MIL88-A as a mediator for the degradation of sulfamethoxazole in PS systems: implication of solar irradiation for process improvement†
Received
2nd August 2022
, Accepted 22nd September 2022
First published on 22nd September 2022
Abstract
The aim of this study was to demonstrate a feasible system that utilizes solar irradiation for the activation of persulfate PS using a mediator, MIL88-A, an iron-based MOF since PS is poorly activated under sunlight. UVA was also used to simulate solar irradiation, which contains both UVA and UVB. A solution containing sulfamethoxazole (SMX) antibiotic (5 mg L−1) placed in continuously stirred reactors, irradiated with either UVA or natural solar light and spiked with PS (2 mM) and MIL88-A slurry was investigated. The study showed the combined effects of UVA or solar/MIL88-A/PS on the degradation of SMX. The total degradation of SMX occurred in 2 h using the UVA/MIL88-A/PS system compared to just 5–20 min in the solar/MIL88-A/PS system; the system was also examined for its recyclability and matrix effect such as MOF load, pH and UV irradiation as well as chloride, phosphate and bicarbonate contents. The results showed that (i) MIL88-A is recyclable up to five successive cycles, whereby 100% SMX degradation is achieved after 2 h; (ii) chlorides slightly enhanced SMX degradation, while phosphates and bicarbonates greatly inhibited it. This study also investigated the SMX degradation mechanism using XPS, EPR and TOF-SIMS techniques. They revealed that the degradation mechanism is mainly based on sulfate radical (SR) and hydroxyl radical (HR) oxidation. Three SMX degradation products were identified using an HPLC coupled with a QToF high-resolution mass spectrometer.
Environmental significance
This research investigated the applicability of indirectly activating persulfate using solar radiation. MIL88-A was used as a mediator to activate persulfate using UVA and UVB irradiation. This was done to overcome the limitation faced in the UVC requirement for PS activation, which can only be produced artificially at the ground level. To achieve such an objective, sulfamethoxazole (SMX) antibiotic was used as a probe. A special reactor was designed and constructed to keep MIL88-A in suspension under solar radiation limiting the inner filter effect. Full SMX degradation was achieved in a short time without interference from common water species. In addition, recyclability experiments showed that MIL88-A can be used for up to five successive cycles without any significant decrease in the SMX degradation rate.
1. Introduction
Pharmaceuticals and personal care products (PPCPs) are a group of various organic and inorganic compounds. These materials include antibiotics, hormones, antimicrobial agents, NSAIDs, cosmetics, fragrances, etc. They were classified as emerging environmental contaminants in the last two decades due to their environmental and potential human health risks.1–3 Many research projects were conducted to study the effect of PPCPs not only on the aquatic environment but also on the terrestrial one including their presence, magnitudes, factors that affect their levels, consequences, and treatment.1 PPCPs tend to enter the environment through different routes. Human and animal waste is one of the main routes, in addition to the uncontrolled disposal of expired medications into wastewater or landfills which may reach surface and ground water as well as soil. Similarly, industrial effluents proved to contain considerable amounts of these PPCPs.4,5 They are mainly made of two components: excipients and active pharmaceutical ingredients (APIs). The presence of APIs in waste water treatment plants had been stated, in many countries over the world, at the levels of ng L−1 to μg L−1,6 such as the US,7 UK,8 Spain,9 Finland,10 and Japan.11 The resistance of PPCPs to the available treatment methods led to the accumulation and possibly the bio-amplification of pharmaceuticals in water, which has a significant negative impact on the ecosystem and possibly on human health.12 Over the last two decades, these categories of PPCPs have been widely used due to their low cost, availability over the counter and minor side effects. Consequently, significant concentrations of these pharmaceuticals and their metabolites reach groundwater, and even surface and drinking water.13,14 Persulfate (PS) has been widely used in the past few years due to the variety of activation mechanisms that can be applied for PS,15 being (i) a cost-effective method compared to other AOPs,10 and (ii) an environmentally friendly method that degrades pharmaceuticals and results in sulfate ions in aqueous medium.16 Upon its activation, PS (E0 = 2.1 V) generates sulfate radicals (SRs) (E0 = 2.6 V) characterized by high reactivity close to that of hydroxyl radicals (HRs) (E0 = 2.7). However, the lifetime of SRs is much greater than that of HRs, t = 20–30 μs versus t = 20 ns, respectively. Moreover, SRs are distinctively more selective and have a high oxidizing ability across a broader range of pH compared to HRs, allowing therefore more efficient and targeted water treatment applications.17 The most common PS activation methods are thermal (eqn (1)),18–20 UV (eqn (2))21–23 and chemical using Fe2+ ions in a homogeneous medium (eqn (3))24,25 or heterogeneous catalyst such as MOFs (Metal–Organic Frameworks) or magnetite.26,27 The aim of this study is to activate PS under solar irradiation using a mediator MIL88-A, an iron-based MOF.| | | S2O82− → 2SO4˙− (thermal activation) 30 °C < T < 99 °C | (1) |
| | | S2O82− + Fe2+ → S042− + SO4˙− + Fe3+ (chemical activation) | (2) |
| | | S2O82− + hv → 2SO4˙− (UV activation) | (3) |
MOFs are a relatively new class of porous 3d materials. They have a modular structure that gives them huge structural diversity and the possibility of creation of materials with tailored characteristics. They are synthesized through different methods such as microwave, ultrasonication, electrochemical and mechanochemical processes. MOFs are also functionalized so that they suit certain applications. They are highly porous and consist of metal ions or clusters connected to organic linkers to form one, two, or three-dimensional structures. This has changed upon discovering that the inorganic part has high dimensionality that can form layers and may extend to frameworks and not only chains.28,29 This combination of organic and inorganic structures leads to materials with unique properties, so MOFs are often characterized by their high surface area reaching around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 g−1 and huge pore volumes around 50% of the total volume or more (0.99 cm3 g−1 10−2 for MIL88A).30,31 These properties make them good candidates for gas adsorption or separation,32–34 drug delivery,35,36 magnetism,37 polymerization,38 catalysis,39 and many other applications. MOFs, especially those having Fe as a metal ion, showed effective photo-catalytic activity for the removal of organic pollutants. This activity is accomplished by the use of a mediator such as PS. Recently, Fe-based MOFs have been investigated for the activation of PS to degrade organic dyes.40,41 Experiments showed promising results especially in recyclability and reproducibility making MOFs a greener alternative.42 For example, Li et al. studied the degradation of acid orange 7 (AO7) by activating PS using four different MOFs.42 The results showed that MIL-101 (Fe) has the best adsorption properties and catalytic activity toward PS activation for the removal of AO7.42 In addition to that, Shen et al. studied the degradation of atrazine with Bi2MoO6 nanosheets using peroxymonsulfate (PMS) as an activator under visible light irradiation. The results showed that full degradation of atrazine was accomplished within 60 min.43 Furthermore, Hu et al. demonstrated the degradation of an organophosphorus flame retardant, tris(2-chloroethyl) phosphate (TCEP).44 The photocatalysis reaction involved the activation of MIL-101(Fe) for the conversion of Fe(III) to Fe(II) (eqn (4)), which induced further transformation of PS into SRs (eqn (3)), allowing the oxidative degradation of TCEP (eqn (5)) as well as other studies using iron-based MOFs.44
000 m2 g−1 and huge pore volumes around 50% of the total volume or more (0.99 cm3 g−1 10−2 for MIL88A).30,31 These properties make them good candidates for gas adsorption or separation,32–34 drug delivery,35,36 magnetism,37 polymerization,38 catalysis,39 and many other applications. MOFs, especially those having Fe as a metal ion, showed effective photo-catalytic activity for the removal of organic pollutants. This activity is accomplished by the use of a mediator such as PS. Recently, Fe-based MOFs have been investigated for the activation of PS to degrade organic dyes.40,41 Experiments showed promising results especially in recyclability and reproducibility making MOFs a greener alternative.42 For example, Li et al. studied the degradation of acid orange 7 (AO7) by activating PS using four different MOFs.42 The results showed that MIL-101 (Fe) has the best adsorption properties and catalytic activity toward PS activation for the removal of AO7.42 In addition to that, Shen et al. studied the degradation of atrazine with Bi2MoO6 nanosheets using peroxymonsulfate (PMS) as an activator under visible light irradiation. The results showed that full degradation of atrazine was accomplished within 60 min.43 Furthermore, Hu et al. demonstrated the degradation of an organophosphorus flame retardant, tris(2-chloroethyl) phosphate (TCEP).44 The photocatalysis reaction involved the activation of MIL-101(Fe) for the conversion of Fe(III) to Fe(II) (eqn (4)), which induced further transformation of PS into SRs (eqn (3)), allowing the oxidative degradation of TCEP (eqn (5)) as well as other studies using iron-based MOFs.44
| |  | (4) |
| | | SO4−˙ + contaminant → byproducts | (5) |
In this study, SMX was the probe selected for the degradation process. It is an antimicrobial agent, which is used to treat a variety of bacterial infections such as middle ear, urine, and respiratory infections. Due to its overuse, its active molecules reach surface and ground water due to different industrial and domestic activities. Studies showed that it is present at low concentrations in surface water around the globe (e.g. 21 ng L−1);45 nevertheless, this low concentration causes certain modifications in the microbial community structure leading to antibiotic resistance in environmental microbial communities.46–48 This entails its elimination at its point sources. Several research groups have investigated the elimination of SMX from waste water by chlorination,49 ozonation50 photo-Fenton and solar photo-Fenton,51,52 and photo-catalysis.53–55 All the mentioned treatment methods are energy and catalyst consuming, sometimes leading to stable and toxic metabolites. Accordingly, AOPs using PS have been widely investigated due to their advantages in terms of non-toxic by-product formation (sulfates) and low cost.16 For instance, SMX removal was demonstrated by Ghauch et al. and Ayoub and Ghauch through PS assisted micrometric Fe0 as well as bimetallic and trimetallic iron-based particles in aqueous solution.24,56 However, all the above-mentioned systems lack the option of recyclability and reproducibility making the systems relatively expensive and can generate sludge due to iron corrosion product (ICP) formation. In this paper, a heterogeneous catalyst (MIL88-A) will be used to activate PS. First, the choice of the MOF was based on several factors. It must contain an activator for PS such as a metal. Iron is considered one of the most suitable metals for PS activation since it is abundant, relatively safe, and easy and affordable to produce. On the other hand, the choice of the ligand, the MOF's second component, is also based on similar criteria such as affordability, safety, and abundance so MIL88-A was chosen since it consists of a safe and cheap ligand and metal source as well. MIL88-A is composed of ferric chloride as the metal source and fumaric acid as the ligand. The synthesis of MIL88-A is based on water as the solvent in contrast to most other MOFs that require organic solvents.57,58 Second, we tried to find a low-cost ligand, which is fumaric acid in our case and a green synthesis method, which is based on water as a solvent instead of an organic one. In fact, Wang et al. studied the catalytic activity of MIL88-A with PS for the degradation of orange G (OG), where MIL88-A synthesized at 85 °C for 2 h gave the highest degradation rate of OG that reached 96.4%.31 These results drove our attention towards the use of MIL88-A as the PS activator. We tried in this work to explore additional factors that can improve the properties of MIL88-A in aqueous solutions and play the role of a PS activator in a heterogeneous medium with the possibility of showing some synergistic effect. For example, we investigated the combination of PS activators in systems such as UVA/MIL88-A/PS or solar/MIL88-A/PS and monitored the impact on SMX removal in terms of the degradation rate and transformation product nature and persistence. This was possible upon using the time of flight secondary ion mass spectrometry (ToF SIMS) technique previously used for the characterization of materials of environmental and pharmaceutical interest59–63 as well as the time of fight high resolution mass spectrometry (ToF HRMS) technique for the identification of transformation products. Moreover, we studied the effect of various factors such as salinity, carbonates, and phosphates on the performance of both systems UVA/MIL88-A/PS/SMX and solar/MIL88-A/PS/SMX toward SMX degradation and the potential application of this system on real pharmaceutical industry effluents.
2. Materials and methods
2.1. Chemicals
Sulfamethoxazole (SMX) (C10H11N3O3S) and sodium persulfate (Na2S2O8, purity ≥ 99%) were obtained from Sigma Aldrich (USA). Potassium iodide (KI) (puriss, 99.0–100.5%) was used for PS quantification and monobasic (H2NaO4P assay ≥ 99.0%) and dibasic phosphate buffer (HNaO4P assay: 98–100.5%) used to study the effect of phosphate were purchased from Sigma Aldrich (China, France, and Germany, respectively). Fumaric acid (C4H4O) and iron(III) chloride (FeCl3) (both reagent grade > 97%) used in the synthesis of MIL88-A were acquired from Sigma Aldrich (France and Switzerland, respectively). Ethanol (absolute) was purchased from Scharlau (Spain). Formic acid and methanol used as HPLC mobile phases were acquired from Loba Chemie (India) and Honeywell (Germany), respectively. Millipore deionized water (DI) was used in the preparation of all solutions. To evaluate the matrix effect, sodium bicarbonate (NaHCO3) and sodium chloride (NaCl) were acquired from Fluka (Netherlands). Furthermore, hydrochloric acid (HCl) and sodium hydroxide used to modify the pH were purchased from Fluka (Switzerland, Germany, respectively).
2.2. Synthesis of MIL88-A
MIL88-A was synthesized using a hydrothermal synthesis technique previously reported.27,41,64 This method was selected since it is greener than the conventional solvothermal method. It was done by adding 1949 mg of fumaric acid and 4544 mg of ferric chloride to a beaker containing 84 mL of DI water. The solution was stirred to homogenize for 1 h using a magnetic stirrer at 300 rpm. Later, the solution was transferred to a 100 mL Teflon-lined stainless-steel autoclave bomb and heated in an oven at 85 °C for 24 h. The autoclave bomb was then removed and left to cool passively before opening. After that, the solution containing suspended MIL88-A crystals was transferred to two 50 mL Falcon tubes to be centrifuged at 4000 rpm (G-force = 2200) for 10 min. The crystals were then collected, transferred to a 400 mL beaker, and then washed three times with 390 mL of ethanol/DI (1:1) solution, followed by two consecutive DI washes. This washing process was followed to ensure the complete removal of all unreacted fumaric acid and ferric chloride. The precipitate was recovered after each wash by centrifugation using the aforementioned conditions. Finally, the precipitate was dried in a vacuum oven at 100 °C for no less than 10 h. Each synthesis yielded 2350 ± 220 mg of MIL88-A, which was characterized to ensure its purity.
2.3. Characterization of MIL88-A
2.3.1. XRD, SEM, BET, TGA, FTIR and XPS analyses.
MIL88-A was characterized according to the common techniques reported in the literature.41 First, to guarantee the crystallinity of the obtained crystals, a D8 Advance (Bruker) X-ray diffractometer (XRD), equipped with a copper anode material (40 mA, 40 kV), was used to obtain the X-ray diffraction pattern. MIL88-A was set on a customized zero-background holder and scanned with a scan rate of 0.02° per second from 5° to 20° (2θ). To identify the morphology of the synthesized material, a scanning electron microscope (SEM), Tescan, Mira III, was used. Also, to determine the accurate surface area and pore size of MIL88-A, a Brunauer–Emmett–Teller (BET) surface area and pore size analyzer (Micromeritics, 3 flex surface area characterization) was used. Furthermore, a Micromeritics, Q2000 dynamic light scattering instrument (DLS) was used to calculate the hydrodynamic diameter, particle size distribution and zeta potential. Moreover, MIL88-A thermogravimetric analysis (TGA) was conducted in a nitrogen atmosphere with a heating frequency of 5 °C min−1 and a temperature of 30 to 900 °C using a TG 209 F1 Iris (Netzsch, Germany). Finally, a Bruker Tensor 27 IR was used to perform Fourier Transform Infrared Spectroscopy (FTIR) of the TGA analyser exhaust under nitrogen. XPS measurements were performed with a VG ESCALAB 220iXL spectrometer (Thermo Fisher Scientific) utilizing focused mono-chromatized AlKα radiation (1486.6 eV) of a beam size of ∼500 μm2 (power of 150 W). The measurements were done on the MOF crystals being mixed with 10% conductive carbon (Super-C 65, Timcal). The pressure in the analysis chamber was approximately 2 × 10−9 mbar. The spectrometer was calibrated on a clean silver surface by measuring the Ag 3d5/2 peak at a binding energy (BE) of 368.25 eV with a full width at half maximum (FWHM) of 0.78 eV. All the spectra were recorded under the conditions of 30 eV pass energy and 50 eV for the surveys in steps of 50 meV and a dwell time of 50 ms. The calibration of the binding energy peak positions is applied on C 1s located at 284.6 eV.65
2.3.2. TOF-SIMS analysis.
The experiments were performed using a TOF-SIMS V mass spectrometer (ION-TOF GmbH, Münster, Germany). Bismuth clusters (Bi3+) at an energy of 25 keV with a low intensity of 0.08 pA were used to keep all the ions under the saturation limits. Negative secondary ions were extracted at an energy of +2 keV passing through a single-stage reflector before hitting a single microchannel plate detector. Charge compensation was provided by 21 eV electrons. The reflector voltage was kept at +50 V. The internal mass calibration of the acquired spectra is done using C−, C2−, C3H3O2− and Fe2O3H−, which permits a mass accuracy of a few ppm. The mass resolution at m/z 24 was 3000. The images of the negative ions, presented in this work, were collected using Bi3+ on a 100 × 100 μm2 size area with a pixel size of 0.4 × 0.4 μm2. The lateral resolution was 800 nm. More details on the analysis conditions can be consulted elsewhere.66
2.4. Experimental procedure
All solutions were prepared on a daily basis using DI water. SMX stock solution (100 ppm) was prepared by dissolving 100 mg of SMX in a 1000 mL volumetric flask. The solution was stirred overnight away from light. PS stock solution (100 mM) was prepared by dissolving 2381 mg of sodium persulfate in a 100 mL volumetric flask. For all the experiments that were carried out in the laboratory in the presence of UVA, the required volume of SMX stock solution was added to a 400 mL beaker along with a specific volume of DI. After that, at t0, the essential amount of MIL88-A was added and left to stir for 1 min. The reaction was initiated by the addition of PS solution and turning on UVA lamps. In order to ensure consistent mixing and accumulation of MIL88-A, continuous stirring was established throughout the 120 min reaction time. Samples were taken immediately before and after the addition of MIL88-A as well as before and after spiking with PS. After that, samples were taken every 20 min till t = 120 min. All samples were filtered using 0.45 μm PTFE 13 mm disc filters (Jaytee Biosciences Ltd, UK) and later stored at 4 °C in amber HPLC vials for a maximum period of 12 h before analysis.
On the other hand, for the experiments that were carried out under solar irradiation, a specific amount of MIL88-A was added to a home-built rotisserie shaker that was placed directly under sun light, and then a required volume of SMX solution was added along with a specific volume of DI water. The reaction was initiated by the addition of PS solution, and the reaction time was set to be 120 min, and samples were taken immediately before and after the addition of MIL88-A as well as before and after spiking with PS. A sample was taken at t = 5 min after which samples were taken every 20 min till t = 120 min following the same procedure as described above. The timing of the sample collection was varied by different experimental criteria. Control experiments were done either without PS and/or MIL88-A and/or UVA. Experiments were conducted in triplicate and each sample was analysed twice for uncertainty measurements.
2.5. Reaction setup
All experiments were done using six 400 mL beakers placed on the top of a solid state multi-magnetic stirrer of capacity equal to six beakers, irradiated with two commercial T5 8 watts near-ultraviolet (UVA) fluorescent lamps, of dosage 700 μW cm−2, placed on the top of the beakers as shown in Fig. S1.† The emission spectrum of the lamps is presented in Fig. S2† and was obtained using a modified spectrophotometer.67 This spectrum shows main emission between 350 and 400 nm lying in the UVA-vis range. However, for the experiments conducted under solar energy, 8 homemade watertight 110 mL borosilicate reactors were attached radially to a Labquake shaker rotisserie of speed 8 revolutions per min. The solar flux measured at the start and at the end of the experiment was equal to 3950 μW cm−2 and 1600 μW cm−2, respectively. This mixing setup was developed to maintain MIL88-A in suspension during irradiation as shown in Fig. S3.†
2.6. Chemical analysis
For the quantification and identification of SMX and its degradation by-products, a high-performance liquid chromatograph (Agilent 1100 HPLC) equipped with a quaternary pump, a vacuum degasser, an auto sampler maintained at 4 °C, and a thermally controlled column compartment set at 30 °C in addition to a DAD detector was used. The elution process was carried out on a C-18 reverse phase column (5 μm; 4.6 mm internal diameter × 250 mm in length) connected to a security guard column HS C-18 (5 μm; 4.0 mm internal diameter 20 mm long). SMX was quantified at its maximum absorbance wavelength of λ = 263 nm. The mobile phase consisted of methanol: 0.1% formic acid solution of (50:50) (v/v) and was kept under a constant flow rate of 0.5 mL min−1. The injection volume was set to 25 μL. Under these conditions, SMX was eluted at a retention time of 6.3 min. The linear dynamic range (LDR) obtained was between 0.1 and 10 mg L−1 with a limit of detection = 0.0009 mg L−1 (Fig. S4†).68 To account for leaching of iron in solution an atomic absorption spectrometer Ice3000 AAS series equipped with a flame atomizer was used. For MS analysis, a high-resolution mass spectrometer detector (HRMS X500R QTOF MS) was used. The HRMS detector along with a DAD detector was coupled to an UPLC (Shimadzu X500R) equipped with a quaternary pump, a vacuum degasser, an auto-sampler with cooling maintained at 4 °C, and a thermally controlled column compartment set at 30 °C. The elution process was carried out on a C-18 column (100 × 2.1 mm). The mobile phase consisted of a mixture of solvent A (99.9% methanol/0.1% formic acid) and solvent B (0.1% formic acid). The chromatographic elution conditions were as follows: 0–16.5 min, gradient 95% to 5% A, 16.5–21.5 min, gradient 95% to 5% B, and the flow rate was kept constant at 0.4 mL min−1 with a run time of 25 min.
3. Results and discussion
3.1. Characterization of MIL88-A
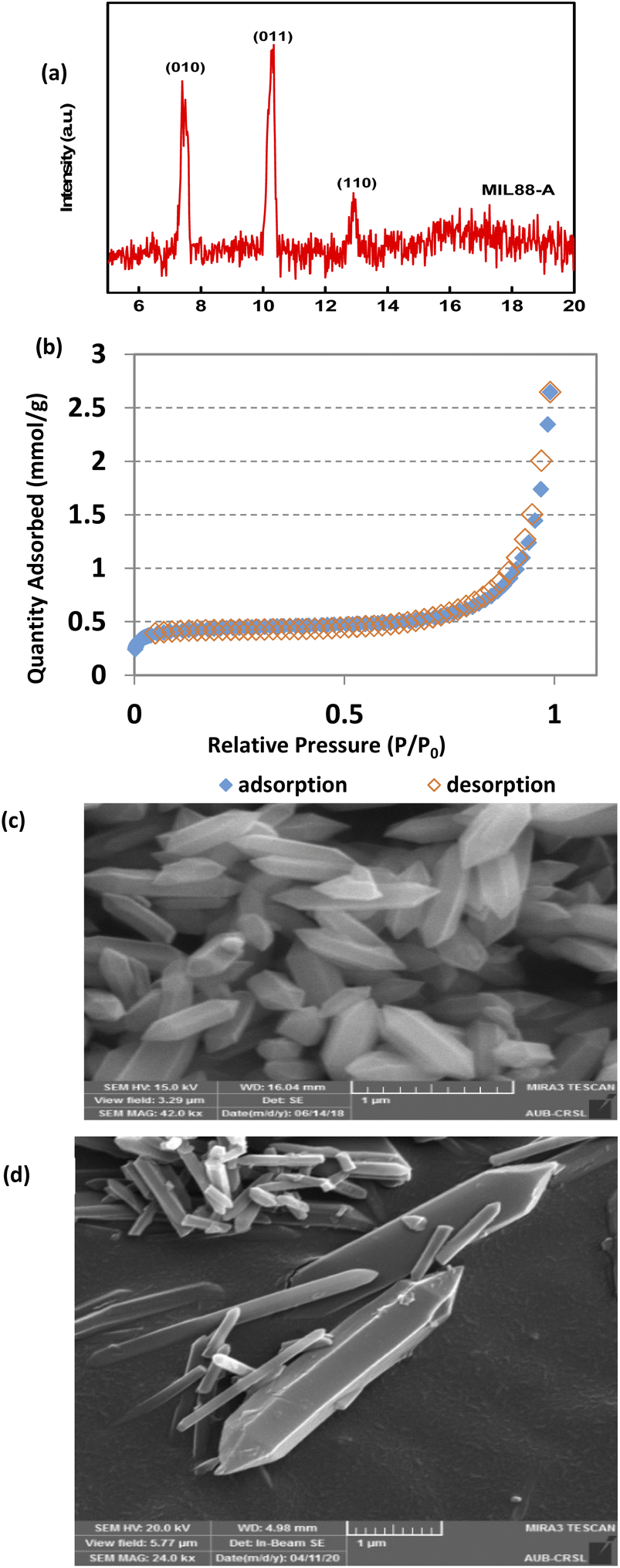
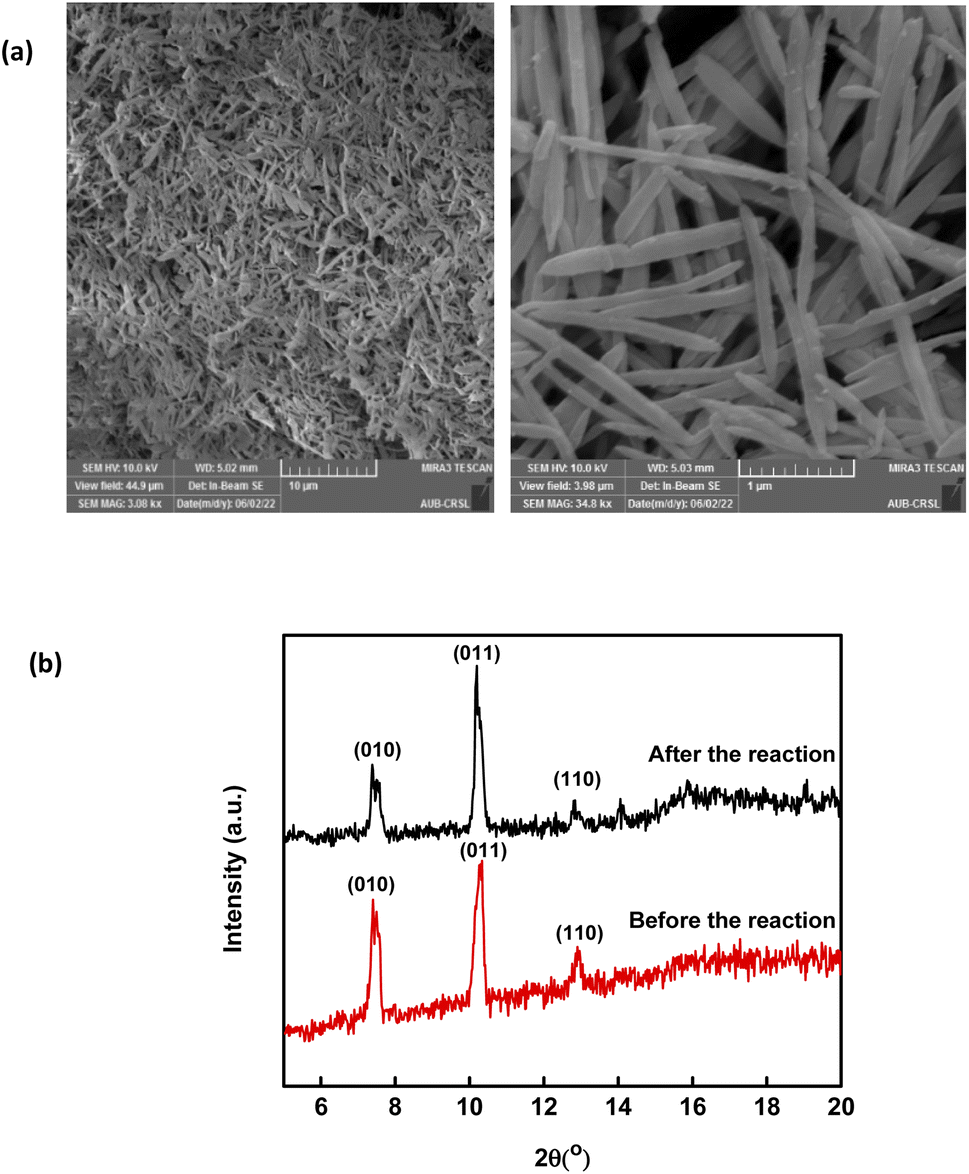
The XRD pattern of MIL88-A showed well-resolved peaks at 2θ positions of 7.7°, 10.6° and 12.9° (Fig. 1a). These peaks, although they don't exactly match with what was reported experimentally in the literature,41,69 are however consistent with the theoretical simulated ones, indicating the crystalline nature of MIL88-A. Furthermore, SEM images show hexagonal crystals of a single phase morphology typical of MIL88-A, with sizes ranging from 100 to 800 nm (Fig. 1c and d). This is similar to the size range previously published.41,69 This was further verified by dynamic light scattering analysis that revealed an average hydrodynamic diameter of 411.7 nm with the distribution profile of the hydrodynamic diameter presented in Fig. S5.† In the DI matrix, zeta potential analysis was carried out as well. The values obtained ranged from −5 mV to + 5 mV, suggesting that MIL88-A crystals are vulnerable to settling and require continuous stirring to remain suspended. Moreover, the nitrogen adsorption desorption isotherm showed a good adsorption capacity of 2.6463 mmol g−1(Fig. 1b). The surface area of MIL88-A was calculated to be equal to 37.3 ± 1.8 m2 g−1, which is relatively better than that reported in the literature ranging from 19 to 30 m2 g−141. Finally, TGA analysis (Fig. S6†) revealed a 6% weight loss at a furnace temperature below 100 °C. This is mostly due to water evaporation from the MIL88-A sample. Then, a second weight loss (14.7%) was observed and attributed to the dissociation of the organic linker (fumaric acid) that is used in the synthesis process of the MIL88-A sample, which proceeded till 400 °C. Then a significant weight loss of 30% is detected, after which a complete breakdown of fumaric acid is observed at around 600 °C resulting in a final weight loss of 14% before ash remains. A different pattern of total dissociation was observed with pure fumaric acid at around 300 °C and was reported before in the literature.70 It is suggested that the thermal stability of fumaric acid increases when it is entrapped within the MIL88-A framework, requiring a higher temperature for its dissociation, which was then validated by FTIR analysis (Fig. S7†). The results showed three significant signals at 3 different temperatures (316, 445 and 613 °C). In fact, the obtained spectra showed bands characteristic of the following functional groups: (i) O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO stretching at 2350 cm−1 at all temperatures; (ii) CCO stretching at 2100–2150 cm−1 at temperature 613 °C confirming the existence of fumaric acid residues at t ≥ 300 °C.
CO stretching at 2350 cm−1 at all temperatures; (ii) CCO stretching at 2100–2150 cm−1 at temperature 613 °C confirming the existence of fumaric acid residues at t ≥ 300 °C.
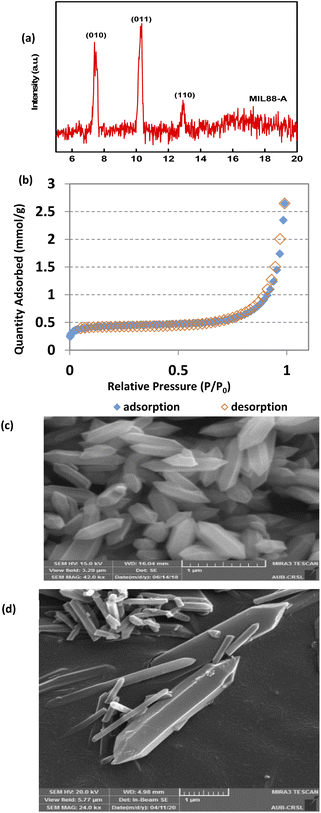 |
| | Fig. 1 SEQ Fig. \* ARABIC Characterization of synthesized MIL88-A: (a) XRD diffraction pattern, (b) BET adsorption/desorption isotherms 71, and (c and d) SEM of crystals at different magnifications.71 | |
Also, XPS analysis of MIL88-A was conducted before and after experiment. As can be noticed from Fig. S8,† the oxidation state of Fe atoms within the MOF powder on the three prepared samples (as synthesized, before exposure to UV, and after exposure to UV) was examined by XPS. The results showed that Fe 2p3/2−1/2 and O 1s core levels as presented show the same features confirming that the oxidation state of Fe remains the same after the experiment. The Fe 2p3/2 and Fe 2p1/2 binding energies are measured at 712.4 eV and 726 eV, respectively. The binding energy position as well as the presence of a satellite peak at 719.4 eV are characteristic of Fe in an oxidation state of +3 like in Fe2O3.
3.2. MIL88-A/PS/SMX in the absence of UVA
A preliminary experiment was conducted to test the capability of MIL88-A for activating PS in the absence of any illumination (in the dark). 50 mg of MIL88-A was added to a 200 mL solution containing [SMX]0 = 10 mg L−1 and [PS]0 = 2 mM. The reaction time was set to be 2 h and samples were taken every 10 min for the first hour followed by a single sample at t = 120 min. Control experiments were also conducted, in which SMX degradation was tested in the presence of MIL88-A as well as PS only under the above-mentioned conditions. As can be noticed (Fig. S9†), both controls showed insignificant drops by 10% and 12% at t = 0 min, respectively, after which the [SMX] remained constant. This shows that PS only and MIL88-A only are not effective in removing SMX from the solution. For the case where MIL88-A and PS are combined (lower curve), the % degradation of [SMX] was around 21% after a period of 2 h, which can be attributed to a slight activation of PS at room temperature at the surface of MIL88-A. Therefore, it can be concluded that the MIL88-A/PS system should require the presence of an assisting agent for the effective elimination of SMX.
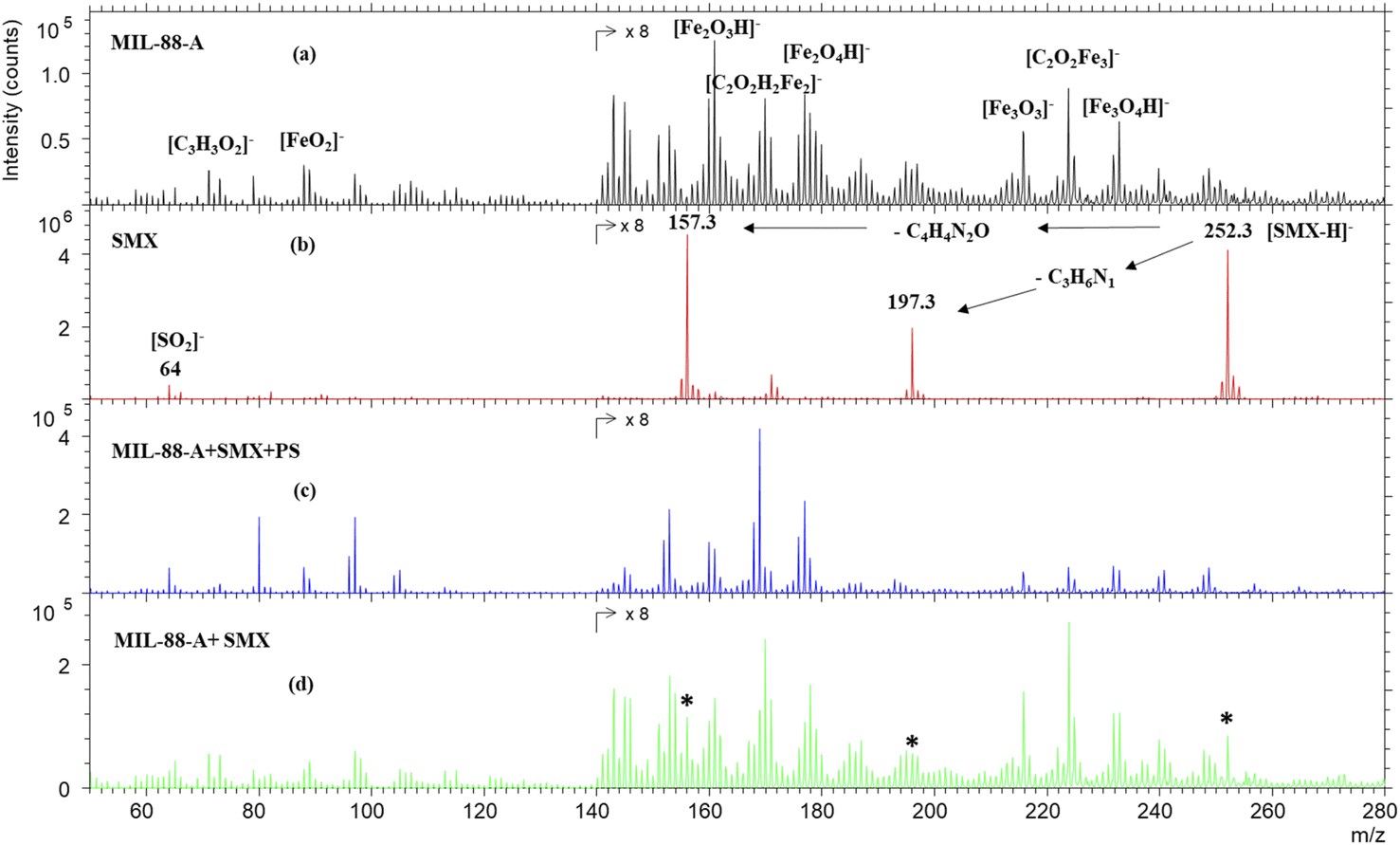
To further investigate the SMX removal process, ToF SIMS analysis was done on pristine MIL88-A and on used MIL88-A in SMX solution. As can be seen from Fig. 2a, the negative mass spectrum of MIL88-A shows a series of ions corresponding to different fragments and recombined fragments including [C3H3O2]−, [FeO2]−, [Fe2O3H]−, [C2O2H2Fe2]−, [Fe2O4H]−, [Fe3O3]−, [C2O2Fe3]− and [Fe3O4H]− at 71.05, 87.84, 160.69, 169.73, 176.64, 215.53, 223.66, and 231.53 m/z, respectively. These ions are attributed to the secondary ions generated by the reactants used for the synthesis of MIL88-A e.g. fumaric acid and iron(III) chloride under Bi3+ cluster beam impact. The mass spectrum of pure SMX powder, as displayed in Fig. 2b, shows mainly the well-known deprotonated SMX molecular ion [SMX–H]− at 252.3 m/z. In addition, the spectrum reveals 197.3 and 157.3 m/z SMX fragments corresponding to the loss of the C3H6N and C4H4N2O moieties, respectively. These fragments have already been identified in previous work.25 However, these fingerprints are missing from the mass spectrum of MIL88-A collected after reaction in the MIL88-A/PS/SMX system (Fig. 2c). In order to check the possibility of identifying SMX on MIL88-A, an analysis was done on a mixture of pristine MIL88-A mixed with SMX powder at 5 ppm. The results show the presence of the corresponding SMX ions (molecular and fragments), which can be considered as a proof of absence of any matrix effect.
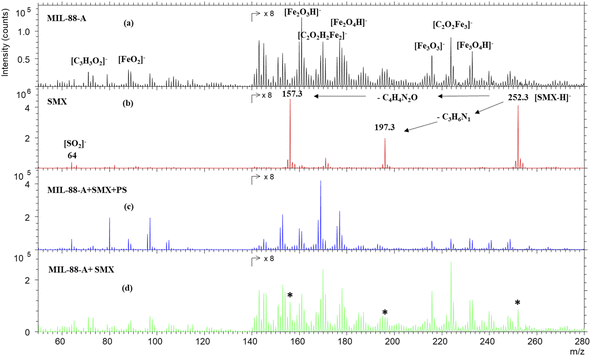 |
| | Fig. 2 TOF-SIMS negative spectra of (a) MIL88-A as prepared, (b) SMX powder, (c) MIL88-A after use in the presence of SMX and PS, and (d) MIL88-A+ 5% SMX powder. The areas of the spectra are multiplied by 8 for visual purposes. * The characteristic peaks of the SMX molecule. | |
Compared to previous work done with a naproxen (NPX) probe, one can deduce that SMX is not adsorbed on MIL88-A in view of the absence of its fingerprint while NPX was clearly present on the surface of MIL88-A used in a similar system e.g. MIL88-A/PS/NPX71 as was attested by the NPX fragment ion at 185.1 m/z.
3.3. MIL88-A/PS/SMX system in the presence of UVA (UVA/MIL88-A/PS/SMX)
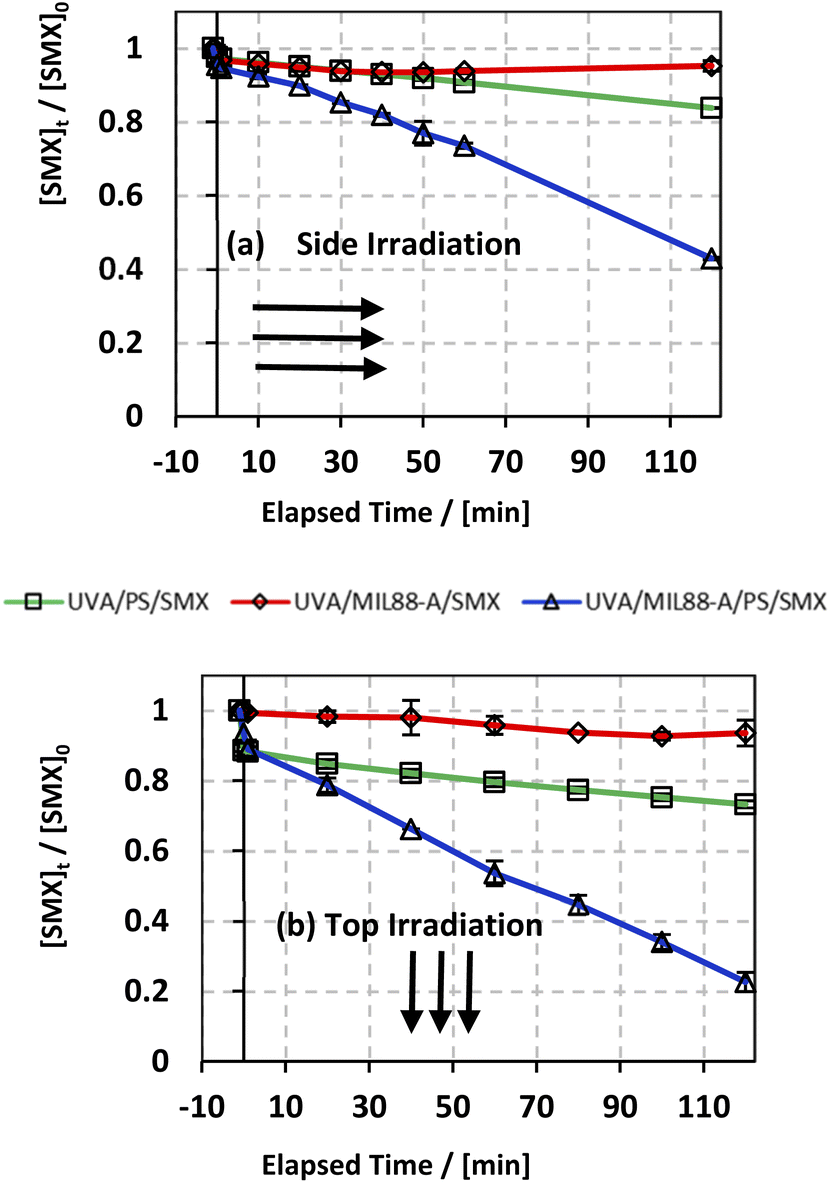
In an attempt to improve the SMX degradation process in the MIL88-A/PS system, experiments were carried out under UVA irradiation either on the side or at the top of the beakers (Fig. S1†). UVA irradiation has been demonstrated in different studies,27,72 as an effective factor to enhance the activation of PS initiated by an iron-based system, which is the case here. The reconversion of Fe(III) into Fe(II) species takes place upon UVA irradiation, therefore forming a redox cycle for a sustained PS activation process in the reactive medium (eqn (4)). In order to study the effectiveness of UVA/MIL88-A/PS for the degradation of SMX, two controls were conducted, one in the presence of MIL88-A only (UVA/MIL88-A/SMX system) and the other in the presence of PS only (PS/UVA/SMX system) (Fig. 3). The results showed slight SMX degradation (5% vs. 7% for side and top UVA irradiation, respectively) after a reaction time of 2 h in the presence of MIL88-A only (UVA/MIL88-A/SMX system) (Fig. 3a and b; two upper curves). This implies that no significant adsorption of SMX on MIL88-A took place since [SMX] slightly decreased with time. On the other hand, the rate of SMX degradation slightly improved to reach 17% and 27% after 2 h in PS/UVA/SMX as can be noticed (Fig. 3a and b; two middle curves). This is mostly due to the presence of a modest number of SRs generated upon PS photolysis in the bulk solution. However, a great improvement in SMX degradation is noticed reaching 58% and 78% when MIL88-A/PS systems were irradiated with UVA (side vs. top) over a reaction time of 2 h (Fig. 3a and b; lower curves). This can be explained by the additional role that UVA can play by activating PS, not only in the bulk solution, but also at the surface of MIL88-A playing the role of a catalyst as demonstrated in our previous work.71 These results suggested an optimization approach to better improve SMX degradation as shown in the next sections.
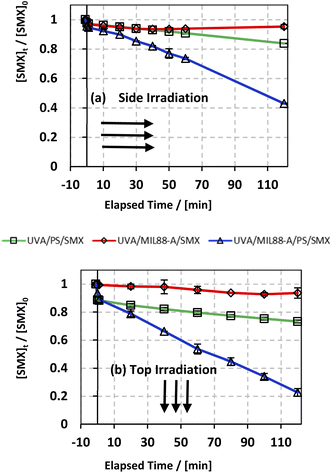 |
| | Fig. 3 The % degradation of SMX irradiated with UVA lamps as a function of time (min) in three different systems: UVA/PS/SMX, UVA/MIL88-A/SMX and UVA/MIL88-A/PS/SMX in (a) UVA lamps are placed on the side of the reactors and (b) UVA lamps are placed on the top of the reactors. Experimental conditions: [SMX]0 = 10 mg L−1, [PS]0 = 2 mM, and [MIL88-A]0 = 250 mg L−1. Vertical bars represent the standard deviations of the means; absent bars fall within symbols. Sample before t = 0 min was taken before the addition of PS and after PS addition at t0. | |
3.4. UVA/MIL88-A/PS/SMX: system optimization
3.4.1. Optimization of UVA irradiation.
As discussed above, one can notice that positioning the UVA lamps on the top of the beakers improved the degradation yield of SMX by a factor of 35% (78% vs. 58% degradation) after a reaction time of 2 h (Fig. 3a and b; lower curves) compared to the side irradiation system. This can be explained by (a) a greater UVA irradiance reaching SMX solutions directly without passing through the borosilicate glass of the beaker attenuating considerably the UVA irradiance and (b) a greater surface area to which the UVA irradiated SMX solution is exposed to. As such this produces a higher number of SRs contributing as well to more effective reconversion of Fe(III) into Fe(II) resulting in greater PS activation.
3.4.2. Effect of [MIL88-A]0.
Three different concentrations of [MIL88-A]0 (10, 125 and 250 mg L−1) were tested at fixed [PS]0 (2 mM) so as to determine the optimum amount of the catalyst that should be added to the reactive medium in order to achieve complete degradation of SMX within 5 h. As can be noticed (Fig. S10†), the results showed that all concentrations of MIL88-A yielded 100% SMX degradation however with different reaction rates. It was witnessed that 125 mg L−1 of MIL88-A showed the highest degradation rate as attested by the lower curve of the graph of Fig. S10.† This suggests that a lower concentration of MIL88-A cannot generate temporally enough active sites for PS activation on hand, while an excessive amount of MIL88-A might produce a high number of SRs resulting in some temporal quenching reactions. Accordingly, 125 mg L−1 was adopted as the optimum concentration for MIL88-A to carry out the rest of this study as shown in the next sections.
3.4.3. Recyclability.
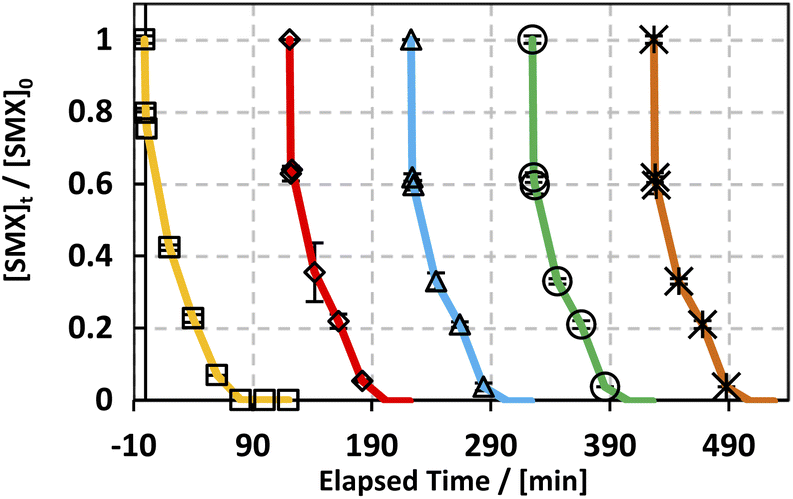
Throughout the scope of catalytic materials such as MIL88-A, recyclability is an important variable for assessing their cost/commercial value since a heterogeneous catalyst can be recovered and used again. In this part, MIL88-A was recovered three times successively after each experiment. The recovery cycle included the collection of MIL88-A used and then its separation using centrifugation followed by drying in a vacuum oven at 90 °C for 24 h. After that, MIL88-A was used in a second, third, fourth and then in a fifth experiment. However, it is worth noting that each time the quantity of MIL88-A recovered decreased due to the difficulty in collecting MIL88-A. This was due to the fact that some of the MIL88-A crystals are stuck in the 0.45 μm PTFE filter used in the sampling process. The results showed that 100% degradation of SMX was obtained in each of the five successive cycles, with the catalytic activity of MIL88-A improved after each cycle (Fig. 4). For every next cycle, the volume of SMX solution (5 mg L−1) was adjusted in a way to keep the concentration of MIL88-A constant equal to 125 mg L−1, in order to perform an accurate and reproducible comparative study of the results obtained from each cycle. The improvement in the catalytic activity of MIL88-A across cycles may be explained first by the fact that the more defects in a heterogeneous catalyst the better its catalytic activity and that was clearly inferred from the data shown in Fig. 4. In fact, the first cycle showed around 58% degradation within the first 20 min of reaction time compared to cycle 2 and cycle 3 where it reached 65% and 68%, respectively. Also, looking at cycle 3 and cycle 4, the catalytic activity of MIL88-A remained the same where the degradation rate of SMX reached 68% in both cycles. Second, these results demonstrated the high potential of PS in reactivating the MIL88-A surface. In order to confirm this hypothesis, SEM images were taken after each cycle. MIL88-A showed a rod-like morphology similar to that obtained with freshly synthesized MIL88-A; however MIL88-A crystals lost some of their homogeneity when the crystals were elongated (Fig. 5a). The XRD pattern of MIL88-A recorded after the experiments showed peaks at two theta positions that comply with the freshly prepared MIL88-A as shown in Fig. 5b.
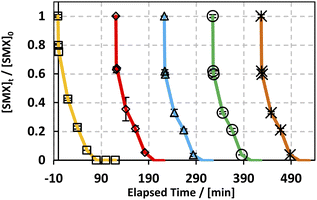 |
| | Fig. 4 Recyclability experiments of MIL88-A in the UVA/MIL88-A/PS/SMX system. Experimental conditions: [SMX]0 = 5 mg L−1; [PS]0 = 2 mM; [MIL88-A] = 125 mg L−1; error bars are calculated as ts/√n, where absent bars fall within the symbols. | |
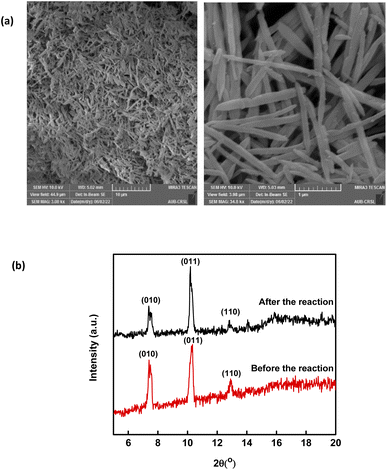 |
| | Fig. 5 (a) SEM images of recycled MIL88-A and (b) XRD pattern of new vs. used MIL88-A crystals. | |
3.5. solar/MIL88-A/PS/SMX system
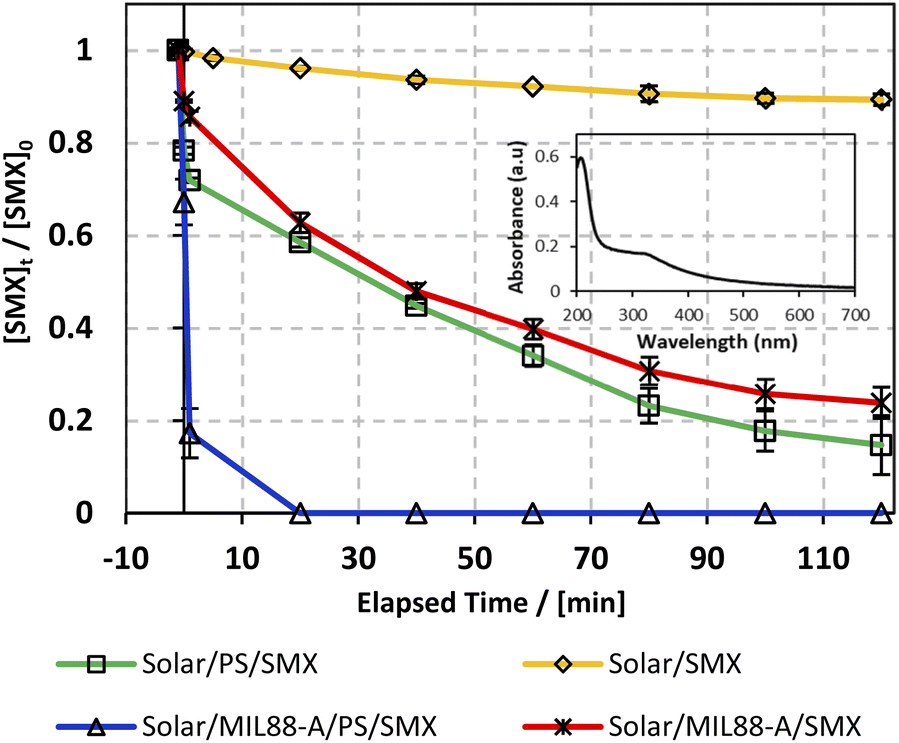
As can be noticed from Fig. 6, different control experiments were done to better understand the effectiveness of the solar/MIL88-A/PS/SMX system on SMX degradation. We witnessed that stand-alone solar energy kept the medium unchanged, whereby the degradation of SMX reached only 10% after 2 h (Fig. 6, upper curve), which can be attributed to the photosensitive properties of SMX toward the solar spectrum.73 It is important to mention that during the conducted experiments, the solar flux reached a maximum of 3900 μW cm−2 initially and decreased to reach a minimum of 1600 μW cm−2 at the end of the reaction as the experiment started at 11:00 am and ended by 1.00 pm. For instance, throughout the day, the sun ray's intensity increases gradually to reach its maximum at noon (12:00 pm), and then decreases at dawn and dusk, and in between other hours of the day. The drop in SMX concentration was less than the one observed in a similar system (20% SMX degradation24) in the presence of PS, at room temperature however in the absence of any kind of irradiation. Accordingly, a successful AOP scenario should be dependent on additional activation agents such as iron particles, UV irradiation22,25 or heat20 under standalone conditions or in combination. This verifies the imperative role of solar energy in the generation of reactive oxidative species (ROS) in a medium containing adequate additives. For example, several studies have also investigated the use of solar energy however with H2O2 as an oxidant in combination with a heterogeneous catalyst and showed promising results of TOC removal in waste water treatment.74 Accordingly, in this part PS and MIL88-A were added separately to the system in an attempt to optimize SMX degradation on hand and to account for the contribution of each additive in SMX degradation on the other hand. Luckily, both additives showed an almost similar trend in the variation of the degradation extent of SMX reaching 82% and 78%, respectively, after a reaction time of 2 h (Fig. 6, middle curves). Moreover, the results showed that when PS and MIL88-A are both combined in the SMX solar irradiated solution, the degradation extent improved drastically to reach 100% in a reaction time of less than 20 min. We recall that the MIL88-A UV/vis absorption spectrum in solution showed much greater absorbance in the UV range (200–380 nm) than in the visible range (400–700 nm) as can be attested by the inset of Fig. 6 showing the UV/vis spectrum of pure MIL88-A solution. This confirms the synergistic role played by the oxidant (PS) and the catalyst (MIL88-A) improving solar PS activated systems (solar/MIL88-A/PS/SMX) toward full degradation of SMX.
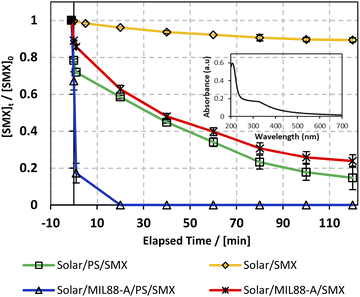 |
| | Fig. 6 The % degradation of SMX under solar irradiation as a function of time (min) in four different systems: solar/SMX, solar/PS/SMX, solar/MIL88-A/SMX and solar/MIL88-A/PS/SMX. The inset shows the UV/vis spectrum of the MIL88-A solution. Reactors were placed under sunlight in a rotisserie shaker. Experimental conditions: [SMX]0 = 5 mg L−1, [PS]0 = 2 mM, and [MIL88-A]0 = 125 mg L−1. Vertical bars represent the standard deviations of the means; absent bars fall within symbols. Sample before t = 0 min was taken before the addition of PS and after PS addition at t0. | |
3.6. Matrix effect: solar vs. UVA activated systems
3.6.1. Case of chlorides.
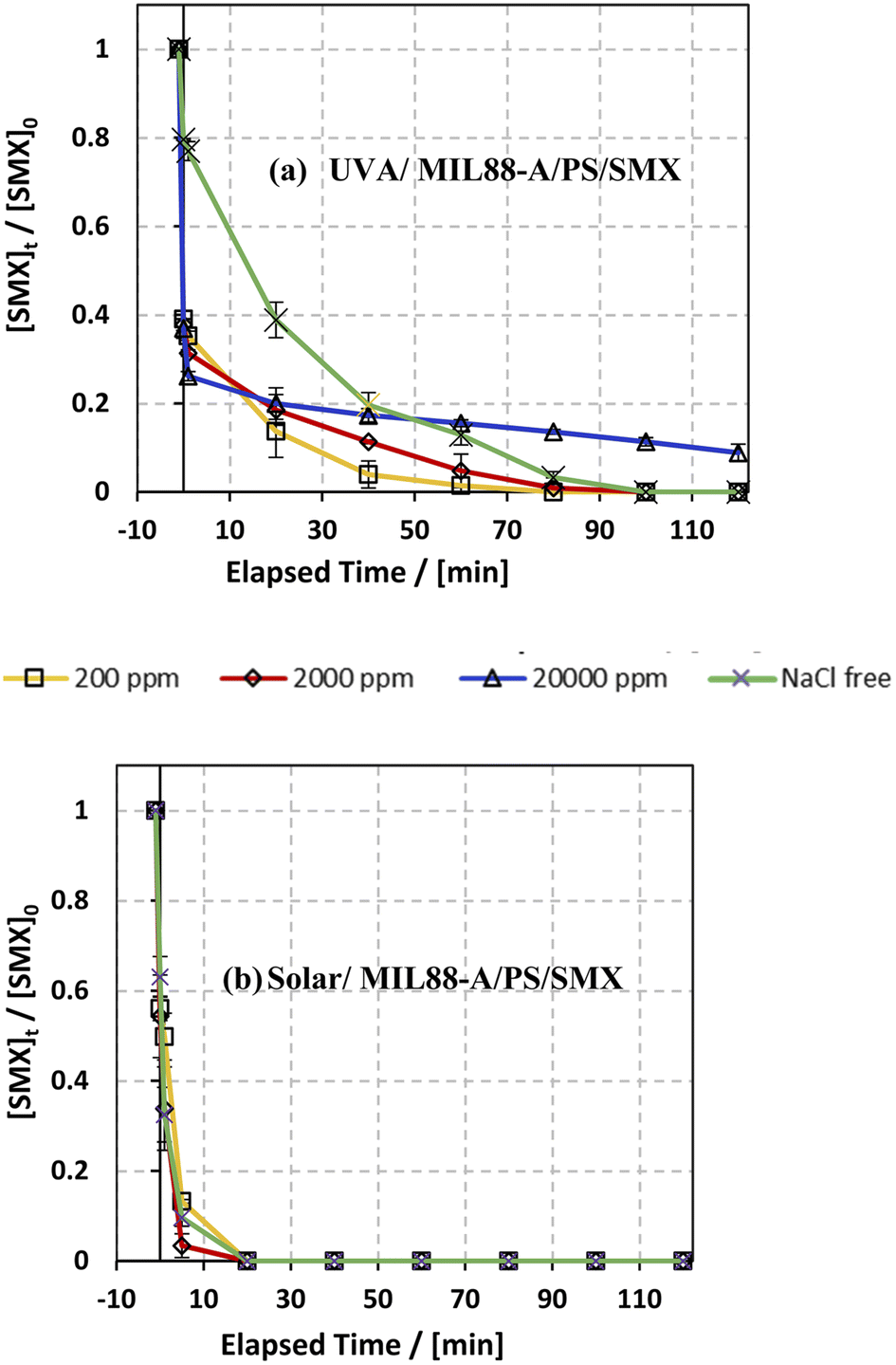
The influence of common anions in natural water was determined on SMX degradation in both UVA/MIL88-A/PS/SMX and solar/MIL88-A/PS/SMX systems. Three different concentrations of chlorides corresponding to fresh water ([NaCl] = 200 mg L−1), brackish water ([NaCl] = 2000 mg L−1), and saline water ([NaCl] = 20000 mg L−1) were tested to mimic natural water conditions.75 As can be noticed from the UVA/MIL88-A/PS system (Fig. 7a), both fresh water and brackish water showed a similar trend of the variation in the % degradation of SMX with slight enhancement of the process, whereby SMX degradation reached 100% after 80 min similar to that obtained with a NaCl-free experiment. This is mainly due to the formation of chlorine radicals (Cl˙) with a redox potential E0 = 2.432 V close to that of SRs (E0 = 2.4 V) as shown in eqn (6) and (7) in addition to the formation of reactive hydroxyl radicals (HO˙). However, SMX degradation was accompanied by a slight inhibition in saline water, whereby it reached around 90% after 2 h (Fig. 7a). However, this was not the case in the solar/MIL88-A/PS system where the three different concentrations of NaCl showed no significant effect on the degradation of SMX (Fig. 7b). This can be explained by the excellent photocatalytic properties of MIL88-A under solar irradiation resulting thereby in heterogeneous PS activation on the surface of the photocatalyst. Moreover, one should not neglect, to a certain extent, thermal PS activation due to a slight increase in the temperature of the solution (from room temperature to ∼40 °C) after 2 h of solar irradiation and that in a close reactor.20,76 These results are in accordance with previous research done on the assessment of the NaCl effect on ketoprofen and chloramphenicol degradation in PS activated systems as demonstrated by Ghauch et al.20,22 In these systems, the degradation of ketoprofen/chloramphenicol was inhibited in brackish and more significantly in saline water where the concentration of Cl− ions is above 10 mM (584 mg L−1 of NaCl) and the chloride quenching effect is more evident in the media.| | | SO4−˙ + Cl− → Cl˙ + SO42− | (6) |
| | | Cl2˙− + Cl2˙− → 2Cl− + Cl2 | (8) |
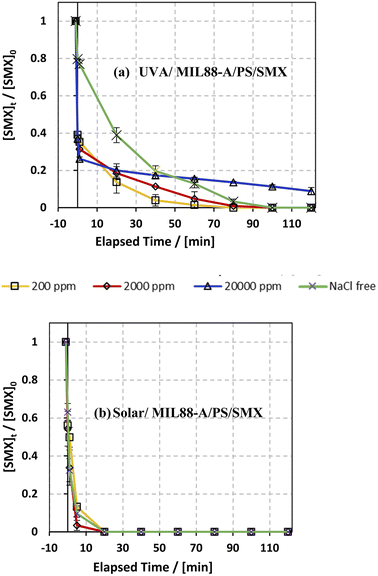 |
| | Fig. 7 Effect of [NaCl] = 200−20000 mg L−1 on the degradation of SMX as a function of time (min): (a) in the UVA/MIL88-A/PS/SMX system and (b) in the solar/MIL88-A/PS/SMX system. Experimental conditions: [SMX]0= 5 mg L−1, [PS]0 = 2 mM, and [MIL88-A]0 = 125 mg L−1. Error bars are calculated as ts/√n, where absent bars fall within the symbols. | |
3.6.2. Case of bicarbonates.
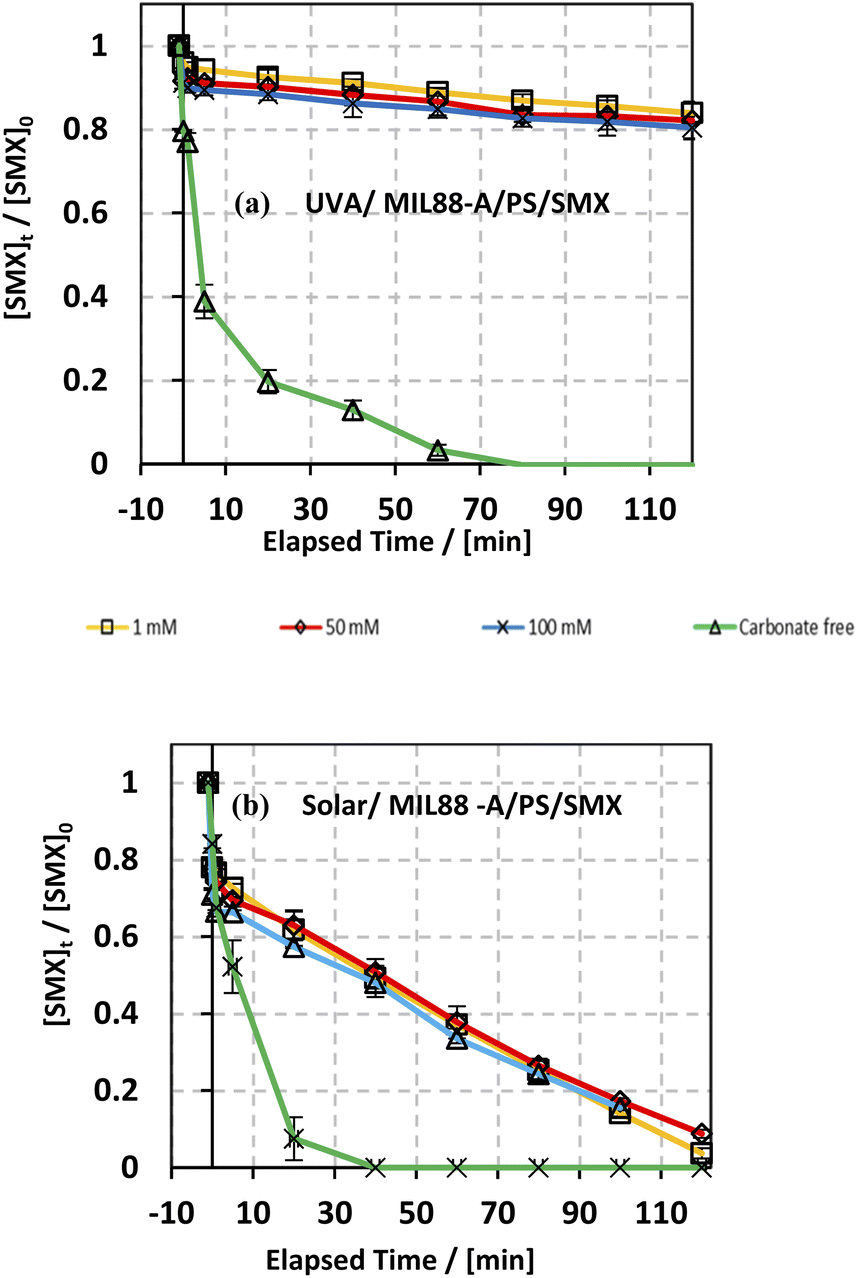
The effect of bicarbonate was also studied in both systems: UVA/MIL88-A/PS/SMX and solar/MIL88-A/PS/SMX as shown in Fig. 8. It is evident that in the first system, the addition of NaHCO3 inhibited greatly the SMX degradation process. In fact, regardless of NaHCO3 concentration, [SMX] dropped by only 10% after a reaction time of 20 min (upper curves) compared to 80% degradation in the case of carbonate-free solution (lower curve). Moreover, one can notice that the SMX degradation rate reached a steady state at 80 min of reaction with a maximum of 20% of degradation reached in all carbonated systems. However, an additional 20% of SMX was degraded in the bicarbonate-free system to reach full degradation at a reaction time of 80 min. On the other hand, the results showed that in the solar/MIL88A/PS/SMX system gradual degradation of SMX at a constant rate occurred in all bi-carbonated systems regardless of [NaHCO3] (Fig. 8b). It also showed that the degradation of SMX is independent of the ionic strength of the solution. This inhibitory effect of NaHCO3 can be explained by the reaction of SRs with products which have moderate oxidative properties (E° = 1.59 V) compared to that of SRs toward SMX (eqn (9)). Moreover, one can admit a less inhibitory effect of bicarbonate on SMX degradation in the solar irradiated system. This can be explained by the role that MIL88-A can play as a more effective photocatalyst when irradiated by a full solar spectrum including more energetic radiation e.g. UVB rather than with only the UVA spectrum of lower energy. As such, SMX degradation is more likely to occur close to the surface of the catalyst rather than in the bulk of the solution. Finally, it is important to mention that for bicarbonate concentration above 50 mM, the pH of the reactive medium increased toward significantly basic pH values e.g. pH > 8.50 affecting thereby the activation process of PS toward a lower SMX oxidation reaction. However, this is not well revealed for the lowest concentration of bicarbonate that did not affect the activation process of PS since the final pH of the solution exhibited values lower than the starting pH conditions e.g. pHi = 6.46 and pHf = 5.31 for the UVA system and pHi = 6.18 and pHf = 5.19 for the solar irradiated system as previously proved by the formation of acidic species such as HS2O8− in solution.20 At low bicarbonate concentration, the ionic strength of the solution is not enough to keep good buffer capacity resulting in breaking the buffer strength (Table S1†). Finally, it is worth noting that the pH of the medium plays an important role in terms of SMX ionization and charge distribution. The pK1 and pK2 values of SMX are about 1.39 and 5.8, respectively. This means that at pH lower than 1.39, SMX exists as a cation, and between pH 1.39 and 5.8 SMX, it exhibits neutral charge, while at pH above 5.8, SMX is negatively charged. This can clearly explain the neutrality of SMX toward MIL88-A under the pH working conditions. For example, the dominant working pH in carbonate-free or slightly containing carbonate solution e.g. 1 mM favors the neutral form of SMX. A previous study showed that acidification of the carbonated solution might reverse the inhibition process because of the removal of carbonate species, through CO2 escape from solution, opening the way toward the quenching-free reaction medium for an improved reaction stoichiometric efficiency.24| | | SO4−˙ + HCO3− + SO42− + CO3−˙ + H+ | (9) |
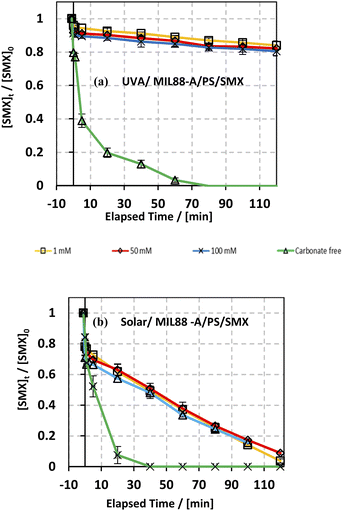 |
| | Fig. 8 Effect of bicarbonate concentration on the degradation of SMX as a function of time in different systems: (a) UVA/MIL88-A/PS/SMX and (b) solar/MIL88-A/PS/SMX systems. Experimental conditions: [HCO3−] = 1–100 mM, [SMX]0 = 5 mg L−1, [PS]0 = 2 mM, and [MIL88-A]0 = 125 mg L−1. Error bars are calculated as ts/√n, where absent bars fall within the symbols. | |
3.6.3. Case of phosphates.
The phosphate effect on SMX degradation was also assessed in both UVA and solar systems to account for phosphate residues that may escape from conventional wastewater treatment stations on hand and to study the pH effect on the degradation of SMX on the other hand. The results are somehow comparable to the bicarbonate case in terms of efficiency drop in the SMX degradation extent (Fig. S11†). As can be noticed (Fig. S11a†), after a sudden drop in SMX upon spiking with PS solution, the kinetics of the reaction changed with phosphate buffer (PB) addition, which leads to an instant drop in SMX by 70% upon addition of PS after which it remained constant throughout the 2 h reaction time (Fig. S11a†). In contrast, this was not the case for the solar/MIL88-A/PS system, where the degradation rate of SMX reached around 80% at the end of the reaction with the different concentrations of PB (5, 10 and 20 mM) (Fig. S11b†). One can assume that PB has an inhibitory effect on the degradation process of SMX, but it varied differently with the two studied systems. This can be attributed to the fact that phosphate species form stable complexes with Fe2+ ions and accumulate on the surface of MIL88-A, thus preventing the chemical activation of PS that is accomplished by free Fe2+ ions in solution or adsorbed on the surface of the MOF. This hypothesis has already been confirmed through our previous work on the degradation of SMX and ranitidine in Fe/PS systems.24,25
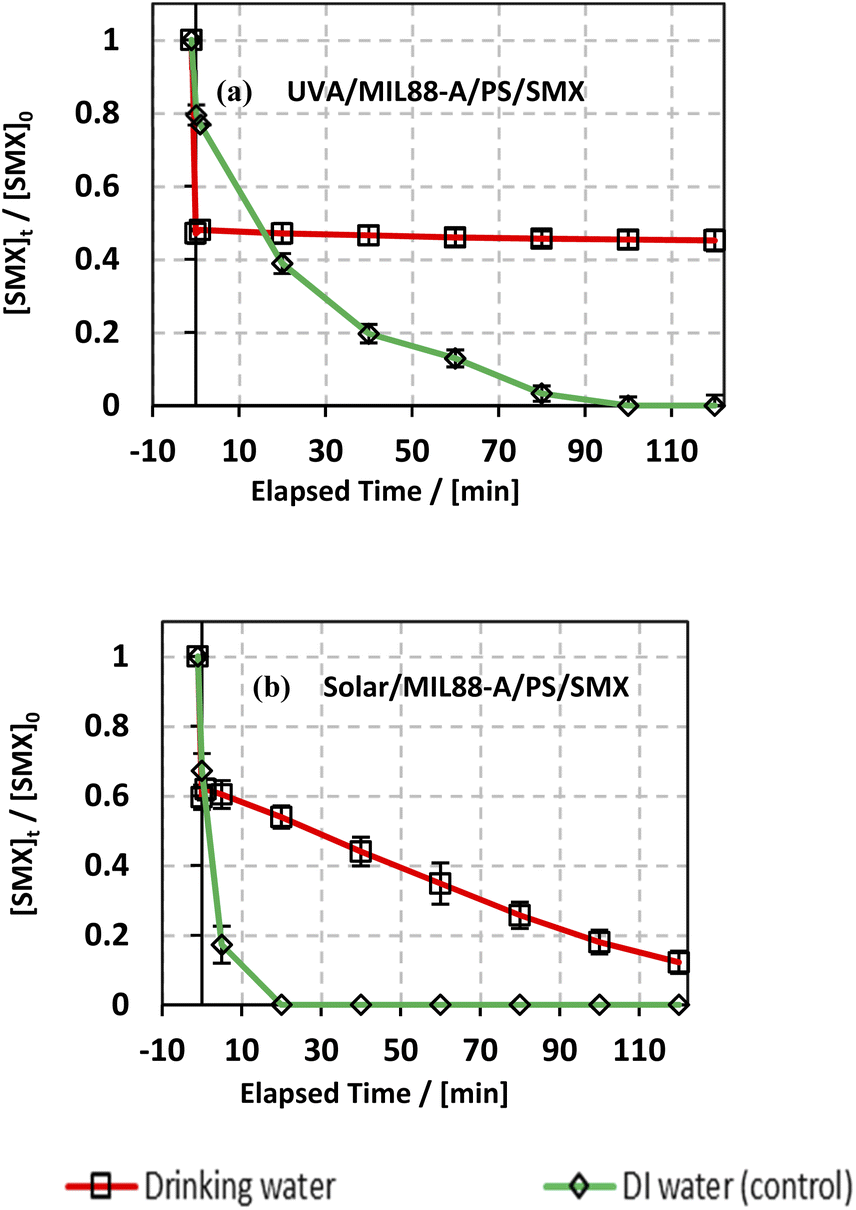
3.6.4. Case of drinking water.
The degradation of SMX was also studied in a drinking water solution to account for the effect of the different species present in a local bottled drinking water brand (Nestle) of pH 7 having the following composition in mg L−1: Mg2+ = 5; HCO3− = 120; SO42− = 50; Cl− = 8; Ca2+ = 52; Na+ = 5; K+ = 0.5; and total dissolved solid = 200. As can be noticed from Fig. 9, the use of drinking water as a matrix instead of DI water led to a significant inhibition of SMX degradation in the UVA/MIL88-A/PS/SMX system, whereby SMX degradation reached around 53% only. This can be explained by the fact that the Nestle water used in this experiment contains enough bicarbonate concentration for quenching SRs (eqn (9)) just upon PS activation either in solution or at the surface of the MIL88-A catalyst. Moreover, one can notice a similar trend in the degradation curves of SMX in DI and in drinking water either for the UVA/MIL88-A/PS/SMX or the solar/MIL88-A/PS/SMX system. For example, considering the UVA/MIL88-A/PS/SMX system, the % degradation yield of SMX reached almost 50% in drinking water [HCO3− = 2 mM]) while it showed only 20% degradation in DI water (Fig. 8a). We recall that the drinking water solution contains Cl− that favors SMX degradation as previously demonstrated (Fig. 7). Accordingly, it is not surprising to have better SMX degradation in drinking water where slight SR quenching takes place in the medium at the same time as an improved oxidation process due to the formation of chlorine radicals contributing to more SMX degradation. However, the solar system showed better results as it is less affected by the presence of bicarbonate as long as the reaction is more driven by photolysis at the surface of MIL88-A with sufficient energy as provided by the solar irradiation. The results in the solar system showed a constant degradation rate of SMX to reach almost full degradation compared to the UVA system showing a steady state, limiting therefore SMX degradation to a maximum of 55% after a reaction time of 2 h (Fig. 9a).
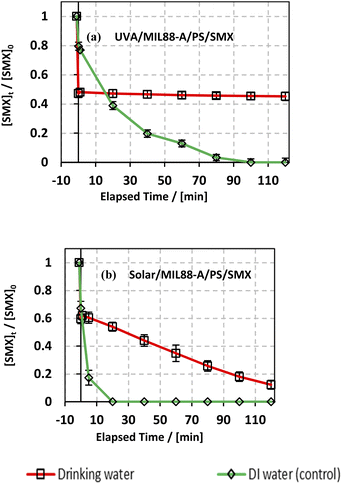 |
| | Fig. 9 Effect of drinking water and deionized water (control) on the degradation of SMX as a function of time (min): (a) in the UVA/MIL88-A/PS/SMX and (b) in the solar/MIL88-A/PS/SMX systems. Experiments were done in a rotisserie shaker. Experimental conditions: [SMX]0 = 5 mg L−1, [PS] = 2 mM, and [MIL88-A]0 = 125 mg L−1. Error bars are calculated as ts/√n, where absent bars fall within the symbols. | |
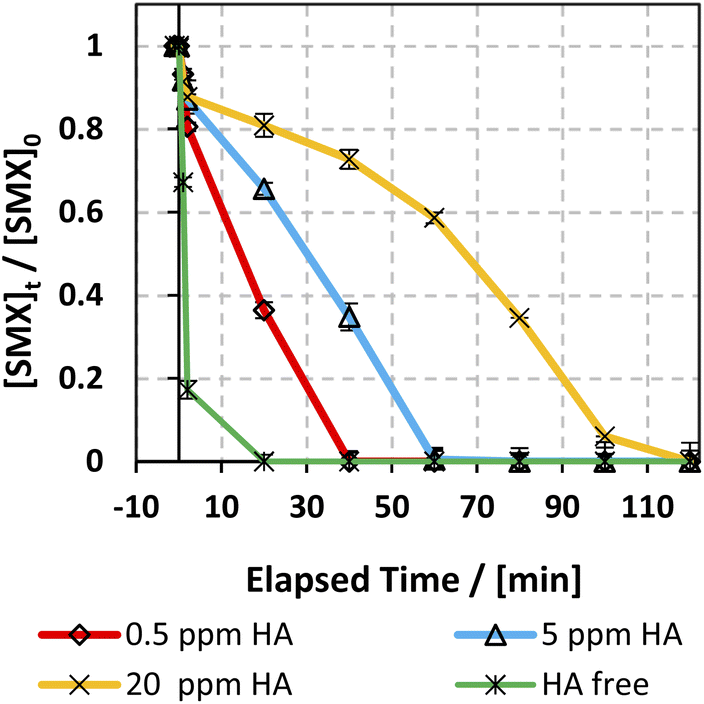
3.6.5. Case of humic acids (HA).
The effect of humic acids (HA) on the degradation efficiency of the applied system has been studied due to their significant abundance in natural water. HA concentration typically varies between 0.1 and 20 mg L−1,77 so three [HA]0 concentrations were selected: 0.5, 5, and 20 mg L−1. Fig. 10 shows the effect of different [HA] on the SMX degradation rate. As can be inferred, HA inhibited the degradation of SMX whereby complete degradation of SMX was attained within 5–20 min in the HA free experiment, while it increased to 40 and 60 min with 0.5 and 5 mg L−1 [HA] respectively. This observation is in accordance with previous work done on KTP degradation using different PS activated systems.20 Analogous results were also expressed in the studies done on the degradation of chloramphenicol in UV-APS by Ghauch et al.22 This inhibition is mainly ascribed to the HA's quenching effect due to the presence of abundant electron-rich sites thereby attracting electrophilic species such as SRs and HRs.78–81 In addition, the considerable decrease in the SMX degradation rate at [HA] = 20 mg L−1 in the solar/MIL88-A/PS system is also attributed to the inner filter effect induced by HA due to its high absorbance at 254 nm.79,82
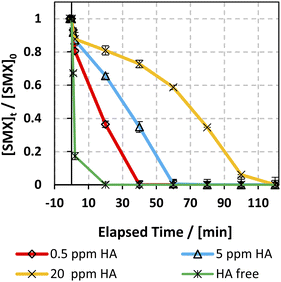 |
| | Fig. 10 Effect of HA on the degradation of SMX as a function of time (min) in the solar/MIL88-A/PS/SMX system. Experiments were done in a rotisserie shaker. Experimental conditions: [HA] = 0.5, 5 and 20 mg L−1 [SMX]0 = 5 mg L−1, [PS] = 2 mM, and [MIL88-A]0 = 125 mg L−1. Error bars are calculated as ts/√n, where absent bars fall within the symbols. | |
3.7. Leaching assessment
In an attempt to account for leached iron (Fe) in solution, AAS measurements was done on sample solution after a 2 h experiment. The sample was filtered through a 0.45 μm filter and then centrifuged to remove any MIL88-A crystals remained suspended in solution. The results showed that [Fe] in solution was 0.002 ppm, which is below the limit of the detection of the AAS instrument (0.02 ppm). This proves that there is a very negligible leaching of iron from MIL88-A in the bulk of the solution. This also verifies that the degradation of SMX is done through iron clusters present on the surface of MIL88-A and not by Fe2+ or Fe 3+ dissolved in the reaction mixture.
3.8. EPR measurements
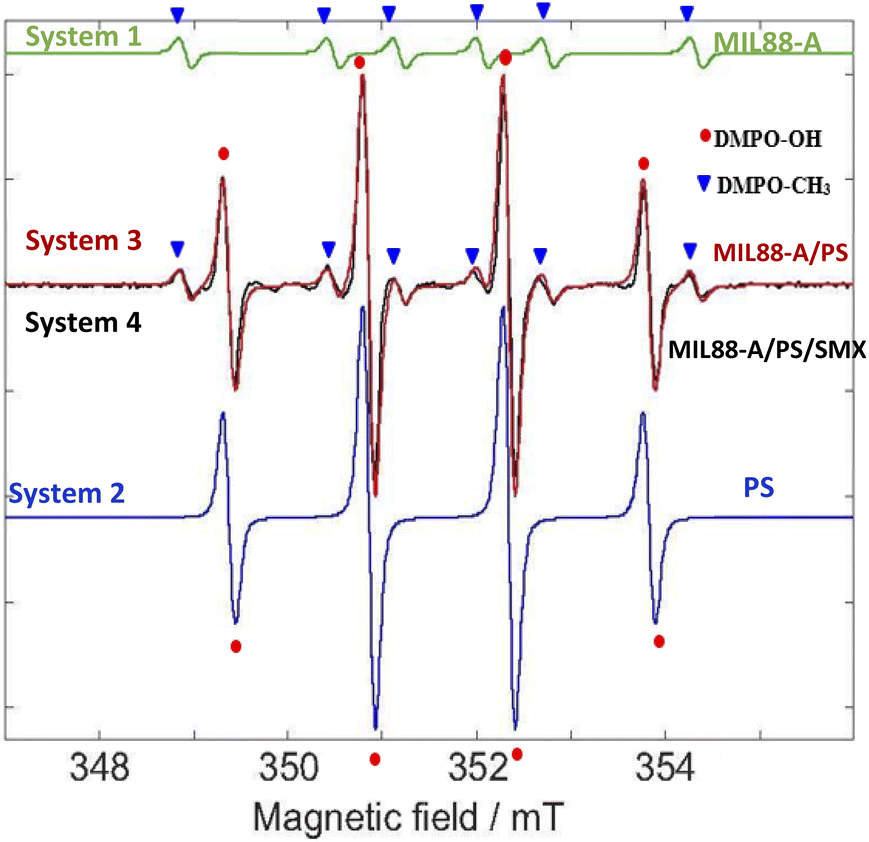
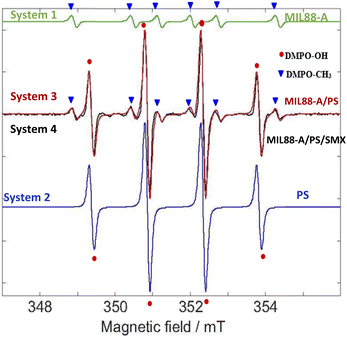
Since MIL88-A is used as a heterogeneous catalyst to activate PS for the degradation of SMX, it is suggested that SRs and HRs are generated. They react with the target molecule (SMX) in an oxidative mechanism to fully degrade and transform SMX into harmless species. In such a heterogeneous system, it was challenging to identify the presence of these species with the common quenching techniques (TBA and MeOH) that were used before with homogeneous catalysts such as Fe2+ ions in PS-based AOPs.20,83 In fact, the identification of radicals relies on the use of quenchers such as MeOH and TBA; however they may highly interfere with MIL88-A active sites. Thus, the EPR technique was selected as an appropriate analytical method to determine the presence of these radicals in the system. This was accomplished by the use of 5,5-dimethyl-L-pyrroline N-oxide (DMPO) as a free radical targeting molecule.84–86
The EPR analysis in this study was done on four different solutions of the following systems: System 1 (MIL88-A), System 2 (PS), System 3 (MIL88-A/PS) and System 4 (MIL88-A/PS/SMX) to conduct a reliable comparative study. As can be noticed from Fig. S12,† there are no detectable signals in System 1 compared to the peaks that are shown in the three investigated systems (2–4). To better understand the results of the EPR spectra obtained, a simulation on the EPR spectra was conducted via the EasySpin library for MATLAB, where the rotational tumbling (5 × 10−11 s) and the ‘chili’ functions were used.87 The results showed the presence of DMPO–OH adducts of intensity 1:2:2:1 and a hyperfine splitting constant of aN = aH = 1.49 mT and another series of six peaks with intensities 1:1:1:1:1:1 that account for the DMPO–CH3 adduct and a hyperfine splitting constant aN = 1.58 mT and aH = 2.58 mT (Fig. 11). The obtained alkyl radical may be generated from the alkyl leaching from the organic linker used in the synthesis process of MIL88-A (e.g. fumaric acid) or from ethanol solvent which is used in the washing process. For system 1, one can estimate that the OH radicals present may be generated from the photo-activation of MIL88-A under the day light as previously investigated. Moreover, the EPR spectrum of DMPO solution in system 2 shows four peaks attributed to the DMPO–OH adduct with an intensity of 1:2:2:1 and a hyperfine splitting constant of aN = aH = 14.9 G that are much more resolved than the ones obtained in system 1 that accounts for the pure formation of HRs. It is important to mention that with time, as the measurements take place, the activation of PS is possible at room temperature, and thus unstable, very short lifetime DMPO–adducts are formed.
 |
| | Fig. 11 EPR spectra. Green – simulated EPR spectrum for trapped methyl radicals. aN =1.58 mT and aH =2.28 mT. Blue – simulated EPR spectrum for trapped hydroxyl radicals. aN = 1.49 mT and aH = 1.49 mT. Red – the sum of the above two simulated trapped radical spectra. Black – experimental EPR spectrum under the following experimental conditions: [PS] = 2.5 mM, [MIL88-A] = 12.5 mg L−1, and [DMPO] = 100 mM. | |
Accordingly, following a rational expectation, the production of DMPO–OH adducts is favored in aqueous medium. This could occur either by the nucleophilic substitution of DMPO– (eqn (11)) or by the trapping of OH˙ formed (eqn (10)) as already investigated.88 Upon spiking with PS (System 4), one can notice, in addition to the DMPO–OH four split lines, six peaks with an intensity of 1:1:1:1:1:1 corresponding to the DMPO–SO4− adduct with hyperfine splitting constants of aN = 13.9 G, aH = 10 G, aH = 1.48 F, and aH = 0.78 G. It is worth noting that measurements done on the same system in the presence of SMX (System 3) showed the presence of the same DMPO adducts however with less improved DMPO–adduct. This can be attributed to the fact that some of the catalytically generated radicals reacted with the SMX probe rather than with DMPO resulting in lower trapping probability. Accordingly, the use of the EPR technique helped in identifying the presence of HRs and SRs and their coexistence in solution. We recall that some of the SRs are being converted into HRs upon reaction with water as previously demonstrated. This is the reason why the intensity of the DMPO–OH adduct always shows a greater amplitude than DMPO–adducts in aqueous systems. In conclusion, one can assume that the degradation process of SMX takes place through the radical mechanism since we were able to identify both SRs and HRs through the EPR measurements done.
| | | SO4−˙ + H2O → OH˙ + H+ + SO42− | (10) |
| | | DMPO − SO4− + H2O → DMPO − OH + H+ + SO42− | (11) |
3.9. Degradation mechanism
3.9.1. Identification of degradation products.
Under the current experimental conditions, SMX showed, through its degradation in a MOF-activated PS system, the presence of three byproducts on HPLC (Fig. S13†). The identity of these byproducts is further investigated in Section 3.9.3 using high-resolution mass spectrometry.
3.9.2. Proposed activation mechanism: surface based activation of PS on MIL88-A.
Based on the EPR measurements, the elimination of SMX in the UVA/MIL88-A/PS system was mainly due to a radical oxidation process. For instance, Fe active species present in the MIL88-A are trivalent since they originate from the ferric chloride salt used in the synthesis process of MIL88-A through which PS chemical activation may occur by a one-electron reduction mechanism (eqn (12) and (13)) as was previously proven.27,71,89| | ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe3+ + S2O82− → Fe2+ + S2O8−˙ Fe3+ + S2O82− → Fe2+ + S2O8−˙ | (12) |
| | | Fe2+ + S2O82− → Fe3+ + SO4−˙ + SO42− | (13) |
For instance, XPS analysis was done on three different samples: pristine MIL88-A, after experiment in the absence of UVA and after experiment in the presence of UVA. As can be inferred from Fig. S8,† the Fe 2p3/2−1/2 and O 1s core levels of the three tested samples showed the same structures validating the same Fe oxidation state after the reaction. The results showed binding energy values at 712.4 eV and 726 eV for Fe 2p3/2−1/2 and O 1s, respectively, and the presence of a satellite peak at 719.4 eV. Its presence along with the position of the binding energy are considered typical characteristics of Fe in a +III oxidation state, which is the case in Fe2O3.90,91 It is important to mention that Fe in a +II oxidation state could be present; however it is quickly oxidized to Fe3+ in the presence of air or oxygen. Moreover, the hydrolysis of PS and SRs may occur in the reactive medium in the presence of water to generate SO4−˙, O2−˙ and OH˙ in the absence of UVA (eqn (14)–(16)). Fe2+ is formed as stated in eqn (12), and generates unstable PS radicals that undergo oxidative reactions quickly in the medium. After that, Fe3+ is generated back through PS activation. So, SRs are produced to effectively degrade SMX through oxidative mechanisms yielding less stable molecules potent to further transformation. Furthermore, in the presence of UVA irradiation, Fe3+ (coordinatively unsaturated metal sites) could undergo photochemical conversion into Fe2+ (eqn (17)) on the surface of MIL88-A as demonstrated by Zhang et al.92 and Jiang et al.93,94 As a result, HRs are produced and attack SMX allowing its degradation. This was clearly noticed by the complete degradation of SMX in the UVA/MIL88-A/PS system compared to only 12% degradation in the MIL88-A/PS system. Our results corroborate those obtained by Zhang et al. on the degradation of tetracycline in MIL88-A/PS systems in the dark and under visible irradiation.
| | | S2O82− + 2H2O → 2SO42− + HO2− + 3H+ | (14) |
| | | S2O82− + HO2− → SO42− + SO4−˙ + O2−˙ + H+ | (15) |
| | | SO4−˙+ H2O → SO42− + OH˙ + H+ | (16) |
| | | Fe(III)OH2+ + hv → Fe(II) + HO˙ | (17) |
3.9.3. Proposed mechanism for SMX degradation.
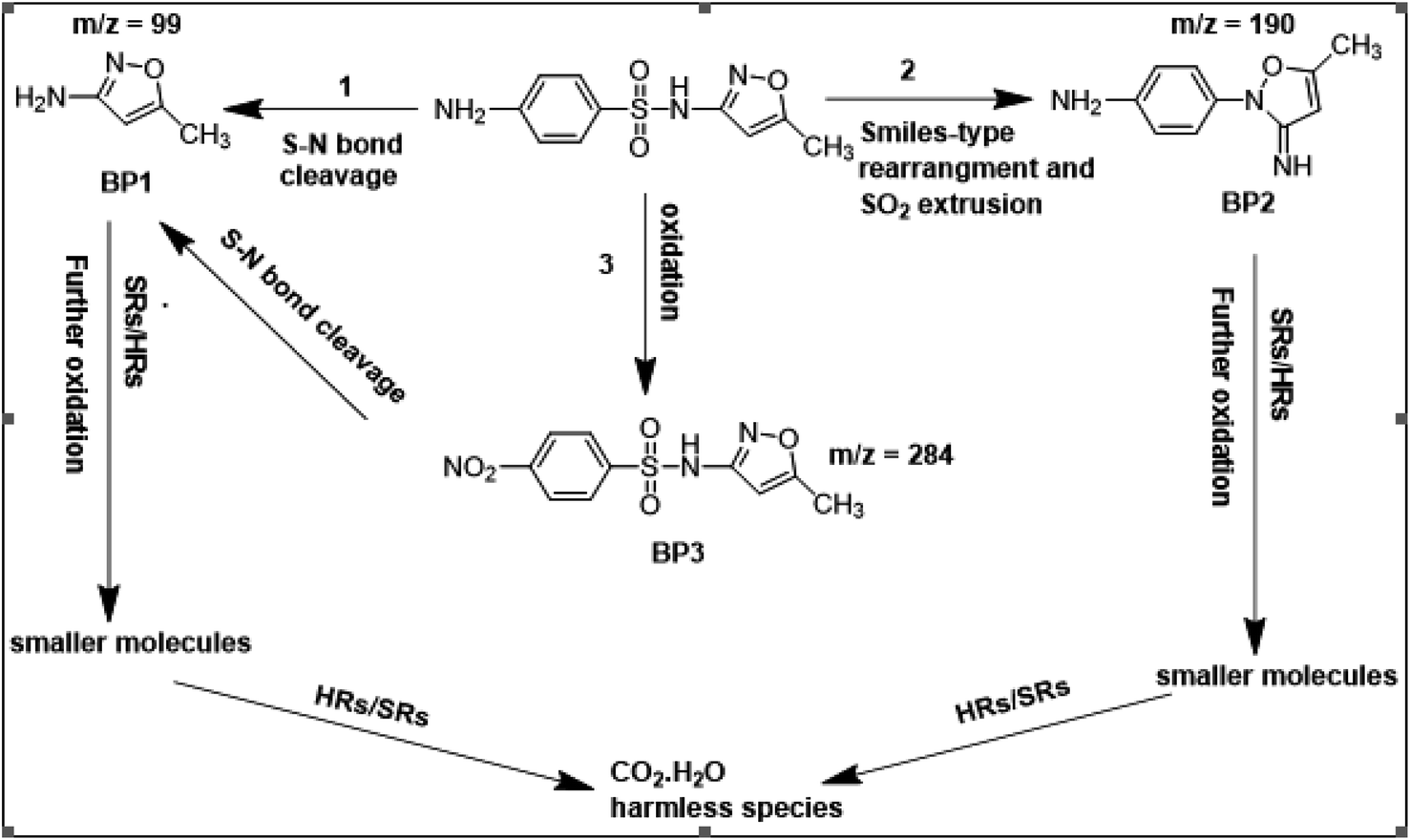
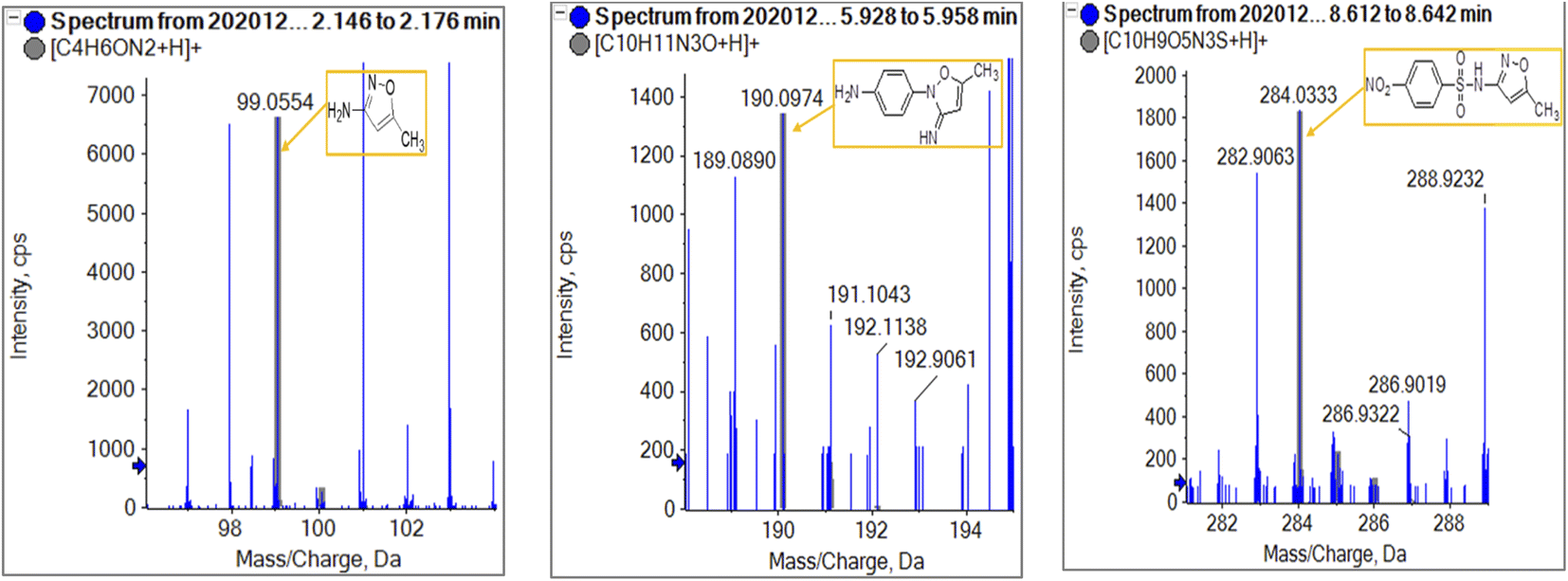
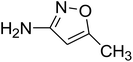
To get more insight into the SMX removal mechanism, mass spectrometry (MS) was conducted in the present study. It is important to mention that some studies worked on identifying SMX degradation products;24,56,95 however, the results obtained varied from one system to another depending on the MS analyzer used. In this study, a high resolution mass spectrometer SCIEX X500R QTOF was used to detect and identify SMX degradation products in the UVA/MIL88-A/PS/SMX system. The HRMS allows accurate determination of the mass of all byproducts obtained with the designed structures (error ≤ 2 ppm). In the above-mentioned system, three byproducts (BP1, BP2 and BP3 at retention times of 7.0, 8.1, and 11.1 min, respectively) were obtained and identified by HPLC/DAD and HPLC/DAD/MS analysis (Fig. S13†). As can be inferred from the overall mechanism shown in Fig. 12, in pathway 1, the sulfonamide moiety was susceptible to an electrophilic attack by OH˙ or SO4˙− leading to S–N bond cleavage, and thus BP1 is formed (m/z = 99) as presented in Fig. 13a. In pathway 2, intermolecular Smiles-type rearrangement of the anilino radical took place by SR attack through the electron transfer mechanism producing a SO2 extrusion product (SEP) BP2 at m/z = 190 as shown in Fig. 13b. Finally, in pathway 3, SMX undergoes direct oxidation to give BP3; specifically SRs and/or HRs attacked N7 of SMX, which is the most electronegative nitrogen in the SMX structure (m/z = 284) (Fig. 13c). It is worth noting that these different BPs obtained (Table 1) disappeared totally at a later stage of the treatment as they were further oxidized toward potential mineralization.
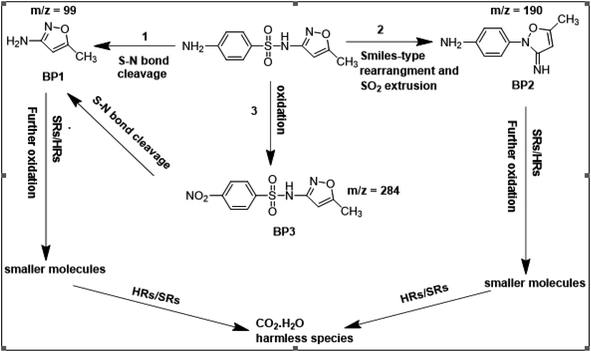 |
| | Fig. 12 Suggested overall degradation mechanism of SMX in the UVA/MIL88-A/PS or solar/MIL88-A/PS systems. | |
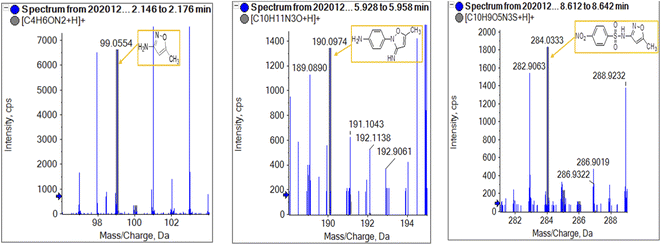 |
| | Fig. 13 HR mass spectrum fragmentation patterns under oxygen conditions of (a) BP1, (b) BP2, and (c) BP3. | |
Table 1 SMX and its byproducts identified by QTOF MS
| Compound |
Molecular formula |
ESI mode |
m/z |
Error (ppm) |
R.T. (min) |
Proposed structure |
| SMX |
C10H11N3O3S |
Positive |
254.0593 |
−0.3 |
5.92 |
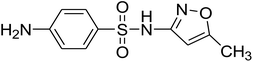
|
| BP1 |
C4H6ON2 |
Positive |
99.0554 |
0.8 |
2.16 |
|
| BP2 |
C10H11N3O |
Positive |
190.0974 |
−0.2 |
5.93 |
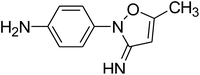
|
| BP3 |
C10H9O5N3S |
Positive |
284.0333 |
−1 |
8.63 |
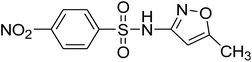
|
4. PS quantification and RSE calculation
[PS] was tracked throughout the reaction time using a modified HPLC method68 previously developed in our laboratory. The results showed that [PS] decreased from 2 mM initially to reach around 0.5 mM after a 2 h reaction time (Fig. S20†). Moreover, the reaction stoichiometric efficiency (RSE) was calculated. The RSE is defined as the ratio of the number of moles of the degraded organic contaminant divided by the number of moles of oxidant (PS) consumed in the process.| | | %RSE = n(SMX)degraded/n(PS)consumed × 100 | (18) |
The RSE showed a relatively high value (6.12%). This value is slightly greater to what was reported by Ghauch et al.24 (5.20%) and Ayoub et al.56 (5.55%) when PS was activated by micrometric Fe0 iron particles in aqueous solution and plated micrometric iron particles, respectively. This proves that SMX degradation was improved in similar chemically activated systems using however MIL88-A as a mediator.
5. Economic feasibility
The economic efficiency was assessed through cost calculation using electrical energy per order (EEO) for UVA/MIL88-A/PS and solar/MIL88-A/PS. EEO is defined as the electric energy in kilowatt hours (kWh) needed to degrade one order of magnitude of the contaminant per unit volume (m3), expressed as per eqn (19).96| | | EEO = P × t × 1000/V × log(Ci/Cf) | (19) |
where P (kW) is the power supplied to the system, V (L) is the volume treated in time t (h), and Ci and Cf are the initial and final concentrations of the pollutant, respectively.
The electrical and total system costs were later obtained according to eqn (20) and (21):
| | | Electrical energy cost ($ per m3) = EEO (kWh per m3) × power cost ($ per kWh) | (20) |
| | | Total system cost ($ per m3) = electrical energy cost + reagent cost | (21) |
Table. S3† summarizes the total energy cost of the studied systems in US dollars in Lebanon corresponding to 0.169 US dollars. The cost of reagents is calculated using wholesale prices for every case (Table S3†). By comparing the costs presented in the table to previous work done on different probes such as theophylline and ketoprofen, we notice that the use of the solar PS activated system is the cheapest one at 2.76 $ per m3. For more clarification, Amasha et al. obtained a total cost of 44.41 $ per m3 upon the degradation of ketoprofen (KTP) in the thermally activated PS system, with [KTP]0 = 7.87 μM (2.00 mg L−1), [PS]0 = 1 mM and T = 60 °C,20 while El Hakim et al. got a value of 24.7. $ per m3 using the UVC/PS activated system for theophylline degradation in a real pharmaceutical effluent with [PS]0 = 25 mM.83 These results present in advance the cost effective solar-based AOP systems and more specifically persulfate application on a larger scale such as artificial wetlands for the treatment of hazardous effluents.
6. Conclusions
In this study, PS-based AOPs proved to be highly efficient in the degradation of emergent contaminants such as pharmaceutical active ingredients e.g. SMX. Herein, a heterogeneous catalyst, MIL88-A, was synthesized with the advantage of using a low-cost, simple and organic solvent free process. The elimination of SMX was demonstrated in two different systems: UVA/MIL88-A/PS and solar/MIL88-A/PS systems. Several parameters were considered to reach a higher efficiency for the degradation process. [MIL88-A] alone showed no significant effect on the degradation of SMX in the absence and in the presence of irradiation (UVA or solar). The combined effect of UVA or solar/MIL88-A/PS showed promising results, whereby the total degradation of SMX was reached within 80 min and 5–20 min in the UVA/MIL88-A/PS and solar/MIL88-A/PS systems, respectively. The results also demonstrated that MIL88-A may be used for five successive cycles of activation experiments with PS without showing a decrease in the catalytic activity of the MOF. The results also showed the superiority of the solar/MIL88A-A/PS system over the UVA/MIL88-A/PS system in terms of additives making the oxidation process more efficient under sunlight irradiation regardless of the solution common ion content, e.g. Cl− and HCO3−. Moreover, HRMS analyses were conducted to identify the degradation products of SMX, and three different byproducts were identified with the proposed formation mechanism of each. EPR analysis showed the presence of HRs as well as SRs to better elucidate the activation mechanism of PS on the surface of the MOF. The calculated %RSE (6.24%) was comparable to those determined in similar persulfate-based systems however using iron particles or Fe2+ as PS activators. Future directions to consider are the investigation into a continuous treatment system capable of improving the %RSE along with the mineralization extent that was not considered in this work. Moreover, this work puts forth in advance the feasibility of using MIL88-A as a mediator to activate PS on a large scale and at a low cost under sunlight for the degradation of organic contaminants in agriculture ponds and artificial wetlands. This can efficiently contribute to reducing pollution stress by pharmaceutical compounds and pesticides for effluents before reaching the regular waste water treatment plants.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded in part by the University Research Board (award number 103603) of the American University of Beirut and USAID-Lebanon through The National Academy of Sciences under PEER project 5–18 (award number 103262/sub-grant 2000007572). The authors are thankful to Joan Younes, Simon Al-Ghawi and Boutros Sawaya for their technical assistance and the personnel of the K. Shair CRSL for their kind help. They are also very thankful to Prof. David Sedlak PEER US partner (UC-Berkeley), Dr Mario El Kazzi (Paul Scherrer Institute) for performing and interpreting the XPS analysis, and Dr Maxim Yilkov and Sahar Naim (ETH, Switzerland) who helped in performing the EPR analysis.
References
- A. J. Ebele, M. A.-E. Abdallah and S. Harrad, Emerging Contam., 2017, 3, 1–16 CrossRef.
-
L. Malaeb, G. M. Ayoub, M. Al-Hindi, L. Dahdah, A. Baalbaki and A. Ghauch, in Energy Procedia, Elsevier Ltd, 2017, vol 119, pp. 723–732 Search PubMed.
- A. Baalbaki, G. M. Ayoub, M. Al-Hindi and A. Ghauch, Sci. Total Environ., 2017, 574, 583–593 CrossRef CAS PubMed.
- P. Bottoni, S. Caroli and A. B. Caracciolo, Toxicol. Environ. Chem., 2010, 92, 549–565 CrossRef CAS.
-
K. E. Arnold, A. R. Brown, G. T. Ankley and J. P. Sumpter, 2014.
- J.-L. Liu and M.-H. Wong, Environ. Int., 2013, 59, 208–224 CrossRef CAS PubMed.
- G. R. Boyd, J. M. Palmeri, S. Zhang and D. A. Grimm, Sci. Total Environ., 2004, 333, 137–148 CrossRef CAS PubMed.
- D. Ashton, M. Hilton and K. V. Thomas, Sci. Total Environ., 2004, 333, 167–184 CrossRef CAS PubMed.
- M. Carballa, F. Omil, J. M. Lema, M. Llompart, C. García-Jares, I. Rodríguez, M. Gómez and T. Ternes, Water Res., 2004, 333, 137–148 Search PubMed.
- N. Lindqvist, T. Tuhkanen and L. Kronberg, Water Res., 2005, 39, 2219–2228 CrossRef CAS PubMed.
- N. Nakada, T. Tanishima, H. Shinohara, K. Kiri and H. Takada, Water Res., 2006, 40, 3297–3303 CrossRef CAS PubMed.
- J. Fick, H. Söderström, R. H. Lindberg, C. Phan, M. Tysklind and D. G. J. Larsson, Environ. Toxicol. Chem., 2009, 28, 2522–2527 CrossRef CAS PubMed.
- T. Heberer, Toxicol. Lett., 2002, 131, 5–17 CrossRef CAS PubMed.
- T. A. Ternes, M. Meisenheimer, D. McDowell, F. Sacher, H. J. Brauch, B. Haist-Gulde, G. Preuss, U. Wilme and N. Zulei-Seibert, Environ. Sci. Technol., 2002, 30, 3855–3863 CrossRef PubMed.
- I. A. Ike, K. G. Linden, J. D. Orbell and M. Duke, Chem. Eng. J., 2018, 338, 651–669 CrossRef CAS.
- Y. Zhou, Y. Xiang, Y. He, Y. Yang, J. Zhang, L. Luo, H. Peng, C. Dai, F. Zhu and L. Tang, J. Hazard. Mater., 2018, 359, 396–407 CrossRef CAS PubMed.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- A. Ghauch, A. M. Tuqan and N. Kibbi, Chem. Eng. J., 2012, 197, 483–492 CrossRef CAS.
- A. Ghauch, A. M. Tuqan, N. Kibbi and S. Geryes, Chem. Eng. J., 2012, 213, 259–271 CrossRef CAS.
- M. Amasha, A. Baalbaki and A. Ghauch, Chem. Eng. J., 2018, 350, 395–410 CrossRef CAS.
- M. Amasha, A. Baalbaki, S. Al Hakim, R. El Asmar and A. Ghauch, J. Adv. Oxid. Technol., 2018, 21, 20170099 Search PubMed.
- A. Ghauch, A. Baalbaki, M. Amasha, R. El Asmar and O. Tantawi, Chem. Eng. J., 2017, 317, 1012–1025 CrossRef CAS.
- Y. Liu, X. He, Y. Fu and D. D. Dionysiou, J. Hazard. Mater., 2016, 305, 229–239 CrossRef CAS PubMed.
- A. Ghauch, G. Ayoub and S. Naim, Chem. Eng. J., 2013, 228, 1168–1181 CrossRef CAS.
- S. Naim and A. Ghauch, Chem. Eng. J., 2016, 288, 276–288 CrossRef CAS.
-
M. G. Antoniou, Mechanistic studies on the degradation of cyanobacterial toxins and other nitrogen containing compounds with hydroxyl and sulfate radical based Advanced Oxidation Technologies, University of Cincinnati, 2010 Search PubMed.
- K.-Y. A. Lin, H.-A. Chang and C.-J. Hsu, RSC Adv., 2015, 5, 32520–32530 RSC.
- G. Férey, Chem. Soc. Rev., 2008, 4732–4734 Search PubMed.
- N. A. Khan, Z. Hasan and S. H. Jhung, J. Hazard. Mater., 2013, 244–245, 444–456 CrossRef CAS PubMed.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef CAS.
- J. Wang, J. Wan, Y. Ma, Y. Wang, M. Pu and Z. Guan, RSC Adv., 2016, 6, 112502–112511 RSC.
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724–781 CrossRef PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2011, 112, 782–835 CrossRef PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2011, 112, 1232–1268 CrossRef PubMed.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 6260–6266 CrossRef CAS PubMed.
- M. Kurmoo, Chem. Soc. Rev., 2009, 9, 5273–5282 Search PubMed.
- T. Uemura, N. Yanai and S. Kitagawa, Chem. Soc. Rev., 2009, 38, 1228–1236 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC.
- F. Ke, L.-G. Qiu, Y.-P. Yuan, F.-M. Peng, X. Jiang, A.-J. Xie, Y.-H. Shen and J.-F. Zhu, J. Hazard. Mater., 2011, 196, 36–43 CrossRef CAS PubMed.
- J. Wang, J. Wan, Y. Ma, Y. Wang, M. Pu and Z. Guan, RSC Adv., 2016, 6, 112502–112511 RSC.
- X. Li, W. Guo, Z. Liu, R. Wang and H. Liu, Appl. Surf. Sci., 2016, 369, 130–136 CrossRef CAS.
- Z. Shen, H. Zhou, Z. Pan, Y. Guo, Y. Yuan, G. Yao and B. Lai, J. Hazard.
Mater., 2020, 400, 123187 CrossRef CAS PubMed.
- H. Hu, H. Zhang, Y. Chen, Y. Chen, L. Zhuang and H. Ou, Chem. Eng. J., 2019, 368, 273–284 CrossRef CAS.
- C. Nguyen Dang Giang, Z. Sebesvari, F. Renaud, I. Rosendahl, Q. Hoang Minh and W. Amelung, PLoS One, 2015, 10, e0131855 CrossRef PubMed.
- J. C. Underwood, R. W. Harvey, D. W. Metge, D. A. Repert, L. K. Baumgartner, R. L. Smith, T. M. Roane and L. B. Barber, Environ. Sci. Technol., 2011, 45, 3096–3101 CrossRef CAS PubMed.
- B. A. Wilson, V. H. Smith, F. deNoyelles and C. K. Larive, Environ. Sci. Technol., 2003, 37, 1713–1719 CrossRef CAS PubMed.
- C. W. Knapp, C. A. Engemann, M. L. Hanson, P. L. Keen, K. J. Hall and D. W. Graham, Environ. Sci. Technol., 2008, 42, 5348–5353 CrossRef CAS PubMed.
- M. C. Dodd and C.-H. Huang, Environ. Sci. Technol., 2004, 38, 5607–5615 CrossRef CAS PubMed.
- R. F. Dantas, S. Contreras, C. Sans and S. Esplugas, J. Hazard. Mater., 2008, 150, 790–794 CrossRef CAS PubMed.
- M. Petrovic and D. Barceló, TrAC, Trends Anal. Chem., 2007, 26, 486–493 CrossRef CAS.
- A. G. Trovó, R. F. P. Nogueira, A. Agüera, C. Sirtori and A. R. Fernández-Alba, Chemosphere, 2009, 77, 1292–1298 CrossRef PubMed.
- M. N. Abellán, B. Bayarri, J. Giménez and J. Costa, Appl. Catal., B, 2007, 74, 233–241 CrossRef.
- M. N. Abellán, J. Giménez and S. Esplugas, Catal. Today, 2009, 144, 131–136 CrossRef.
- D. Klauson, M. Krichevskaya, M. Borissova and S. Preis, Environ. Technol., 2010, 31, 1547–1555 CrossRef CAS PubMed.
- G. Ayoub and A. Ghauch, Chem. Eng. J., 2014, 256, 280–292 CrossRef CAS.
- S. Bhattacharjee, J.-S. Choi, S.-T. Yang, S. B. Choi, J. Kim and W.-S. Ahn, J. Nanosci. Nanotechnol., 2010, 10, 135–141 CrossRef CAS PubMed.
- H. Li, J. Wan, Y. Ma, Y. Wang, X. Chen and Z. Guan, J. Hazard. Mater., 2016, 318, 154–163 CrossRef CAS PubMed.
- J. P. Thomas, A. Bejjani, B. Nsouli, A. Gardon and J. M. Chovelon, Int. J. Mass Spectrom., 2009, 279, 59–68 CrossRef CAS.
- J.-P. Thomas, A. Bejjani, B. Nsouli, A. Gardon and J.-M. Chovelon, Rapid Commun. Mass Spectrom., 2008, 22, 2429–2435 CrossRef CAS PubMed.
- B. Nsouli, A. Bejjani, S. Della Negra, A. Gardon and J.-P. Thomas, Anal. Chem., 2010, 82, 7309–7318 CrossRef CAS PubMed.
- M. Maaz, T. Elzein, D. Dragoe, A. Bejjani, N. Jarroux, C. Poulard, N. Aubry-Barroca, B. Nsouli and P. Roger, J. Mater. Sci., 2019, 54, 1184–1196 CrossRef CAS.
- E. Martínez-Campos, T. Elzein, A. Bejjani, M. J. García-Granda, A. Santos-Coquillat, V. Ramos, A. Muñoz-Bonilla and J. Rodríguez-Hernández, ACS Appl. Mater. Interfaces, 2016, 8, 6344–6353 CrossRef PubMed.
- T. Chalati, P. Horcajada, R. Gref, P. Couvreur and C. Serre, J. Mater. Chem., 2011, 21, 2220–2222 RSC.
- G. Ferraresi, C. Villevieille, I. Czekaj, M. Horisberger, P. Novák and M. El Kazzi, ACS Appl. Mater. Interfaces, 2018, 10, 8712–8720 CrossRef CAS PubMed.
- A. Bejjani, M. Noun, S. Della-Negra, R. Tannous, G. Chalhoub, M. Hamdan and B. Nsouli, Anal. Chem., 2019, 91, 8864–8872 CrossRef CAS PubMed.
-
A. Ghauch and A. Ammouri, US Pat., 10354469, 2019 Search PubMed.
- A. Baalbaki, N. Zein Eddine, S. Jaber, M. Amasha and A. Ghauch, Talanta, 2018, 178, 237–245 CrossRef CAS PubMed.
- K. Y. Andrew Lin, H. A. Chang and C. J. Hsu, RSC Adv., 2015, 5, 32520–32530 RSC.
- E. Y. Ionashiro, F. J. Caires, A. B. Siqueira, L. S. Lima and C. T. Carvalho, J. Therm. Anal. Calorim., 2012, 108, 1183–1188 CrossRef CAS.
- R. El Asmar, A. Baalbaki, Z. Abou Khalil, S. Naim, A. Bejjani and A. Ghauch, Chem. Eng. J., 2021, 405, 126701 CrossRef CAS.
- W. Huang, C. Jing, X. Zhang, M. Tang, L. Tang, M. Wu and N. Liu, Chem. Eng. J., 2018, 349, 603–612 CrossRef CAS.
- C. P. Silva, C. Oliveira, A. Ribeiro, N. Osório, M. Otero, V. I. Esteves and D. L. D. Lima, Chemosphere, 2020, 238, 124613 CrossRef CAS PubMed.
- J. M. Monteagudo, A. Durán, R. Culebradas, I. San Martín and A. Carnicer, J. Environ. Manage., 2013, 128, 210–219 CrossRef CAS PubMed.
- H. A. Gorrell, Am. Assoc. Pet. Geol. Bull., 1958, 42, 2513 Search PubMed.
- S. Al Hakim, A. Baalbaki, O. Tantawi and A. Ghauch, RSC Adv., 2019, 9, 33472–33485 RSC.
-
S. J. Boggs, D. Livermore and M. G. Seitz, Humic substances in natural waters and their complexation with trace metals and radionuclides: a review, [129 references], 1985 Search PubMed.
- M. E. Lindsey and M. A. Tarr, Water Res., 2000, 34, 2385–2389 CrossRef CAS.
- P. Westerhoff, S. P. Mezyk, W. J. Cooper and D. Minakata, Environ. Sci. Technol., 2007, 41, 4640–4646 CrossRef CAS PubMed.
- P. M. D. Gara, G. N. Bosio, M. C. Gonzalez, N. Russo, M. del Carmen Michelini, R. P. Diez and D. O. Mártire, Photochem. Photobiol. Sci., 2009, 8, 992–997 CrossRef CAS PubMed.
- Q. Zhang, J. Chen, C. Dai, Y. Zhang and X. Zhou, J. Chem. Technol. Biotechnol., 2015, 90, 701–708 CrossRef CAS.
- C. S. Uyguner and M. Bekbolet, Catal. Today, 2005, 101, 267–274 CrossRef CAS.
- S. Al Hakim, S. Jaber, N. Z. Eddine, A. Baalbaki and A. Ghauch, Chem. Eng. J., 2020, 380, 122478 CrossRef CAS.
- Z. Wei, F. A. Villamena and L. K. Weavers, Environ. Sci. Technol., 2017, 51, 3410–3417 CrossRef CAS PubMed.
- Y. Liu, X. Chen, Y. Yang, Y. Feng, D. Wu and S. Mao, Chem. Eng. J., 2019, 358, 408–418 CrossRef CAS.
- X. Duan, C. Su, J. Miao, Y. Zhong, Z. Shao, S. Wang and H. Sun, Appl. Catal., B, 2018, 220, 626–634 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- M. J. Davies, B. C. Gilbert, J. K. Stell and A. C. Whitwood, J. Chem. Soc., Perkin Trans., 1992, 2, 333–335 RSC.
- H. Liu, T. A. Bruton, F. M. Doyle and D. L. Sedlak, Environ. Sci. Technol., 2014, 48, 10330–10336 CrossRef CAS PubMed.
- Y. Gao and S. A. Chambers, J. Cryst. Growth, 1997, 174, 446–454 CrossRef CAS.
- H. Bao, X. Chen, J. Fang, Z. Jiang and W. Huang, Catal. Lett., 2008, 125, 160–167 CrossRef CAS.
- Y. Zhang, J. Zhou, X. Chen, L. Wang and W. Cai, Chem. Eng. J., 2019, 369, 745–757 CrossRef CAS.
- S. Jiang, Z. Zhao, J. Chen, Y. Yang, C. Ding, Y. Yang, Y. Wang, N. Liu, L. Wang and X. Zhang, Surf. Interfaces, 2022, 101843 CrossRef CAS.
- C. Y. Kwan and W. Chu, Water Res., 2003, 37, 4405–4412 CrossRef CAS PubMed.
- T. Luo, J. Wan, Y. Ma, Y. Wang and Y. Wan, Environ. Sci.: Processes Impacts, 2019, 21, 1560–1569 RSC.
- J. R. Bolton, K. G. Bircher, W. Tumas and C. A. Tolman, Pure Appl. Chem., 2001, 73, 627–637 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Antoine
Ghauch
*b and
Antoine
Ghauch