
Highly efficient triplet–triplet annihilation upconversion in polycaprolactone: application to 3D printable architectures and microneedles†
Jeong-Min
Park
a,
Haklae
Lee
a,
Hyun-Seok
Choe
a,
Suk-kyun
Ahn
b,
Keum-Yong
Seong
c,
Seung Yun
Yang
c and
Jae-Hyuk
Kim
 *a
*a
aDepartment of Chemical and Environmental Engineering, Pusan National University, Busan 46241, Republic of Korea. E-mail: jaehyuk.kim@pusan.ac.kr
bDepartment of Polymer Science and Engineering, Pusan National University, Busan, 46241, Republic of Korea
cDepartment of Biomaterials Science (BK21 Four Program), Pusan National University, Miryang 50463, Republic of Korea
First published on 31st December 2021
Abstract
This research reports the next generation of solid-state triplet–triplet annihilation upconversion (TTA-UC) host material (polycaprolactone, PCL) for highly efficient, processable, and biocompatible solid-state TTA-UC. UC PCL was successfully fabricated using a drop-casting method and showed intense UC emission, moderate photostability, and an enhanced UC quantum yield (3.1%). After the characterization of its photochemical properties, the UC PCL was manipulated to form 3D UC structures and UC microneedles by using a commercially available 3D printer and thermal press molding method, respectively, and exhibited strong UC emission even after being subjected to heat manipulation. This is the first report describing an effective and processable solid-phase UC host material, thus paving the way for various applications in solar and biophotonic devices by integrating UC materials with complex 3D shapes.
Photon upconversion (UC) has been widely studied in photonic devices and biomedical applications (e.g., photocatalysts,1–5 photovoltaic cells,6–8 bioimaging,9–13 and biotherapy14,15) owing to its unique ability to convert lower energy photons into a single photon with higher energy.16–18 Among the various known UC mechanisms, triplet–triplet annihilation upconversion (TTA-UC) has attracted considerable attention owing to its advantages such as the low-power excitation, wide absorption band, and a relatively high UC quantum yield.19,20 TTA-UC can be achieved through a series of energy transfers between two types of organic chromophores known as a sensitizer and acceptor: (1) the sensitizer is first excited to the singlet state via photon absorption, and then the triplet excited state is generated through intersystem crossing (ISC). (2) This triplet energy is transferred to the acceptor and the acceptor's triplet excited state is generated via triplet–triplet energy transfer (TTET). (3) TTA between two or more excited acceptors produces one singlet excited state in an acceptor, which then emits upconverted delayed fluorescence.21 These processes are based on Dexter energy transfer, which occurs as a result of electron exchange between molecules (orbital overlap) within 10 Angstroms; therefore, UC host materials should provide good diffusivity.22,23 In addition, oxygen protection should be considered because oxygen molecules can restrict the TTA-UC process by quenching the triplet energy of chromophores (i.e., the sensitizer and acceptor).
In earlier studies, TTA-UC was generally achieved in deoxygenated organic solvents (e.g., toluene, tetrahydrofuran, dimethylformamide, and benzene) to maximize molecular diffusivity and provide sufficient chromophore solubility.24–26 However, without the deoxygenation process, TTA-UC in organic solvents cannot be achieved owing to the quenching of triplet-excited chromophores by triplet state oxygen molecules that normally exist under ambient conditions. Therefore, a deoxygenation process (e.g., inert gas purging or freeze–pump–thaw method) is necessary for successful TTA-UC in organic solvents. In addition to this drawback, their volatility, toxicity, and flammability place serious obstacles in the way of their practical applicability. To overcome these disadvantages, solid-state TTA-UC (using polymers or hydrogels as a UC host material) has been widely researched.27–32 Among various UC host materials for solid-state TTA-UC, flexible polyurethane (PU) has been selected as a viable solid-state UC host material owing to its fair molecular diffusivity, mechanical stability, and oxygen protection. Previously, we reported a new multilayer thin-film architecture consisting of UC PU and an oxygen barrier film (i.e., polyvinyl alcohol, PVA) that showed satisfactory long-term photostability and the highest UC quantum yield (3.5%; using PdTPBP and perylene as the sensitizer and acceptor, respectively).33 Although these results are interesting, there were several inherent problems: (1) the fabrication of this multilayer thin-film architecture required several processes to provide oxygen protection for efficient TTA-UC. (2) Only molded, 2-dimensional, and relatively simple shapes could be fabricated owing to an irreversible polymerization process in PU (crosslinking between two monomers). As a result, it was not possible to re-access or transform the PU matrix once it became fully polymerized, and thus fabricating complex shapes or 3D structures became difficult.
In this study, we overcome these problems by selecting polycaprolactone (PCL) as a solid-state UC host material (UC PCL), which has a low glass-transition temperature (ca. −60 °C) and low oxygen permeability. These characteristics enable chromophore diffusion and offer robust protection from ambient oxygen molecules; therefore, UC PCL can successfully achieve intense, highly efficient, and long-term photostable solid-state TTA-UC. In addition to these characteristics, PCL is a biocompatible and thermoplastic polymer with a low melting point (∼60 °C); thus, PCL has been widely used in biomedical applications and 3D printing.34–38 By integrating these characteristics with TTA-UC, to the best of our knowledge, we were the first to fabricate 3D UC structures and UC microneedles (UC MNs) by using a hot-melt extrusion (HME)-type 3D printer and thermal press molding method, respectively. The 3D UC structures can be readily fabricated into the desired shapes, and they exhibit strong UC emission even under low-power 635 nm LED irradiation. UC MNs were easily inserted into porcine skin without any breakage owing to their strong mechanical stability, and they upconverted blue light after deeply penetrating the skin. This result indicates that the UC MNs can be effectively used in photodynamic therapy or can help the activation of photoinitiators for drug delivery systems. Consequently, UC PCL, which is highly efficient, photostable, and processable, has considerable potential in various photonic devices and biophotonics.
To analyze the photochemical properties, we first fabricated UC PCL films using a drop-casting method (Fig. S1, ESI†).39 PdTPBP and perylene, which have frequently been utilized for red-to-blue UC, were selected as the sensitizer and acceptor, respectively (Fig. 1A). PdTPBP has an intense absorption band at 360–480 nm (Soret band) and 600–640 nm (Q-band), which can effectively capture red photons and emit phosphorescence centered at 800 nm, whereas perylene has an absorption band at 420–450 nm and emits fluorescence centered at 470 nm (Fig. 1B). With these characteristics, the captured red photons were successfully converted into blue photons through the TTA-UC process (Fig. S2, ESI†). For the analysis of the UC properties, the emission spectra of four samples (PCL, PdTPBP PCL, perylene PCL, and UC PCL) were measured under 635 nm laser irradiation (Fig. 2A). While PCL and perylene PCL showed no emission, PdTPBP PCL and UC PCL showed phosphorescence centered at 800 nm owing to the presence of PdTPBP. Compared to the phosphorescence of PdTPBP PCL, the decreased phosphorescence of UC PCL indicates energy transfer from the excited triplet sensitizer to the ground-state singlet acceptor to generate an excited triplet acceptor. The intense UC emission centered at 470 nm also indicates efficient triplet–triplet annihilation between the two triplet acceptors. Moreover, this intense UC emission of UC PCL was observed with the naked eye through a 500 nm shortpass filter, even under excitation using a low-power 625 nm LED (23.3 mW cm−2, Fig. 2B).
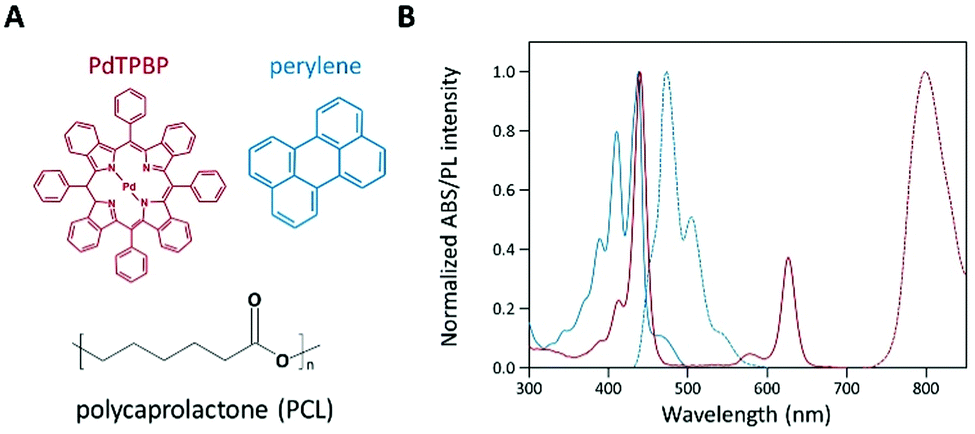 | ||
| Fig. 1 (A) Molecular structures of PdTPBP (sensitizer), perylene (acceptor), and PCL (UC host material). (B) Normalized absorption (solid) and emission (dashed) spectra: sensitizer (PdTPBP, red) and acceptor (perylene, blue) in a PCL film. | ||
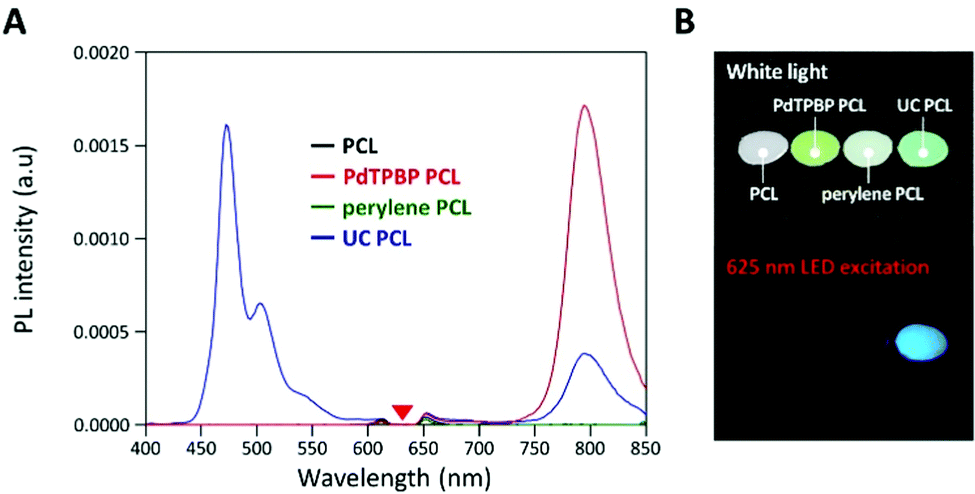 | ||
| Fig. 2 (A) Emission spectra of each sample under 635 nm laser irradiation. A 632 nm notch filter was used to eliminate the scattered incident light. (B) Photographs of PCL, PdTPBP PCL, perylene PCL, and UC PCL under white light (top images) and 625 nm LED excitation (bottom images, 23.3 mW cm−2), acquired through a 500 nm shortpass filter. | ||
To verify the TTA-UC phenomenon, we analyzed the lifetimes of phosphorescence and UC emission, UC emission intensity, and UC quantum yield of UC PCL (Fig. 3A–D). In the phosphorescence lifetime analysis (Fig. 3A), the phosphorescence of PdTPBP PCL monoexponentially decayed with a lifetime (τ0) of 245 μs, which corresponds to the reported result of PdTPBP's phosphorescence lifetime in deoxygenated tetrahydrofuran.33 This result ensures that PCL has inherent oxygen protection and it is a competent UC host material even without additional coating to prevent oxygen quenching; therefore, the UC emission of the UC PCL retained 86% of its original value even after 120 h (Fig. S3, ESI†). Unlike the monoexponential decay of the PdTPBP PCL owing to the radiative decay of the triplet excited state of PdTPBP, the UC PCL showed biexponential decay with a short lifetime (by TTET; τ1) and a long lifetime by radiative decay of the triplet excited state of PdTPBP (τ2) owing to the coexistence of PdTPBP and perylene. τ1 gradually decreased with increasing concentrations of perylene, allowing us to calculate the TTET efficiency (90%) and the bimolecular quenching constant (kq = 6.61 × 106 M−1 s−1) by analyzing the Stern–Volmer plot (Fig. S4, ESI†); these results were found to be comparable to those of previous studies on solid-state UC host materials.33 The analysis of the UC lifetime also confirmed that the TTA-UC process was successful (Fig. 3B). As the concentration of perylene increased, the UC rising time shortened from 346 μs to 120 μs, indicating that the populating rate of triplet excited acceptors is accelerated by increasing the acceptor concentration, which is similar to the previous studies.40,41 On the other hand, an increased triplet lifetime of perylene in UC PCL is obtained (1.98 to 3.27 ms) as the acceptor concentration increases by the tail fitting of the UC emission decay at 470 nm according to the relationship, IUC(t) ∝ exp(−t/τUC) = exp(−2t/τT), where τUC is the UC emission lifetime. This is because the back energy transfer from triplet acceptor to triplet sensitizer is reduced as the acceptor concentration increases.42 Further evidence of the TTA-UC process was obtained by analyzing the power-dependence of the UC emission intensity. The UC emission intensity gradually transitioned from a quadratic (slope = 2.0) to linear (slope = 1.0)-dependence with the increase in laser intensity in a double logarithmic plot of UC emission intensity against power density (Fig. 3C). This characteristic transition is consistent with the established kinetics of typical TTA-UC systems, which indicates that the TTA process becomes increasingly dominant for depopulation of the acceptor's triplet excited state with the increase in excitation power, whereas pseudo-first-order decay is dominant under low excitation power. In our film, the threshold excitation intensity (crossover point between these two sections; Ith), which indicates that the TTA process is more dominant than the pseudo-first-order decay, was 35 mW cm−2. In Fig. 3D, the UC quantum yield of the UC PCL was measured as a function of power density (details in Text S1). Theoretically, the maximum UC quantum yield (ΦUC) is 50% because TTA-UC is a bimolecular process that converts two or more lower-energy photons into one higher-energy photon. The highest ΦUC value of UC PCL was measured to be 3.1%, which is more efficient or comparable with those of other UC host materials using the same chromophores, such as micellar carriers in water (1.2%), oleic acid (1.7%), and an oxygen-barrier-coated polymer film (3.5%).10,33,43 The successful development of UC host materials introduced the opportunity for integration with solar cells, which is a major TTA-UC application. In earlier research, the performance of solar cells could be enhanced by applying the TTA-UC layer to the bottom of the solar cells because wasted long-wavelength light is converted into short-wavelength light by the TTA-UC layer, thereby extending the absorption range of the solar cells.6–8 Under ambient conditions, however, it became difficult to achieve effective and photostable TTA-UC owing to oxygen quenching; thus, deoxygenated UC solutions in sealed glass cuvettes or glass-encased UC films were selected as TTA-UC layers. In contrast, UC PCL shows a high UC quantum yield and enhanced photostability without additional deoxygenation under ambient conditions. As a result, employing UC PCL as the TTA-UC layer for the enhancement of the solar cell efficiency enables us to fabricate the TTA-UC layer with less effort and time. Hence, UC PCL can be potentially employed as the next-generation solid-state UC host material in a wide range of applications.
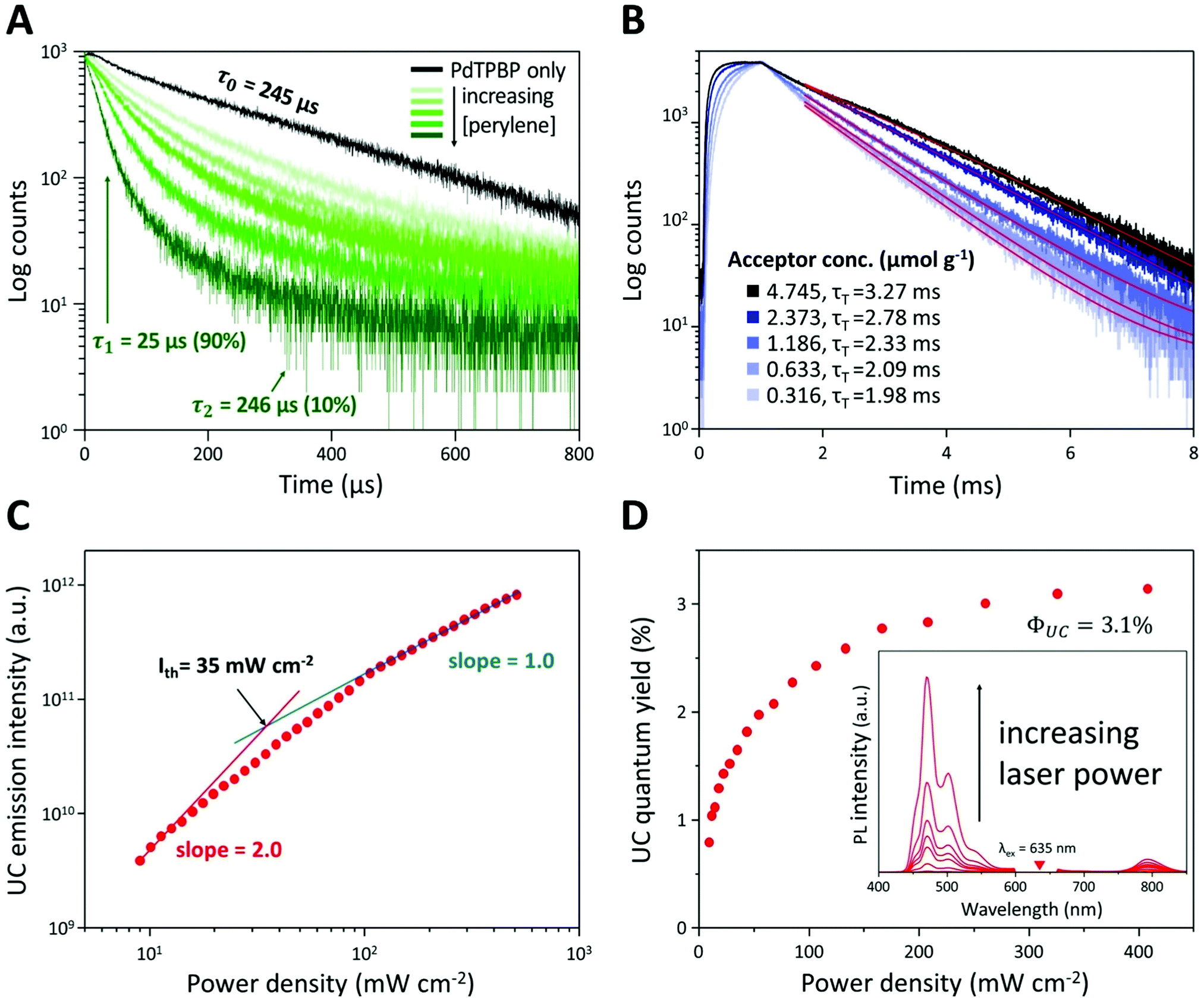 | ||
| Fig. 3 (A) Phosphorescence lifetimes of PdTPBP with increasing concentrations of perylene ([PdTPBP] = 0.03 μmol g−1 and [perylene] = 0–4.7 μmol g−1 in PCL). (B) UC emission decay profiles of the UC PCL at 470 nm following excitation at 635 nm ([PdTPBP] = 0.03 μmol g−1 and [perylene] = 0–4.7 μmol g−1 in PCL). (C) Integrated UC emission intensity and (D) UC quantum yields of the UC PCL as a function of the power density of 635 nm laser excitation ([PdTPBP] = 0.03 μmol g−1 and [perylene] = 4.7 μmol g−1 in PCL). The inset in (D) shows UC emission spectra of UC PCL with increasing 635 nm laser excitation. A 632 nm notch filter was used to eliminate the scattered incident light. | ||
The uncomplicated processability of PCL meant that we could extend its applications. PCL has been used for 3D printing based on HME; therefore, we can readily fabricate various desired shapes or structures and overcome the limitations of UC PU by employing the 3D printing process for the fabrication of solid-state UC host materials. As shown in Fig. 4A, the UC PCL in the syringe was output through a high-temperature extruder under constant pressure conditions that allowed us to fabricate a 3D UC structure with the desired shape (Fig. 4B). This 3D UC structure showed strong UC emission even under a low-power excitation, such as a 625 nm commercial LED (23.3 mW cm−2, Fig. 4C), and it was visible to the naked eye through a 500 nm shortpass filter. Furthermore, The UC quantum yield of the 3D UC structure was measured to be 2.9%, which is almost identical to the UC quantum yield of the drop-cast UC PCL film (3.1%, Fig. S5, ESI†), indicating that 3D-printed UC PCL maintains the UC quantum yield even after the HME process of a 3D printer. From these results, we expect that TTA-UC can be achieved using complex structures as well, and the UC PCL can be recycled into various shapes with outstanding UC properties using a HME-type 3D printer.
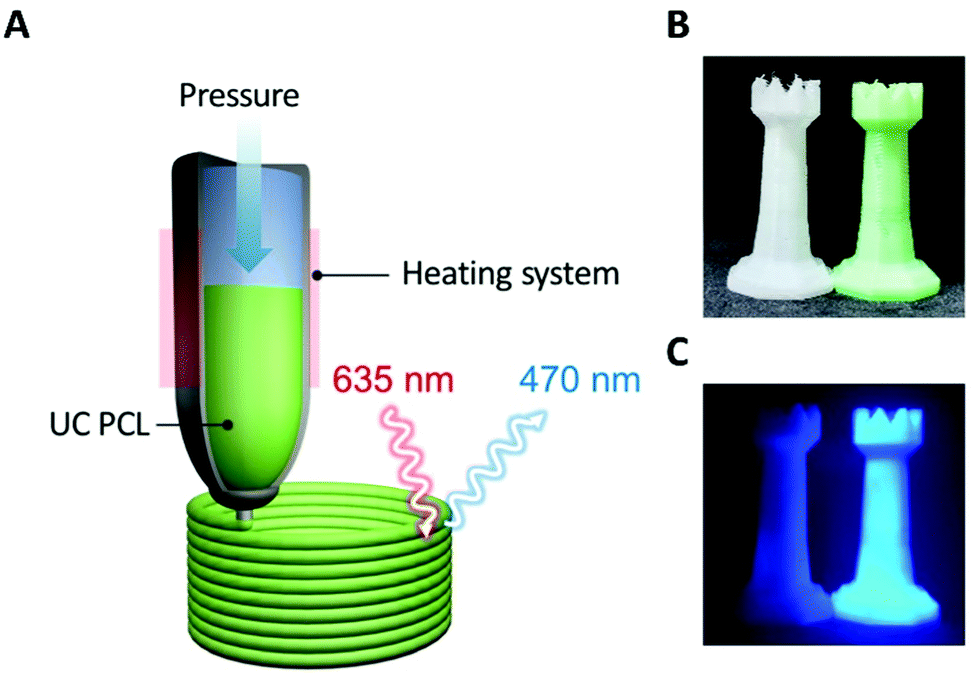 | ||
| Fig. 4 (A) Schematic illustration of the fabrication of a 3D UC structure using a HME-type 3D printer. Photographs of 3D structures with and without the UC dye under (B) white light and (C) 625 nm LED excitation (23.3 mW cm−2), acquired through a 500 nm shortpass filter. | ||
UC PCL can further be manipulated in the form of microneedles (MNs) for biophotonic applications. As shown in Fig. 5A and B, UC MNs were successfully fabricated from the mold using the thermal press molding method. The UC emission was clearly observed even at the minuscule MN tips under 635 nm laser irradiation owing to the excellent photostability of the UC PCL (Fig. 5B, center; 44 mW cm−2). To demonstrate that UC MNs can be used in the practical biophotonic field, an insertion test and UC test after insertion were performed using porcine skin. As a result, the porcine skin was easily pierced by the UC MNs, which had sufficient mechanical strength to penetrate the skin (ESI,† Fig. S6, left). Subsequently, under 635 nm laser irradiation, it was confirmed that the UC emission deeply penetrated the porcine skin (ESI,† Fig. S6, right). To confirm that the TTA-UC can be achieved at the tip of the UC MNs, different MNs (wherein only tips were fabricated by using UC PCL with the backing layer being fabricated using pristine PCL, denoted as tip UC MNs) were prepared (Fig. 5C). The tip UC MNs also easily pierced the porcine skin and exhibited deeply penetrated UC emission (Fig. 5D), confirming that TTA-UC can be achieved even at the minuscule tips.
 | ||
| Fig. 5 (A) Schematic illustration of the fabrication of UC MNs by using the thermal press molding method. (B) Photographs of UC MNs under white light (left), 635 nm laser excitation with a diverging lens, acquired through a 600 nm shortpass filter (center, 44 mW cm−2) and magnified image of the MN tip (right, scale bar = 200 μm). The inset shows the scanning electron microscopy (SEM) image of the MN tip (scale bar = 10 μm). (C) Photograph of tip UC MNs. (D) Cross-sectional optical image of tip UC MNs under white light (top panel) and 635 nm laser excitation (bottom panel, 44 mW cm−2), acquired through a 600 nm shortpass filter. Scale bars in (D) represent 500 μm. | ||
By combining the outstanding UC properties and processability of UC PCL, 3D UC structures and UC MNs were successfully fabricated. Despite being subjected to heat during their fabrication (i.e., HME-type 3D printing or thermal press molding method), they exhibited moderate UC emission. Accordingly, the solid-state TTA-UC system can be expanded to conventional photonic fields (e.g., solar cells, photocatalysts, and anticounterfeiting systems). Furthermore, TTA-UC in PCL can be applied in biophotonic fields owing to the biocompatibility of PCL.
In summary, we selected and characterized PCL, which is affordable, biocompatible, and can be readily restructured by using a commercially available 3D printer and thermal press molding as a solid-state UC host material to overcome the existing limitations of solid-state TTA-UC, such as oxygen protection and chromophore diffusion. UC PCL showed intense UC emission even under low-power excitation, such as a commercial LED light source. Under 635 nm laser irradiation, UC PCL exhibited a high UC quantum yield of approximately 3.1% and moderate long-term photostability under ambient conditions without any additional oxygen protection coating owing to its low oxygen permeability. The superiority of UC PCL as a solid-state UC host material is expected to further advance research into integrating solar cells with solid-state UC host materials. Moreover, the processability and biocompatibility of UC PCL allow the fabrication of desired shapes and complex structures using a 3D printer or thermal press molding. As a proof-of-concept, UC MNs were fabricated, and we demonstrated that UC MNs had sufficient mechanical strength to pierce porcine skin; the UC emission successfully penetrated the porcine skin. To the best of our knowledge, this is the first report describing a versatile upconverting polymeric host, thus paving the way for various applications in solar and biophotonic devices by integrating UC materials with the desired 3D shapes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF-2021M3H4A4079509 and NRF- 2021R1I1A3A04037015).References
- J.-H. Kim and J.-H. Kim, J. Am. Chem. Soc., 2012, 134, 17478–17481 CrossRef CAS PubMed.
- H.-i. Kim, O. S. Kwon, S. Kim, W. Choi and J.-H. Kim, Energy Environ. Sci., 2016, 9, 1063–1073 RSC.
- H.-i. Kim, S. Weon, H. Kang, A. L. Hagstrom, O. S. Kwon, Y.-S. Lee, W. Choi and J.-H. Kim, Environ. Sci. Technol., 2016, 50, 11184–11192 CrossRef CAS PubMed.
- J. Fang, W. Wang, C. Zhu, L. Fang, J. Jin, Y. Ni, C. Lu and Z. Xu, Appl. Catal., B, 2017, 217, 100–107 CrossRef CAS.
- J. Fang, Y. Chen, W. Wang, L. Fang, C. Lu, C. Zhu, J. Kou, Y. Ni and Z. Xu, Appl. Catal., B, 2019, 258, 117762 CrossRef CAS.
- C. Li, C. Koenigsmann, F. Deng, A. Hagstrom, C. A. Schmuttenmaer and J.-H. Kim, ACS Photonics, 2016, 3, 784–790 CrossRef CAS.
- D. Choi, S. K. Nam, K. Kim and J. H. Moon, Angew. Chem., Int. Ed., 2019, 58, 6891–6895 CrossRef CAS PubMed.
- Y. Zhou, C. Ruchlin, A. J. Robb and K. Hanson, ACS Energy Lett., 2019, 4, 1458–1463 CrossRef CAS.
- S. H. Askes, W. Pomp, S. L. Hopkins, A. Kros, S. Wu, T. Schmidt and S. Bonnet, Small, 2016, 12, 5579–5590 CrossRef CAS PubMed.
- O. S. Kwon, H. S. Song, J. O. Conde, H.-I. Kim, N. Artzi and J.-H. Kim, ACS Nano, 2016, 10, 1512–1521 CrossRef CAS PubMed.
- S. Mattiello, A. Monguzzi, J. Pedrini, M. Sassi, C. Villa, Y. Torrente, R. Marotta, F. Meinardi and L. Beverina, Adv. Funct. Mater., 2016, 26, 8447–8454 CrossRef CAS.
- J. Park, M. Xu, F. Li and H.-C. Zhou, J. Am. Chem. Soc., 2018, 140, 5493–5499 CrossRef CAS PubMed.
- M. Xu, X. Zou, Q. Su, W. Yuan, C. Cao, Q. Wang, X. Zhu, W. Feng and F. Li, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed.
- L. Huang, Y. Zhao, H. Zhang, K. Huang, J. Yang and G. Han, Angew. Chem., Int. Ed., 2017, 56, 14400–14404 CrossRef CAS PubMed.
- R. Zhang, Y. Guan, Z. Zhu, H. Lv, F. Li, S. Sun and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 37479–37490 CrossRef CAS PubMed.
- M. Pollnau, D. R. Gamelin, S. Lüthi, H. Güdel and M. P. Hehlen, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337 CrossRef CAS.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, J. Org. Chem., 2011, 76, 7056–7064 CrossRef CAS PubMed.
- P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- C. B. Murphy, Y. Zhang, T. Troxler, V. Ferry, J. J. Martin and W. E. Jones, J. Phys. Chem. B, 2004, 108, 1537–1543 CrossRef CAS.
- N. Yanai and N. Kimizuka, Acc. Chem. Res., 2017, 50, 2487–2495 CrossRef CAS PubMed.
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776–3778 RSC.
- T. N. Singh-Rachford and F. N. Castellano, Inorg. Chem., 2009, 48, 2541–2548 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. Lett., 2010, 1, 195–200 CrossRef CAS.
- Z. Jiang, M. Xu, F. Li and Y. Yu, J. Am. Chem. Soc., 2013, 135, 16446–16453 CrossRef CAS PubMed.
- T. N. Singh-Rachford, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2009, 131, 12007–12014 CrossRef CAS PubMed.
- J. Peng, X. Guo, X. Jiang, D. Zhao and Y. Ma, Chem. Sci., 2016, 7, 1233–1237 RSC.
- P. Bharmoria, S. Hisamitsu, H. Nagatomi, T. Ogawa, M.-A. Morikawa, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2018, 140, 10848–10855 CrossRef CAS PubMed.
- Y. Sasaki, M. Oshikawa, P. Bharmoria, H. Kouno, A. Hayashi-Takagi, M. Sato, I. Ajioka, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2019, 58, 17827–17833 CrossRef CAS PubMed.
- C. Ye, J. Ma, S. Chen, J. Ge, W. Yang, Q. Zheng, X. Wang, Z. Liang and Y. Zhou, J. Phys. Chem. C, 2017, 121, 20158–20164 CrossRef CAS.
- A. L. Hagstrom, H.-L. Lee, M.-S. Lee, H.-S. Choe, J. Jung, B.-G. Park, W.-S. Han, J.-S. Ko, J.-H. Kim and J.-H. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 8985–8992 CrossRef CAS PubMed.
- P.-T. Ko, I.-C. Lee, M.-C. Chen and S.-W. Tsai, J. Taiwan Inst. Chem. Eng., 2015, 51, 1–8 CrossRef CAS.
- K. Nigoghossian, S. Saska, L. M. Christovam, F. Coelho, C. A. G. Beatrice, A. A. Lucas, P. I. Neto, J. V. L. D. Silva, A. Tercjak and M. S. Baptista, J. Braz. Chem. Soc., 2020, 31, 638–652 CAS.
- E. Malikmammadov, T. E. Tanir, A. Kiziltay, V. Hasirci and N. Hasirci, J. Biomater. Sci., Polym. Ed., 2018, 29, 863–893 CrossRef CAS PubMed.
- J. F. Mano, D. Koniarova and R. Reis, J. Mater. Sci.: Mater. Med., 2003, 14, 127–135 CrossRef CAS PubMed.
- H. N. Chia and B. M. Wu, J. Biol. Eng., 2015, 9, 1–14 CrossRef CAS PubMed.
- Y. Sano, Drying Technol., 1992, 10, 591–622 CrossRef CAS.
- E. Radiunas, S. Raišys, S. Juršėnas, A. Jozeliūnaitė, T. Javorskis, U. Šinkevičiūtė, E. Orentas and K. Kazlauskas, J. Mater. Chem. C, 2020, 8, 5525–5534 RSC.
- E. M. Gholizadeh, S. K. K. Prasad, Z. L. Teh, T. Ishwara, S. Norman, A. J. Petty, J. H. Cole, S. Cheong, R. D. Tilley, J. E. Anthony, S. Huang and T. W. Schmidt, Nat. Photonics, 2020, 14, 585–590 CrossRef CAS.
- D. Meroni, A. Monguzzi and F. Meinardi, J. Chem. Phys., 2020, 153, 114302 CrossRef CAS PubMed.
- A. Turshatov, D. Busko, S. Baluschev, T. Miteva and K. Landfester, New J. Phys., 2011, 13, 083035 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc04834a |
| This journal is © The Royal Society of Chemistry 2022 |