
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical and quantum chemical studies of peculiarities of 2,5-di-Me-pyrazine-di-N-oxide oxidation in the presence of methanol at single-walled and multi-walled carbon nanotube paper electrodes
S. I.
Kulakovskaya
*a,
A. V.
Kulikov
ab,
T. S.
Zyubina
a,
A. S.
Zyubin
a,
L. N.
Sviridova
b,
E. V.
Stenina
b,
A. G.
Ryabenko
a,
E. V.
Zolotukhina
a and
Yu. A.
Dobrovolskiy
a
aInstitute of Problems of Chemical Physics, Russian Academy of Sciences, 142432, Chernogolovka, Moscow Region, Russia. E-mail: kulsi@icp.ac.ru
bM.V. Lomonosov Moscow State University, Leninskie Hills, 119991, Moscow, Russia
First published on 16th December 2021
Abstract
The use of methanol (MeOH) in direct methanol fuel cells has increased the interest in the search for new electrode materials and catalysts that allow the oxidation of MeOH to be carried out under conditions that satisfy their practical applications: low cost, stability, and high catalytic activity. In this work, electrochemical and quantum chemical methods were used to study peculiarities of the electrocatalytic system 2,5-di-Me-pyrazine-di-N-oxide–methanol at single-walled and multi-walled carbon nanotube (CNT) paper electrodes in comparison with a glassy carbon (GC) electrode in 0.1 M Bu4NClO4 solution in acetonitrile (MeCN). The adsorption energies of MeOH, CH3COOH and H2O on the CNT surface were determined by quantum chemical modeling; this opened the door for the explanation of effects found in the Pyr1–MeOH catalytic system and for the ascertainment of factors affecting the catalytic efficiency of the process at CNT electrodes. We believe that this research will be helpful in using this process in electrocatalysis, sensors and direct methanol fuel cells, since the deactivation of aromatic di-N-oxides (as opposed to processes involving noble metals or noble metal oxides) is insignificant.
1. Introduction
The study of methanol oxidation is of particular interest in connection with its use in direct methanol fuel cells (DMFCs).1,2 In DMFCs, the most commonly used catalysts for methanol oxidation are noble metals and noble metal oxides. However, the high cost of noble metals and the poisoning of catalysts by irreversibly adsorbed carbon monoxide (CO) generated during methanol electrooxidation are driving the search for non-noble metals, metal free materials, and catalysts that are inexpensive, stable and active in catalysis.Carbon nanomaterials made of reduced graphene oxide, single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs), modified with additives of noble metals (Pt, Ir, Ru, and Pd) or their oxides (IrO2 and RuO2), are used in the electrocatalytic processes of methanol oxidation.3–8 An alternative approach could be the use of metal-free materials and organic mediators.
It was shown9–15 that the aromatic di-N-oxide radical cations of phenazine-di-N-oxide, pyrazine-di-N-oxide (Pyr0) and its substituted derivatives, electrochemically generated at GC and Pt electrodes, are carriers of active oxygen that is capable of activating the C–H bond of substrates: alcohols, ethers and cyclohexane. It was assumed that the activation process is accompanied by the formation of a complex of radical cations with a substrate that was confirmed by the registration of radical intermediates by EPR electrolysis at oxidation of phenazine-di-N-oxide in MeOH and its deuterated derivatives.11 The detection of the same radical intermediate in CH3OH and CH3OD proved the participation of the CH3 group of alcohol in the formation of the intermediate.
In previous studies,16–19 in the presence of aromatic di-N-oxide, a several-fold increase in the catalytic efficiency of oxidation of organic compounds at SWCNT or MWCNT paper electrodes in comparison with the GC electrode was found. It was noted16–19 that in the absence of the substrate the oxidation current of phenazine-di-N-oxide, 2,3,5,6-tetra-Me-pyrazine-di-N-oxide, at SWCNT or MWCNT paper electrodes is by several times higher than the oxidation current of the ferrocene (Fc) reference. Quantum chemical modeling of the adsorption energies of Pyr1, Fc and MeCN both on the CNT surface and in grooves between two CNTs using the cluster model simulating the surface of conducting and non-conducting carbon nanotubes20 can explain this effect.
In this work the peculiarities of oxidation of Pyr1 in the presence of MeOH were studied in 0.1 M Bu4NClO4 solution in MeCN at SWCNT and MWCNT paper electrodes in comparison with the GC electrode using cyclic voltammetry, EPR electrolysis, differential capacitance and quantum chemical modeling. The study of electrocatalytic oxidation of methanol at the electrodes of CNTs in the presence of aromatic di-N-oxides as mediators is of interest for using this process in electrocatalysis, sensors and direct methanol fuel cells.
2. Materials and methods
Materials and experimental techniques for obtaining cyclic voltammograms, EPR spectra during electrolysis at controlled potentials and dependence of the differential capacitance of the electrical double layer on potentials were described in previous studies.18,20 The quantum chemical modeling technique was described in detail in our previous work.203. Results and discussion
3.1. Quantum-chemical modeling of the adsorption of CH3OH, CH3COOH and H2O on the surface of CNTs
In this paper simulation of the adsorption of MeOH, CH3COOH and H2O at bundles of CNTs with a diameter of 1.4 nm was carried out. The C54H18(c) cluster (structure 1, Fig. 1) was chosen to simulate single-walled conductive (10, 10) CNTs because in our previous work20 it was shown to be a suitable model. For the modeling of the adsorption of CH3OH, CH3COOH and H2O, on the surface of CNTs, the functional wB97XD21 was used in the software package GAUSSIAN-09.22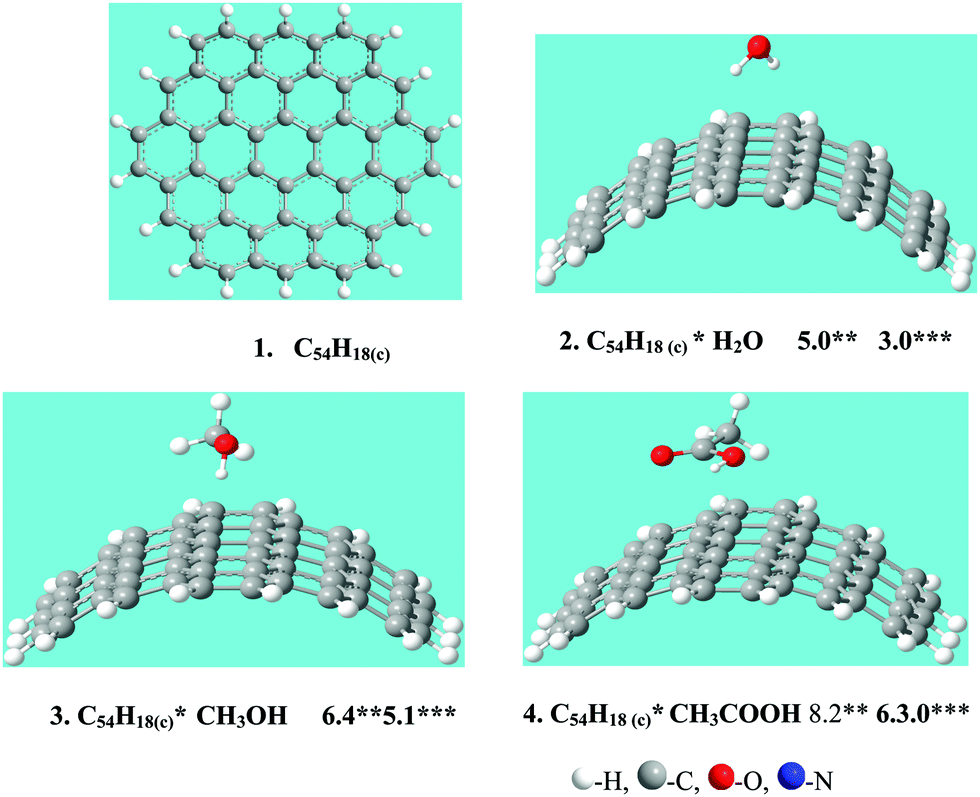 | ||
| Fig. 1 Structure 1 of the cluster C54H18(c) modeling surface of single-walled conductive (10,10) CNTs; structures of clusters C54H18(c)*X, X = H2O, CH3OH and CH3COOH (structures 2–4). Adsorption energies (in kcal mol−1) calculated at different basis sets are given: ** – 6-311G(d,p), *** – 6-311G(d,p) (BSSE). | ||
The obtained values of the adsorption energies of H2O, CH3OH and CH3COOH on the carbon cluster C54H18(c), modeling surfaces of the isolated conductive (10, 10) carbon nanotubes, are presented in Fig. 1 and Table 1. The adsorption energy decreases from CH3COOH (6.3 kcal mol−1) to MeCN (5.9 kcal mol−120), CH3OH (5.1 kcal mol−1) and H2O (3.0 kcal mol−1). An insignificant difference between the obtained values of the adsorption energies of MeCN and MeOH on the CNT surface indicates the possibility of their simultaneous adsorption on the CNT surface.
| N | Structure C54H18(c)*X, X = | E ad | E ad – BSSE |
|---|---|---|---|
| 8 | H2O | 5.0 | 3.0 |
| 9 | CH3OH | 6.4 | 5.1 |
| 10 | MeCN | 6.8 | 5.9 |
| 11 | CH3COOH | 8.2 | 6.3 |
| 12 | Fc | 12.6 | 10.9 |
| 13 | Pyr1 | 18.2 | 14.5 |
3.2. Oxidation of Pyr1 at GC, SWCNT and MWCNT paper electrodes in MeCN
Oxidation of Pyr1 at MWCNT and SWCNT paper electrodes in 0.1 M Bu4NClO4 solution in MeCN occurs at a potential of 1.78 V,20 that is, at higher by 220 mV as compared with oxidation at the GC electrode. The process is irreversible and not diffusion-controlled. In contrast to the GC electrode, the oxidation currents of 1 mM Pyr1 at MWCNT and SWCNT paper electrodes are several times higher than the diffusion oxidation current of 1 mM Fc (as the reference).
In the absence of Pyr1 no oxidation of MeOH is observed on the CV curves obtained at the GC electrode in 0.1 M Bu4NClO4 solution in MeCN (Fig. 2a). In the presence of 1 mM Pyr1 with the addition of MeOH a second anodic peak appears at higher anodic potentials (Fig. 2b). The current of the second anodic peak increases with an increase in the MeOH concentration and shifts to the first anodic peak. At a concentration of 0.5 M MeOH, anodic peaks merge.
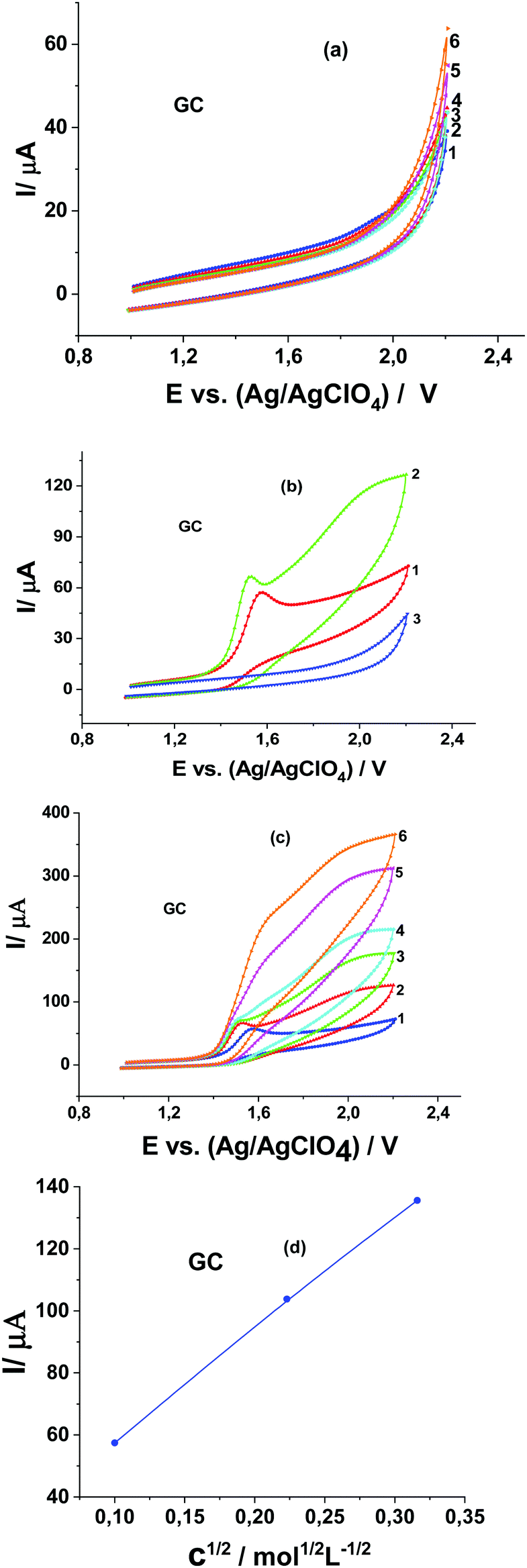 | ||
| Fig. 2 CVs of 0.1 M Bu4NClO4 solution in MeCN at the GC electrode at a potential scan rate of 20 mV s−1 in the presence of MeOH: (a) (1) 0, (2) 0.01, (3) 0.05, (4) 0.1, (5) 0.5, and (6) 1.0 M; (b) (1) 1 mM Pyr1, (2) 1 mM Pyr1 and 0.01 M MeOH, and (3) 0.01 M MeOH; (c) 1 mM Pyr1 and MeOH: (1) 0, (2) 0.01, (3) 0.05, (4) 0.1, (5) 0.5, and (6) 1.0 M; and (d) dependence of the anodic peak current on the square root of the MeOH concentration. | ||
The study of the dependence of the currents of these peaks on the potential scan rate at a MeOH concentration below 0.1 M shows that, as in 0.1 M LiClO4 solution in MeCN,15 the first anodic peak is diffusion (increases directly proportional to the square root of the scan rates; Fig. 3a), and the second anodic peak has a kinetic nature (decreases with an increase of the potential scan rates; Fig. 3a). This dependence of anodic peak current on the scan rate corresponds to the E1C1E2 mechanism of the electrode process when two stages of electron transfer are separated by a chemical stage C1.23,24
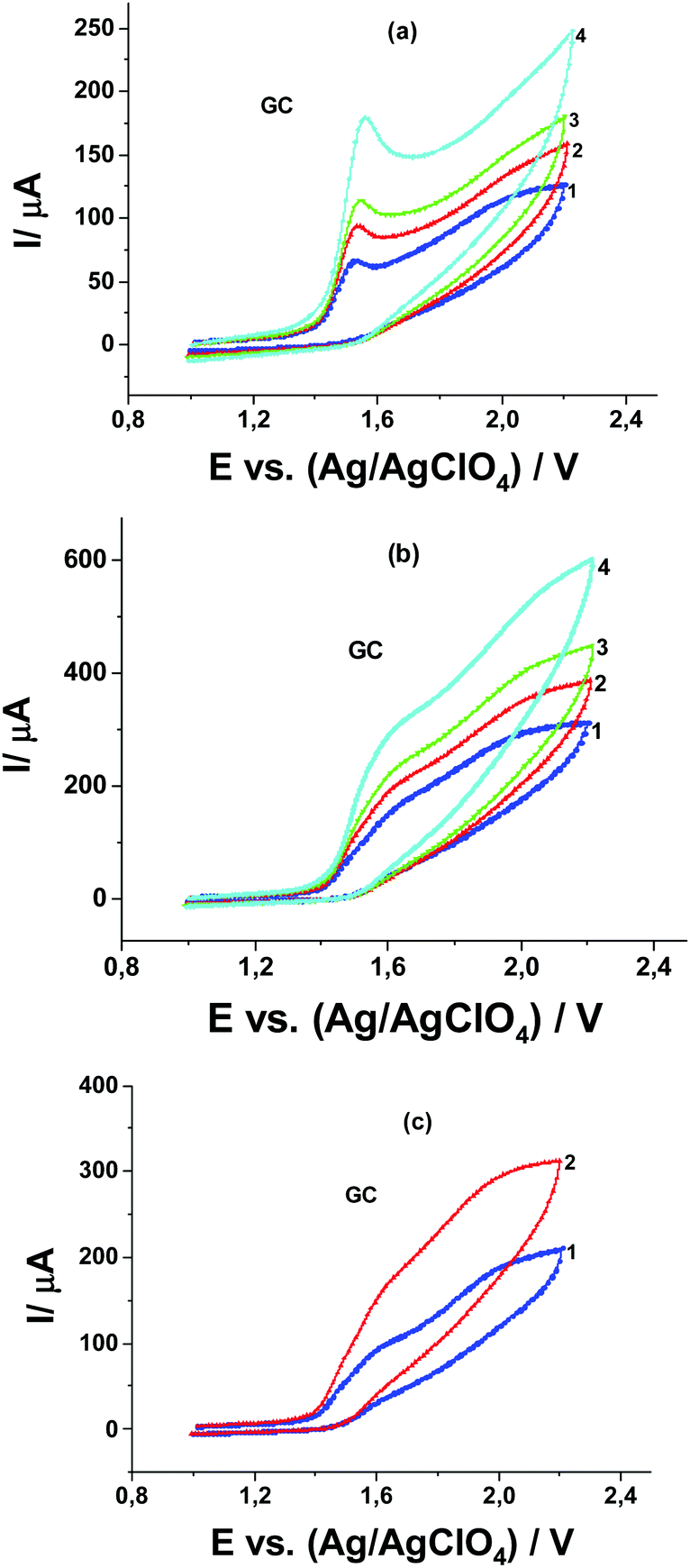 | ||
Fig. 3 CVs of 0.1 M Bu4NClO4 solution in MeCN at the GC electrode in the presence of![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : (a) 1 mM Pyr1 and 0.01 M MeOH at potential scan rates of: (1) 20, (2) 50, (3) 80, and (4) 200 mV s−1; (b) 1 mM Pyr1 and 0.5 M MeOH at potential scan rates of : (1) 20, (2) 50, (3) 80, and (4) 200 mV s−1; and (c) 0.5 M MeOH and Pyr1: (1) 0.5 mM and (2) 1 mM at a potential scan rate of 20 mV s−1. : (a) 1 mM Pyr1 and 0.01 M MeOH at potential scan rates of: (1) 20, (2) 50, (3) 80, and (4) 200 mV s−1; (b) 1 mM Pyr1 and 0.5 M MeOH at potential scan rates of : (1) 20, (2) 50, (3) 80, and (4) 200 mV s−1; and (c) 0.5 M MeOH and Pyr1: (1) 0.5 mM and (2) 1 mM at a potential scan rate of 20 mV s−1. | ||
The second anodic peak at a MeOH concentration above 0.1 M is proportional to the square root of its concentration (Fig. 2d), weakly depends on the potential scan rate (Fig. 3b) and is proportional to the Pyr1 concentration (Fig. 3c). According to a previous study,25 this indicates the catalytic nature of the second anodic peak. The ratio of the catalytic oxidation current of Pyr1 in the presence of 0.5 M MeOH to the current recorded in the absence of MeOH (the catalytic efficiency of the process) is 6.0. It should be noted that in the presence of 2,3,5,6-tetra-Me-pyrazine-di-N-oxide (Pyr2) the catalytic efficiency of MeOH oxidation was 4.0.19 Thus, the catalytic efficiency of the methanol oxidation increased by 1.5 times with an increase of the oxidation potential of di-N-oxide.
It was shown that the dependence of the CV curves of Pyr1 oxidation at the GC electrode in 0.1 M Bu4NClO4 solution in MeCN on the MeOH concentration and the potential scan rate are identical to those obtained in ref. 15 in 0.1 M LiClO4 solution in MeCN. Therefore, the nature of the background cation does not affect the electrocatalytic oxidation of MeOH in the presence of Pyr1 at the GC electrode.
Previously,15 it was established that the EPR spectra of radical cations pyrazine-di-N-oxide and its substituted derivatives were not found in the presence of MeOH in 0.1 M LiClO4 solution in MeCN. The EPR spectra of radical cations could have been detected during electrolysis at the Au electrode in deuterated methanol at a temperature close to the freezing point of the solvent at −85 °C.15 The EPR spectrum of the radical cation of Pyr1 in 0.1 M LiClO4 solution in CD3OD is presented in Fig. 4b and c. The registration of the EPR spectrum of the radical cation of Pyr1 in 0.1 M LiClO4 solution in CD3OD confirms that the Pyr1 structure remains unchanged during the alcohol catalytic oxidation. The registration of the EPR spectra of the Pyr1 radical anion at −35 °C during electrolysis at a controlled potential after alcohol catalytic oxidation at positive potentials (Fig. 4d–f) is further evidence that the structure and concentration of Pyr1 remain unchanged after the catalytic process. Since the nature of the background cation does not affect the electrocatalytic oxidation of MeOH in the presence of Pyr1, these data can be used as evidence for the invariability of the structure and concentration of Pyr1 in the catalytic process in 0.1 M Bu4NClO4 solution in MeCN in the presence of MeOH.
 | ||
| Fig. 4 EPR spectra recorded during electrolysis of 1 mM Pyr1 solution in the presence of 0.1 M LiClO4 at different temperatures and potentials.15 (a) In MeCN at −45 °C and +1.6 V, the dotted line is simulation using a Bruker software Symfonia taking into account hyperfine splitting on all nuclei of Pyr1: aN = 0.047 mT (2), aH1 = 0.042 mT (2), and aH2 = 0.113 mT (6); the numbers in parentheses are the numbers of equivalent nuclei, the half-width at half-height of the Lorentz line is 0.012 mT, (b) in CD3OD at −97 °C and +1.8 V, (c) in CD3OD at −85 °C and +1.7 V, (d) in CH3OH at −35 °C and −0.9 V, (e) in CH3OD at −35 °C and −0.9 V, and (f) in CD3OD at −35 °C and −0.8 V. The spectrum (a) was recorded at the Pt electrode, and the other spectra were obtained at the Au electrode. | ||
In the absence of Pyr1 at CV curves recorded at SWCNT and MWCNT paper electrodes in 0.1 M Bu4NClO4 solution in MeCN in the presence of MeOH from 0.01 to 1.0 M, as in the case of the GC electrode, the MeOH oxidation is not observed (Fig. 5a and b). The oxidation current of Pyr1 at SWCNT and MWCNT paper electrodes in 0.1 M Bu4NClO4 solution in MeCN increases with the addition of MeOH from 0.01 to 1.0 M, and an increase of the anodic current shifts to lower positive potentials (Fig. 5c and d).
 | ||
| Fig. 5 CVs of 0.1 M Bu4NClO4 solution in MeCN at a potential scan rate of 20 mV s−1 in the presence of MeOH: (1) 0, (2) 0.01, (3) 0.05, (4) 0.1, and (5) 0.5 M at (a) SWCNT and (b) MWCNT paper electrodes, (c) at the SWCNT paper electrode in the presence of 1 mM Pyr1 and MeOH: (1) 0, (2) 0.01, (3) 0.05, (4) 0.5, and (5) 1.0, (d) at the MWCNT paper electrode in the presence of 1 mM Pyr1 and MeOH: (1) 0, (2) 0.01, (3) 0.05, (4) 0.1, (5) 0.5, and (6) 1.0 M; and dependence of the anodic peak current on the square root of the MeOH concentration at (e) SWCNT and (f) MWCNT paper electrodes. | ||
Unlike oxidation at the GC electrode, at the CV curves of Pyr1 oxidation at SWCNT and MWCNT paper electrodes in 0.1 M Bu4NClO4 solution in MeCN in the presence of MeOH, one anodic wave is observed. This can be explained by the adsorption of Pyr1 molecules on the surface of SWCNT and MWCNT paper electrodes, which leads to a 220 mV shift towards higher positive potentials of the Pyr1 oxidation potential as compared to the oxidation potential of the unadsorbed Pyr1 molecules on the GC electrode. This shift results in the merging of the first and second anodic waves of Pyr1 oxidation at SWCNT and MWCNT paper electrodes in the presence of MeOH.
The linear dependence of the anodic peak of Pyr1 oxidation on the square root of the concentration of MeOH at SWCNT and MWCNT paper electrodes (Fig. 5e and f) at a MeOH concentration above 0.05 M corresponds to the catalytic nature of this peak.25 The ratio of the catalytic current at 0.5 M MeOH to the diffusion current of ferrocene (as the reference) is 16. Thus the catalytic efficiency of MeOH oxidation at SWCNT and MWCNT paper electrodes is ∼2.7 times higher than at the GC electrode.
It should be noted that in contrast to the GC electrode, on which the catalytic current (Ia) increases proportionally to the concentration of Pyr1 (Fig. 3c), at SWCNT and MWCNT paper electrodes, it increases by a factor of 1.3 (Fig. 6a and b) with a two-fold magnification of the Pyr1 concentration. This can be explained by the fact that in the absence of MeOH the oxidation current of Pyr1 is several times higher than the oxidation current of Fc as a reference and weakly depends on the Pyr1 concentration.20 Since the adsorption energy of Pyr1 (14.5 kcal mol−1, Table 1) on the CNT surface significantly exceeds the adsorption energies of MeCN and MeOH, the presence of MeOH in solution will not affect the adsorption of Pyr1 on the CNT surface and retains a weakly dependent anodic current of Pyr1 oxidation on its concentration.
 | ||
| Fig. 6 CVs of 0.1 M Bu4NClO4 solution in MeCN in the presence of 0.5 M MeOH and Pyr1: (1) 0.5 mM and (2) 1 mM at a potential scan rate of 20 mV s−1 at (a) SWCNT and (b) MWCNT paper electrodes and in the presence of 1 mM Pyr1 and 0.5 M MeOH at potential scan rates of: (1) 20, (2) 50, and (3) 80 mV s−1 at (c) SWCNT and (d) MWCNT paper electrodes. | ||
The adsorption of Pyr1 at the MWCNT paper electrode in 0.1 M Bu4ClO4 solution in MeCN was studied in a previous study20 by measuring the C,E-dependence of the differential double layer capacitance of the electrode on potential. It was found that the differential double layer capacitance of the electrode recorded in 0.1 M Bu4ClO4 solution in MeCN at the MWCNT paper electrode is not changed when 1 mM Pyr1 is added to the solution (Fig. 7). It has been established by quantum chemical modeling20 that the preferred arrangement is the parallel arrangement of the planar part of the Pyr1 molecule adsorbed on the surface of carbon nanotubes. It is known26 that the differential double layer capacitance of the electrode does not decrease at the flat arrangement of the adsorbed aromatic molecules on the electrode surface. With the addition of 0.5 M MeOH to the solution of 1 mM Pyr1 in 0.1 M Bu4ClO4 solution in MeCN the differential double layer capacitance of the electrode decreases. This indicates the formation of the adsorption layer on the surface of the MWCNT paper electrode, including Pyr1, MeOH and MeCN molecules. This conclusion is confirmed by quantum chemical modeling (Fig. 1 and Table 1).
 | ||
| Fig. 7 Dependences of the differential capacitance C of the MWCNT paper electrode at a potential E of 0.1 M Bu4ClO4 solution in MeCN in the presence of Pyr1: 0 (curves 1 and 2) and 1 mM (curves 3–5), 1 mM Pyr1 and 0.5 M MeOH (curve 6–8). Curves 1 and 2 were recorded just after recording the cyclic voltammetry curves, and curves 3, 4 and 5 are settled curves. | ||
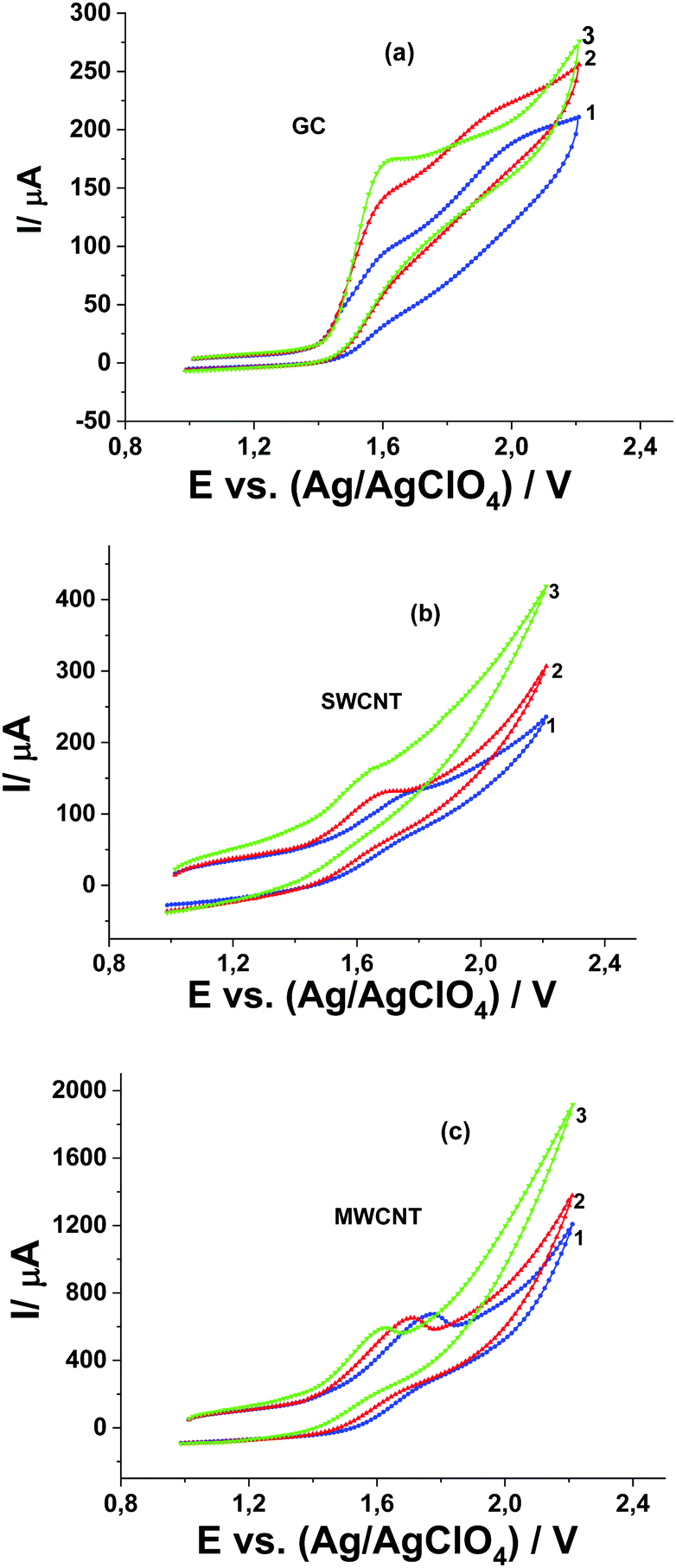
Studying the influence of CH3COOH and water (as a basis) on the potential and current of the catalytic wave of Pyr1 oxidation at GC, SWCNT, and MWCNT paper electrodes in the presence of MeOH makes it possible to establish the mechanism of the electrode process. A decrease of the catalytic current and the shifts of the catalytic wave and the first oxidation wave of Pyr1 towards higher positive potentials (Fig. 8a–c) are observed at the GC electrode with the addition of 1.0 and 2.0 M CH3COOH to solution 1.0 mM Pyr1 and 0.5 M MeOH in 0.1 M Bu4ClO4 solution in MeCN. It should be noted that with the addition of 2.0 M CH3COOH, the catalytic current at the GC electrode decreases by 1.4 times and at SWCNT and MWCNT paper electrodes by 3.8 and 3.1 times, respectively (Fig. 9).
 | ||
| Fig. 8 CVs of 1.0 mM Pyr1 and 0.5 M MeOH in 0.1 M Bu4NClO4 solution in MeCN in the presence of CH3COOH: (1) 0, (2) 1.0, and (3) 2.0 M at (a) (GC), (b) SWCNT and (c) MWCNT paper electrodes at a potential scan rate of 50 mV s−1. | ||
 | ||
| Fig. 9 CVs of 0.5 mM Pyr1 and 0.5 M MeOH in 0.1 M Bu4NClO4 solution in MeCN in the presence of H2O: (1) 0, (2) 0.5, and (3) 2.0 M at (a) (GC), (b) SWCNT and (c) MWCNT paper electrodes at a potential scan rate of 20 mV s−1. | ||
The shift of the catalytic wave at the GC electrode to potentials of the anodic wave of Pyr1 oxidation (Fig. 8a) is recorded with the addition of 0.5 and 2.0 M H2O to a solution of 0.5 mM Pyr1 and 0.5 M MeOH in 0.1 M Bu4ClO4 solution in MeCN (Fig. 8a). This indicates that the oxidation potentials of the reaction product of the radical cation with MeOH and the initial di-N-oxide are close. At SWCNT and MWCNT paper electrodes the influence of the addition of H2O is less pronounced.
The difference in the manifestation of the inhibiting effect of CH3COOH and of the catalytic effect of H2O at SWCNT and MWCNT paper electrodes as compared with the GC electrode can be explained by the performed quantum chemical modeling and the determination of the adsorption energies of MeCN20 and MeOH, CH3COOH and H2O (this work) on the CNT surface (Table 1). The adsorption energy of CH3COOH (6.3 kcal mol−1) on the CNT surface is sufficient to displace MeCN with an adsorption energy of 5.9 kcal mol−1 from the electrode surface. As a result of the adsorption of CH3COOH, its concentration on the CNT surface increases and the inhibiting effect is more pronounced compared to the GC electrode. The adsorption energy of H2O (3 kcal mol−1) on the CNT surface (Table 1) is much lower than the adsorption energy of MeCN and therefore its catalytic effect is less pronounced compared to the GC electrode. A similar difference in the manifestation of the inhibiting effect of CH3COOH and of the catalytic effect of H2O at SWCNT or MWCNT paper electrodes as compared with the GC electrode was observed in ref. 17–19. Quantum chemical modeling has provided an explanation for this effect. The effect of acid and water additions on the catalytic process indicates that the catalytic process is accompanied by proton elimination.
| PyrDNO+˙ + CH3OH → PyrDNOH+ + HOCH2˙ |
Based on the obtained experimental data and quantum chemical modeling the following mechanism of Pyr1 oxidation at SWCNT and MWCNT paper electrodes in MeCN in the presence of MeOH is proposed.
At the GC electrode. At the first electrode stage (E1), Pyr1 is oxidized to the radical cation. This process is one-electron, irreversible, and diffusion-controlled. The charge transfer is followed by an irreversible chemical reaction.
At SWCNT and MWCNT paper electrodes. At the first electrode stage (E1), Pyr1, adsorbed on the CNT surface, is oxidized to the radical cation. This process is irreversible and not controlled by diffusion.
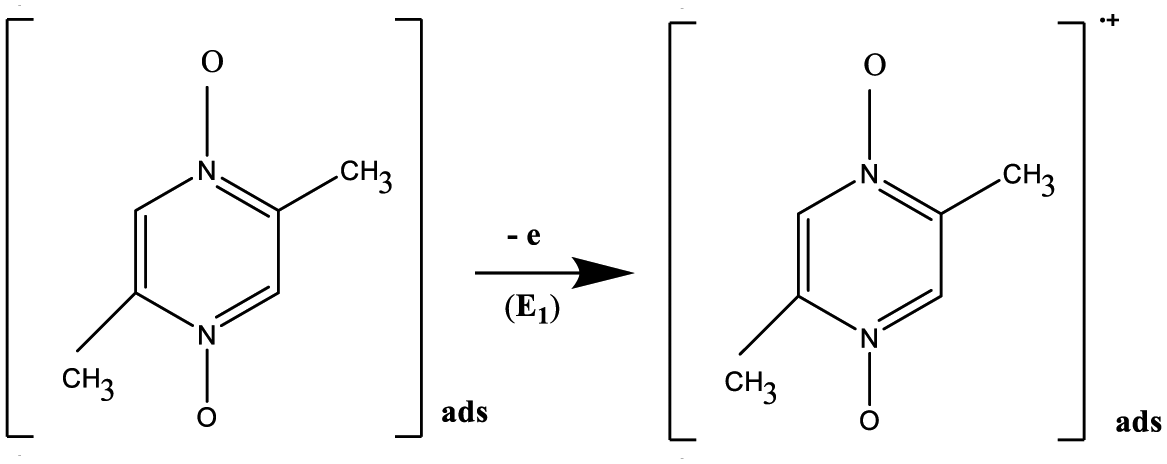
At GC, SWCNT and MWCNT paper electrodes. At the first chemical stage (C1), two competitive chemical reactions of the electrophilic addition of the oxygen atom of Pyr1 radical cations to the C–H bonds of MeCN and MeOH occur. The reactions are accompanied by proton elimination and the formation of radical intermediates (complexes with an N–O–C structure). Compounds of this structure and methods for their preparation are known.27–33
At SWCNT and MWCNT paper electrodes. The chemical stage proceeds with the participation of the Pyr1 radical cations, as well as MeCN and MeOH, adsorbed on the surface of the SWCNT and MWCNT paper electrodes.

At the GC electrode. At the second electrode stage (E2), the radical intermediates are oxidized to the cations.
At SWCNT and MWCNT paper electrodes. The radical intermediates adsorbed on the surface of the SWCNT and MWCNT paper electrodes are oxidized to the cations of (A) and (B).
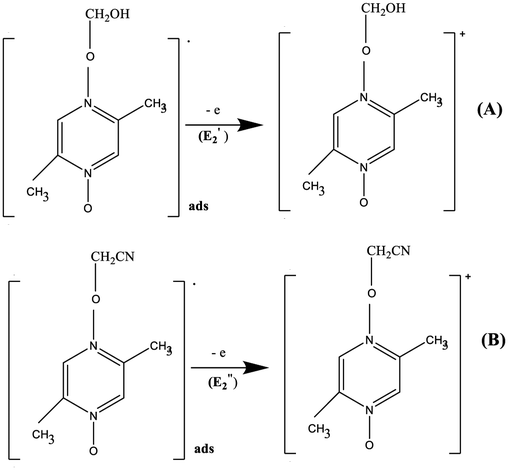
At GC, SWCNT and MWCNT paper electrodes. The second chemical stage (C2) corresponds to the interaction of the cations of (A) and (B) with a nucleophile or a base (a water admixture in the solution). It is assumed that the cation (A) decomposes at the stage
 with the formation of the initial aromatic di-N-oxide and the product of two-electron oxidation of MeOH (formaldehyde). The regenerated aromatic di-N-oxide is immediately oxidized at the electrode to a radical cation, and the cycle is repeated; the catalytic current of the total two-electron oxidation of MeOH is recorded. In a competitive chemical stage
with the formation of the initial aromatic di-N-oxide and the product of two-electron oxidation of MeOH (formaldehyde). The regenerated aromatic di-N-oxide is immediately oxidized at the electrode to a radical cation, and the cycle is repeated; the catalytic current of the total two-electron oxidation of MeOH is recorded. In a competitive chemical stage  , the cation (B) is attached to a base or a nucleophile to form an adduct.20
, the cation (B) is attached to a base or a nucleophile to form an adduct.20
4. Conclusions
In this work, electrochemical and quantum chemical methods were used to study the peculiarities of the behavior of the electrocatalytic system of 2,5-di-Me-pyrazine-di-N-oxide – methanol at single-walled and multi-walled carbon nanotube paper electrodes in comparison with a glassy carbon (GC) electrode. The cluster model describing the surface of conducting carbon nanotubes (10, 10) was used to simulate the adsorption of MeOH, CH3COOH, and H2O on the surface of CNTs. The observed effects in the catalytic system are explained by quantum chemical modeling of the non-covalent interaction of the components of the studied system with the CNT surface. It is concluded that, in the course of the catalytic process at SWCNT and MWCNT electrodes, in contrast to the GC electrode, the adsorption of the components of the catalytic system on their surface is of decisive importance. This research may be helpful in using this process in electrocatalysis, sensors and direct methanol fuel cells.This work was performed in accordance with the state task, state registration no. AAAA-A19-119061890019-5.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Calculations were performed at the Computer Center, Institute of Problems of Chemical Physics, RAS.References
- X. Li and A. Faghri, Review and advances of direct methanol fuel cell (DMFCs) part 1: Design, fabrication, and testing with high concentration methanol solutions, J. Power Sources, 2013, 226, 223–240 CrossRef CAS.
- Y. Feng, H. Liu and J. Yang, A selective electrocatalyst-based direct methanol fuel cell operated at high concentrations of methanol, Sci. Adv., 2017, 3, e1700580 CrossRef PubMed.
- T. Radhakrishnan and N. Sandhyarani, Three dimensional assembly of electrocatalytic platinum nanostructures on reduced graphene oxide – An electrochemical approach for high performance catalyst for methanol oxidation, J. Hydrogen Energy, 2017, 42, 7014–7022 CrossRef CAS.
- Y. Gan, H. Huang and W. Zhang, Electrocatalytic oxidation of methanol on carbon-nanotubes/graphite electrode modified with platinum and molybdenum oxide nanopaticles, Trans. Nonferrous Met. Soc. China, 2007, 17, 214–219 CrossRef CAS.
- S. Wang, X. Wang and S. P. Jiang, PtRu Nanoparticles supported on 1-aminopyrene functionalized multi-walled carbon nanotudes and their electrocatalytic activity for methanol oxidation, Lungmuir, 2008, 24, 10505–10512 CrossRef CAS PubMed.
- Y. Zhao, X. Yang, J. Tian, F. Wang and L. Zhan, Methanol electro-oxidation on Ni@Pd core-shell nanoparticles supported on multi-walled carbon nanotubes in alkaline media, Int. J. Hydrogen Energy, 2010, 35, 3249–3257 CrossRef CAS.
- H. An, L. Pan, H. Cui, D. Zhou, B. Wang, J. Zhai, Q. Li and Y. Pan, Electrocatalytic performance of Pd nanoparticles supported on TiO2-MWCNTs for methanol, ethanol, and isopropanol in alkaline media, J. Electroanalyt. Chem., 2015, 741, 56–63 CrossRef CAS.
- H. Zhang, X. Han and Y. Zhao, Pd–TiO2 nanoparticles supported on reduced graphene oxide: green synthesis and improved electrocatalytic performance for methanol oxidation, J. Electroanal. Chem., 2017, 799, 84–91 CrossRef CAS.
- S. I. Kulakovskaya, V. M. Berdnikov, A. Y. Tikhonov, L. B. Volodarskii and V. E. Maier, A cyclic voltametric study of the mechanism responsible for the oxidation of organic compounds on a glass-carbon electrode in the presence of aromatic N,N′-dioxides as organic mediators, Russ. J. Electrochem., 1993, 29, 40–45 Search PubMed.
- S. I. Kulakovskaya, V. M. Berdnikov, A. A. Vasilenko, A. Y. Tikhonov and L. B. Volodarskii, The mechanism of electrochemical activation of the C–H bond during oxidation of organic compounds in the presence of aromatic N-oxides as the intermediates, Russ. J. Electrochem., 1996, 32, 784–790 CAS.
- S. I. Kulakovskaya, A. V. Kulikov, V. M. Berdnikov, N. T. Ioffe and A. F. Shestakov, Electrochemical and EPR study of the C–H bond activation. Electrocatalytic oxidation with participation of radical cation of phenazine-di-N-oxide, Electrochim. Acta, 2002, 47, 4245–4254 CrossRef CAS.
- S. I. Kulakovskaya, A. V. Kulikov and A. F. Shestakov, Radical cation of pyrazine-di-N-oxyde as a mediator in electrocatalytic oxidation of organic compounds, Russ. J. Electrochem., 2004, 40, 1035–1044 CrossRef CAS.
- S. I. Kulakovskaya, A. V. Kulikov and A. F. Shestakov, Substituted pyrazine-di-N-oxydes as the mediators of catalytic oxidation of organic compounds, Russ. J. Electrochem., 2007, 43, 1156–1163 CrossRef CAS.
- S. I. Kulakovskaya, A. V. Kulikov and A. F. Shestakov, Electrochemical and EPR-study of the mechanism of organic compound oxidation in the presence of mediators – radical cations of substituted pyrazine-di-N-oxydes, Russ. J. Electrochem., 2007, 43, 1234–1242 CrossRef CAS.
- S. I. Kulakovskaya, A. V. Kulikov and A. F. Shestakov, Electrochemical and EPR Studies of the Oxidation Mechanism of Pyrazine-di-N-oxides in the Presence of Methanol and its Deuterated Derivatives, Russ. J. Electrochem., 2012, 48, 1023–1036 CrossRef CAS.
- S. I. Kulakovskaya, A. G. Krivenko, N. S. Komarova, A. V. Kulikov and A. F. Shestakov, Electrochemical and EPR study of the mechanism of oxidation of phenazine-di-N-oxide in the presence of cyclohexanol on glass carbon and single-walled carbon nanotube electrodes, Russ. J. Electrochem., 2014, 50, 1–12 CrossRef CAS.
- S. I. Kulakovskaya, A. V. Kulikov, L. N. Sviridova and E. V. Stenina, Electrochemical and electron paramagnetic resonance study of the mechanism of oxidation of phenazine-di-N-oxide in the presence of isopropyl alcohol at glassy carbon and single-walled carbon nanotube electrodes, Electrochim. Acta, 2014, 146, 798–808 CrossRef CAS.
- S. I. Kulakovskaya, A. V. Kulikov, L. N. Sviridova and E. V. Stenina, Electrochemical and electron paramagnetic resonance study of the mechanism of oxidation of 2,3,5,6-tetra-Me-pyrazine-di-N-oxide as a mediator of electrocatalytic oxidation of isopropyl alcohol at glassy carbon and single-walled carbon nanotube electrodes, J. Electroanal. Chem., 2017, 795, 81–89 CrossRef CAS.
- S. I. Kulakovskaya, A. V. Kulikov, L. N. Sviridova, E. V. Stenina, A. G. Ryabenko and V. G. Basu, Comparative electrochemical and electron paramagnetic resonance study of the mechanism of oxidation of 2,3,5,6-tetra-Me-pyrazine-di-N-oxide as a mediator of electrocatalytic oxidation of metanol at glassy carbon and multi-walled carbon nanotube paper electrodes, J. Adv. Electrochem., 2018, 4(1), 162–167 CrossRef.
- S. I. Kulakovskaya, A. V. Kulikov, T. S. Zyubina, A. S. Zyubin, D. V. Konev, L. N. Sviridova, E. V. Stenina, A. G. Ryabenko and E. V. Zolotukhina, Role of non-covalent interactions at oxidation of 2,5-di-Me-pyrazine-di-N-oxide at glassy carbon, single-walled and multi-walled carbon nanotube paper electrodes, Carbon trends, 2021, 4, 100057, DOI:10.1016/j.cartre.2021.100057.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Fcisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- R. S. Nicholson and J. Shain, Theory of stationary electrode polarography. Single scan and cyclic methods applied to reversible, irreversible, and kinetic systems, Anal. Chem., 1964, 36, 706–723 CrossRef CAS.
- R. S. Nicholson and J. Shain, Theory of stationary electrode polarography for a chemical reaction coupled between two charge transfers, Anal. Chem., 1965, 37, 178–189 CrossRef CAS.
- Z. Galus, Fundamentals of Electrochemical Analysis, New York, Harwoood, 1976 Search PubMed.
- A. N. Frumkin and B. B. Damaskin, Adsorption of organic compounds at electrodes, in Modern Aspects of Electrochemistry, ed. J. O. M. Bockris and B. E. Conway, London, Butterworth, 1964, vol. 3, pp. 149–223 Search PubMed.
- V. A. Golubev, R. V. Miklyush and E. G. Rozantsev, Metod sinteza ketoefirov N-oksipiperidina, (Method of synthesis of ketoether of N-oxypiperidine), Izv. Akad. Nauk SSSR, Ser. Khim., 1972, 3, 656–658 Search PubMed.
- V. A. Golubev and R. V. Miklyush, Novyi preparativnyi metod okisleniya aktivirovannoii metilenovoii gruppy do karbonil’noii, (New preparative method of the oxydation of methylene group to carbonyl), Zh. Org. Khim., 1972, 8, 1356–1357 CAS.
- A. R. Katritzky, The preparation of some substituted pyridine 1-oxides, J. Chem. Soc., 1956, 2404–2408 RSC.
- T. Okamoto and H. Tani, Synthesis of 2- and 4-cyanopyridincs, Chem. Pharm. Bull., 1959, 7, 130–131 CrossRef CAS.
- T. Okamoto and H. Tani, Reaction mechanism in aromatic heterocyclic compound. The reactions of N-alkoxypyridinium derivatives (1), Chem. Pharm. Bull., 1959, 7, 925–930 CrossRef.
- A. R. Katritzky and E. Lunt, N-oxides and related compounds-XXXV. Reactions of N-alkoxy-pyridinium and quinolinium cations with nucleophiles, Tetrahedron, 1969, 25, 4291–4305 CrossRef CAS.
- R. Eisenthal and A. R. Katritzky, The ring-opening of N-methoxypyridinium perchlorate by hydroxide ion, Tetrahedron, 1965, 21, 2205–2213 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |