
Ultralong room-temperature phosphorescence from polycyclic aromatic hydrocarbons by accelerating intersystem crossing within a rigid polymer network†
Xiao
Meng‡
,
Qian
Hu‡
,
Xi
Wang‡
,
Tengfei
Ma
,
Weimin
Liu
 ,
Xingjun
Zhu
* and
Chunhong
Ye
*
,
Xingjun
Zhu
* and
Chunhong
Ye
*
School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: yechh@shanghaitech.edu.cn; zhuxj1@shanghaitech.edu.cn
First published on 1st November 2022
Abstract
Polymer-based room temperature phosphorescence has attracted great attention for potential applications in organic light-emitting diodes, sensors, data encryption, and anti-counterfeiting. One of the most applied strategies to realize efficient room-temperature phosphorescence (RTP) is to suppress the non-radiative transition by introducing secondary interactions (hydrogen bonding, ionic bonding, etc.) between phosphors and the polymer matrix. However, water and moisture will turn off the phosphorescence emission by damaging the hydrogen bonding network. Besides, the role of the polymer matrix in the photophysical process is still unknown. Herein, we successfully achieve robust long-lived room temperature phosphorescence materials based on purely polycyclic aromatic hydrocarbons (PAHs) by in situ doping with a rigid polymer network during polymerization. The materials exhibited an ultralong phosphorescence lifetime of up to 1.27 s, a large Stokes shift of 233 nm and a remarkable duration of afterglow for over 12 s, which are extremely stable even under aqueous conditions. Photophysical analysis and transient absorption spectroscopy reveal that phosphorescence is facilitated by accelerating the intersystem crossing (ISC) from singlet to triplet states and suppressing the non-radiative transition of the triplet excitons within the rigid polymer matrix. Furthermore, 3D phosphorescent structures and 2D large-scale, transparent displays with re-writable encoded information are fabricated with the facilely processed PAHs@polymer RTP materials.
Introduction
Room temperature phosphorescence (RTP) with persistent luminescence has drawn increasing attention for application in the fields of organic light emitting diodes, sensors, data encryption, anti-counterfeiting, bioimaging, and so forth.1–3 RTP is generated by the radiative transitions from the lowest triplet state (T1) to the ground state (S0) at room temperature and the long-lived excited T1 state.4,5 Generally, emission from the triplet state can be easily quenched via intermolecular collision or energy transfer to oxygen.6 Classical RTP chromophores usually include heavy metal atoms, such as halogen, for facilitating spin-orbit interactions or metal–ligand intersystem crossing, thus populating the triplet excitons.7For overcoming the potential toxicity of heavy metal atoms, pure organic-based RTP materials have gained increasing attention due to their environmentally friendly, feasible multi-chemical modification, mechanical flexibility, and usually optical transparency.8,9 However, it is a challenge to obtain both these advantages and excellent phosphorescence performance. Currently, the reported organic RTP systems have lifetimes in the range of hundreds of micro-seconds to seconds and an afterglow durability of dozens of seconds, which has plenty of space for improvement especially in comparison with those of inorganic RTP materials. Therefore, developing new strategies and new organic RTP materials with high afterglow durability and long life time is highly needed. The phosphorescence performance is mainly influenced by the population of triplet excitons, triplet oxygen quenching, and non-radiative dissipations.10 Therefore, it is crucial to boost the population of triplet excited states and minimize non-radiative dissipation for obtaining long lived RTP materials.11–15 A common approach to reducing the non-radiative dissipation is utilizing a polymer matrix that provides a rigid environment to confine the vibration of the phosphors. Within the polymer network, varied intermolecular interactions are usually introduced to enhance the interaction between phosphors and polymers, including H-bonding,16,17 ionic bonding,1 halogen bonding,12,18,19 and so on. However, a weak intermolecular interaction is usually sensitive to environments. For instance, high humidity could weaken the H-bonding and ionic interaction so that phosphorescence would be quenched, which limits its bio-related applications, such as bioimaging and in situ sensing. Therefore, it is urgent and challenging to obtain robust RTP materials with an ultralong lifetime in a purely organic system under harsh environments.20
It is well-known that many polycyclic aromatic hydrocarbons (PAHs) have a long triplet lifetime at low temperature.21 For example, naphthalene, as a typical chromophore, has been actively studied for a couple of years in fluorescent sensors and electron transport materials, but has not been used as a phosphor. The Jablonski diagram at 77 K revealed that naphthalene has a long lifetime triplet state (τ(T1) = 2.3 s).22 PAHs have no heteroatoms, which can be an advantage to overcome the environmental sensitivity issue of RTP materials based on intermolecular interactions. However, to date, there have been only a few reported PAH based organic RTP systems, especially under development for solid polymer based pure PAH RTPs. For instance, inclusion of cyclodextrin with PAHs, deoxygenation and additional solvents containing halogens,23,24 or doping pyrene in amorphous polymers (poly(vinyl acetate-co-acrylic acid) was needed in these works.25
Herein, we demonstrate pure organic RTP materials with robust and ultralong phosphorescence lifetime by using PAHs (including naphthalene, phenanthrene and pyrene) as phosphors and polymethyl methacrylate (PMMA) with a rigid backbone chain as the matrix. Phosphorescence with an afterglow duration of up to 12 s and a lifetime of 1.27 s is achieved. For the first time, it was revealed that the PMMA matrix significantly enhanced the population rate from the singlet to the triplet exited states for the PAH based phosphors. Along with the minimized non-radiative dissipation of phosphors by rigid confinement of the PMMA chain, the PAHs@PMMA system explored here can generate ultralong room temperature phosphorescence without the formation of secondary phosphor–matrix interactions. Moreover, the phosphorescence showed strong environmental adaptability, which is persistent even after immersing in aqueous surroundings. The as-prepared PAHs-RTP material can be used for fabricating large-scaled 2D re-writable displays and transparent 3D phosphorescent objects. This work not only provides a promising RTP material for applications in organic light emitting diodes, optics and data encryption under extreme environments, but also points to an alternative principle to construct an RTP system.
Results and discussion
Naphthalene (Nap), phenanthrene (Phe) or pyrene (Pyr) with long triplet lifetimes at 77 K was chosen as phosphors to achieve room temperature phosphorescence materials with an ultra-long afterglow. Phosphors were doped with the monomers (methyl methacrylate, MMA) followed by in situ polymerization to obtain well-dispersed PAHs@PMMA systems (see the Experimental section for details). This procedure can reduce the quenching of solvents and devote good processing feasibility for large-scale fabrication (Fig. 1a). After UV light (λ = 275 nm) irradiation for 3 min, a bright RTP emission was observed with the naked eye even at a low phosphor concentration of 10−2 mg mL−1. The color of afterglow can be tailored with different PAH phosphors, in which Nap@PMMA and Phe@PMMA exhibited green phosphorescence emission, while Pyr@PMMA did an orange red one, indicating an afterglow duration lasting for up to 10 s, 12 s, and 2 s, respectively (Fig. 1b).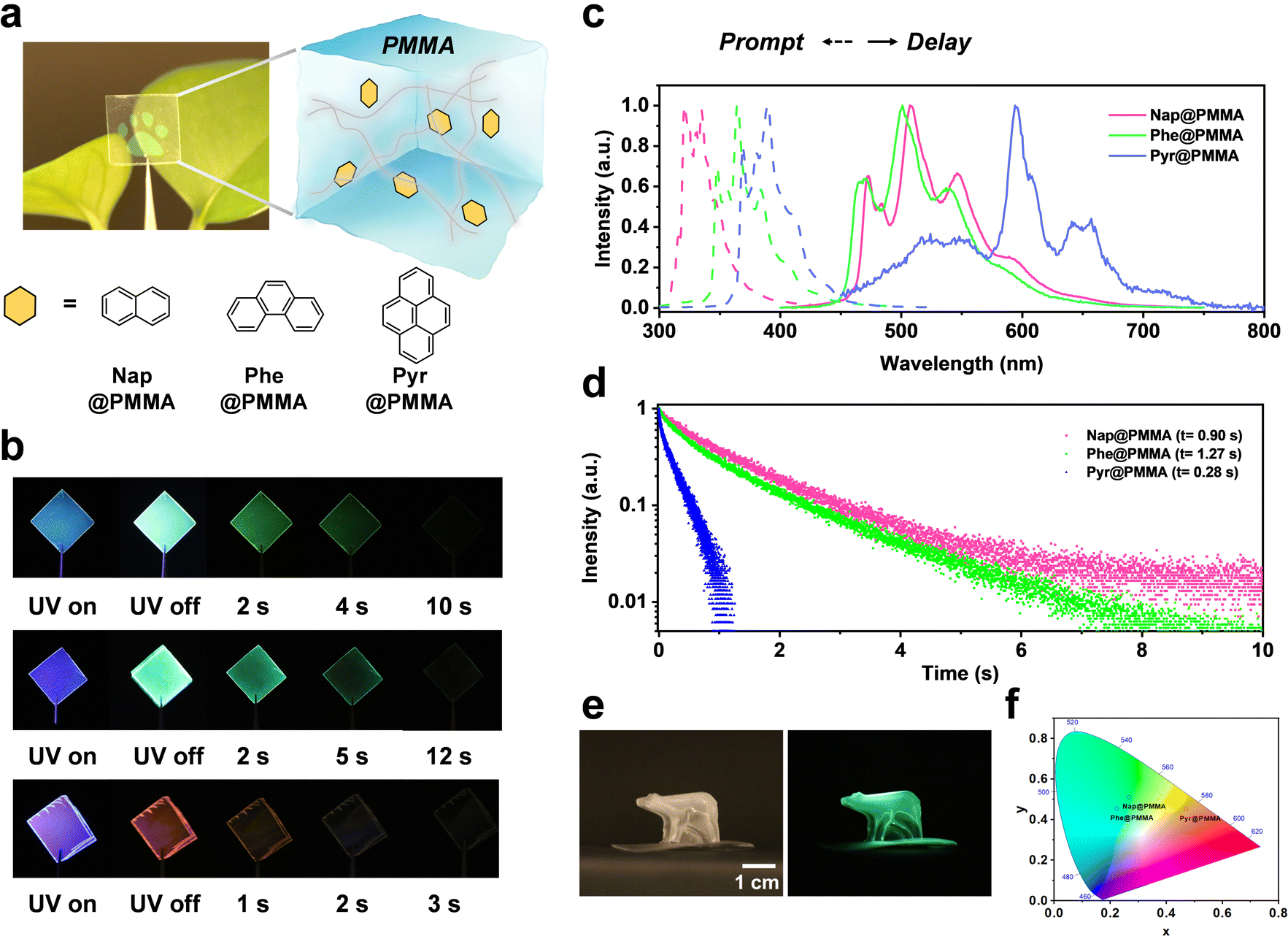 | ||
| Fig. 1 Ultralong afterglow of PAHs doped in PMMA at room temperature. (a) schematic illustration of the phosphor in rigid polymer surroundings; (b) luminescence photographs under 275 nm UV irradiation and at different time internals after UV irradiation (from up to down: Nap@PMMA, Phe@PMMA, and Pyr@PMMA); (c) steady-state fluorescence and delayed RTP spectra (delay time: 10 ms); (d) time-resolved RTP decay curves of Nap@PMMA at 508 nm, Phe@PMMA at 500 nm and Pyr@PMMA at 596 nm (λex = 275 nm); (e) transparent 3D bear of Nap@PMMA showing long-lived RTP; (f) CIE coordinate diagrams of Nap@PMMA, Phe@PMMA, and Pyr@PMMA. | ||
As shown in the steady-state photoluminescence spectra (Fig. 1c), Nap@PMMA, Phe@PMMA and Pyr@PMMA have the main fluorescence emission bands at 320, 364, and 392 nm, respectively. According to the RTP spectra (Fig. 1c), they indicated dominant phosphorescence emission bands at 508, 500, and 596 nm, respectively. Large Stokes shifts of above 200 nm wavelength are demonstrated. Correspondingly, a long life-time up to 0.90 s, 1.27 s, and 0.28 were obtained by fitting the time-dependent RTP luminescence decay (Fig. 1d). These materials displayed obvious afterglow emission with high brightness, which was identifiable with the naked eye even in daylight (photograph in Fig. 1a). Notably, macroscopic 3D objects (e.g. bear- or flower-shaped structures) with long life phosphorescence can also be obtained by in situ polymerization in 3D molds (Fig. 1e and Fig. S1, ESI†). In comparison, the naphthalene sample was doped in PMMA solution, and the film was obtained by drop casting in a PTFE mold. Similar phosphorescence spectra could be detected but the emission was very weak (Fig. S2, ESI†). This suggested that doping the phosphor before the in situ polymerization process was more efficient than simple blending. As observed from the CIE coordinates (Fig. 1f), the samples can display different colors by using Nap@PMMA, Phe@PMMA and Pyr@PMMA.
Next, the photophysical properties of the Nap@PMMA were investigated as an example for understanding the RTP emission of the PAH@PMMA systems. As shown in Fig. 2a and Fig. S3 (ESI†), the absorption and fluorescence excitation spectra in the wavelength range of 250–300 nm were similar, and revealed that the fluorescence originated from naphthalene. The doped naphthalene sample was in a dispersed state at the RTP film instead of forming aggregation, according to the well overlapped spectra collected from its good solvent (hexane) and PMMA film. The steady-state fluorescence spectrum exhibited emission peaks from 310 nm to 390 nm (λmax = 320 nm), and no excitation-dependent fluorescence behavior was observed (Fig. 2b and Fig. S3 and S4, ESI†). As demonstrated by the time-resolved phosphorescence spectra with a maximum emission at λmax = 508 nm, a large Stokes shift of more than 220 nm was obtained, featuring the RTP characteristics. On decreasing the delay time, an obvious increase in emission covering from 400 to 470 nm was detected (Fig. S5, ESI†). The lifetime at this region of Nap@PMMA at room temperature was much shorter than that at 473, 508, and 573 nm (Fig. S6, ESI†). The excitation spectra of phosphorescence showed a very similar peak pattern and wavelength at 473, 508, and 573 nm, indicating the emission peaks came from the same excitation center. The emission peak covering from 400 to 470 nm was reported from phosphorescence from naphthalene monomers, while the peaks at 470 to 700 nm were assigned to the radiative transition from the excited triplet dimer.26 Furthermore, the emission intensity indicated an exponential decreasing over time after turning off the UV light (Fig. 2c).
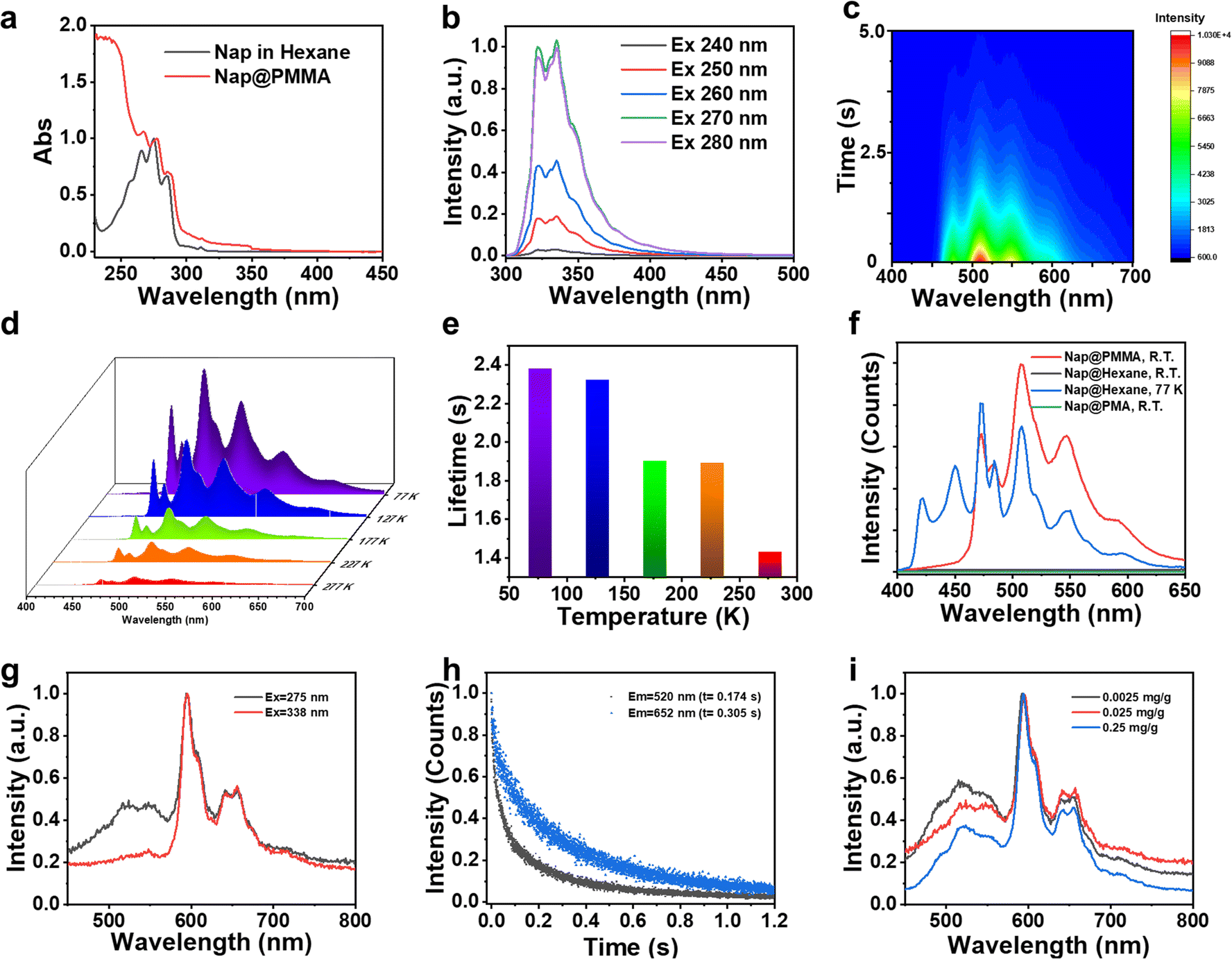 | ||
| Fig. 2 Photophysical properties of phosphor@PMMA. (a) Absorption spectra of Nap@PMMA and naphthalene in hexane solution (c = 10−3 M); (b) fluorescence spectra with different wavelength excitation; (c) time-dependent afterglow luminance delay of Nap@PMMA at 508 nm after the excitation with 275 nm UV light; (d) phosphorescence spectra of Nap@PMMA at different temperatures; (e) lifetime of Nap@PMMA at different temperatures monitoring at 508 nm excited with 275 nm; (f) phosphorescence spectra of Nap@PMMA, Nap@PMA and naphthalene in hexane solution; (g) comparison of normalized phosphorescence spectra of Pyr@PMMA excited at 275 nm and 338 nm; (h) lifetime of Pyr@PMMA at different phosphorescence emission peaks; (i) normalized phosphorescence spectra with different pyrene concentrations. | ||
The phosphorescence emission of Nap@PMMA with an extended lifetime and enhanced emission intensity was observed when decreasing the temperature to 77 K. The peaks of the phosphorescence spectra over the range of 400 nm to 600 nm were all presented at varied temperatures from 77 K to 277 K. The calculated lifetime of 2.38 s at 77 K obtained by exponentially fitting the decay curve is in great agreement with a previous report (Fig. 2d and e).22 The reduced emission intensity and lifetime with elevated temperatures are a result of the increasing non-radiation transition at high temperature. In which, the motion (rotation, vibration, and collision) of the PMMA chain and naphthalene increased, resulting in partial suppression of the triplet excitons through increased non-radiative dissipation.20
Polymethacrylate (PMA) is used as a control matrix, which also has a similar chemical structure but lower Tg, to study the confinement effect of polymer surroundings on RTP. No obvious afterglow is detected from Nap@PMA even under vacuum (Fig. 2f and Fig. S7, ESI†), confirming that rigid confinement12 from a stiff polymer chain is important for RTP. Besides, it is worth mentioning that no afterglow was detected from naphthalene solution in hexane at room temperature. But at 77 K, the phosphorescence spectrum of naphthalene solution in hexane could be obtained in the range of 400 to 600 nm, consistent with that of Nap@PMMA (Fig. 2f). Therefore, the rigid surroundings make a great contribution to afterglow emission.
Notably, Pyr@PMMA showed the excitation and concentration-dependent character of phosphorescence. The phosphorescence emission of Pyr@PMMA was located at 520 nm, 596 nm, and 652 nm. The excitation spectra exhibit that the excited center of 520 nm emission was different from that of the 596 nm and 652 nm peaks, while the 596 nm and 652 nm emission presented an increased excitation at 338 nm (Fig. S8, ESI†). When changing the excitation wavelength to 338 nm, the intensity ratio of the peak located at 520 nm to that at 596 nm drastically decreased (Fig. 2g). Different emission peaks had different phosphorescence lifetimes, and the related phosphorescence lifetimes at 520, 596, and 652 nm were 0.17, 0.28 and 0.31s, respectively (Fig. 1d and 2h). It was proposed that bands at 520 nm and 600 nm of Pyr@PMMA were assigned to the triplet state from different Pyr excimers or aggregates.25 This can also be demonstrated in phosphorescence spectra with different pyrene concentrations from 0.0025 mg g−1 to 0.25 mg g−1. The phosphorescence spectra with different pyrene concentrations all showed multiple bands from 450 nm to 750 nm (Fig. 2i). With the increase of Pyr concentration, the intensity ratio of emission at 520 nm and 596 nm increased correspondingly, indicating that the longer wavelength emission should come from larger aggregate emission. But when increasing the concentration to 2.5 mg mL−1, the phosphorescence was too weak to be detected under the same test conditions (Fig. S9, ESI†). Correspondingly, in the fluorescence spectra, the increased ratio in a higher wavelength was observed (Fig. S10, ESI†). This indicates that aggregates at a high concentration led to the quenching of phosphorescence.
To further understand the role of PMMA, femtosecond time-resolved transient absorption spectra were obtained on Nap@PMMA (Fig. 3a) and naphthalene in hexane at room temperature (Fig. S11, ESI†). The characterized absorption bands of the triplet excitons located in the range of 370–430 nm were detected, which are assigned to the triplet–triplet transition of naphthalene upon photoexcitation.27,28 For the Nap@PMMA system, the absorbance reached its maximum value after ca. 101 ps, which was three orders of magnitude faster than that in hexane solution (101 ns) (Fig. 3b). The phenomenon demonstrated strong evidence that the PMMA surroundings facilitated the population rate of the triplet excitons by accelerating intersystem crossing.
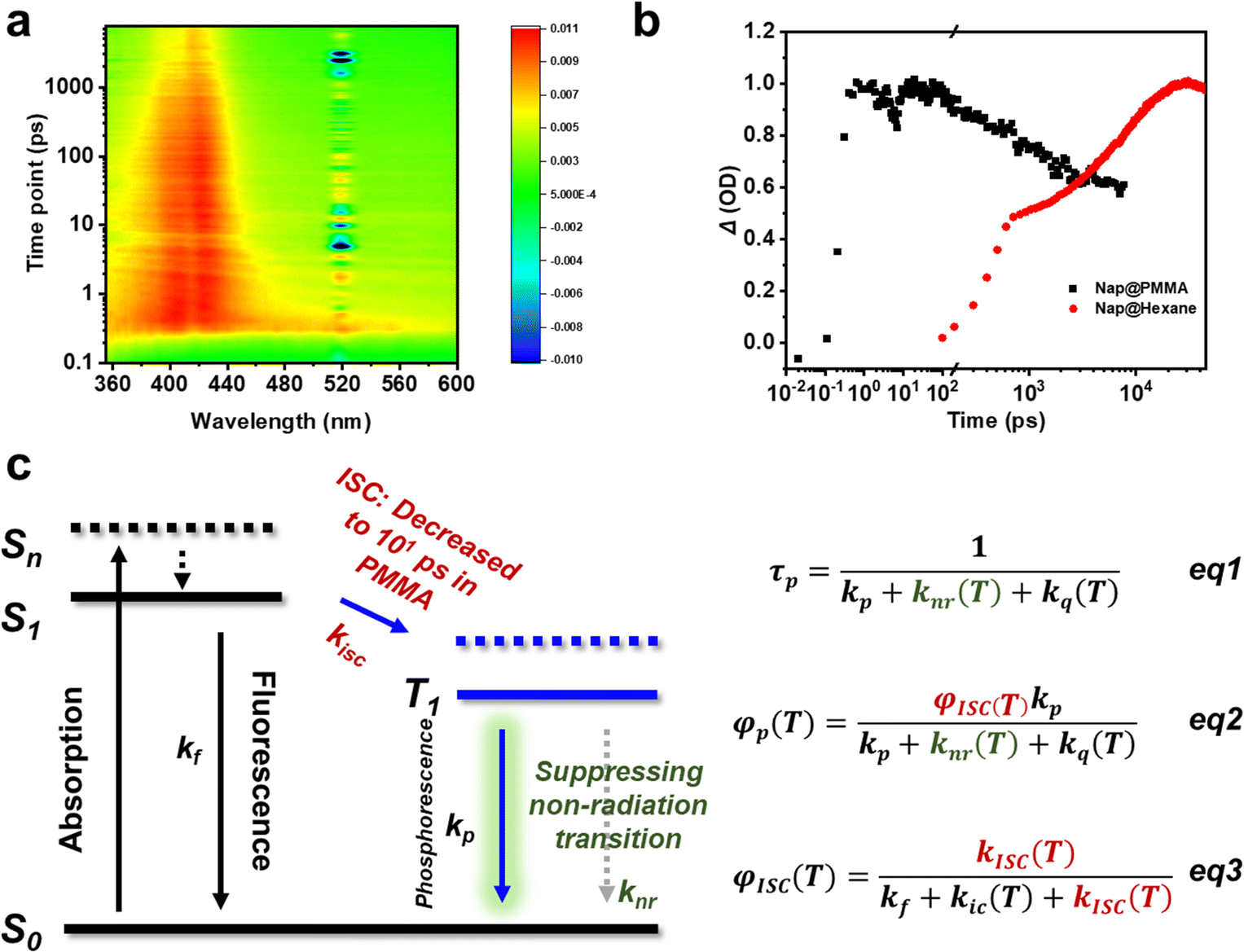 | ||
| Fig. 3 (a) Transient absorption two-dimensional temporal evolution spectra at room temperature of Nap@PMMA; (b) transient absorption traces of Nap@PMMA at 421 nm and naphthalene in hexane at 412 nm; (c) schematic illustration of a Jablonski diagram for long-lived RTP of the phosphor in our system. | ||
Generally, faster ISC might be a result of the enhanced spin-orbit coupling, decreased energy gap between two electronic states of different multiplicities or the formation of exciton clusters.29 Experimentally, only photophysical dynamics of naphthalene was changed when doped in PMMA. We are not sure whether there are any C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Ar and/or other interactions in the doping systems. The concentration was too low to be detected in the solid state by spectroscopy characterization. However, no obvious energy level change of the singlet and triplet state was detected. From the emission spectrum of naphthalene and absorption of PMMA, the energy transfer from PMMA to naphthalene might not be the main reason. With reference to a previous report,28 cluster excitons might serve as the transient species to accelerate the ISC process.
O⋯Ar and/or other interactions in the doping systems. The concentration was too low to be detected in the solid state by spectroscopy characterization. However, no obvious energy level change of the singlet and triplet state was detected. From the emission spectrum of naphthalene and absorption of PMMA, the energy transfer from PMMA to naphthalene might not be the main reason. With reference to a previous report,28 cluster excitons might serve as the transient species to accelerate the ISC process.
Therefore, the polymer surroundings here played a key role in the ultra-long afterglow (Fig. 3c). First, the PMMA acted as highly rigid surroundings for the phosphors, suppressing the non-radiative dissipations. The decreased non-radiation rate (knr) promoted the phosphorescence lifetime (τp) and quantum yield (φp) from eqn (1) and (2), and satisfied kr > knr + kq,30,31 enabling the remarkable long-lived phosphorescence at room temperature. Besides, transient absorption indicated that the PMMA matrix significantly promoted the intersystem crossing (ISC) transition from the singlet to triplet state. The ISC process was accelerated to achieve the condition kisc > kf, resulting in an efficient φisc without using heavy atoms (eqn (3)). Accordingly, φp was significantly enhanced.
The secondary interactions between phosphors and the polymer matrix, such as hydrogen bonding, ionic bonding,32 are not prerequisite in the current system, which is different from the more common polymer-based RTP materials reported previously. Therefore, the PAHs@PMMA demonstrated robust humidity adaptability. The afterglow is persistent without any noticeable reduction of phosphorescence intensity and duration time even by immersing in water (Fig. 4a–c). It is believed that the as-prepared RTP materials have great potential for applications in a highly humid environment.
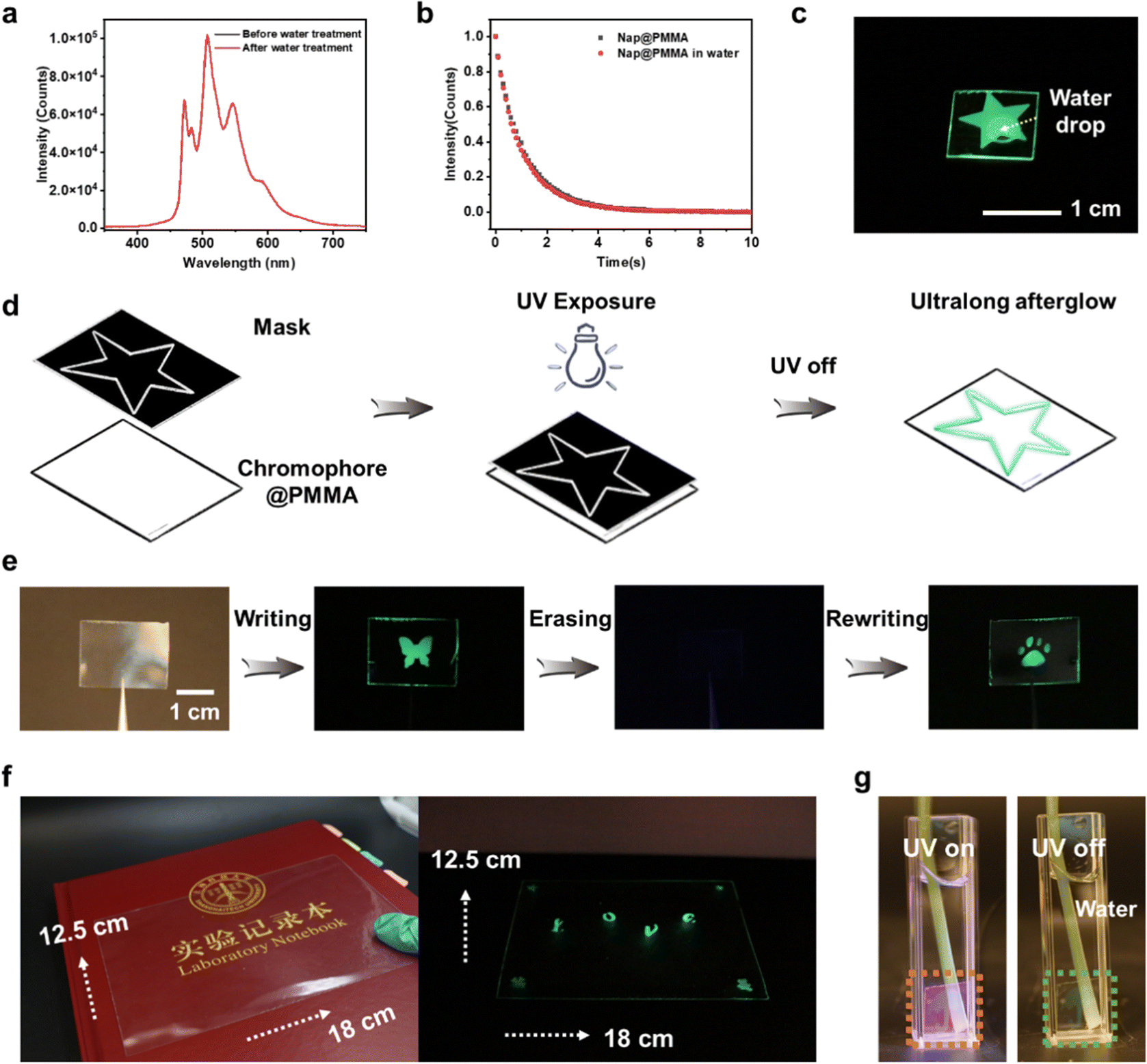 | ||
| Fig. 4 Long-lived RTP with water resistance. (a) Delayed RTP spectra (delay time: 10 ms) before and after water treatment; (b) time-dependent afterglow luminance delay of Nap@PMMA before and after in water at 508 nm (excited with 275 nm UV light); (c) photographs of photo-activated long-lived RTP of transparent Nap@PMMA film with a water drop on it; (d) schematic illustration of the process for the writing film using lithography; (e) application of photo-activated RTP of Nap@PMMA of the rewritable and erasable display for data storage and encryption (writing by UV irradiation with photo-mask; erasing under ambient conditions for 2 days); (f) photograph of the photo-activated long-lived RTP of transparent Nap@PMMA film with patterned (marked with “love” in the center and four different patterns in the corners) UV exposure; (g) photographs of Nap@PMMA (with umbrella patterned UV exposure) soaked in water after the UV light was turned off. | ||
In addition, only very weak RTP emission was detected with a flashed UV irradiation. However, two orders of magnitude enhancement of the afterglow intensity were observed with an extended UV-irradiation of 3 min, showing an efficient UV light irradiation-dependent behavior (Fig. S12a, ESI†). The enhanced RTP emission can still be detected after exposure under an ambient environment for 2 days or purging oxygen for 10 min (Fig. S12b and c, ESI†). It was proposed that when electrons of naphthalene were excited to the singlet state, some of them would jump to the excited triplet state through an intersystem crossing process. Excited triplet states in organic molecules can be readily quenched by oxygen molecules via triplet–triplet interactions, leading to non-radiative relaxation of the emitter.33 When the Nap@PMMA film was irradiated by UV light, the triplet oxygen in excited states was continuously converted to excited singlet state oxygen (Fig. S12c, ESI†).34 The depletion of triplet oxygen by UV irradiation and the low oxygen permeation rate into the PMMA matrix also contributed to the photo-activated RTP emission.35,36
Next, by utilizing the photo-activated RTP properties, different phosphorescent patterns were successively written onto the same transparent film with a photomask (Fig. 4d). For instance, the transparent Nap@PMMA film was irradiated by UV light through a mask with a butterfly-shaped opening, and the corresponding pattern with glowing green phosphorescence emission was obtained. Then, the pattern could be erased by keeping under ambient conditions for ∼2 days or continuously purging oxygen for 10 minutes, as the oxygen permeated into the polymer matrix and quenched the triplet excitons. Next, the second-round writing process can be repeated with another mask, and a new glowing pattern was obtained (a bear footprint) (Fig. 4e). Furthermore, taking advantage of the in situ polymerization strategy, a large-scale RTP film can also be fabricated with writable and erasable properties (Fig. 4f). The pattern could not be recognized with the naked eye. However, the embedded image and information can be decoded after the excitation of UV light. The as-prepared pattern in Nap@PMMA was maintained even when soaked in water (Fig. 4g), which showed the potential for water resistant applications. The rewritable feature of the as-prepared material makes it promising in applications such as encryption, anti-counterfeit and information storage.
Conclusion
In conclusion, pure organic RTP materials without heavy atoms were successfully achieved by simply in situ doping polycyclic aromatic hydrocarbons (PAHs) as phosphors in the PMMA matrix during polymerization. The afterglows of PAHs with a large Stokes shift of more than 200 nm and an ultralong duration of over 12 secs were achieved at room temperature. The rigid PMMA matrix played a key role in promoting the intersystem crossing of phosphors from the singlet to triplet state and suppressing non-radiative dissipation. Extremely fast intersystem crossing in the order of tens of picoseconds was experimentally observed for the first in the PMMA RTP system. Furthermore, the PAHs@PMMA system demonstrated robust water-resistant phosphorescence emissions due to the absence of secondary interactions between phosphors and the matrix. The potential applications of RTP materials in 2D displays and 3D devices featuring ultra-long afterglow and writable/erasable properties were demonstrated simply by utilizing programmed molds. The findings here provide a guideline for developing phosphor-doped polymer RTP materials with ultralong phosphorescence emission and good moisture-tolerance, which present a promising platform for designing optics, light emitting diodes, and data encryption technology working at high humidity.Experimental section
Materials
Methyl methacrylate (MMA) and pyrene were purchased from Sigma-Aldrich, 2,2′-azobis(2-methylpropionitrile) (AIBN) was purchased from Sinopharm Chemical Reagent Co., Ltd, naphthalene was purchased from Damas-beta and phenanthrene was purchased from Aladdin, respectively. Naphthalene, phenanthrene, pyrene and AIBN were purified by recrystallization. Chemicals were purchased commercially and used without further purification unless otherwise noted.Fabrication of PMMA-PAH phosphorescence materials
The phosphor (0.02 mg mL−1) and AIBN (0.6%) were added into MMA. The mixture was degassed by a freeze–thaw cycle and then pre-polymerized at 80 °C for 25 min. Then the pre-polymer mixture was poured into the mold with 2D (e.g. film) or 3D (e.g. flower and bear) shapes for post-polymerization (40 °C for 10 hours, 70 °C for 8 hours and then 100 °C for 2 hours) to obtain the target materials, defined as Nap@PMMA, Phe@PMMA and Pyr@PMMA, respectively. The thickness of the films is ca. 0.12 mm. If there are no special instructions, the power of the 275 nm LED UV light for sample treatment is 0.5 W and the irradiation time is 3 min.Photophysical analysis
UV-Vis absorption spectra were obtained on an Agilent Cary5000 UV-Vis spectrophotometer. The fluorescence properties were determined using a HORIBA Fluorolog-3 fluorescence spectrophotometer with a xenon lamp. Phosphorescence spectra were obtained using a Horiba FluoroMAX+ with a microsecond flash lamp and a delay time of 10 ms. Afterglow spectra were obtained using a HORIBA Duetta with a CCD detector. The photos were recorded using a Canon EOS 70D camera.Transient absorption (TA) spectra were collected using a commercial transient absorption spectrometer (HELIOS, Ultrafast System) at room temperature. The 800 nm fundamental pulses were generated from a Ti:sapphire laser system (Coherent, Astrella, 35 fs, 7 mJ per pulse, 1 kHz repetition rate.). A 260 nm pulse was used as the actinic pump for our TA experiment, which was generated by 800 nm fundamental pulses via an optical parametric amplifier (OPerA Solo, Coherent Inc.). The actinic pump power was then reduced to ∼50 μW via a neutral density filter. Broadband supercontinuum white light with a wavelength ranging from ∼340 nm to ∼650 nm serves as the probe pulse.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 21975160, No. 82001945), the Shanghai Pujiang Program (No. 20PJ1410700), the Shanghai Sailing Program (No. 22YF1428000), and the Double First-Class Initiative Fund of ShanghaiTech University (No. SYLDX006, SYLDX028). The authors are grateful to Prof. Yong Yang and Dr Junyu Lang from ShanghaiTech University for the discussion. Photo-physical characterization was performed at the center of the Analytical Instrumentation Center (AIC) at the School of Physical Science and Technology (SPST), ShanghaiTech University and Horiba lab.References
- Z. Cai, H. Ma, H. Shi, H. Wang, X. Wang, L. Xiao, W. Ye, K. Huang, X. Cao, N. Gan, C. Ma, M. Gu, L. Song, H. Xu, Y. Tao, C. Zhang, W. Yao, Z. An and W. Huang, Nat. Commun., 2019, 10, 4247 CrossRef PubMed.
- M.-M. Fang, J. Yang and Z. Li, Chin. J. Polym. Sci., 2019, 37, 383–393 CrossRef CAS.
- L. Gu, X. Wang, M. Singh, H. Shi, H. Ma, Z. An and W. Huang, J. Phys. Chem. Lett., 2020, 11, 6191–6200 CrossRef CAS PubMed.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- Y. Li, M. Gecevicius and J. Qiu, Chem. Soc. Rev., 2016, 45, 2090–2136 RSC.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- X. Yang and D. Yan, Chem. Sci., 2016, 7, 4519–4526 RSC.
- H. Thomas, D. L. Pastoetter, M. Gmelch, T. Achenbach, A. Schlögl, M. Louis, L. Feng and S. S. Reineke, Adv. Mater., 2020, 32, 2000880 CrossRef CAS PubMed.
- P. Ceroni, Chem, 2016, 1, 522–530 Search PubMed.
- X. Jiang, J. Peng, J. Wang, X. Guo, D. Zhao and Y. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 3591–3600 CrossRef CAS PubMed.
- H. Gao and X. Ma, Aggregate, 2021, 2, e38 Search PubMed.
- Y. Li, L. Jiang, W. Liu, S. Xu, T.-Y. Li, F. Fries, O. Zeika, Y. Zou, C. Ramanan, S. Lenk, R. Scholz, D. Andrienko, X. Feng, K. Leo and S. Reineke, Adv. Mater., 2021, 33, 2101844 CrossRef CAS PubMed.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Nat. Mater., 2021, 20, 1539–1544 CrossRef CAS PubMed.
- Z. Wang, Y. Zheng, Y. Su, L. Gao, Y. Zhu., J. Xia, Y. Zhang, C. Wang, X. Zheng, Y. Zhao, C. Yang and Y. Li, Sci. China Mater., 2022, 65, 2160–2168 CrossRef CAS.
- Y. Zhang, Y. Su, H. Wu, Z. Wang, C. Wang, Y. Zheng, X. Zheng, L. Gao, Q. Zhou, Y. Yang, X. Chen, C. Yang and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 13675–13685 CrossRef CAS PubMed.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., 2014, 126, 11359–11363 CrossRef.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- J. Wang, X.-Y. Lou, Y. Wang, J. Tang and Y.-W. Yang, Macromol. Rapid Commun., 2021, 42, 2100021 CrossRef CAS PubMed.
- Y. Zhao, X. Chen, J. Xu, Q. Zhang, L. Gao, Z. Wang, L. Qu, K. Wang, Y. Li, Z. Cai, Y. Zhao and C. Yang, J. Am. Chem. Soc., 2022, 144, 6107–6117 CrossRef PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081–2093 RSC.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- J. L. Kropp and W. R. Dawson, J. Phys. Chem., 1967, 71, 4499–4506 CrossRef CAS.
- Z. Wang, L. Gao, Y. Zheng, Y. Zhu, Y. Zhang, X. Zheng, C. Wang, Y. Li, Y. Zhao and C. Yang, Angew. Chem., Int. Ed., 2022, 61, E202203254 CAS.
- T. Takemura, H. Baba and Y. Shindo, Chem. Lett., 1974, 1091–1096 CrossRef CAS.
- V. A. Svetlichnyi, T. N. Kopylova, G. V. Mayer and I. N. Lapin, Russ. Phys. J., 2005, 8, 901–906 CrossRef.
- X. Wang, W. G. Kofron, S. Kong, C. S. Rajesh, D. A. Modarelli and E. C. Lim, J. Phys. Chem. A, 2000, 104, 1461–1465 CrossRef CAS.
- X. Zhang, L. Du, W. Zhao, Z. Zhao, Y. Xiong, X. He, P. F. Gao, P. Alam, C. Wang, Z. Li, J. Leng, J. Liu, C. Zhou, J. W. Y. Lam, D. L. Phillips, G. Zhang and B. Z. Tang, Nat. Commun., 2019, 10, 5161 CrossRef PubMed.
- X. Ma, C. Xu, J. Wang and H. Tian, Angew. Chem., 2018, 130, 11020–11024 CrossRef.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed.
- L. Gu, H. Wu, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944 CrossRef CAS PubMed.
- Y. Yu, M. S. Kwon, J. Jung, Y. Zeng, M. Kim, K. Chung, J. Gierschner, J. H. Youk, S. M. Borisov and J. Kim, Angew. Chem., Int. Ed., 2017, 56, 16207–16211 CrossRef CAS PubMed.
- M. Louis, H. Thomas, M. Gmelch, A. Haft, F. Fries and S. Reineke, Adv. Mater., 2019, 31, 1807887 CrossRef PubMed.
- L. Zang, W. Shao, M. S. Kwon, Z. Zhang and J. Kim, Adv. Opt. Mater., 2020, 8, 2000654 CrossRef CAS.
- F. Gu, T. Jiang and X. Ma, ACS Appl. Mater. Interfaces, 2021, 13, 43473–43479 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc02557d |
| ‡ Xiao Meng, Qian Hu and Xi Wang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |