DOI:
10.1039/D1YA00078K
(Paper)
Energy Adv., 2022,
1, 99-112
Production of jet fuel-range hydrocarbon biofuel by hydroxyalkylation–alkylation of furfural with 2-methylfuran and hydrodeoxygenation of C15 fuel precursor over a Ni/γ-Al2O3 catalyst: a reaction mechanism†
Received
16th December 2021
, Accepted 15th January 2022
First published on 19th January 2022
Abstract
This study presents manufacturing jet fuel-range (C9–C15) hydrocarbon biofuels from 2-methylfuran and furfural. The process involves a hydroxyalkylation–alkylation reaction, followed by hydrodeoxygenation of the C15 fuel precursor. The hydroxyalkylation–alkylation reaction was investigated under various cation exchange resin loadings, furfural/2-methylfuran molar ratios, and reaction temperatures. The hydroxyalkylation–alkylation reaction results were further corroborated by an appropriate kinetic model. Hydrodeoxygenation proceeds via the sequential furan ring-hydrogenation and ring-opening reactions, followed by the combination of dehydroformylation, hydrodeoxygenation, and cracking reactions. The dehydroformylation reaction was the leading pathway over a Ni/γ-Al2O3 catalyst, forming mainly C14H30 alkane. The oxygenate conversion was boosted with rising hydrogen pressure, Ni metal content on γ-Al2O3, and reaction temperatures. Both hydrodeoxygenation and hydrogenation reactions proliferated at elevated hydrogen pressure with the enrichment of the C15 alkane and ring-opening and ring-hydrogenation products. The cracking, ring-opening, and ring-hydrogenation reactions were promoted at elevated reaction temperatures with a significant amount of lighter alkanes.
1. Introduction
At the moment, greater than 80% of the world's energy is derived from non-renewable coal, natural gas, and petroleum. With the rapid growth of the population and gradual expansion of economics, the energy demand in the world is continuously mounting and anticipated to increase by about 25% by 2040.1 At present, diesel, jet fuel, and gasoline are the major transportation fuels, which account for nearly 28% of the primary energy consumption in the world.1 Currently, jet fuel and diesel account for around 7% and 35% of the energy consumption in the transportation sector, and their demands are projected to rise by 55% and 45% in the coming 20 years, respectively.1 Currently, transportation fuels are mainly produced by petroleum refining. Petroleum is, however, limited and depleting rapidly. Moreover, the use of petroleum poses environmental issues owing to the release of poisonous and greenhouse gases. The quest for renewable automobile fuels is thus growing throughout the globe. Petroleum originates from the gradual decay of biomass for millions of years under intense heat and pressure. At present, biomass is the sole renewable source of carbon in our biosphere. Therefore, biomass is considered the potential substitute for petroleum to produce renewable transportation fuels, called biofuels. Similar to a petroleum refinery, the biomass is refined in a biorefinery to obtain various biofuels and value-added chemicals.
The lignocellulose biomass is the most abundant on the earth. It includes agriculture wastes, residues from forest management exercises, and various cellulosic energy crops. Significant progress has been made in the past to convert this biomass to biofuels, such as biobutanol, bioethanol, etc.2 However, these are oxygen-containing biofuels with reduced fuel mileage and incongruity with refinery infrastructures. Continuous efforts are thus made for manufacturing biofuels comparable to present transportation fuels, called hydrocarbon biofuels.3 Fast pyrolysis,4 hydrothermal liquefaction,5,6 and biomass-to-liquid7 are some of the possible technologies for manufacturing hydrocarbon biofuels from lignocellulose biomass. Alternatively, the carbohydrate fraction of the lignocellulose biomass can be converted to hydrocarbon biofuels via various intermediate and platform chemicals.8 Furfural finds a special place among the platform chemicals owing to its exceptional derivative potentials, including value-added chemicals, hydrocarbon biofuels, and fuel additives.9 Furfural is produced from the low-value hemicellulose fraction of lignocellulose biomass by a dehydrocyclization reaction using an acid catalyst. Furfural is a highly reactive oxygen-containing compound with poor fuel properties. The deoxygenation of furfural, however, produces C4–C5 hydrocarbons, which are inappropriate as transportation fuels. Therefore, the carbon–carbon coupling reaction, such as hydroxyalkylation–alkylation (HAA), is necessary to obtain transportation fuel precursors with a longer carbon chain length (Reaction 1). In the present study, the HAA reaction of furfural with 2-methylfuran (2-MF) was explored to produce the C15 fuel precursor. 2-MF is generally derived from furfural by a selective hydrogenation reaction.10 Subsequent hydrodeoxygenation (HDO) of this C15 fuel precursor produces jet fuel-range hydrocarbon biofuels.
The idea of the aforementioned carbon–carbon coupling reaction was pioneered by Huber's and Dumesic's groups.11,12 Their research groups first reported multi-step reactions to produce hydrocarbon biofuels from carbohydrates. Their process includes dehydration of sugars to furfural/5-hydroxymethylfurfural (5-HMF), conversion of furfural/5-HMF by the aldol-condensation reaction with acetone, and dehydration/hydrogenation of the condensed product. Recently, Corma et al. and Zhang et al. reported a new pathway for hydrocarbon biofuels from furanic platform chemicals.13,14 This process involves the HAA reaction between a carbonyl compound (furfural/5-HMF) with α-hydrogen-containing furan (2-MF), followed by HDO of the high molecular weight fuel precursors (Scheme 1). The α-hydrogen-containing furanic platform chemical, furfural, however, exhibited no reactivity with carbonyl compounds owing to the existence of the electronegative aldehyde group attached to the furan ring. The protective methyl group in the furanic structure of 2-MF, however, makes it very much reactive for HAA reaction. Therefore, the HAA reaction of 2-MF was investigated with numerous carbonyl compounds, for instance, furfural, 5-HMF, butanal, acetone, angelica lactone, ethyl levulinate, mesityl oxide, acetoin, and hydroxyl acetone.13–22 Furthermore, 2-MF was oligomerized to a trimer followed by HDO to produce diesel-range biofuel.23
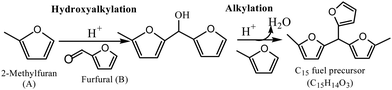 |
| | Scheme 1 Hydroxyalkylation–alkylation reaction of 2-MF with furfural. | |
HAA reaction proceeds in the presence of both homogeneous and heterogeneous acid catalysts.18,24 Homogeneous acid catalysts are, however, unsuitable for industrial processes owing to their corrosiveness and complexity in separation and recycling. On the contrary, the solid-acid catalysts can be easily separated from the reaction mixtures and reused. The solid-acid catalysts, such as heteropolyacids, zeolites, acidic resins, and functionalized metal oxides, were thus widely used for this reaction.20,25 Among these, acidic resins demonstrated high catalytic activity with a stable catalytic performance towards HAA reaction.25,26 Nafion-212 displayed the maximum catalytic activity with 75% 2-MF conversion and a 67% carbon yield under mild reaction conditions.14,25 The sulfonated nanostructured carbon showed more than 90% 2-MF conversion.27 In this study, the HAA reaction of furfural with 2-MF was investigated using a cation exchange resin (CER) as the catalyst. The role of various reaction variables, such as CER loading, reaction temperature, and 2-MF/furfural ratio, were further studied to obtain optimum reaction conditions. An appropriate kinetic model was further proposed to validate the experimental data.
HDO of the fuel precursors obtained by HAA reaction was reported over C, SiO2, and SiO2–Al2O3 supported noble metal (Pd, Pt, Ru, etc.) catalysts.14,18,28 The Pt/ZrP showed the best catalytic activity with a 94% carbon yield and C15 alkane as the primary product.14 The noble metal-based catalysts are, however, costly with limited industrial applications. The focus has been shifted to bifunctional catalysts for one-pot synthesis of fuel-range alkanes, such as Pd loaded NbOPO4/SBA-15, Nb2O5/SBA-15, and FeOx/SiO2 and Ru/HAP combined with HZSM-5.29–31 However, the multifunctional catalysts involve complex preparation methods.32 Recently, a new strategy of mechanical mixing of acid catalysts with supported metal catalysts was assessed.32,33 In the present study, we proposed a γ-Al2O3 supported low-cost transition metal, nickel, as the catalyst for HDO of the C15 fuel precursor. The extensive characterization of the catalysts was performed to establish the structure-catalytic activity relation. The understanding of the reaction mechanism is of profound importance in designing suitable catalysts for HDO reaction and to obtain desired hydrocarbons by tuning appropriate process parameters. However, the comprehensive mechanistic study of HDO of the C15 fuel precursor is limited in the published literature. The examination of the HDO reaction mechanism for the C15 fuel precursor is thus another objective of the present study. A systematic experimental investigation was carried out for HDO of the C15 fuel precursor over Ni/γ-Al2O3 catalysts with different Ni metal contents on γ-Al2O3 under a wide range of temperatures and hydrogen partial pressures. The detailed quantification of individual hydrocarbons and oxygenated intermediate compounds were carried out to corroborate a suitable HDO reaction mechanism.
2. Experimental
2.1 Preparation of catalysts
The Ni/γ-Al2O3 catalysts were synthesized by the incipient wetness impregnation method. At first, a saturated solution of the requisite amount of nickel nitrate hexahydrate (99% purity, Loba Chemie) was prepared using deionized water. The saturated solution of the precursor was then added to γ-Al2O3 and mixed continuously for two hours. The resulting wet catalyst was dried for twelve hours in an oven at 373 K and calcined at 823 K for six hours. This catalyst was referred to as the calcined catalyst. The NiO/γ-Al2O3 (calcined) catalyst was finally activated by reduction using hydrogen at 923 K for six hours. The obtained catalyst was denoted as the reduced catalyst. The catalyst with x wt% Ni metal on γ-Al2O3 was denoted as xNiAl.
2.2 Characterization of the catalysts
The specific surface areas and pore-volumes of the Ni/γ-Al2O3 catalysts were obtained from multipoint nitrogen adsorption–desorption isotherms (77 K) using the Brunauer–Emmett–Teller (BET) equation and Barret–Joyner–Halenda (BJH) method, respectively (Micromeritics, ASAP 2020 system). The catalysts were initially degassed at 623 K for three hours under a vacuum to remove moisture, impurities, and adsorbed gases. The adsorption isotherm was measured by the gradual rise of partial pressure of nitrogen up to the saturation pressure for complete adsorption of nitrogen on the catalyst. The desorption isotherm was then measured by reducing the partial pressure of nitrogen slowly to desorb nitrogen from the catalyst. Temperature-programmed reduction (TPR), temperature-programmed desorption of ammonia (NH3-TPD), and pulse chemisorption experiments were performed using an Autochem II 2920, Micromeritics system. The instrument was equipped with TCD for continuous monitoring of hydrogen consumption and NH3 desorption during the experiment. TPR study elucidates the presence of reducible species, reduction temperature, ease of reduction, and interaction between different species. For the TPR study, the metal oxide catalyst (calcined) was first pretreated at 523 K for one hour with argon (20 ml per min) to remove moisture and adsorbed gases and then cooled to room temperature. Following pretreatment, the catalysts were exposed to 10 vol% H2 in Ar (50 ml per min), and the temperature was then raised linearly from room temperature to 1223 K with a 10 K per min heating rate. The amount of hydrogen consumed in the TPR study was calculated from the peak area using pulse calibration data. For hydrogen pulse chemisorption, the reduction of the metal oxide catalyst (calcined) was first performed at 923 K under a flow of 10 vol% H2 in Ar (20 ml per min). The reduced catalyst was then flushed with Ar (20 ml per min) with simultaneous cooling up to 323 K. Following cooling, successive pulses of 10 vol% H2–Ar were introduced. The pulses were continued until the complete chemisorption of hydrogen. The percentage dispersion of nickel and nickel surface area per g nickel were estimated using the quantity of chemisorbed hydrogen. NH3-TPD discloses the acidity of the catalyst and its strength. The catalyst was first degassed at 523 K for one hour under an Ar flow (10 ml per min) and then cooled to 323 K. Ammonia (20 ml per min) was then introduced at 323 K and equilibrated for 30 min. The physisorbed ammonia was then removed by sending Ar (50 ml per min) for one hour, and the catalyst was then heated linearly up to 1073 K with 10 K per min heating rate to quantify the desorbed ammonia at different temperatures. The powder X-ray diffraction (XRD) pattern of the NiO/γ-Al2O3 and Ni/γ-Al2O3 catalysts was obtained using a Rigaku smart lab X-ray diffractometer at 2θ of 10°–90° and 2° per min scanning rate. The morphological information was obtained by transmission electron microscopy (TEM) using a JEOL JEM 2100FX system with a filament of LaB6 at 200 kV. The fine powder sample was first dissolved in ethanol using ultrasonication for 2 h. One drop of the prepared specimen was then deposited on a carbon-coated grid and the ethanol was evaporated before analysis.
2.3 Hydroxyalkylation–alkylation (HAA) reaction study
HAA reaction was investigated in a batch-reactor assembly consisting of a round bottom flask, condenser, and magnetic stirrer. The reactor was submerged in a temperature-controlled oil bath. The oil bath was placed on the hot plate of a magnetic stirrer. A PID controller was used to control the temperature of the oil bath. After the attainment of the set point temperature of the oil bath, known quantities of furfural (98% purity, Alfa Aesar), 2-MF (>98% purity, Alfa Aesar), and a CER catalyst (min 4.5 meq per g, Sisco Res. Lab. Pvt. Ltd) were charged into the round bottom flask and stirred at a constant stirring speed. The reaction samples were acquired at different time intervals. These samples were filtered using a syringe filter and diluted with 1-octanol (external standard, Spectrochem Pvt. Ltd, 98% purity). The products were identified by GC-MS (Shimadzu, GC-2010 system) and quantified by GC-FID using nitrogen as the carrier gas (Fig. S1, ESI†).
2.4 Hydrodeoxygenation (HDO) reaction of C15 fuel precursor
The C15 fuel precursor was first prepared by the HAA reaction of 2-MF with furfural using the CER catalyst with excess 2-MF under otherwise optimal reaction conditions. The reaction mixture was filtered to separate the CER catalyst and then diluted with acetone. The unreacted reactants (mainly 2-MF) and acetone were then removed from the product mixture using a rotary evaporator to obtain the C15 fuel precursor with more than 95% purity. HDO of the C15 fuel precursor was then carried out at high hydrogen pressure in a stirred autoclave reactor. The reactor was heated using an electrical heater, whose temperature was maintained at ±1 K of a set point temperature using a PID controller. At first, 3 g of C15 fuel precursor, 69 g of iso-octane (solvent and internal standard, Sisco Res. Lab. Pvt. Ltd, 99% purity), and a measured quantity of Ni/γ-Al2O3 catalyst were loaded into the reactor. The air from the reactor was removed by hydrogen flushing for about 10 min. The pressure of the reactor was then raised using hydrogen, and the reactor was heated to the set point temperature, while maintaining a constant stirring speed of 1000 rpm. The high agitation speed was maintained to eradicate the external mass transfer resistance in this reaction. After attaining the set point temperature, the first sample was acquired, and it was designated as the initial (zero min) reaction sample. Additional reaction mixtures were acquired at several time intervals. The collected samples were first quenched with acetone, and the suspended catalyst particles were then separated using a centrifuge. The identification of the chemical formulae and quantification of the products were then performed by GC-MS and GC-FID, respectively, using a ZB-5HT capillary column (30 m × 0.32 mm × 0.10 μm) and nitrogen as the carrier gas. The oven temperature was initially kept at 313 K for five minutes, increased to 498 K with a ramp rate of 30 K per min, maintained there for two minutes, then raised to 508 K with a heating rate of 20 K per min, and held there for four minutes. The detector and injector were kept at 653 K and 613 K, respectively. Following the completion of the reaction, the gas-phase samples were analysed by GC-FID (Fig. S2, ESI†) and GC-TCD (Fig. S3, ESI†) to examine the possible formation of volatile hydrocarbons and gases.
3. Results and discussion
3.1. Hydroxyalkylation–alkylation reaction
The 2-MF conversion was only about 20% at 300 min of reaction time for 0.5 g of the CER loading (Fig. 1A). The 2-MF conversion was, however, increased to around 39% by raising the CER loading to 1.0 g. The enhanced catalytic activity of the CER was because of the rise in catalytically active acid sites. The 2-MF conversion was, however, not increased much by increasing the catalyst loading to 1.5 g owing to the presence of excess catalytic sites than required for the reaction. The CER loading of 1.0 g was thus considered as the optimum. In our study, the intermediate products from the hydroxyalkylation reaction were not observed (Scheme 1). This might be due to the slower rate of hydroxyalkylation reaction than the alkylation reaction.27 The hydroxyalkylation reaction was thus considered as the rate-determining step in the kinetic model. Moreover, we observed only small quantities of other products (less than 1%), especially at longer reaction times. These products were, therefore, excluded from the results. All reactions were studied at a high agitation rate to eliminate the external mass transfer resistance. The reaction was assumed to be first order for both the reactants: 2-MF (A) and furfural (B). Accordingly, the rate of formation of A was expressed by the following rate equation considering X as the conversion of A and θB as the ratio of initial moles of B to that of A (CB0/CA0).| | 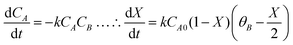 | (1) |
The following equation was obtained after integrating the above equation.| | 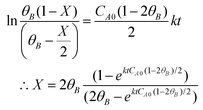 | (2) |
The kinetic study for the HAA reaction was executed in the temperature range of 308–328 K (Fig. 1B). The 2-MF conversion at 6 h of reaction time was around 33% at 308 K and enhanced to about 39%, 42%, and 49% at 313 K, 323 K, and 328 K, respectively. The experimental 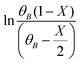 vs. time (t) data at different temperatures fitted well by linear lines passing through the origin (Fig. S4, ESI†). This result confirms that the reaction is second order. The rate constant of this reaction was calculated at different temperatures from the slope of
vs. time (t) data at different temperatures fitted well by linear lines passing through the origin (Fig. S4, ESI†). This result confirms that the reaction is second order. The rate constant of this reaction was calculated at different temperatures from the slope of 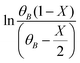 vs. time (t) (Fig. 1B). The 2-MF conversion was then estimated based on eqn (2) using the calculated rate constants at various reaction temperatures and matched with 2-MF conversions obtained from experiments. 2-MF conversions calculated using the above kinetic model fitted very well with the experimental 2-MF conversions, as observed from the figure. The activation energy (15.09 kcal mol−1) and pre-exponential factor (1.8 × 105 (kmol m−3)−1 s−1) were further obtained from the Arrhenius plot of 1/temperature vs. ln(k) (Fig. S5, ESI†). The high activation energy further elucidates the absence of external mass transfer resistance. The study was then extended to various 2-MF/furfural mole ratios to validate the established kinetic model. The 2-MF conversion was decreased with the increase in the 2-MF/furfural molar ratio from 1
vs. time (t) (Fig. 1B). The 2-MF conversion was then estimated based on eqn (2) using the calculated rate constants at various reaction temperatures and matched with 2-MF conversions obtained from experiments. 2-MF conversions calculated using the above kinetic model fitted very well with the experimental 2-MF conversions, as observed from the figure. The activation energy (15.09 kcal mol−1) and pre-exponential factor (1.8 × 105 (kmol m−3)−1 s−1) were further obtained from the Arrhenius plot of 1/temperature vs. ln(k) (Fig. S5, ESI†). The high activation energy further elucidates the absence of external mass transfer resistance. The study was then extended to various 2-MF/furfural mole ratios to validate the established kinetic model. The 2-MF conversion was decreased with the increase in the 2-MF/furfural molar ratio from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 3:1 (Fig. 1C).27 For different 2-MF/furfural mole ratios, the 2-MF conversions were estimated using the calculated rate constants at 313 K and compared with the 2-MF conversions obtained from experiments. The calculated 2-MF conversions for various 2-MF/furfural molar ratios matched the corresponding experimental 2-MF conversions quite well.
:1 to 3:1 (Fig. 1C).27 For different 2-MF/furfural mole ratios, the 2-MF conversions were estimated using the calculated rate constants at 313 K and compared with the 2-MF conversions obtained from experiments. The calculated 2-MF conversions for various 2-MF/furfural molar ratios matched the corresponding experimental 2-MF conversions quite well.
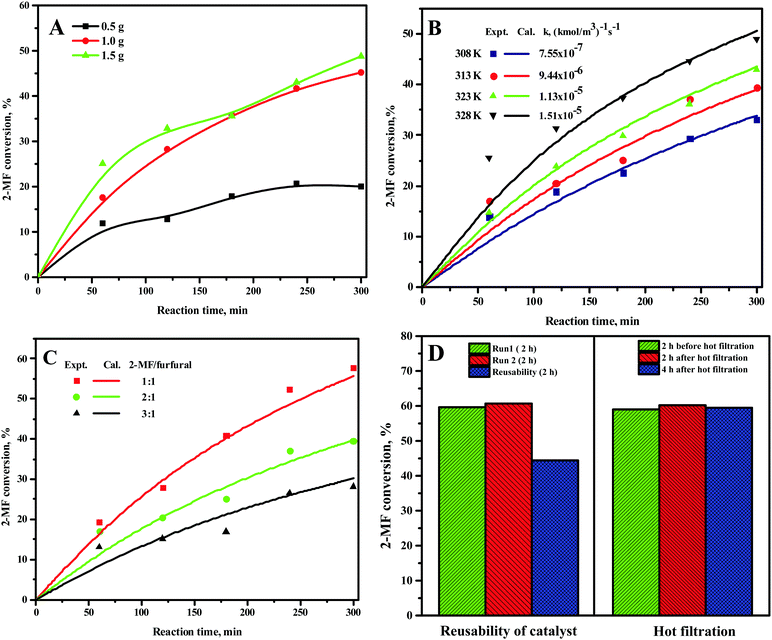 |
| | Fig. 1 Effect of (A) CER loading, (B) reaction temperature, and (C) 2-MF/furfural mole ratio on 2-MF conversion, and (D) reusability and hot filtration study. Reaction conditions: (A) 0.1997 mol 2-MF, 0.1004 mol furfural, and 313 K. (B) 0.1997 mol 2-MF, 0.1004 mol furfural, and 1 g CER. (C) 313 K and 1 g CER. (D) 328 K, 1.5 g CER, and 2:1 2-MF/furfural. | |
The reproducibility study was further performed under identical reaction conditions, and the reaction results were found to be reproducible (Fig. 1D). The spent CER from these reactions was separated, washed with ethanol, and then dried at 373 K overnight. The activity of the spent CER was tested under the same reaction conditions. The spent CER exhibited a slightly lower 2-MF conversion compared to the fresh catalyst (Fig. 1D). The hot-filtration test was further performed to check the possible catalyst deactivation by leaching of –SO3H groups. The reaction was initially carried out for two hours, the catalyst was separated from the reaction mixture under hot conditions, and the reaction was then continued for an additional four hours without the catalyst. However, the conversion of 2-MF remained unchanged after hot filtration. This result eliminates the possibility of deactivation of CER by leaching of –SO3H groups. The optimum HAA reaction conditions were found to be 1 g CER loading, 1:1 2-MF/furfural, and 328 K. About 66% 2-MF conversion and 96% selectivity towards C15 fuel precursor was observed under the optimum conditions at 5 h of reaction time. However, the C15 fuel precursor was prepared using 2:1 2-MF/furfural mole ratio (0.43 mol 2-MF and 0.21 mol furfural) under otherwise optimum reaction conditions. The conversion of furfural was more than 95% under these reaction conditions. A slight excess of 2-MF was used for the preparation of the C15 fuel precursor for an easy elimination of excess volatile 2-MF from the reaction mixture after the reaction.
3.2. Characterization of the Ni/γ-Al2O3 catalysts
The specific surface area and pore volume of both the NiO/γ-Al2O3 (calcined) and Ni/γ-Al2O3 (reduced) catalysts were lower than those of γ-Al2O3 (Table 1). Moreover, the specific surface area and pore volume of these catalysts was decreased with increasing Ni metal content on γ-Al2O3.34,35 This was due to the blockage of γ-Al2O3 pores and coverage of the γ-Al2O3 surface by Ni/NiO particles. It was further observed that the Ni/γ-Al2O3 (reduced) catalysts exhibited a lower specific surface area than the NiO/γ-Al2O3 (calcined) catalysts. This might be due to the agglomeration/sintering of nickel metal during high-temperature reduction at 973 K.
Table 1 Physicochemical properties of the Ni/γ-Al2O3 catalysts
| Catalyst |
BET |
Chemisorption |
dc, nm |
Acidity, mmol g−1 |
TPR |
| cal |
red |
MD |
SM |
cal (NiO) |
red (Ni) |
red |
MH2 |
DR |
| SSA |
PV |
SSA |
PV |
| red = Reduced catalyst; cal = calcined catalyst; PV = pore volume, cm3 g−1; SSA = specific surface area, m2 g−1; MD = dispersion of nickel, %, SM = nickel surface area, m2 g−1 nickel; dc = Ni/NiO crystallite size; MH2 = mmol of H2 consumed per g catalyst; DR = degree of reduction of NiO, %. |
| γ-Al2O3 |
237 |
0.83 |
— |
— |
— |
— |
— |
— |
NIL |
— |
— |
| 10NiAl |
198 |
0.61 |
180 |
0.63 |
1.16 |
7.74 |
13.8 |
10.53 |
0.15 |
1.32 |
77.5 |
| 15NiAl |
193 |
0.60 |
177 |
0.64 |
0.90 |
6.00 |
14.24 |
12.37 |
0.39 |
1.78 |
69.8 |
| 20NiAl |
189 |
0.59 |
169 |
0.62 |
0.80 |
5.45 |
14.59 |
14.04 |
0.50 |
2.41 |
71.1 |
| 25NiAl |
169 |
0.52 |
156 |
0.55 |
0.77 |
5.17 |
19.16 |
20.04 |
0.44 |
2.84 |
66.7 |
| 20NiAl (spent) |
— |
— |
142 |
0.59 |
0.62 |
4.38 |
— |
20.65 |
0.43 |
2.10 |
62.0 |
TPR study exhibited three distinct reduction peaks for NiO/γ-Al2O3 (calcined) catalysts at 590–618 K, 670–685 K, and 700–850 K (Fig. 2A).34 The bulk NiO showed a reduction peak at 618 K. The NiO/γ-Al2O3 catalysts also displayed a similar reduction peak at 590-618 K. The lowest temperature reduction peak for the NiO/γ-Al2O3 catalysts was thus assigned to bulk NiO having a weak interaction with the γ-Al2O3 support. The peak observed at 670–685 K corresponded to the dispersed NiO reduction. The interaction of dispersed NiO with the γ-Al2O3 support was slightly stronger compared to that of bulk NiO. The strong metal-support interaction for dispersed NiO was reflected by the slightly higher reduction temperatures. The reduction peak at 700–850 K was associated with NiAl2O4 species.34 This species has the strongest interaction with the support with the highest reduction temperature. The hydrogen consumption was further calculated from TPR experiments, as shown in Table 1. The hydrogen consumption was improved with the growing nickel metal content on γ-Al2O3. However, the degree of NiO reduction was slightly lower for higher nickel metal content on γ-Al2O3. The dispersion of nickel and nickel surface area per gm nickel were reduced slightly for rising Ni metal content on γ-Al2O3 (Table 1).34 This was due to the increased amount of bulk NiO with increasing Ni metal content on γ-Al2O3 (Fig. 2A).
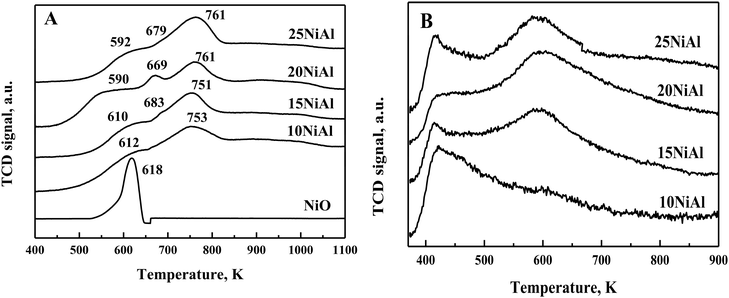 |
| | Fig. 2 (A) TPR profiles of the bulk NiO and NiO/γ-Al2O3 (calcined) catalysts. (B) NH3-TPD profiles of the Ni/γ-Al2O3 (reduced) catalysts. | |
The NH3-TPD results exhibited two types of acid sites for the Ni/γ-Al2O3 (reduced) catalysts (Fig. 2B). The first NH3 desorption peak at a lower temperature (400–517 K) represents the weak acid sites. These acid sites were due to the surface hydroxyl groups.36 The second NH3 desorption peaks at 517–678 K represent the moderate acid sites. The under-coordinated Lewis acidic aluminium cations were responsible for these acid sites.36 The moderate acid sites were absent in the 10 wt% Ni/γ-Al2O3 catalyst. Besides, the intensity of the moderate acid sites was increased with rising Ni metal content on γ-Al2O3 from 15 to 25 wt%. The total acidity was calculated from these NH3 desorption peaks, as shown in Table 1. The total acidity was increased with rising Ni metal content on γ-Al2O3 up to 20 wt% and decreased slightly for 25 wt%. This was due to a slight decrease in moderate acid sites for 25 wt% Ni/γ-Al2O3. The drop in acidity for 25 wt% nickel metal content on γ-Al2O3 was owing to the existence of a large amount of bulk NiO, as observed from the TPR results (Fig. 2A).
The powder XRD pattern of both the NiO/γ-Al2O3 (calcined) and Ni/γ-Al2O3 (reduced) catalysts with various Ni metal contents on γ-Al2O3 are presented in Fig. 3. The powder XRD pattern of the bulk NiO and support (γ-Al2O3) were additionally acquired to distinguish the diffraction peaks corresponding to NiO and γ-Al2O3. The NiO diffraction peaks for the NiO/γ-Al2O3 catalyst were observed at 2θ of 37.28° (1 1 1), 43.28° (2 0 0), and 62.87° (2 2 0). Similarly, the Ni/γ-Al2O3 (reduced) catalysts revealed the Ni species at 2θ of 37.09° (1 1 1) and 44.30° (2 0 0). The crystallite size of the catalysts was also determined using the Scherrer equation, as shown in Table 1. The NiO and Ni crystallite sizes were enlarged with the rising nickel metal contents on γ-Al2O3 for the calcined and reduced catalysts, respectively. The enlarged Ni crystallite sizes resulted in a drop in nickel dispersion and the nickel surface area per g nickel (Table 1).
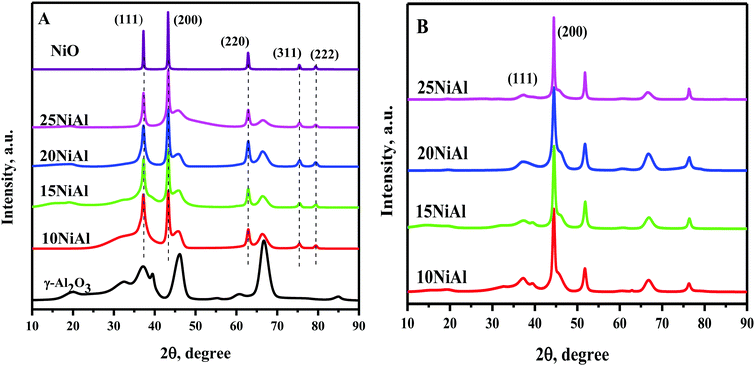 |
| | Fig. 3 Powder XRD pattern of the (A) Ni/γ-Al2O3 (calcined) and (B) Ni/γ-Al2O3 (reduced) catalysts. | |
3.3. Reaction mechanism for HDO of the C15 fuel precursor
The C15 fuel precursor is a complex chemical compound with multiple functionalities. The reaction mechanism for HDO of such a complex molecule is thus quite intricate. In this study, we made an effort to delimit a possible HDO reaction mechanism to elucidate the product distribution over a wide range of reaction conditions. C9–C15 alkanes were detected as the hydrocarbon products over the Ni/γ-Al2O3 catalysts (Fig. 4). We also perceived a trivial quantity of greater than C15 alkanes during this study. However, the C13–C15 alkanes were the leading hydrocarbon products, with C14H30 being the dominant one. Besides, many oxygenated intermediate compounds were perceived. However, the GC peaks of these oxygenated intermediate compounds were overlapping, posing a great difficulty in their clear identification. The chemical formulae of a few of these oxygenated intermediate compounds were identified, as shown in Fig. 4 and Fig. S6 (ESI†).
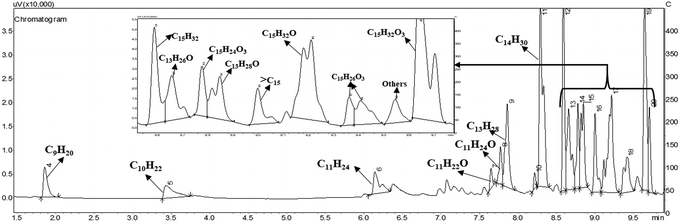 |
| | Fig. 4 A typical GC chromatogram for HDO of the C15 fuel precursor. Reaction conditions: 0.5 g 20NiAl, 573 K, 15 min of reaction time, and 30 bar H2. | |
Typical concentration profiles for hydrocarbons and oxygenated intermediate compounds are displayed in Fig. 5. The C15 fuel precursor was not detected in the initial (zero min) reaction sample. On the contrary, a high wt% of C15H26O3 (I) was observed in the initial reaction sample (Scheme 2). Furthermore, the wt% C15H26O3 (I) was declined rapidly with the progress of the reaction. These results revealed that the furan ring hydrogenation was the initial step of the reaction. Liu et al. observed three isomers of C15H26O3 as the major products during the initial reaction time.37 We further observed GC peak splitting for C15H26O3. Therefore, we cannot disregard the possibility of these isomers completely. Furthermore, XIV–XVI compounds in Scheme 2 bear the same chemical formula, C15H26O3. The furan rings generally undergo sequential hydrogenation of individual double bonds. However, we detected only a trivial amount of a partial ring hydrogenation product (C15H24O3) during this study. Dutta et al. observed partial and complete furan ring hydrogenation products only at 298 K and 50 bar hydrogen pressure over the Pd/C-Hf(OTf)4 catalyst.38 However, the furan ring hydrogenation products were absent at 498 K with the concurrent appearance of furan ring-opening products. The furan ring hydrogenation reaction occurs on metallic sites. Therefore, this reaction was rapid over the Ni/γ-Al2O3 catalyst with a high wt% of complete furan ring hydrogenation product (C15H26O3, I) at the initial reaction time. On the other hand, furan ring-opening products were absent in the initial reaction sample. These results endorse the faster rate of furan ring hydrogenation than the ring-opening reaction under the prevailing reaction conditions. The furan ring hydrogenation was reported to be competitive with the furan ring-opening reaction at high hydrogen pressure.39 Besides, the hydrogenation of double bonds of furan was reported to be thermodynamically favorable with the formation of tetrahydrofuran.40 The furan ring hydrogenation product was found to be dominant during HDO of 2,5-dimethylfuran.37,38,41
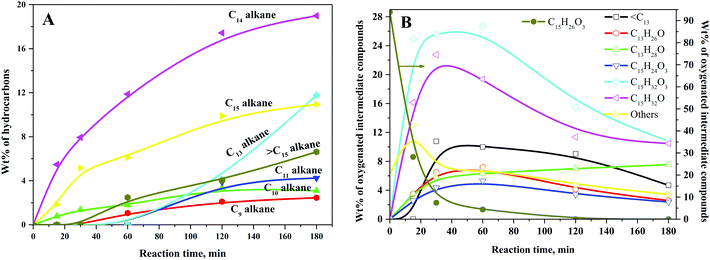 |
| | Fig. 5 Typical concentration profiles of (A) hydrocarbons and (B) oxygenated intermediate compounds for HDO of the C15 fuel precursor. Reaction conditions: 0.5 g 20NiAl, 543 K, and 30 bar H2. | |
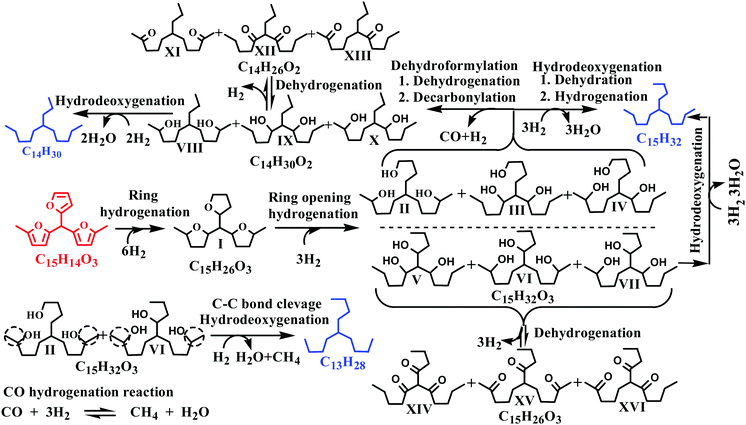 |
| | Scheme 2 Proposed reaction mechanism for hydrodeoxygenation of the C15 fuel precursor. | |
We observed a noticeable quantity of C15H32O3 isomers (II–VII) at 15 min of reaction time. The wt% of this product was further increased up to 30 min of reaction time and decreased gradually beyond this reaction time. C15H32O3 isomers (II–VII) were formed by the furan ring opening of the full ring hydrogenation product (C15H26O3, I) of the C15 fuel precursor. The formation of the C15H32O3 (II–VII) isomers was reflected by the splitting of the corresponding GC peaks (Fig. 4). However, we could not confirm the formation of partial furan ring opening compounds. The furan ring opening isomers (C15H32O3, II–VII) subsequently underwent deoxygenation following two primary reaction routes: dehydroformylation and HDO. The three isomers of C15H32O3 (II–IV) have a terminal hydroxyl (OH) group. Following the former route, these C15H32O3 (II–IV) isomers followed dehydroformylation reaction, i.e., dehydrogenation, followed by decarbonylation, to form C14H30O2 (VIII–X). This route was endorsed by the formation of CO in the gas-phase (Fig. S3, ESI†). Palla et al. studied the HDO of aliphatic alcohols with the terminal OH group over the Ni/γ-Al2O3 catalyst and observed alkanes and alkenes with one carbon less than the alcohols as the product.42 These products were reported to be formed by dehydrogenation, followed by either decarbonylation or dehydroformylation reactions.42 The C14H30O2 (VIII–X) isomers then underwent HDO, i.e., dehydration, followed by hydrogenation reaction, to form C14H30. The C14H30O2 (VIII–X) isomers can also undergo the dehydrogenation reaction to form the corresponding ketones. Yang et al. reported the HDO of 5-nonanone using aluminosilicate supported Ni and Co catalysts, and perceived 5-nonanol as the intermediate product.43 The HDO reaction was proposed to follow via hydrogenation to 5-nonanol, followed by dehydration and hydrogenation reaction. HDO of 2-hexanone and 3-hexanone was reported to follow via initial hydrogenation to the corresponding alcohols.44 These alcohols then underwent HDO following dehydration and hydrogenation reaction. The dehydrogenation of C14H30O2 (VIII–X) isomers to the corresponding C14H26O2 (XI–XIII) compounds was thus considered a reversible reaction.
Following the latter route (HDO), the C15H32O3 (II–VII) isomers underwent deoxygenation through dehydration, followed by the hydrogenation reaction, forming C15H32 alkane. The C15H32O3 (V–VII) compounds can also undergo a reversible dehydrogenation reaction to the corresponding ketones (XIV–XVI). The dehydroformylation reaction occurs over metallic centers of the catalyst, while acidic sites are responsible for the HDO reaction. The Ni/γ-Al2O3 is a weakly acidic metal catalyst (Table 1 and Fig. 2B). The dehydroformylation reaction was thus relatively faster compared to the HDO reaction over the Ni/γ-Al2O3 catalyst with C14 alkane as the dominant hydrocarbon. The dehydroformylation reaction was also reported to be dominant for the C15 fuel precursor over the Pd/C-Hf(OTf)4 catalyst, with C14 alkane as the leading product.38 Li et al. studied the HDO of the C14 fuel precursor derived from 2-MF and butanal by the HAA reaction over zeolite (HZSM-5 and HMOR)-supported nickel as a bifunctional (metal and acid) catalyst.19 The HDO reaction was found to be the dominant route, with C14 alkanes as the major product.
We further studied the HDO of 2-octanol over the Ni/γ-Al2O3 catalyst to explore the HDO reaction mechanism of secondary alcohols. n-Octane was found to be the primary product with only a small amount of n-heptane and 2-octanone. We envisioned the formation of n-heptane by the cracking reaction of the terminal carbon–carbon bond attached to the OH group. Based on these results, we proposed the terminal carbon–carbon bond cleavage of C15H32O3 (II and VI) isomers, followed by HDO of the resulting compounds, to form C13H28 alkane. The wt% of C13 alkane was negligible at the early stages of the reaction and became prominent only after 60 min of reaction time. These results further support the formation of C13 alkane by a relatively slower cracking reaction. Similarly, the C9–C12 hydrocarbons were not observed up to 30 min of reaction time, and only a trivial quantity of these hydrocarbons appeared at 60 min of reaction time (Fig. 5A). The C9–C12 hydrocarbons were possibly formed by a cracking reaction at various positions of II–X compounds under severe reaction conditions. The above hypothesis was further supported by the formation of the cracked oxygenated intermediate compounds (Fig. S4, ESI†) and lighter hydrocarbons in the gas-phase (Fig. S2, ESI†) under extreme reaction conditions. Li et al. reported that the carbon–carbon bond cleavage between the furan moiety and tertiary carbocation was responsible for the formation of lower hydrocarbons by a series of hydrogenation and dehydration reactions.45,46 We observed a small quantity of greater than C15 hydrocarbons. These hydrocarbons were perhaps formed by self- or cross-condensation of ketones, followed by HDO of the condensed compounds. Gurbuz et al. also reported the condensation of 2-hexanone using palladium-supported ceria-zirconia mixed oxides.47 For the C15 fuel precursor, the decarbonylation of ketones could be a possible reaction route for the deoxygenation. Morioka et al. reported nickel/N-heterocyclic carbene-mediated decarbonylation of simple aromatic ketones.48 However, the success of ketone decarbonylation is limited due to the difficulty in activation of two inert carbon–carbon bonds. The decarbonylation of ketones was thus not included in our proposed HDO reaction mechanism.
3.4. Role of hydrogen partial pressure
HDO of the C15 fuel precursor was carried out at various hydrogen pressures for a fixed total pressure of 30 bar to elucidate the roles of hydrogen partial pressure on the conversion of oxygenates and wt% of hydrocarbons and oxygenated intermediate compounds. At a 20 bar hydrogen pressure, the oxygenate conversion was only about 50% at 180 min of reaction time and increased to about 95% at 180 min of reaction time for both 25 bar and 30 bar hydrogen pressures (Fig. 6A). The enhanced conversion of oxygenates at elevated hydrogen pressures was due to the greater availability of hydrogen for the conversion of oxygenates to hydrocarbons via hydrogenation and HDO reaction. Conversely, the wt% of C14 alkane was high at a 20 bar hydrogen pressure and reduced slightly at higher hydrogen pressures (Fig. 6B). However, the wt% of C15 alkane was low at a 20 bar hydrogen pressure and increased significantly at higher hydrogen pressures. While a high pressure is unsuitable for dehydroformylation reaction, the HDO reaction is favorable at a high hydrogen pressure. The wt% of C15 alkane was thus enhanced with increasing hydrogen partial pressure with a concurrent decline in the wt% of C14 alkane. According to Zhang's group, C14H30 alkanes have a high cetane number and low pour point and can be considered as diesel or fuel additives.17 At a 20 bar hydrogen pressure, the wt% of furan ring hydrogenated products, i.e., C15H24O3 and C15H26O3, was quite high and decreased at higher hydrogen pressures (Fig. 6C). However, the combined wt% of furan ring opening products, i.e., C15H32O3 and C15H32O, was lower at a 20 bar hydrogen pressure compared to 25 and 30 bar (Scheme 2). These results disclosed enhanced furan ring hydrogenation and ring-opening reactions at elevated hydrogen pressures. The cracking reaction is favorable at a low hydrogen pressure. The wt% of the C8, C10, and C11 oxygenated intermediate compounds was thus quite significant at a 20 bar hydrogen pressure and reduced at higher hydrogen pressures. Since the conversion of oxygenates was not increased much beyond a 25 bar hydrogen pressure, all subsequent studies were carried out at a 30 bar hydrogen pressure.
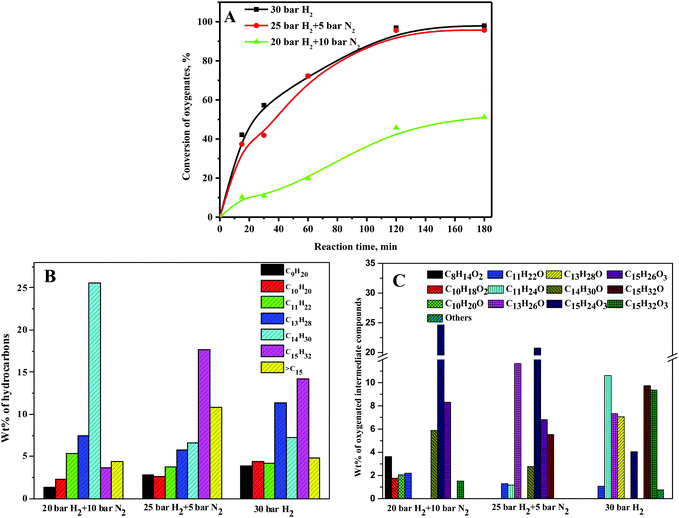 |
| | Fig. 6 Role of partial pressure of hydrogen on (A) conversion of oxygenated compounds, (B) wt% of hydrocarbons, and (C) wt% of oxygenated intermediate compounds. Reaction conditions: 0.5 g 20NiAl and 573 K. B and C: 50% conversion of oxygenated compounds. | |
3.5. Effect of nickel metal content on γ-Al2O3
The conversion of the C15 fuel precursor was almost absent in the absence of the catalyst, demonstrating insignificant thermal cracking under the reaction conditions. The reaction was also carried out in the presence of γ-Al2O3 to comprehend the role of the support in this reaction. The conversion of the C15 fuel precursor was nearly absent, implying an insignificant role of γ-Al2O3 alone for this reaction. The conversion of oxygenates at 5 h of reaction time was about 20% over 10NiAl (Fig. 7A). The conversion of oxygenates was improved with the rising nickel metal content on γ-Al2O3 up to 20NiAl and decreased slightly for 25NiAl. The conversion of oxygenates at 300 min of reaction time was about 86% over 20NiAl and dropped to about 76.4% for 25NiAl. The increase in catalytic activity with the rising nickel metal content on γ-Al2O3 until 20 wt% was owing to the rise in the amount of metallic sites. This was reflected by the metallic surface area results, i.e., [nickel surface area per g nickel] × [wt% of nickel] (Table 1). However, a slight decrease in catalytic activity for 25NiAl was due to the existence of a large quantity of catalytically less-active bulk nickel metal species, as observed from the TPR results (Fig. 2).35 The nickel crystallite size was also bigger for 25NiAl compared to 20NiAl (Table 1). Moreover, the acidity of 25NiAl was less compared to 20NiAl (Table 1).49
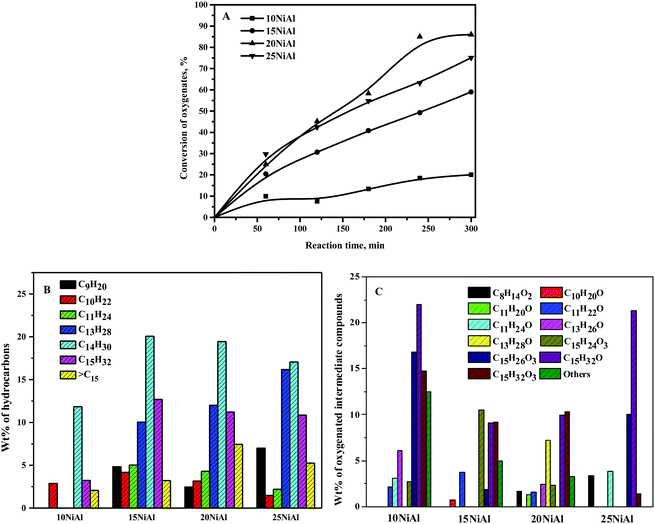 |
| | Fig. 7 Effect of nickel metal content on γ-Al2O3 on the (A) conversion of oxygenates and (B) wt% of hydrocarbons, and (C) wt% of oxygenated intermediate compounds. Reaction conditions: 0.5 g catalyst, 543 K, and 30 bar H2. B and C: 20% conversion of oxygenates for 10NiAl and 60% conversion of oxygenates for 15NiAl, 20NiAl, and 25NiAl. | |
The influence of nickel metal content on γ-Al2O3 on the wt% of hydrocarbons and oxygenated intermediate products is displayed in Fig. 7B and C, respectively. C14 alkane was detected as the main hydrocarbon product, followed by C15 and C13 alkane for all the catalysts. The wt% of C9–C11 alkanes was relatively lower compared to that of C13–C15 alkanes. The cumulative wt% of hydrocarbon products was improved with rising Ni metal content on γ-Al2O3 up to 20 wt% (about 86% at 300 min). The wt% values of individual alkane products were, however, not affected much for the increasing nickel metal content on γ-Al2O3. It is obvious as the nature of active sites is similar for all the catalysts. Conversely, a vast range of oxygenated intermediate compounds was observed as the products. A significant quantity of C15H26O3 (I of Scheme 2) was observed for 10NiAl. The wt% of C15H26O3 was, however, quite small for higher nickel metal content on γ-Al2O3. The catalytic activity of the Ni/γ-Al2O3 catalysts was boosted with the growing nickel metal content on γ-Al2O3. The low selectivity to C15H26O3 at 15 wt% and higher nickel content on γ-Al2O3 was due to its rapid conversion to C15H32O3. On the other hand, the wt% of C15H32O3 was quite significant for all the catalysts. Since 20NiAl showed the highest catalytic activity, all other studies were carried out in the presence of 20NiAl.
3.6. Effect of reaction temperature
The conversion of oxygenates was boosted by the elevation of the reaction temperature, as shown in Fig. 8A. At 4 h of reaction time, the conversion of oxygenates was around 17% at 523 K and reached around 85% at 543 K. However, almost complete conversion of oxygenates was achieved after 2 h of reaction time at 573 K. At 523 K, C14 and C15 alkanes were perceived as the primary hydrocarbons with only a small amount of lighter hydrocarbons (Fig. 8B). Besides, C13 alkane was absent at 523 K. However, a significant quantity of the C13 alkane was observed at 543 K and 573 K. At 573 K, C13 alkane was, however, the dominating hydrocarbon. Furthermore, the wt% values of lighter hydrocarbons were quite significant at 543 K and 573 K. These results exhibited the enhancement of cracking reaction at elevated reaction temperatures.45,50 Moreover, only a trivial quantity of >C15 alkanes was observed at 523 K. However, a significant quantity of >C15 alkanes was observed at 543 K and 573 K. The condensation of oxygenated intermediate compounds (ketones) is favorable at elevated reaction temperatures with high wt% of >C15 alkanes. At 523 K, the wt% of the furan ring hydrogenation product (C15H26O3) was relatively higher compared to the furan ring opening products (C15H32O3) (Fig. 8B). The furan ring hydrogenation product was, however, almost absent at 543 K and 573 K. On the contrary, a significant quantity of furan rings opening product was observed at 543 K and 573 K. This was due to the faster rate of ring hydrogenation and ring-opening reaction at elevated temperatures. At 573 K and 2 h of reaction time, wt% values of alkanes at 100% conversion of oxygenates were as follows: 19.6% C9H20, 3.9% C10H22, 14.3% C11H24, 0.8% C12H26, 33.8% C13H28, 16.1% C14H30, 9.0% C15H32, and 2.5% >C15. High wt% of C13 alkane was due to the dominating cracking reaction at the high reaction temperature. At 543 K and 6 h of reaction time, the wt% values of alkanes at 85.8% conversion of oxygenates were as follows: 9.4% C9H20, 7.2% C10H22, 11.0% C11H24, 10.8% C13H28, 20.5% C14H30, 21.4% C15H32, and 5.4% >C15.
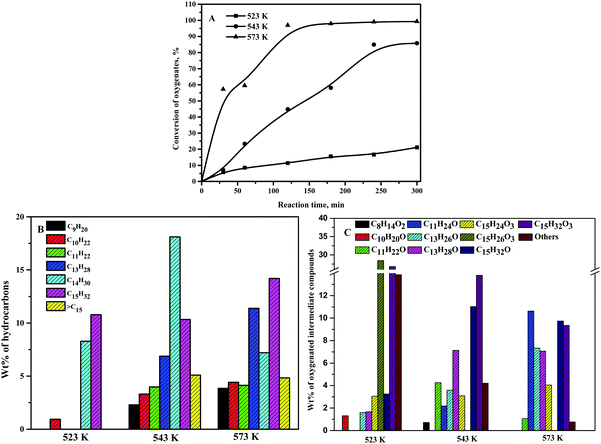 |
| | Fig. 8 Effect of reaction temperature on the (A) conversion of oxygenated compounds, (B) wt% of hydrocarbons, and (C) wt% of oxygenated intermediate compounds. Reaction conditions: 0.5 g 20NiAl and 30 bar H2. B and C: 20% conversion of oxygenates for 523 K and 50% conversion of oxygenates at other temperatures. | |
3.7. Reproducibility and reusability of Ni/γ-Al2O3
The reproducibility of experimental results for HDO of the C15 fuel precursor was confirmed by conducting two reactions under similar reaction conditions over 20NiAl catalyst. The conversion of oxygenates and wt% of hydrocarbons and oxygenated intermediate compounds were almost identical for both runs (Table 2). At 300 min of reaction time, almost complete conversions of oxygenates (99.3% and 97.6%) with negligible wt% of oxygenated intermediate compounds were perceived for both the runs. In order to investigate the reusability of 20NiAl, the spent catalyst was filtered from both of these runs, and the adsorbed liquid was then removed by repeated ethanol wash. The separated catalyst was dried overnight in an oven at 373 K, followed by calcination at 823 K and activation by H2 at 973 K. We performed a fresh HDO reaction using the regenerated catalyst under reaction conditions similar to that of the freshly prepared catalyst. The regenerated catalyst exhibited a somewhat lower conversion of oxygenates (82%) compared to that of the freshly prepared catalyst. The BET surface area of the regenerated catalyst (142 m2 g−1) was lower than that of the freshly prepared catalyst (169 m2 g−1) (Table 1). Furthermore, the metal dispersion of the regenerated catalyst was somewhat lesser with a slightly bigger nickel crystallite size compared to the freshly prepared catalyst. The HR-TEM image of the regenerated catalyst (54.8 nm) also exhibited a larger nickel particle size compared to that of the freshly prepared catalyst (27.8 nm) (Fig. S7, ESI†). Similarly, the regenerated catalyst exhibited slightly reduced acidity and lesser degree of reduction than those of the freshly prepared catalyst (Table 1 and Fig. S8, ESI†). The change in these catalyst characteristics was responsible for a slightly reduced catalytic activity of the regenerated catalyst than that of the freshly prepared catalyst.50 However, the wt% values of hydrocarbons for regenerated catalysts were comparable to those of the freshly prepared catalyst (Table 2).
Table 2 Reproducibility and reusability of the 20NiAl catalyst
|
|
Conversion of oxygenates at 300 min, % |
Wt% of hydrocarbons at 60% conversion of oxygenates |
| C9H20 |
C10H22 |
C11H24 |
C12H26 |
C13H28 |
C14H30 |
C15H32 |
| Reaction conditions: 0.5 g 20NiAl, 573 K, and 30 bar H2. |
| Run 1: fresh catalyst |
99.3 |
5.8 |
2.1 |
3.4 |
6.7 |
20.6 |
15.6 |
5.8 |
| Run 2: fresh catalyst |
97.6 |
5.1 |
2.8 |
4.2 |
6.8 |
19.4 |
13.4 |
8.3 |
| Run 3: regenerated catalyst |
81.8 |
7.6 |
1.7 |
3.5 |
6.7 |
21.4 |
14.8 |
4.3 |
4. Conclusions
The present study provides a systematic investigation of two-step conversion of furfural and 2-MF to jet fuel range (C9–C15 alkanes) hydrocarbon biofuel: (i) HAA reaction over the CER catalyst and (ii) HDO of the C15 fuel precursor over the Ni/γ-Al2O3 catalyst. The optimum HAA reaction conditions were 1 g CER, 1:1 2-MF/furfural, and 328 K. The empirical kinetic model fairly corroborated the experimental HAA reaction data. The HAA reaction exhibited an activation energy of 15.09 kcal mol−1. HDO of the C15 fuel precursor occurred through sequential furan ring hydrogenation and ring-opening reactions. The deoxygenation of furan ring-opening products then succeeded through a combination of dehydroformylation and HDO reactions. The dehydroformylation reaction was the dominating pathway with C14 alkane as the major hydrocarbon product. The HDO reaction was, however, enhanced at elevated hydrogen pressure with improved wt% of C15 alkane. The catalytic activity was enhanced with rising Ni metal content on γ-Al2O3 without much effect on the wt% values of the products. The cracking reaction was substantial at elevated reaction temperatures with high wt% values of lighter hydrocarbons. Nearly complete conversion of oxygenates with 38.6 wt% <C13, 33.8 wt% C13, 16.1 wt% C14, 9.0 wt% C15, and 2.5 wt% >C15 alkanes was observed over 20NiAl at 573 K and 2 h of reaction time.
Author contributions
Alekhya Kunamalla: Investigation and writing – original draft. Bhushan S. Shrirame: investigation. Sunil K. Maity: conceptualization, project administration, supervision, and writing – review and editing.
Nomenclatures
| BET | Brunauer–Emmett–Teller |
| BJH | Barret–Joyner–Halenda |
| CER | Cation exchange resin |
| FID | Flame ionization detector |
| GC | Gas chromatography |
| HAA | Hydroxyalkylation–alkylation |
| HDO | Hydrodeoxygenation |
| 2-MF | 2-Methylfuran |
| MS | Mass spectrometry |
| NH3-TPD | Temperature programmed desorption of ammonia |
| TEM | Transmission electron microscopy |
| TPR | Temperature programmed reduction |
| XRD | X-ray diffraction |
Conflicts of interest
There is no conflict of interest.
Notes and references
-
N. Jones, The outlook for energy: A view to 2040. https://www.connaissancedesenergies.org/sites/default/files/pdf-actualites/2016_outlook_for_energy_exxonmobil.pdf.
- M. N. A. M. Yusoff, N. W. M. Zulkifli, B. M. Masum and H. H. Masjuki, RSC Adv., 2015, 5, 100184–100211 RSC.
- S. K. Maity, Renewable Sustainable Energy Rev., 2015, 43, 1446–1466 CrossRef CAS.
- P. Kumar, P. Kumar, P. V. C. Rao, N. V. Choudary and G. Sriganesh, Fuel, 2017, 199, 339–345 CrossRef CAS.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- F. Li, Y. Yuan, Z. Huang, B. Chen and F. Wang, Appl. Catal., B, 2015, 165, 547–554 CrossRef CAS.
- M. J. Tijmensen, A. P. Faaij, C. N. Hamelinck and M. R. van Hardeveld, Biomass Bioenergy, 2002, 23, 129–152 CrossRef CAS.
- S. K. Maity, Renewable Sustainable Energy Rev., 2015, 43, 1427–1445 CrossRef CAS.
- K. Yan, G. Wu, T. Lafleur and C. Jarvis, Renewable Sustainable Energy Rev., 2014, 38, 663–676 CrossRef CAS.
- A. S. May, S. M. Watt and E. J. Biddinger, React. Chem. Eng., 2021, 6, 2075–2086 RSC.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed.
- R. M. West, Z. Y. Liu, M. Peter and J. A. Dumesic, ChemSusChem, 2008, 1, 417–424 CrossRef CAS PubMed.
- A. Corma, O. de la Torre and M. Renz, ChemSusChem, 2011, 4, 1574–1577 CrossRef CAS PubMed.
- G. Li, N. Li, Z. Wang, C. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, ChemSusChem, 2012, 5, 1958–1966 CrossRef CAS PubMed.
- T. Wang, K. Li, Q. Liu, Q. Zhang, S. Qiu, J. Long, L. Chen, L. Ma and Q. Zhang, Appl. Energy, 2014, 136, 775–780 CrossRef CAS.
- A. Corma, O. de la Torre, M. Renz and N. Villandier, Angew. Chemie, 2011, 123, 2423–2426 CrossRef.
- G. Li, N. Li, J. Yang, A. Wang, X. Wang, Y. Cong and T. Zhang, Bioresour. Technol., 2013, 134, 66–72 CrossRef CAS PubMed.
- W. Wang, N. Li, S. Li, G. Li, F. Chen, X. Sheng, A. Wang, X. Wang, Y. Cong and T. Zhang, Green Chem., 2016, 18, 1218–1223 RSC.
- S. Li, N. Li, G. Li, L. Li, A. Wang, Y. Cong, X. Wang, G. Xu and T. Zhang, Appl. Catal., B, 2015, 170, 124–134 CrossRef.
- C. Zhu, T. Shen, D. Liu, J. Wu, Y. Chen, L. Wang, K. Guo, H. Ying and P. Ouyang, Green Chem., 2016, 18, 2165–2174 RSC.
- G. Li, N. Li, S. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, Chem. Commun., 2013, 49, 5727–5729 RSC.
- S. Li, N. Li, W. Wang, L. Li, A. Wang, X. Wang and T. Zhang, Sci. Rep., 2016, 6, 4–10 CrossRef PubMed.
- I. Yati, M. Yeom, J. W. Choi, H. Choo, D. J. Suh and J. M. Ha, Appl. Catal., A, 2015, 495, 200–205 CrossRef CAS.
- Q. Deng, P. Han, J. Xu, J.-J. Zou, L. Wang and X. Zhang, Chem. Eng. Sci., 2015, 138, 239–243 CrossRef CAS.
- S. Li, N. Li, G. Li, L. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, Green Chem., 2015, 17, 3644–3652 RSC.
- X. Zhang, Q. Deng, P. Han, J. Xu, L. Pan, L. Wang and J.-J. Zou, AIChE J., 2017, 63, 680–688 CrossRef CAS.
- D. Damodar, A. Kunamalla, M. Varkolu, S. K. Maity and A. S. Deshpande, ACS Sustainable Chem. Eng., 2019, 7, 12707–12717 CrossRef CAS.
- S. Liu, T. R. Josephson, A. Athaley, Q. P. Chen, A. Norton, M. Ierapetritou, J. I. Siepmann, B. Saha and D. G. Vlachos, Sci. Adv., 2019, 5, 1–9 Search PubMed.
- C. Wen, E. Barrow, J. Hattrick-Simpers and J. Lauterbach, Phys. Chem. Chem. Phys., 2014, 16, 3047–3054 RSC.
- K. Li, F. Zhou, X. Liu, H. Ma, J. Deng, G. Xu and Y. Zhang, Catal. Sci. Technol., 2020, 10, 1151–1160 RSC.
- A. Samikannu, L. J. Konwar, K. Rajendran, C. C. Lee, A. Shchukarev, P. Virtanen and J. P. Mikkola, Appl. Catal., B, 2020, 272, 118987 CrossRef CAS.
- D.-H. Liu, H.-L. He, J.-J. Wang, S.-Y. Zhou, T. Zeng, X.-Y. Gao, Y. Xiao, X. Yi, A. Zheng, Y.-B. Zhang and Z. Li, Green Chem., 2021, 23, 9974–9981 RSC.
- H. Li, Z. Gui, S. Yang, Z. Qi, S. Saravanamurugan and A. Riisager, Energy Technol., 2018, 6, 1060–1066 CrossRef CAS.
- S. R. Yenumala, S. K. Maity and D. Shee, Catal. Sci. Technol., 2016, 6, 3156–3165 RSC.
- P. Kumar, S. R. Yenumala, S. K. Maity and D. Shee, Appl. Catal., A, 2014, 471, 28–38 CrossRef CAS.
- R. Kumar, K. Kumar, N. V. Choudary and K. K. Pant, Fuel Process. Technol., 2019, 186, 40–52 CrossRef CAS.
- S. Liu, S. Dutta, W. Zheng, N. S. Gould, Z. Cheng, B. Xu, B. Saha and D. G. Vlachos, ChemSusChem, 2017, 10, 3225–3234 CrossRef CAS PubMed.
- S. Dutta and B. Saha, ACS Catal., 2017, 7, 5491–5499 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 1983, 6, 159–164 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- S. Liu, W. Zheng, J. Fu, K. Alexopoulos, B. Saha and D. G. Vlachos, ACS Catal., 2019, 9, 7679–7689 CrossRef CAS.
- V. C. S. Palla, D. Shee and S. K. Maity, RSC Adv., 2014, 4, 41612–41621 RSC.
- X. Yang, R. W. Jenkins, J. H. Leal, C. M. Moore, E. J. Judge, T. A. Semelsberger and A. D. Sutton, ACS Sustainable Chem. Eng., 2019, 7, 14521–14530 CrossRef CAS.
- B. Hočevar, M. Grilc, M. Huš and B. Likozar, Appl. Catal., B, 2017, 218, 147–162 CrossRef.
- G. Li, N. Li, J. Yang, L. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, Green Chem., 2014, 16, 594–599 RSC.
- S. Li, N. Li, G. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, Catal. Today, 2014, 234, 91–99 CrossRef CAS.
- E. I. Gürbüz, E. L. Kunkes and J. A. Dumesic, Appl. Catal., B, 2010, 94, 134–141 CrossRef.
- T. Morioka, A. Nishizawa, T. Furukawa, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2017, 139, 1416–1419 CrossRef CAS PubMed.
- J. Yang, N. Li, S. Li, W. Wang, L. Li, A. Wang, X. Wang, Y. Conga and T. Zhang, Green Chem., 2014, 16, 4879–4884 RSC.
- Q. Xia, Y. Xia, J. Xi, X. Liu, Y. Zhang, Y. Guo and Y. Wang, ChemSusChem, 2016, 10, 747–753 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: GC-FID chromatograms for HAA reaction and gas-phase sample for HDO, GC-TCD chromatogram for HDO, kinetic and Arrhenius plots for HAA reaction, GC-FID chromatogram for HDO, HR-TEM images, and TPR and NH3-TPD of the regenerated catalyst. See DOI: 10.1039/d1ya00078k |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Bhushan S.
Shrirame
and
Sunil K.
Maity
,
Bhushan S.
Shrirame
and
Sunil K.
Maity



 vs. time (t) data at different temperatures fitted well by linear lines passing through the origin (Fig. S4, ESI†). This result confirms that the reaction is second order. The rate constant of this reaction was calculated at different temperatures from the slope of
vs. time (t) data at different temperatures fitted well by linear lines passing through the origin (Fig. S4, ESI†). This result confirms that the reaction is second order. The rate constant of this reaction was calculated at different temperatures from the slope of  vs. time (t) (Fig. 1B). The 2-MF conversion was then estimated based on eqn (2) using the calculated rate constants at various reaction temperatures and matched with 2-MF conversions obtained from experiments. 2-MF conversions calculated using the above kinetic model fitted very well with the experimental 2-MF conversions, as observed from the figure. The activation energy (15.09 kcal mol−1) and pre-exponential factor (1.8 × 105 (kmol m−3)−1 s−1) were further obtained from the Arrhenius plot of 1/temperature vs. ln(k) (Fig. S5, ESI†). The high activation energy further elucidates the absence of external mass transfer resistance. The study was then extended to various 2-MF/furfural mole ratios to validate the established kinetic model. The 2-MF conversion was decreased with the increase in the 2-MF/furfural molar ratio from 1
vs. time (t) (Fig. 1B). The 2-MF conversion was then estimated based on eqn (2) using the calculated rate constants at various reaction temperatures and matched with 2-MF conversions obtained from experiments. 2-MF conversions calculated using the above kinetic model fitted very well with the experimental 2-MF conversions, as observed from the figure. The activation energy (15.09 kcal mol−1) and pre-exponential factor (1.8 × 105 (kmol m−3)−1 s−1) were further obtained from the Arrhenius plot of 1/temperature vs. ln(k) (Fig. S5, ESI†). The high activation energy further elucidates the absence of external mass transfer resistance. The study was then extended to various 2-MF/furfural mole ratios to validate the established kinetic model. The 2-MF conversion was decreased with the increase in the 2-MF/furfural molar ratio from 1







