery cancer therapy | 73 |
| PEG–PMT | DTX | Nanoparticles | ROS (100 μM H2O2), pH (7.4–5.0) | Drug delivery colon cancer therapy | 71 |
| PEG–P(PDL-co-MDEA-co-TDP) | DTX | Nanoparticles | ROS (100 μM H2O2), pH (7.4–5.0) | Dual delivery cervical and colon cancer therapy | 47 |
| PEG-b-PMNT | — | Nanoparticles | ROS (0.5 μM H2O2, 0.1 mM FeSO4), pH | Radioprotective agent | 46 |
| P(PDL-co-MS-co-TS) | — | Hydrogel | ROS (30–70 mM H2O2), pH (7.2–5.4) | Dynamic scaffolds redox environments | 67 |
| mPEG-b-575EDT-b-mPEG | Nile red | Nanoparticles | ROS (300 mM H2O2), pH, temperature (37 °C) | Drug delivery | 44 |
| P(α-ethylthio-CL), P(α-phenylthio-CL) | DOX, Ce6 | Nanoparticles | ROS (5 mM H2O2), light (650 nm, 0.1 W cm−2) | Chemo-phototherapy for cancer | 41 |
| mPEG-b-PMSPEP | PTX, Ce6 | Nanoparticles | ROS (1.2–18.8 mM H2O2), light (660 nm, 1.0 W cm−2) | Dual chemo-photodynamic cancer therapy | 37 |
| MTPA + TMPTGE | PTX, Ce6 | Nanoparticles | ROS (25.7 mM H2O2), light (660 nm) | Dual chemo-photodynamic cancer therapy | 39 |
| S–Eu(III)–CPs | — | Nanoparticles | ROS (0.5 mM H2O2), light (617 nm) | Biosensors and bio-imaging agents | 48 |
| PC-g-PEG–LAC | DOX, 6-MP | Nanoparticles | ROS, photo-thermal | Dual chemo-photothermal cancer therapy | 74 |
| P(EDDT–DAP), P(EDDT–DAA) | — | Nanoparticles | ROS (3 M H2O2), HCl (pH 2) | Dual responsive scaffolds | 33 |
| PEG-b-PGRCPT, PEG-b-PGCPT, PEG-b-PRCPT | CPT | Nanoparticles | ROS (10 mM H2O2), GSH (10 mM) | Dual drug delivery cervical cancer therapy | 51 |
| mPEG-b-P(Des-a-Cys) | CPT | Nanoparticles | ROS (1–100 μM H2O2), GSH (1–20 μM) | Dual drug delivery lung, gastric, and colon cancer therapy | 49 |
| P(EGDMA–DSH) | — | Nanoparticles | ROS (300 mM H2O2), enzymes (10 U esterase) | Dual responsive scaffolds | 34 |
In addition to this, the thioether functionality is a promising candidate in the biomaterials field as it is present in the amino acid L-methionine and in proteins through alkylation of L-cysteine. Interestingly, the incorporation of L-methionine amino acids within the polymer chain allows the production of polypeptides with reversibility properties after oxidation to methionine sulfoxide by the action of reductase enzymes present in the human body, which allows the cell functions to be regulated maintaining normal operation. Therefore, thioether-containing amino acids can be used for chemoselective modification and conjugation of functional polypeptides giving rise to new materials with additional functionalities taking advantage of the features of both components.
Apart from single ROS responsive materials, the design of dual responsive polymers plays a key role in the development of multiresponsive materials allowing the functionality and final applications of the developed nanoparticles and hydrogels to be enhanced and/or enlarged. Among them, those combining dual ROS- and thermo-, or pH-, or light-, or enzyme-responsive behaviours have started to be investigated although it is a less explored field opening the route for future investigations. In this sense, multiresponsive thioether-based polypeptide materials could be ideal candidates as they can combine polymer and peptides properties giving rise to a plethora of ROS responsive materials for target therapeutic applications.
On the other hand, most applications are focused on the encapsulation of anticancer drugs, i.e., DOX, DTX, and PTX, within the self-assembled NPs and hydrogels for cancer therapies, and only a few examples show the employment of these materials as carriers of other specific molecules or enzymes such as glucose oxidase (GOx) to widen the employment of these materials for other diseases like diabetes and osteoporosis, as well as for bio-imaging and bio-sensing purposes for early detection and treatment of diseases.
Overall, although significant progress has been accomplished in this field, the scarce literature on this topic makes it very attractive opening a myriad of opportunities to explore and paving the way for the development of the next-generation of multiresponsive materials for target biomedical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by EU Horizon 2020 FETOPEN-2018–2020 Programme “LION-HEARTED”, Grant No. 828984.Notes and references
- Y. Cheng, X. Jiao, T. Xu, W. Wang, Y. Cao, Y. Wen and X. Zhang, Small, 2017, 13, 1701942 CrossRef PubMed.
- L. Romero-Azogil, E. Benito, N. Iglesias, E. Galbis, M. V. de-Paz and M. G. García-Martín, in Redox Polymers for Energy and Nanomedicine, The Royal Society of Chemistry, 2021, pp. 415–453 Search PubMed.
- J. Y. Oh and Z. Bao, Adv. Sci., 2019, 6, 1900186 CrossRef PubMed.
- A. Gelmi and C. E. Schutt, Adv. Healthcare Mater., 2021, 10, 2001125 CrossRef CAS PubMed.
- M. Criado-Gonzalez, L. Corbella, B. Senger, F. Boulmedais and R. Hernández, Langmuir, 2019, 35, 11397–11405 CrossRef CAS PubMed.
- L. C. Tomé, L. Porcarelli, J. E. Bara, M. Forsyth and D. Mecerreyes, Mater. Horiz., 2021, 8, 3239–3265 RSC.
- E. Bihar, T. Roberts, E. Ismailova, M. Saadaoui, M. Isik, A. Sanchez-Sanchez, D. Mecerreyes, T. Hervé, J. B. De Graaf and G. G. Malliaras, Adv. Mater. Technol., 2017, 2, 1600251 CrossRef.
- G. Kocak, C. Tuncer and V. Bütün, Polym. Chem., 2017, 8, 144–176 RSC.
- Y. Kotsuchibashi, Polym. J., 2020, 52, 681–689 CrossRef CAS.
- M. Criado-Gonzalez, J. Rodon Fores, D. Wagner, A. P. Schröder, A. Carvalho, M. Schmutz, E. Harth, P. Schaaf, L. Jierry and F. Boulmedais, Chem. Commun., 2019, 55, 1156–1159 RSC.
- M. Criado-Gonzalez, B. Loftin, J. Rodon Fores, D. Vautier, L. Kocgozlu, L. Jierry, P. Schaaf, F. Boulmedais and E. Harth, J. Mater. Chem. B, 2020, 8, 4419–4427 RSC.
- C. Maerten, L. Jierry, P. Schaaf and F. Boulmedais, ACS Appl. Mater. Interfaces, 2017, 9, 28117–28138 CrossRef CAS PubMed.
- N. Tyagi, K. Gambhir, S. Kumar, G. Gangenahalli and Y. K. Verma, J. Mater. Sci., 2021, 56, 16790–16823 CrossRef CAS.
- A. Görlach, E. Y. Dimova, A. Petry, A. Martínez-Ruiz, P. Hernansanz-Agustín, A. P. Rolo, C. M. Palmeira and T. Kietzmann, Redox Biol., 2015, 6, 372–385 CrossRef PubMed.
- B. D'Autréaux and M. B. Toledano, Nat. Rev. Mol. Cell Biol., 2007, 8, 813–824 CrossRef PubMed.
- A. N. Kolodkin, R. P. Sharma, A. M. Colangelo, A. Ignatenko, F. Martorana, D. Jennen, J. J. Briedé, N. Brady, M. Barberis, T. D.-G. A. Mondeel, M. Papa, V. Kumar, B. Peters, A. Skupin, L. Alberghina, R. Balling and H. V. Westerhoff, NPJ Syst. Biol. Appl., 2020, 6, 34 CrossRef PubMed.
- R. L. Auten and J. M. Davis, Pediatr. Res., 2009, 66, 121–127 CrossRef CAS PubMed.
- M. J. Donahue, A. Sanchez-Sanchez, S. Inal, J. Qu, R. M. Owens, D. Mecerreyes, G. G. Malliaras and D. C. Martin, Mater. Sci. Eng., R, 2020, 140, 100546 CrossRef.
- M. Criado-Gonzalez, E. Espinosa-Cano, L. Rojo, F. Boulmedais, M. R. Aguilar and R. Hernández, ACS Appl. Mater. Interfaces, 2022, 14, 10068–10080 CrossRef CAS PubMed.
- D. Chow, M. L. Nunalee, D. W. Lim, A. J. Simnick and A. Chilkoti, Mater. Sci. Eng., R, 2008, 62, 125–155 CrossRef PubMed.
- Q. Xu, C. He, C. Xiao and X. Chen, Macromol. Biosci., 2016, 16, 635–646 CrossRef CAS PubMed.
- D. M. Townsend, K. D. Tew and H. Tapiero, Biomed. Pharmacother., 2004, 58, 47–55 CrossRef CAS PubMed.
- T. J. Deming, Bioconjugate Chem., 2017, 28, 691–700 CrossRef CAS PubMed.
- W. Vogt, Free Radical Biol. Med., 1995, 18, 93–105 CrossRef CAS PubMed.
- A. R. Rodriguez, J. R. Kramer and T. J. Deming, Biomacromolecules, 2013, 14, 3610–3614 CrossRef CAS PubMed.
- C.-C. Song, F.-S. Du and Z.-C. Li, J. Mater. Chem. B, 2014, 2, 3413–3426 RSC.
- C.-J. Zhang, T.-C. Zhu, X.-H. Cao, X. Hong and X.-H. Zhang, J. Am. Chem. Soc., 2019, 141, 5490–5496 CrossRef CAS PubMed.
- A. Napoli, M. Valentini, N. Tirelli, M. Müller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed.
- B. Yan, Y. Zhang, C. Wei and Y. Xu, Polym. Chem., 2018, 9, 904–911 RSC.
- A. Dominguez-Alfaro, M. Criado-Gonzalez, E. Gabirondo, H. Lasa-Fernández, J. L. Olmedo-Martínez, N. Casado, N. Alegret, A. J. Müller, H. Sardon, A. Vallejo-Illarramendi and D. Mecerreyes, Polym. Chem., 2022, 13, 109–120 RSC.
- O. S. Manoukian, M. R. Arul, N. Sardashti, T. Stedman, R. James, S. Rudraiah and S. G. Kumbar, J. Appl. Polym. Sci., 2018, 135, 46068 CrossRef PubMed.
- W.-X. Wu, X.-L. Yang, B.-Y. Liu, Q.-F. Deng, M.-M. Xun, N. Wang and X.-Q. Yu, RSC Adv., 2016, 6, 11870–11879 RSC.
- F. Jasinski, A. Rannée, J. Schweitzer, D. Fischer, E. Lobry, C. Croutxé-Barghorn, M. Schmutz, D. Le Nouen, A. Criqui and A. Chemtob, Macromolecules, 2016, 49, 1143–1153 CrossRef CAS.
- S. H. Hong, T. Patel, S. Ip, S. Garg and J. K. Oh, Langmuir, 2018, 34, 3316–3325 CrossRef CAS PubMed.
- M. Geven, R. d'Arcy, Z. Y. Turhan, F. El-Mohtadi, A. Alshamsan and N. Tirelli, Eur. Polym. J., 2021, 149, 110387 CrossRef CAS.
- G. Wang, P. Huang, M. Qi, C. Li, W. Fan, Y. Zhou, R. Zhang, W. Huang and D. Yan, ACS Omega, 2019, 4, 17600–17606 CrossRef CAS PubMed.
- J. Wang, D. Li, W. Tao, Y. Lu, X. Yang and J. Wang, Biomacromolecules, 2019, 20, 1740–1747 CrossRef CAS PubMed.
- J. Herzberger, K. Fischer, D. Leibig, M. Bros, R. Thiermann and H. Frey, J. Am. Chem. Soc., 2016, 138, 9212–9223 CrossRef CAS PubMed.
- G. Wang, P. Huang, L. Wang, X. Chen, Y. Zhou, W. Huang and D. Yan, SmartMat, 2022, 1–10 Search PubMed.
- J. Y. Quek, P. R.-L. Dabare, R. Bright, A. Postma and K. Vasilev, Mater. Today Chem., 2021, 20, 100444 CrossRef CAS.
- L. Yu, M. Zhang, F.-S. Du and Z.-C. Li, Polym. Chem., 2018, 9, 3762–3773 RSC.
- F. El Mohtadi, R. d’Arcy, J. Burke, J. M. Rios De La Rosa, A. Gennari, R. Marotta, N. Francini, R. Donno and N. Tirelli, Biomacromolecules, 2020, 21, 305–318 CrossRef CAS PubMed.
- M. Tang, P. Hu, Q. Zheng, N. Tirelli, X. Yang, Z. Wang, Y. Wang, Q. Tang and Y. He, J. Nanobiotechnol., 2017, 15, 39 CrossRef PubMed.
- C. Xiao, J. Ding, L. Ma, C. Yang, X. Zhuang and X. Chen, Polym. Chem., 2015, 6, 738–747 RSC.
- P. C. Mattos dos Santos, P. E. Feuser, P. B. Cardoso, B. T. Steiner, E.-d S. Córneo, R. Scussel, A.-d C. Viegas, R. A. Machado-de-Ávila, C. Sayer and P. H. Hermes de Araújo, J. Biomater. Sci., Polym. Ed., 2018, 29, 1935–1948 CrossRef CAS PubMed.
- C. P. Feliciano, K. Tsuboi, K. Suzuki, H. Kimura and Y. Nagasaki, Biomaterials, 2017, 129, 68–82 CrossRef CAS PubMed.
- Y.-h Gong, M. Shu, J.-h Xie, C. Zhang, Z. Cao, Z.-z Jiang and J. Liu, J. Mater. Chem. B, 2019, 7, 651–664 RSC.
- H.-S. Wang, W.-J. Bao, S.-B. Ren, M. Chen, K. Wang and X.-H. Xia, Anal. Chem., 2015, 87, 6828–6833 CrossRef CAS PubMed.
- Y.-T. Chiang, Y.-W. Yen and C.-L. Lo, Biomaterials, 2015, 61, 150–161 CrossRef CAS PubMed.
- Y. Yang, B. Sun, S. Zuo, X. Li, S. Zhou, L. Li, C. Luo, H. Liu, M. Cheng and Y. Wang, Sci. Adv., 2020, 6, eabc1725 CrossRef CAS PubMed.
- W. Yin, W. Ke, N. Lu, Y. Wang, A. A.-W. M.-M. Japir, F. Mohammed, Y. Wang, Y. Pan and Z. Ge, Biomacromolecules, 2020, 21, 921–929 CrossRef CAS PubMed.
- Y. Du, W. He, Q. Xia, W. Zhou, C. Yao and X. Li, ACS Appl. Mater. Interfaces, 2019, 11, 37411–37420 CrossRef CAS PubMed.
- C. G. Pappas, R. Shafi, I. R. Sasselli, H. Siccardi, T. Wang, V. Narang, R. Abzalimov, N. Wijerathne and R. V. Ulijn, Nat. Nanotechnol., 2016, 11, 960–967 CrossRef CAS PubMed.
- H. Wang, Z. Feng and B. Xu, Angew. Chem., Int. Ed., 2019, 58, 10423–10432 CrossRef CAS PubMed.
- L. Grassi and C. Cabrele, Amino Acids, 2019, 51, 1409–1431 CrossRef CAS PubMed.
- B. Sjöberg, S. Foley, B. Cardey, M. Fromm and M. Enescu, J. Photochem. Photobiol., B, 2018, 188, 95–99 CrossRef PubMed.
- B. P. Nowak, L. Schlichter and B. J. Ravoo, Angew. Chem., Int. Ed., 2022, 134, e202201791 CrossRef.
- N. Makukhin, V. Havelka, E. Poláchová, P. Rampírová, V. Tarallo, K. Strisovsky and J. Míšek, FEBS J., 2019, 286, 4024–4035 CrossRef CAS PubMed.
- Q. Xu and C.-C. Chu, J. Biomed. Mater. Res., Part A, 2021, 109, 524–537 CrossRef CAS PubMed.
- J. Yoo, N. Sanoj Rejinold, D. Lee, S. Jon and Y.-C. Kim, J. Controlled Release, 2017, 264, 89–101 CrossRef CAS PubMed.
- X. Fan, S. Zhu, L. Yan and H. Zhu, J. Appl. Polym. Sci., 2021, 138, 51386 CrossRef CAS.
- S. Kumar, G. Hause and W. H. Binder, ACS Omega, 2020, 5, 19020–19028 CrossRef CAS PubMed.
- A. Duro-Castano, L. Rodríguez-Arco, L. Ruiz-Pérez, C. De Pace, G. Marchello, C. Noble-Jesus and G. Battaglia, Biomacromolecules, 2021, 22, 5052–5064 CrossRef CAS PubMed.
- S. Yu, C. Wang, J. Yu, J. Wang, Y. Lu, Y. Zhang, X. Zhang, Q. Hu, W. Sun, C. He, X. Chen and Z. Gu, Adv. Mater., 2018, 30, 1801527 CrossRef PubMed.
- D. Zhao, Q. Zhou, K. Yang, H. Yang, Q. Tang and X. Zhang, Macromol. Chem. Phys., 2019, 220, 1900106 CrossRef.
- Q. Xu, C. He, K. Ren, C. Xiao and X. Chen, Adv. Healthcare Mater., 2016, 5, 1979–1990 CrossRef CAS PubMed.
- D. Spitzer, L. L. Rodrigues, D. Straßburger, M. Mezger and P. Besenius, Angew. Chem., Int. Ed., 2017, 56, 15461–15465 CrossRef CAS PubMed.
- X. Fu, Y. Ma, Y. Shen, W. Fu and Z. Li, Biomacromolecules, 2014, 15, 1055–1061 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2012, 134, 4112–4115 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2014, 136, 5547–5550 CrossRef CAS PubMed.
- M. Su, S. Xiao, M. Shu, Y. Lu, Q. Zeng, J. Xie, Z. Jiang and J. Liu, Colloids Surf., B, 2020, 193, 111067 CrossRef CAS PubMed.
- Y.-S. Kim, S. Kim, H. C. Kang and M. S. Shim, J. Ind. Eng. Chem., 2019, 75, 238–245 CrossRef CAS.
- X. Zhang, T. Zhu, Y. Miao, L. Zhou and W. Zhang, J. Nanobiotechnol., 2020, 18, 136 CrossRef CAS PubMed.
- X. Wu, L. Zhou, Y. Su and C.-M. Dong, Biomacromolecules, 2016, 17, 2489–2501 CrossRef CAS PubMed.
- J. Yu, C. Qian, Y. Zhang, Z. Cui, Y. Zhu, Q. Shen, F. S. Ligler, J. B. Buse and Z. Gu, Nano Lett., 2017, 17, 733–739 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |