
Spatiotemporal control over 3D protein nanocage superlattices for the hierarchical encapsulation and release of different cargo molecules†
Xiaorong
Zhang
,
Ruiqi
Zeng
,
Tuo
Zhang
 ,
Chenyan
Lv
,
Jiachen
Zang
* and
Guanghua
Zhao
*
,
Chenyan
Lv
,
Jiachen
Zang
* and
Guanghua
Zhao
*
College of Food Science & Nutritional Engineering, China Agricultural University, Beijing Key Laboratory of Functional Food from Plant Resources, Beijing 100083, China. E-mail: zangjiachen@cau.edu.cn; gzhao@cau.edu.cn
First published on 25th November 2022
Abstract
Taking inspiration from Nature, we have constructed a two-compartment system based on 3D ferritin nanocage superlattices, the self-assembly behavior of which can be spatiotemporally controlled using two designed switches. One pH switch regulates the assembly of the ferritin subunit into its shell-like structure, whereas the other metal switch is responsible for assembly of the 3D superlattices from ferritin nanocages as building blocks. Consequently, this system holds great promise for the hierarchical encapsulation and release of two different cargo molecules.
Compartmentalization is one of the intelligent principles of Nature that is critical to the evolution of eukaryotic cells, and instrumental in the origin of life.1,2 Compartmentalization, also known as “site isolation”, enables the spatial separation of biomolecules and processes, leading to the possibility of spatially separating reaction pathways or incompatible components.3,4 Eukaryotic cells are multi-compartmentalized structures where each compartment performs different life activities, thus providing the cell with spatiotemporal control over informational and metabolic processing, which is of vital significance for cell complexity and efficiency.5 Following Nature's inspiration of constructing compartmentalized structures, chemists have attempted to use colloidosome- and emulsion-based systems to either build artificial cell mimics for compartmentalizing compounds in order to isolate incompatible reagents or to create artificial multicellular systems.6–10 Although these attempts have led to encouraging results, these methods have been applied only in particular cases and are not very versatile. So far, the supramolecular self-assembly of biomolecules has provided a powerful bottom-up strategy for building multi-compartment materials. However, the use of 3D protein frameworks for constructing artificial cell mimics, to compartmentalize incompatible reagents, has been largely unexplored.
Cage-like proteins such as viral capsids, ferritins, sHSP and Dps are hollow, well-defined nanoparticles that are widely distributed in animals, plants, and microorganisms, and which serve as multipurpose containers for nucleic acid transport and delivery, iron mineralization and CO2 fixation.11–13 Nowadays, protein nanocages have been repurposed by using their interior cavities, intended for the encapsulation of cargo molecules.14–17 Furthermore, recently these protein nanocages have been utilized as nanoscale building blocks to implement biomimetic approaches directed toward the fabrication of 3D superlattices using various strategies, such as key interface redesign regulated protein assembly,18–21 metal-directed protein assembly,22–25 effector-mediated protein assembly,26–30 and crystallization mediated protein assembly.31 Importantly, these reported 3D protein nanocage superlattices contain two different types of spaces, i.e., the inherent inner cavity of the protein nanocage and the interglobular cavities of the lattice, and are therefore referred to as two-compartment systems. Recently, the interglobular lattice cavities have also been used to encapsulate guest molecules.32 Furthermore, protein nanocage superlattices have served as a platform for encapsulating different molecules, either both in the inner cavity,33 or one in the inner cavity and the other in interglobular cavity.34 By contrast, the hierarchical encapsulation of two different cargoes through spatiotemporal control over the self-assembly behavior of the two-compartment systems remains a challenge.
To overcome these shortcomings, here we have built a two-compartment system using ferritin nanocages as the building blocks. By introducing two switches (pH and metal switches) to the system nearby the C4 interfaces, the self-assembly behavior can be spatiotemporally controlled for the hierarchical encapsulation and release of two different cargo molecules. First, via pH-induced reversible self-assembly of the ferritin nanocage, one molecule, rhodamine 6G (R6G), was encapsulated within its inherent inner cavity. Second, via the metal switch, the R6G-containing ferritin nanocages self-assemble into 3D superlattices; during this process, another molecule, BHQ-2 (BHQ = black hole quencher), which is the quencher of R6G, can be entrapped within the interglobular cavity of the superlattice. Conversely, via removal of the binding metal ions with EDTA, followed by adjusting the pH to 3.5, these two molecules can be released in a step-by-step manner (Fig. 1). The whole process demonstrates a hierarchical encapsulation and release procedure, while the choice of R6G and BHQ-2 enables the whole process to be visible.
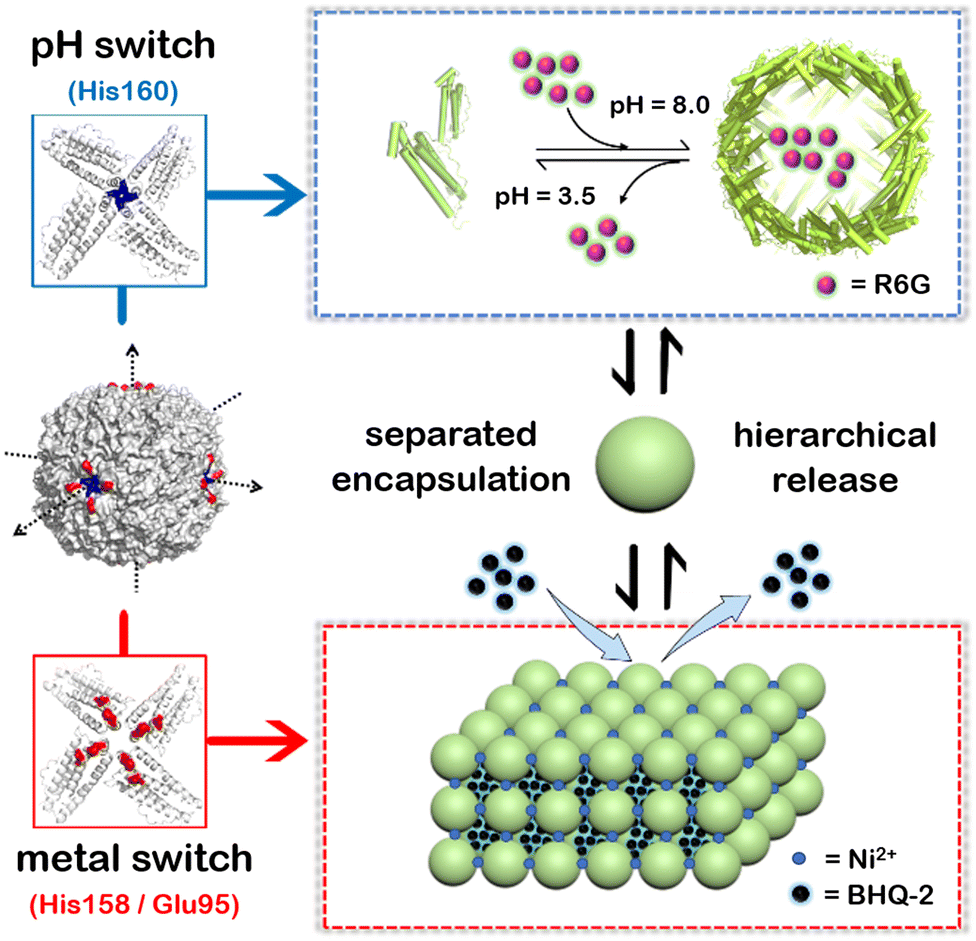 | ||
| Fig. 1 Schematic illustration of a two-compartment system based on 3D protein nanocage lattices for the hierarchical encapsulation and release of two different cargoes through the design of pH and metal switches. The pH switch is composed of the His160 motif located in the C4 channel of ferritin, while the metal switch consists of Glu95 and His158. R6G molecules are first loaded within the inner cavity of the ferritin nanocage through the pH switch, and then BHQ-2 molecules are encapsulated in the interglobular cavities of the ferritin lattice controlled via the metal switch. Conversely, by eliminating metal ions using EDTA and then adjusting the pH to 3.5, the BHQ-2 and R6G molecules are released successively. | ||
In this work, we selected recombinant Marsupenaeus japonicus ferritin (MjFer) as the building blocks due to its easy purification, high yield, and known crystal structure. Like other ferritins, MjFer comprises 24 identical subunits assembling into a quasi-spherical shell with an outer diameter of 12 nm and an inner diameter of 8 nm.35,36 Recently, we made an MjFer mutant via single mutation, in which Thr158 was replaced by histidine (T158HMjFer), that can self-assemble into 3D protein nanocage arrays in response to Ni2+.37 Since the 3D protein assembly is mainly controlled by Ni2+ coordination, it is hardly affected by pH changes in solution; by contrast, the pH has a considerable effect on the assembly of our other recently constructed 3D protein nanocage frameworks via hydrophobic or π–π stacking interactions.18,19 In addition, the designed His158 in the mutant T158HMjFer is located on the outer surface nearby the C4 channels, which is also involved in the above 3D protein assembly through π–π interactions.37 To simplify the design challenge, we used T158HMjFer as the starting material for further design.
First, we planned to design a pH switch to regulate the assembly of the ferritin nanocage itself. Although a pH of ∼2.0 or 11.0 has been widely utilized to control the disassembly and reassembly of ferritin nanocages for the encapsulation of cargo(es) within the inner cavity of ferritin, up to ∼60% of the total protein is denatured or damaged during this process because of such harsh pH conditions, as demonstrated by our group and other research groups.38–40 Inspired by the versatile properties of histidine (His) motifs (pH-controlled protonation and coordination with transition metal ions such as Ni2+) and the higher symmetry of the C4 channels, we attempted to insert a His motif as a pH switch into the C4 channels of ferritin based on the following considerations. First, the incorporation of a His motif into the C4 channels would cause the ferritin molecules to disassemble at a pH value higher than 2.0 due to electrostatic repulsion, thereby avoiding acidic denaturation of the protein. Second, the designed His motifs could also serve as ligands to coordinate with metal ions, bridging four subunits together along the C4 rotation channels, thereby strengthening the ferritin shell-like structure for its further 3D assembly upon treatment with Ni2+. Thus, the inserted His residue(s) could play a dual role in the designed 3D protein assembly. Third, the ferritin nanocage with, octahedral symmetry, usually comprises three kinds of channel (C2, C3, and C4). Among them, C4 has the largest symmetry. More importantly, three C4 axes of ferritin closely resemble the X–Y–Z coordinate axes when the cavity center is set as the origin of the coordinates, and hence such symmetry enables the ferritin nanocages to assemble into 3D protein lattices with a simple cubic structure, as shown in Fig. 1.
Analysis of the crystal structure of T158HMjFer (PDB: 6LRX) revealed that Leu160 is an ideal position for inserting the His motif, because the side chain of Leu160 protrudes into the C4 channels of MjFer, thereby causing electrostatic repulsion to trigger a protein disassociation reaction. To this end, one new mutant was prepared, in which Leu160 was replaced by His, and is referred to as H158/H160MjFer (Fig. S1, ESI†). After overexpression in E. coli, we purified it to homogeneity as suggested by native-PAGE and SDS-PAGE (Fig. S2a and b, ESI†). Similarly, results showed that the designed H158/H160MjFer nanocages stay in a monodisperse state at pH 8.0 (Fig. S2c, ESI†). Subsequently, the pH-controlled self-assembly properties of H158/H160MjFer were investigated. Interestingly, dynamic light scattering (DLS) analysis showed that the shell-like structure of H158/H160MjFer disassembled at pH 3.5, while T158HMjFer remained stable under such conditions (Fig. S3, ESI†). Thus, the newly fabricated mutant is able to disassemble under relatively benign conditions.
Next, we evaluated the efficiency of the reconstituted protein nanocages of H158/H160MjFer with recombinant human H-chain ferritin (rHuHF) and T158HMjFer as two control samples. As displayed in Fig. S4 (ESI†), upon disassembly at pH 3.5, followed by adjusting the pH back to 8.0, 81.4 ± 2.3% of the H158/H160MjFer nanocages were recovered. By contrast, upon disassembly at pH 2.3, rHuHF, T158HMjFer and H158/H160MjFer showed a markedly lower reconstituted efficiency of 56 ± 1.4%, 53.7 ± 4.2%, 61.7 ± 3.1%, respectively. Thus, it appears that the pH can act as a switch to control the disassembly and reassembly of the designed ferritin nanocage H158/H160MjFer with high efficiency under relatively benign conditions.
To shed light on whether or not Ni2+ could serve as the second switch to trigger the above reconstituted protein nanocages to further assemble into 3D superlattices, their assembly behavior in solution was analyzed using TEM and small-angle X-ray scattering (SAXS) upon treatment with Ni2+ (Fig. S5, ESI†). As expected, well-organized 3D protein arrays were formed, as evidenced via TEM (Fig. S5a and b, ESI†). Integrated one-dimensional SAXS curves revealed that the identified q values at 0.053, 0.076, 0.091, 0.107 corresponded to Bragg reflections of the (100), (110), (111), (200) planes, respectively, which is indicative of a simple cubic structure (Fig. S5c, ESI†).
To elucidate the detailed structure of the 3D protein arrays of H158/H160MjFer mediated by Ni2+, we obtained qualified cubic protein crystals (Fig. 2a) with good diffraction and solved the crystal structure at a resolution of ∼2.1 Å (Table S1, ESI†). The crystal structure revealed that the Ni2+-mediated ferritin nanocages arranged into a simple cubic structure. Importantly, intermolecular associations between the ferritin molecules are bridged through Ni2+ involved metal coordination with Glu95/Glu95′ (Fig. 2b). The distance between the oxygen from Glu95 and Ni2+ is measured to be 2.5 Å (Fig. 2c). Furthermore, His158 also participates in the ferritin assembly via π–π stacking interactions (Fig. 2d). In total, there are four metal coordination bonds and four pairs of π–π stacking interactions formed between neighboring ferritin molecules, confirming that the nickel ions play a key role in the formation of 3D protein frameworks. Besides, at the C4 axis of the protein nanocage, four His residues at position 160 coming from four subunits form coordination bonds with one nickel ion, and the shortest distance between each histidine and the nickel ion is 2.1 Å (Fig. 2e). Thus, it appears that the coordinated nickel ion stabilizes the shell-like structure of ferritin. Such a stabilized structure of the H158/H160MjFer nanocage by nickel ions is conducive to its further assembly into three-dimensional protein packing (Fig. 2f). All these results demonstrate that nickel ions can serve as the second switch for driving the pH-treated protein nanocages to self-assemble into 3D protein nanocage superlattices. Thus, using pH and metal switches, the assembly of the ferritin nanocage itself and the assembly of the 3D protein nanocage superlattices are spatiotemporally separated from each other, a property that is favourable for the hierarchical encapsulation and release of two distinct cargo molecules.
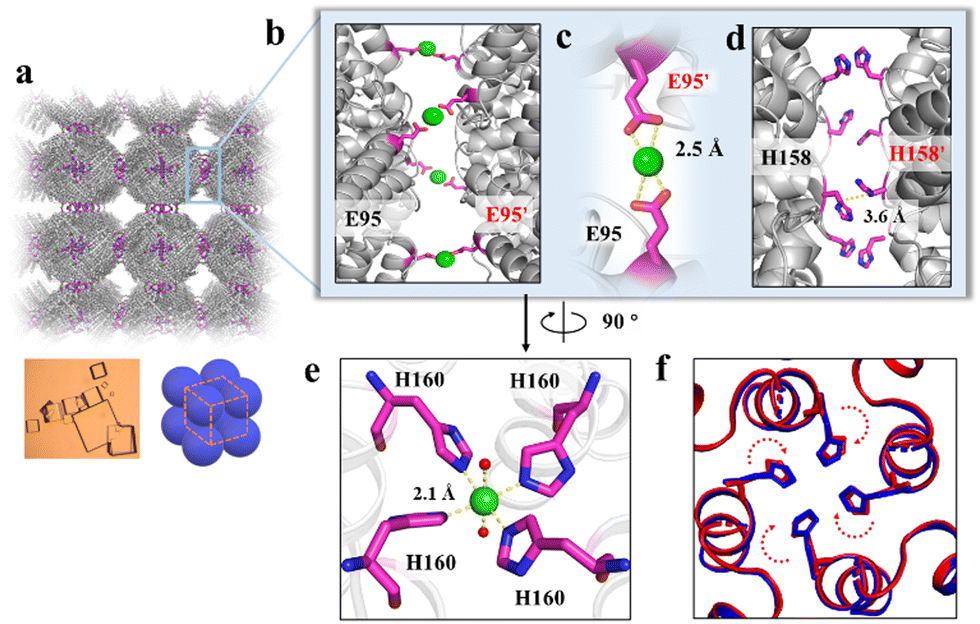 | ||
| Fig. 2 Crystal structure of the 3D protein nanocage arrays. (a) Assembly of ferritin nanocages into simple cubic packing in the crystal structure (PDB ID: 7XRG). (b) Two neighbouring ferritin molecules are bridged by Ni2+ (green spheres) involved coordination at the C4 interfaces. (c) Close-up views of the metal coordination, where Ni2+ is coordinated with Glu95 and Glu95' from two adjacent ferritin moieties. (d) His-induced π–π stacking interactions at the C4 interfaces. (e) An expanded view of one Ni2+ complex at the 4-fold channel, where Ni2+ is coordinated to two H2O molecules (red spheres) and four His160 residues from each subunit. (f) Superposition of the C4 interface with (blue) and without (red) Ni2+. | ||
To confirm the above view, we selected R6G and its quencher BHQ-2 (Fig. S6, ESI†) as two model cargoes to be encapsulated and released separately by the above constructed two-compartment system so that the two processes could be traced by fluorescence. In the first encapsulation experiment, we first disassembled the H158/H160MjFer nanocage at pH 3.5, followed by mixing with R6G molecules and then adjusting the pH to 8.0, as shown in Fig. 3a. The resultant solution showed a visually distinguishable red colour compared with the apo H158/H160MjFer sample (Fig. 3b). To confirm this observation, the resultant solutions were examined via UV/vis spectroscopy. As shown in Fig. 3b, the R6G-loading ferritin solution showed two major absorptions, namely one is the protein maximal absorption at about 280 nm, and another appears at about 525 nm, which corresponds to the characteristic absorption of R6G. More evidence obtained via TEM analysis showed that black uranium-containing cores can be seen in the cavity of apoferritin alone due to the diffusion of uranium into the ferritin cavity (Fig. 3c), while such discrete electron-dense cores disappeared, and instead, white nanoparticles were observed with most of the ferritin molecules upon H158/H160MjFer being treated with R6G (Fig. 3d). All these results suggested that R6G molecules were encapsulated within the ferritin cavity through the aforementioned pH-induced disassembly and reassembly process. After encapsulation, we determined the amount of R6G molecules within the cavity of ferritin spectrophotometrically, and found an average of ∼33.4 R6G molecules loaded inside each H158/H160MjFer molecule, with a loading efficiency of 8.4%.
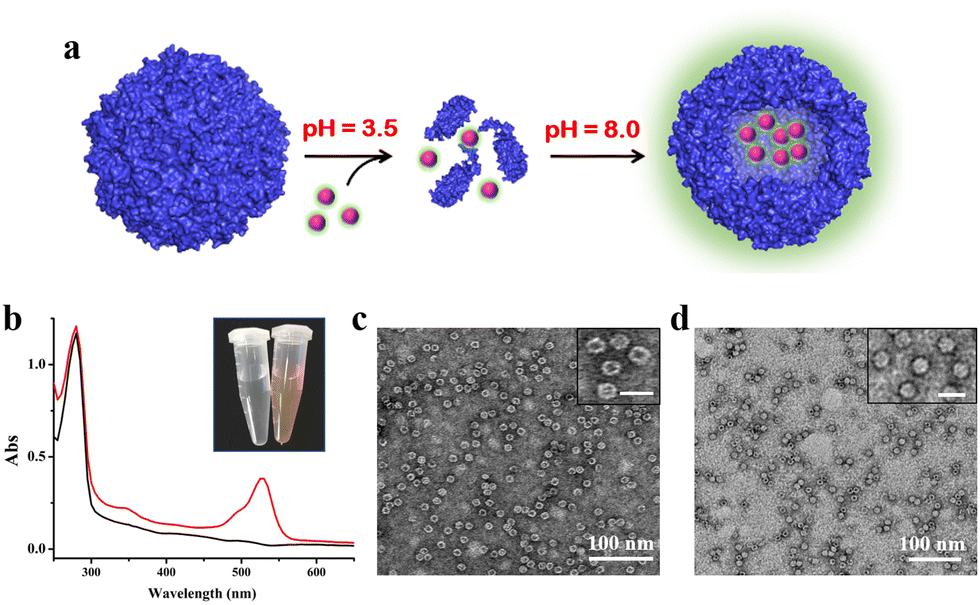 | ||
| Fig. 3 Encapsulation of cationic R6G inside the inner cavity of the ferritin nanocage. (a) Encapsulation of R6G within apo H158/H160MjFer molecules by taking advantage of the reversible disassembly and reassembly characteristics controlled by pH 3.5 and 8.0 conditions. (b) UV/vis spectra of the R6G-loaded ferritin nanocomposites (red line) and ferritin itself (black line). Inset: pictures of untreated ferritin (left), and the R6G-loaded ferritin nanocomposites (right). (c and d) TEM images of untreated ferritin molecules (c) and R6G-loaded ferritin molecules (d). The insets show magnifications, in which scale bars represent 20 nm. | ||
Having successfully encapsulated R6G in the cavity of H158/H160MjFer, we moved forward to encapsulate another type of cargo molecule in the interglobular cavities of the 3D ferritin nanocage lattices. BHQ-2 was selected because the fluorescence resonance energy transfer (FRET) between R6G and BHQ-2 is closely associated with their distance, which enables the encapsulation process to be visualized. To this end, BHQ-2 was mixed with the R6G-loaded H158/H160MjFer molecules at a protein/BHQ-2 ratio of 1/200, and subsequently 0.7 mM of Ni2+ plus 500 mM NaCl was added to the solution to trigger 3D protein assembly. After shaking gently overnight, the resulting ferritin superlattices were monitored using TEM and SAXS. TEM analysis revealed that the ferritin molecules assemble into well-organized 3D arrays (Fig. 4a and b). In parallel, SAXS was performed to determine whether or not the assembly of the R6G-loaded H158/H160MjFer molecules was a structurally simple cubic lattice in solution. The intense (hkl) = (100) signal can be seen at q = 0.053 Å−1, and the next clear signals are at 0.076, 0.091, and 0.107 Å−1. When the q = 0.053 Å−1 position is given the value 1, the following signals fall in the positions √2, √3, and √4, which correspond to the plane reflections of the Miller indices of (110), (111), and (200), indicative of a simple cubic structure (Fig. 4c). Thus, the R6G-loaded ferritin molecules still assemble into simple cubic lattices even in the presence of BHQ-2. After standing for a period of time, the above-formed large protein assemblies as precipitates settles at the bottom with the colour of BHQ-2 (Fig. 4d), demonstrating that the BHQ-2 molecules are encapsulated within the interglobular cavities of the 3D protein nanocage superlattices.
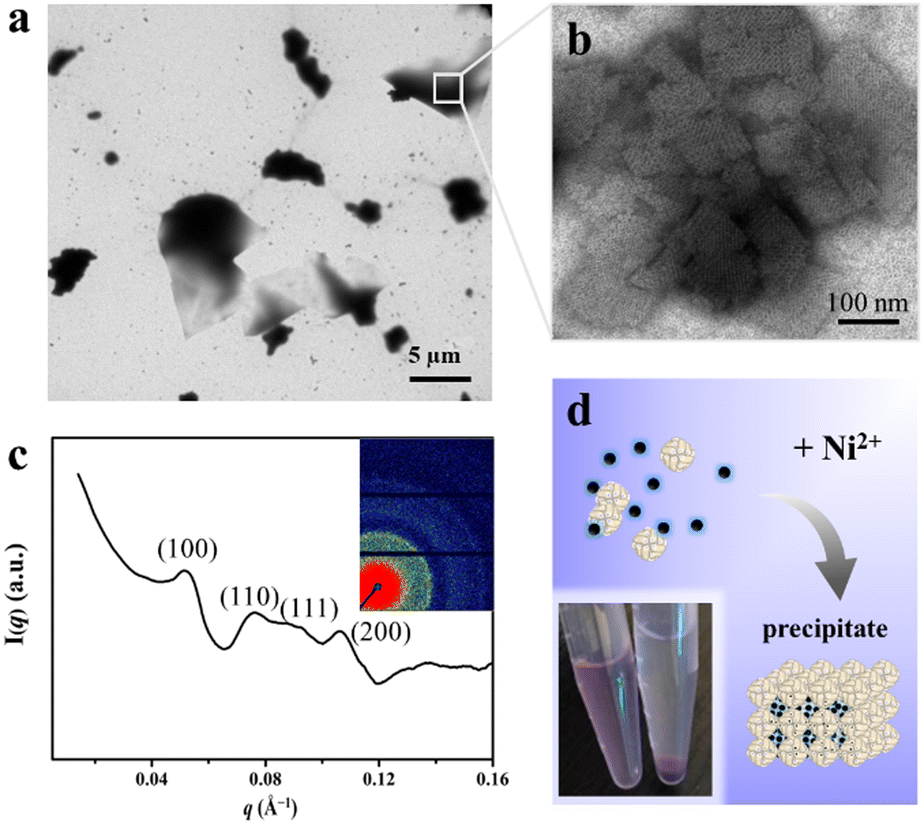 | ||
| Fig. 4 Encapsulation of BHQ-2 within the interglobular cavities of the 3D ferritin superlattice. (a) Low-magnification TEM image of the 3D ferritin superlattices formed in the presence of BHQ-2. (b) High- magnification image of the 3D ferritin superlattices formed in the presence of BHQ-2. (c) Miller indexed curves measured from BHQ-2 encapsulated ferritin superlattices. The inset image shows the 2D SAXS pattern of the ferritin assemblies. (d) Microphotographs of the prepared samples before (left) and after (right) standing. | ||
To further confirm the physically separated encapsulation of R6G and BHQ-2 within two different compartments, FRET experiments were performed. In the absence of BHQ-2, the fluorescence of R6G decreased by ∼15% upon self-assembly of the R6G-loaded H158/H160MjFer molecules into 3D protein nanocage lattices induced by Ni2+ (Fig. 5a, spectra 1 and 2). After the above 3D lattices had disassembled into discrete ferritin nanocages in the presence of EDTA, the fluorescence of R6G was recovered to 90% of its original value (Fig. 5a, spectrum 3).
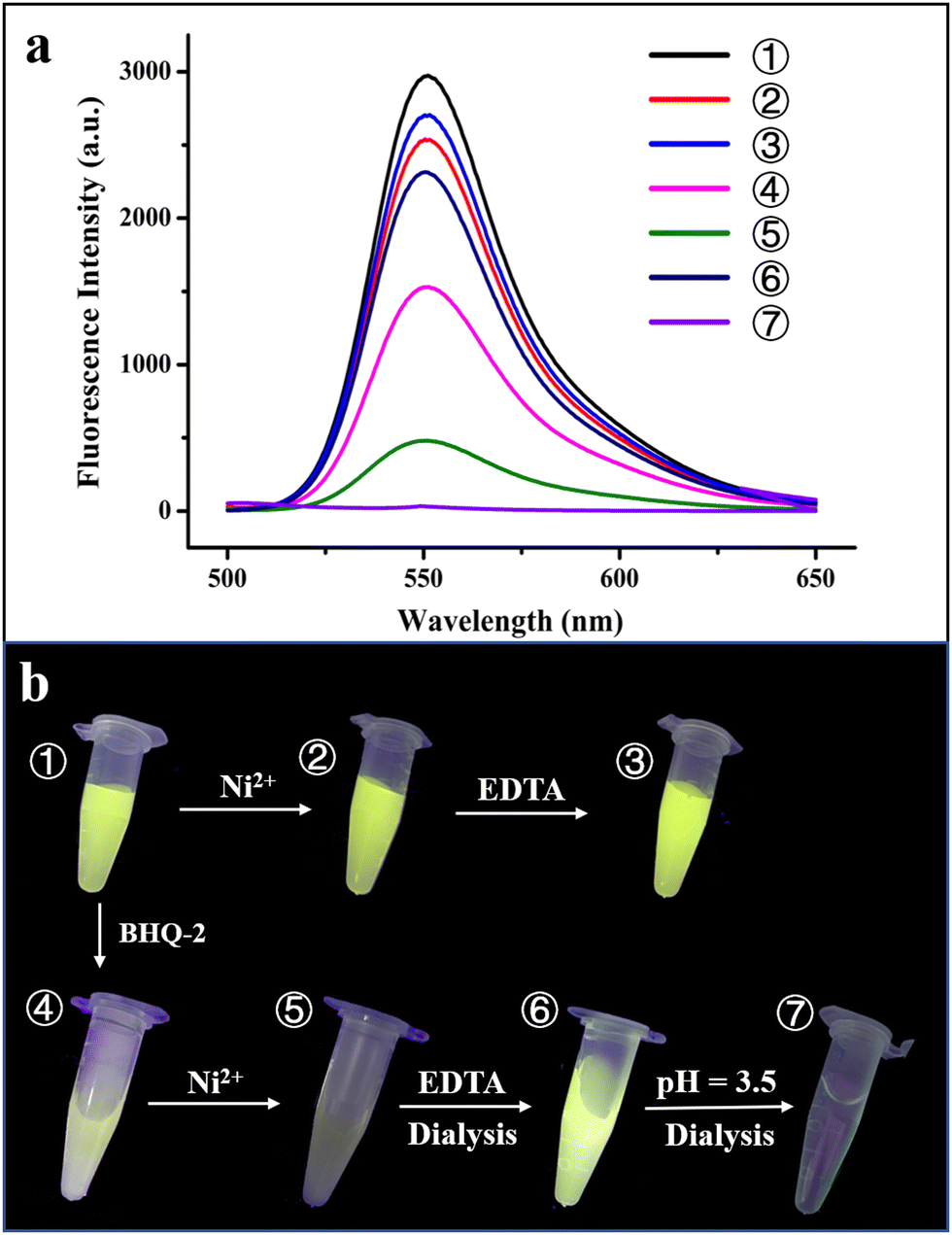 | ||
| Fig. 5 Verification of the hierarchical encapsulation and release process. (a) Fluorescence intensity changes during the course of the hierarchical encapsulation and release of R6G and BHQ-2 by the designed 3D ferritin lattices. (b) Photographs of seven different samples under UV irradiation. ① R6G-loaded ferritin nanocage; ② 3D assembly of R6G-loaded ferritin induced by Ni2+; ③ treatment of the R6G-loaded ferritin 3D assembly by EDTA; ④ mixture of R6G-loaded ferritin and BHQ-2; ⑤ physically separated encapsulation of R6G and BHQ-2 within the 3D ferritin lattices induced by Ni2+; ⑥ and ⑦ hierarchical release of BHQ-2 and R6G from the 3D lattices via EDTA and acidic treatment at pH 3.5, respectively. | ||
Thus, the 3D assembly and disassembly of ferritin in the absence of BHQ-2 has only a slight effect on the R6G fluorescence. In agreement with this view, it is hard to see a colour change among samples 1–3 under UV irradiation (Fig. 5b). Differently, the fluorescence decreased by about 50% upon addition of BHQ-2 to the R6G-loaded ferritin solution, which is indicative of an energy transfer from the donor R6G to the acceptor BHQ-2 to a certain extent (Fig. 5a, spectrum 4). This result was confirmed by the observation that the colour of sample 4 partly disappeared under UV light comparted with that of sample 1 (Fig. 5b). By contrast, upon the successive encapsulation of R6G and BHQ-2 by the designed 3D protein nanocage superlattices, nearly 90% of the R6G fluorescence was quenched (Fig. 5a, spectrum 5), confirming that the BHQ-2 molecules were locked within the interglobular cavities of the lattices. Such larger quenching is most likely derived from closer contact between R6G and BHQ-2 within the 3D ferritin lattices. Support for this idea comes from the observation, under UV light, of a marked change from the green colour to the dark sample that occurs between sample 1 (the R6G-loaded ferritin) and sample 5 (the Ni2+-mediated 3D ferritin lattices), as shown in Fig. 5b, which provides strong evidence for the hierarchical encapsulation behavior. All these findings demonstrate that the designed 3D ferritin nanocage assemblies can be utilized for the physically separated encapsulation of two different cargo molecules.
Another important characteristic of the physically separated encapsulation of two different cargos by the 3D ferritin nanocage assemblies is their controlled release. We hypothesized that EDTA has the ability to chelate Ni2+, causing disassembly of the ferritin superlattice into discrete protein nanocages, thereby releasing the quencher BHQ-2. To this end, the R6G-loaded and BHQ-2-loaded 3D ferritin lattices were preincubated overnight with 100 mM EDTA (Fig. S7, ESI†), followed by dialysis against 50 mM Tris-HCl to remove the released BHQ-2. Accompanied by the above EDTA treatment and dialysis, the fluorescence of the R6G was largely recovered (Fig. 5a, spectrum 6), which is indicative of the release of BHQ-2 (Fig. 5a). This result was confirmed by the visible colour change in the solution under UV light from sample 5 to sample 6 (Fig. 5b). Finally, the disassembly of the ferritin nanocage was carried out by altering the solution pH from 8.0 to 3.5, as shown in Fig. S7(ESI†). It can be seen that the removal of free R6G molecules by dialysis against Tris-HCl results in the complete disappearance of fluorescence (Fig. 5a, spectrum 7). Indeed, almost no colour was observed under UV light (Fig. 5b, sample 7). Taken together, these results demonstrated the capacity of the ferritin superlattice as a platform to release encapsulated cargo molecules in a spatiotemporally controlled manner.
Conclusions
Based on different strategies, many types of 3D protein superstructure have been generated so far through the elaborate design of fairytale protein building blocks. However, it remains a challenge to achieve spatiotemporal control over these protein assemblies. In this study, by designing both pH and metal switches, we have presented the first instance that the self-assembly of 3D ferritin superlattices can be spatiotemporally controlled; consequently, they are able to serve as a scaffold to realize the hierarchical encapsulation and controlled release of two different cargo molecules. Unlike previous co-encapsulation approaches, we harnessed the separate self-assembly of ferritin molecules to encapsulate two different cargo molecules within the inner cavity of the ferritin nanocage and the interglobular cavities of the 3D superlattices. Although 3D ferritin superlattices have been used as a two-compartment system before, it depends on the ability of ferritin to host ionic metals.34 By contrast, the designed 3D ferritin superlattices in this study have the ability to encapsulate different organic molecules in a spatiotemporal manner in solution. This brings about a step-by-step encapsulation, and spatially disassociated state for two types of molecules, but at the same time the release of the entrapped molecules can be spatiotemporally controlled. This represents a general encapsulation and release strategy that could be extended to diverse enzyme couples, synergistic drugs, or other entities, paving the way for the creation of artificial organelles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31972018). The Shanghai Synchrotron Radiation Facility (SSRF) is especially acknowledged for beam time. We thank the staff from the BL17U1/BL18U1/19U1 beamline of the National Center for Protein Sciences Shanghai (NCPSS) at the Shanghai Synchrotron Radiation Facility for assistance during data collection.Notes and references
- J. W. Szostak, D. P. Bartel and P. L. Luisi, Nature, 2001, 409, 387–390 CrossRef CAS PubMed.
- A. Y. Mulkidjanian, A. Y. Bychkov, D. V. Dibrova, M. Y. Galperin and E. V. Koonin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E821–E830 Search PubMed.
- C. M. Agapakis, P. M. Boyle and P. A. Silver, Nat. Chem. Biol., 2012, 8, 527–535 CrossRef CAS PubMed.
- J. E. Dueber, G. C. Wu, G. R. Malmirchegini, T. S. Moon, C. J. Petzold, A. V. Ullal, K. L. J. Prather and J. D. Keasling, Nat. Biotechnol., 2009, 27, 753–759 Search PubMed.
- A. H. Chen and P. A. Silver, Trends Cell Biol., 2012, 22, 662–670 Search PubMed.
- Z. Wang, M. C. M. van Oers, F. P. J. T. Rutjes and J. C. M. van Hest, Angew. Chem., Int. Ed., 2012, 51, 10746–10750 Search PubMed.
- L. Rodríguez-Arco, M. Mann and S. Li, Nat. Mater., 2017, 16, 857–863 CrossRef.
- L. Rodríguez-Arco, B. V. V. S. P. Kumar, M. Li, A. J. Patil and S. Mann, Angew. Chem., Int. Ed., 2019, 58, 6333–6337 Search PubMed.
- A. Dupin and F. C. Simmel, Nat. Chem., 2019, 11, 32–39 Search PubMed.
- Y. Elani, R. V. Law and O. Ces, Nat. Commun., 2014, 5, 5305 Search PubMed.
- T. Douglas and M. Young, Science, 2006, 312, 873–875 CrossRef CAS PubMed.
- N. D. Chasteen and P. M. Harrison, J. Struct. Biol., 1999, 126, 182–194 CrossRef CAS.
- S. Tanaka, C. A. Kerfeld, M. R. Sawaya, F. Cai, S. Heinhorst, G. C. Cannon and T. O. Yeates, Science, 2008, 319, 1083–1086 Search PubMed.
- L. Chen, G. Bai, S. Yang, R. Yang, G. Zhao, C. Xu and W. Leung, Food Res. Int., 2014, 62, 1147–1153 Search PubMed.
- L. Chen, G. Bai, R. Yang, J. Zang, T. Zhou and G. Zhao, Food Chem., 2014, 149, 307–312 Search PubMed.
- K. Fan, X. Jia, M. Zhou, K. Wang, J. Conde, J. He, J. Tian and X. Yan, ACS Nano, 2018, 12, 4105–4115 CrossRef CAS PubMed.
- S. Tetter and D. Hilvert, Angew. Chem., Int. Ed., 2017, 56, 14933–14936 CrossRef CAS.
- K. Zhou, J. Zang, H. Chen, W. Wang, H. Wang and G. Zhao, ACS Nano, 2018, 12, 11323–11332 Search PubMed.
- B. Zheng, K. Zhou, T. Zhang, C. Lv and G. Zhao, Nano Lett., 2019, 19, 4023–4028 Search PubMed.
- H. Chen, Y. Liu, T. Zhang and G. Zhao, J. Colloid Interface Sci., 2020, 582, 1–11 CrossRef.
- H. Chen, T. Zhang, X. Tan, Y. Wang, Y. Liu and G. Zhao, Food Chem., 2021, 349, 129089 Search PubMed.
- P. A. Sontz, J. B. Bailey, S. Ahn and F. A. Tezcan, J. Am. Chem. Soc., 2015, 137, 11598–11601 Search PubMed.
- J. B. Bailey, L. Zhang, J. A. Chiong, S. Ahn and F. A. Tezcan, J. Am. Chem. Soc., 2017, 139, 8160–8166 CrossRef CAS.
- C. Gu, H. Chen, Y. Wang, T. Zhang, H. Wang and G. Zhao, Chem. – Eur. J., 2020, 26, 3016–3021 Search PubMed.
- H. Chen, K. Zhou, Y. Wang, J. Zang and G. Zhao, Chem. Commun., 2019, 55, 11299–11302 RSC.
- S. Chakraborti, A. Korpi, M. Kumar, P. Stepien, M. A. Kostiainen and J. G. Heddle, Nano Lett., 2019, 19, 3918–3924 CrossRef CAS PubMed.
- V. Liljestrom, J. Mikkil and M. A. Kostiainen, Nat. Commun., 2014, 5, 4445 Search PubMed.
- K. McCoy, M. Uchida, B. Lee and T. Douglas, ACS Nano, 2018, 12, 3541–3550 CrossRef CAS PubMed.
- M. A. Kostiainen, P. Ceci, M. Fornara, P. Hiekkataipale, O. Kasyutich, R. J. M. Nolte, J. J. L. M. Cornelissen, R. D. Desautels and J. van Lierop, ACS Nano, 2011, 5, 6394–6402 CrossRef CAS PubMed.
- P. Cigler, A. K. Lytton-Jean, D. G. Anderson, M. G. Finn and S. Y. Park, Nat. Mater., 2010, 9, 918–922 CrossRef CAS.
- M. Künzle, T. Eckert and T. Beck, J. Am. Chem. Soc., 2016, 138, 12731–12734 Search PubMed.
- K. Han, Y. Na, L. Zhang and F. A. Tezcan, J. Am. Chem. Soc., 2022, 144, 10139–10144 CrossRef CAS.
- M. Lach, C. Strelow, A. Meyer, A. Mews and T. Beck, ACS Appl. Mater. Interfaces, 2022, 14, 10656–10668 Search PubMed.
- A. Shaukat, E. Anaya-Plaza, N. K. Beyeh and M. A. Kostiainen, Chem. – Eur. J., 2022, 28(11), e202104341 CAS.
- T. Masuda, J. Zang, G. Zhao and B. Mikami, Protein Sci., 2018, 27, 1955–1960 CrossRef CAS PubMed.
- G. Jutz, P. van Rijn, B. Santos Miranda and A. Böker, Chem. Rev., 2015, 115, 1653–1701 Search PubMed.
- X. Tan, H. Chen, C. Gu, J. Zang, T. Zhang, H. Wang and G. Zhao, Commun. Chem., 2020, 3, 1–9 CrossRef.
- M. A. Kilic, E. Ozlu and S. Calis, J. Biomed. Nanotechnol., 2012, 8, 508–514 CrossRef CAS.
- Z. Yang, X. Wang, H. Diao, J. Zhang, H. Li, H. Sun and Z. Guo, Chem. Commun., 2007, 3453–3455 Search PubMed.
- C. Gu, T. Zhang, C. Lv, Y. Liu, Y. Wang and G. Zhao, ACS Nano, 2020, 14, 17080–17090 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb01961b |
| This journal is © The Royal Society of Chemistry 2022 |