
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced lifetime of the zinc–iodine batteries using hydrocarbon cation-exchange polymer-protected zinc anodes†
Qiliang
Wei
 ab,
Eric
Schibli
c,
Binyu
Chen
a and
Steven
Holdcroft
*a
ab,
Eric
Schibli
c,
Binyu
Chen
a and
Steven
Holdcroft
*a
aDepartment of Chemistry, Simon Fraser University, Burnaby, BC V5A 1S6, Canada. E-mail: holdcrof@sfu.ca
bInstitute of Micro/Nano Materials and Devices, Ningbo University of Technology, Ningbo, 315211, Zhejiang, China
cDepartment of Physics, Simon Fraser University, Burnaby, BC V5A 1S6, Canada
First published on 28th July 2022
Abstract
The zinc–iodine battery is a cost-effective, safe and scalable energy-storage device, which is nevertheless hindered by dendrite formation on the Zn anode and crossover of triiodide anions to the anode. We report that deposition of a fluorine-free cation-exchange polymer, sulfophenylated poly(phenylene)biphenyl (sPPB), onto the Zn anode serves as a protection layer that significantly suppresses Zn dendrite growth and restricts unwanted reaction of triiodide ions at the anode. sPPB-protected Zn anodes possess a relatively low charging polarization overpotential, which is attributed to a lower energy barrier for the desolvation of Zn ions, as supported by DFT calculations. sPPB-protected Zn anodes exhibit a >10× lifetime compared to bare Zn anodes in the Zn//Zn symmetric cells. Aqueous sPPB-protected zinc–iodine batteries deliver an initial capacity of 174 mA h g−1 at 5C and retain 131 mA h g−1 after 4000 cycles.
Introduction
Lithium-ion batteries (LIBs) are a dominant energy carrier of choice for many applications, ranging from portable electronics and smart grid energy storage to electric vehicles.1–3 However, the employment of flammable organic electrolytes in LIBs imposes safety issues.4 Moreover, the cost of LIBs is relatively high due to the cost of lithium resources and transition metals. The development of low-cost, safe, aqueous electrolyte-based metal-ion batteries is experiencing a renewed interest because of these challenges.5–8 In this regard, aqueous Zn anode-based devices, with their inherent safety, high theoretical capacity (820 mA h gZn−1) and useful redox potential of Zn (−0.76 V vs. SHE) hold promise for development of clean energy storage devices.9–12 On the complementary cathode side, significant research has been performed on manganese oxides,13–15 vanadium oxides,16,17 Prussian blue,18–20 quinone derivatives,21,22 and polyanion compounds,23,24 reporting great progress in each type. Zn-Anode based flow batteries are another class of Zn energy storage device,25–28 where redox electrochemistry is achieved under circulating solutions. Among these, the Zn–I2 flow battery is attracting attention due to the fast reversible kinetics of the I−/I3− redox couple, its high theoretical capacity (211 mA h g−1) and potential (0.54 V).25,29–33 Moreover, iodine is an abundant element, with sea water holding ∼55 μg per liter.34On the downside, the iodide or polyiodide (triiodide) species formed during charge/discharge processes dissolve into electrolytes and diffuse to the anode side, resulting in unwanted side reactions, self-discharge and capacity decay of the battery, not dissimilar to the deleterious ‘shuttling’ process of polysulfide ion species in Li–S batteries.35,36 To address these issues, porous carbon materials have been used at the cathode to confine iodide ions and localize the reversible reaction of I− to I2.37–39 Nonetheless, due to the weak interaction between iodine and the host materials, this strategy alone cannot satisfactorily suppress the shuttling and reaction of iodide species to/at the anode, which is exacerbated by Zn dendrite growth at the anode. In an attempt to resolve both these issues, dendrite formation and reaction of iodide species at the anode, Yang et al.34 recently investigated a metal–organic framework (MOF)-based multifunctional membrane possessing ultrasmall pore size to restrict access of iodide ion species at the Zn anode. Despite an observable improvement in the lifetime of the Zn–I2 cells and proof of principle, the MOFs used are prohibitively expensive to be employed at scale.
An alternate triiodide ion blocking material is a negatively-charged cation exchange polymer. Perfluorosulfonic acid (PFSA)-based Nafion, is a well-studied cation exchange polymer of this kind. Cui and co-workers40 have employed Nafion to protect the Zn anode, however, due to its internal 4 nm ion channels, it does not guarantee good electrochemical performance, and there are growing environmental concerns associated with the use of fluorinated compounds. A chemically-stable, fluorine-free cation exchange membrane is sought to improve the performance of aqueous Zn batteries by way of an anode protective layer to suppresses dendrite growth and suppress deleterious reactions of anions at the anode.
Our research group has been actively engaged in the design and synthesis of hydrocarbon membranes with high chemical stability for nearly three decades, and has recently focused on ion-containing polyphenylenes using pre-sulfonated monomers to yield structurally-defined cation exchange polymers which possess high degrees of sulfonation without dissolution.41 The strategy enables incorporation of a wide range property-modifying groups into the main chain. These allow for modifying the length and bulkiness of the phenylene spacer that separates the acid-baring units along the chain,42,43 branching,44 and for controlling the rigidity of the main chain.44 Herein, we investigate a fluorine-free hydrocarbon ionomer, sulfophenylated poly(phenylene)biphenyl (sPPB),42 as a protective layer on the Zn anode, and study its effect in both Zn//Zn symmetric cells and Zn//I2 cell devices. The sPPB layer is proposed to simultaneously suppress reaction of I3− species at the anode (Fig. 1) and enhance the uniform plating/stripping of the Zn anode so as to limit dendrite formation.
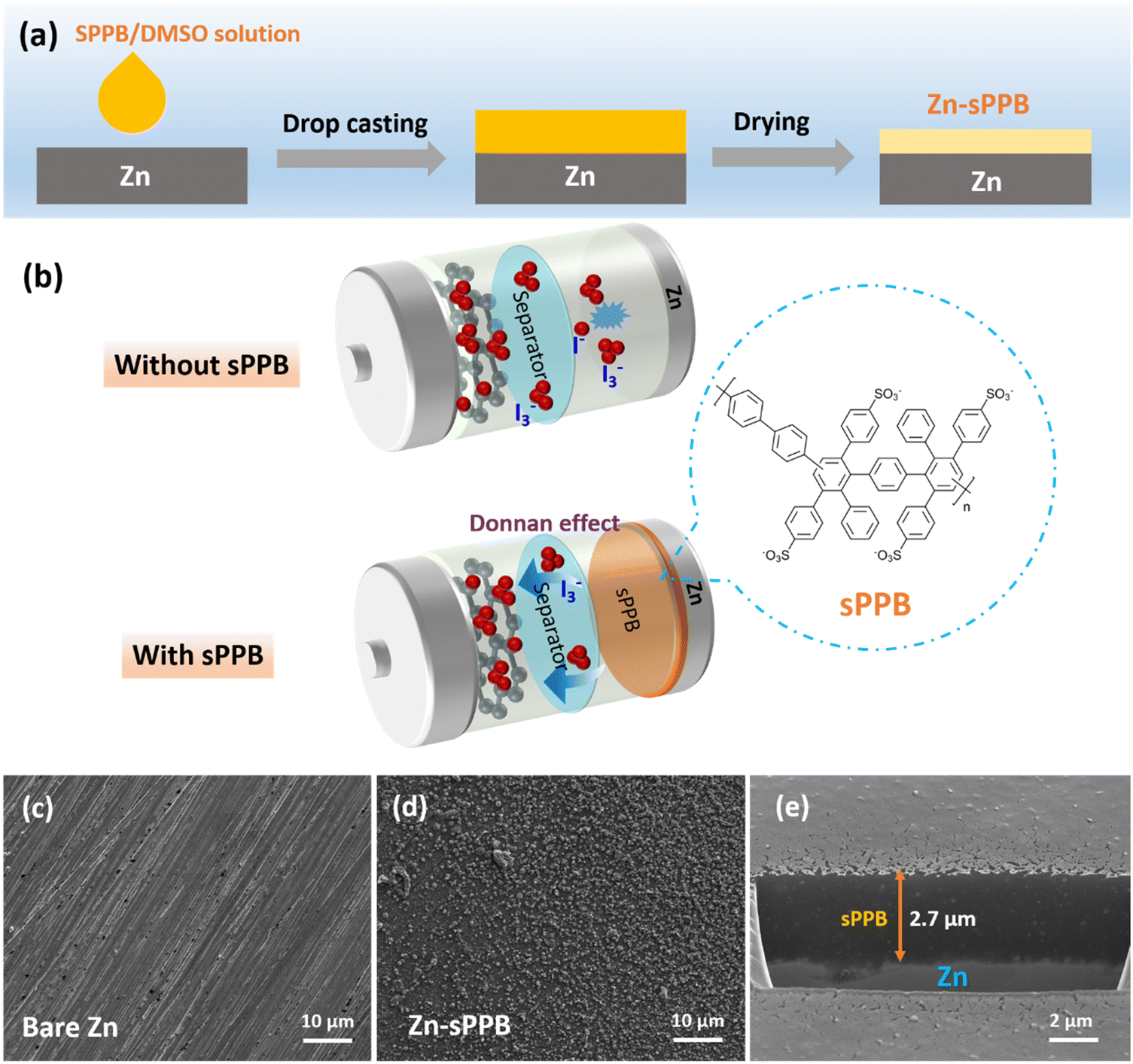 | ||
| Fig. 1 (a) Scheme of the sPPB coating process on Zn metal; (b) mechanism of Zn–I2 batteries with bare Zn and sPPB protected Zn as anodes. SEM images of (c) the bare Zn, (d) top-view of sPPB protected Zn, and (e) cross-section-view of sPPB coated Zn. | ||
Results and discussion
In this work, sulfophenylated poly(phenylene)biphenyl (sPPB) is deposited on the Zn surface to form Zn-sPPB as illustrated in Fig. 1a and described in the Experimental section (ESI†). The protection mechanism of sPPB to repel the triiodide (I3−) crossover for Zn//I2 battery is based on the Donnan effect, as illustrated in Fig. 1b. The morphology of bare Zn and Zn-sPPB was characterized by SEM. After coating, the rough and uneven surface of bare Zn (Fig. 1c) was rendered relatively uniform by the sPPB film (Fig. 1d). Upon closer inspection, (see Fig. S1, ESI†) sPPB exists in globular form with particle sizes in the nm range. With the assistance of a focused ion beam (FIB), the cross-section view of the Zn-sPPB can be observed (Fig. 1e), and a thickness of the sPPB is ∼2.7 μm determined. The low content of Zn (∼1 at%) by EDX indicates the successful coating of sPPB (Table S1, ESI†). XRD diffractograms of both bare Zn and Zn-sPPB (Fig. S3, ESI†) reveals only characteristic peaks of Zn. Attenuated total reflection (ATR) spectra of sPPB and Zn-sPPB (Fig. S4, ESI†) reveal multiple absorption peaks for sPPB between ∼1000–1030 cm−1, assigned to R–SO3−.Electrochemical cells with bare Zn and Zn-sPPB were assembled in symmetric coin cells with 1 M ZnSO4 as supporting electrolyte, and subjected to repeated plating/stripping at 1 mA cm−2. The corresponding performance curves are presented in Fig. 2a. Cells constructed with Zn-Nafion were also examined for comparison. Failure of the cell due to short-circuiting of the electrodes occurred after 40 h of testing for the cells containing bare Zn electrodes, and after 130 h testing for the Zn-Nafion cells; whereas Zn-sPPB cells were stable for >400 h without any sign of short circuit failure. Even under a higher current density, a similar stability can be achieved (Fig. S5, ESI†). Notably, the polarization voltages values of these symmetric cells are subtly different. From Fig. 2b and c, the polarization voltage of the Zn-sPPB symmetric cell is determined to be 37 mV, which is lower than that of the bare Zn cell (60 mV) and Zn-Nafion cell (47 mV). The smaller polarization indicates a lower energy barrier for electrochemical reduction and oxidation processes associated with Zn plating and Zn dissolution. SEM images of the electrodes were taken on deconstructed cells after 40 h for bare Zn cells and after 80 h for Zn-sPPB. As shown in Fig. 2d and f, bare Zn had formed randomly dendritic or rod-like structures, while Zn-sPPB electrodes possessed a dimpled but uniform structure (Fig. 2e and g). The surface topography was analyzed by the profilometry, (Fig. 2h and i). The surface of the bare Zn exhibited a 95 μm differential from valley to peak; Zn-sPPB electrodes exhibited only a 27 μm differential (after twice the length of the cycling time). Detailed profiles can be found in Fig. S6 and S7 (ESI†). These results demonstrate that dendrite growth causes short circuiting of the bare Zn-based cell, and that the sPPB layer reduces dendrite growth. Insight into the protective function of the sPPB layer on Zn deposition behavior can also be proved by a different substrate Ti. Here, the Zn nucleation process was studied in a three-electrode system with bare Ti (or Ti-sPPB coated Ti) serving as the working electrode, Zn as the counter electrode, and SCE as the reference electrode. 1 M ZnSO4 was used as the electrolyte. As shown in the cyclic voltammetry (CV) in Fig. S8 (ESI†), the crossing point P is the crossover potential where the current is zero, and Q (Q′) is the point where Zn starts to deposit. The distance between P and Q (or Q′) (termed as nucleation overpotential45) can be used to gain insights into the polarization behavior of the electrode.46,47 The nucleation overpotential of the sPPB coated Ti is 24 mV lower than that of the bare Ti, indicating the depolarizing effect of sPPB. Chronoamperometry (CA) was conducted to study the Zn deposition growth mechanism (Fig. S9, ESI†). The rapidly-increasing current density for the bare Ti suggests a faster dendrite growth process after 100 s. While for the sPPB coated Ti electrode, after ∼55 s, a stable current density of ∼18 mA cm−2 was observed, indicating a uniform rate of Zn deposition. The influence of the sPPB is to coordinate the deposition of Zn2+ uniformly under a reduced rate leading to an increased number of nucleation sites and formation of a uniform Zn layer.
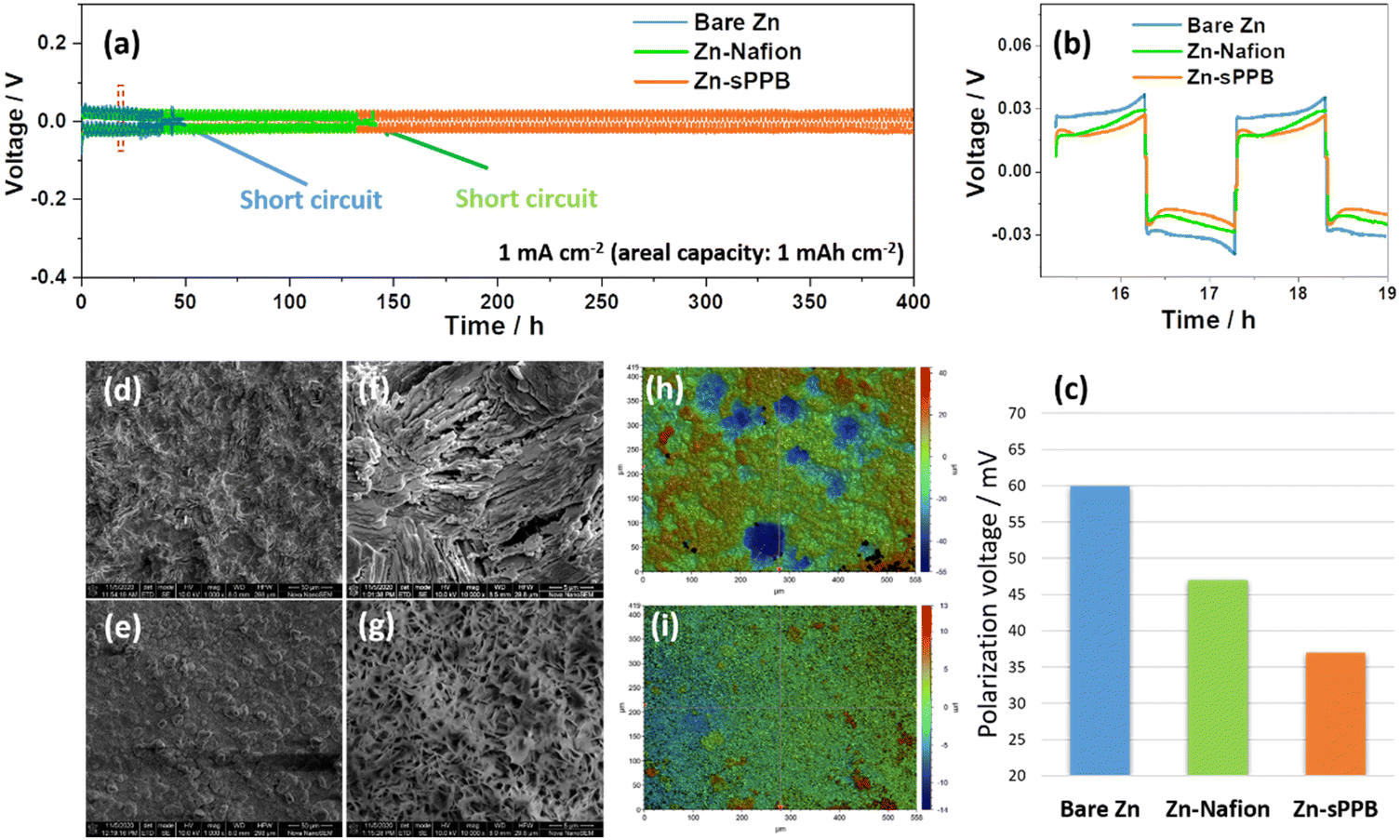 | ||
| Fig. 2 (a) The cycling performance of bare Zn, Zn-Nafion, Zn-sPPB at 1 mA cm−2 with areal capacity of 1 mA h cm−2; (b and c) the comparison of Zn plating/stripping profiles and the polarization voltage values for bare Zn, Zn-Nafion, and Zn-sPPB electrodes between 15–19 h. (d–i) SEM, and height mapping images of (d, f and h) bare Zn after 40 h cycling and (e, g and i) Zn-sPPB after 80 h cycling in the symmetrical cells. | ||
In order to investigate rejection of anionic triiodide species by films of sPPB, a H-type cell was assembled as shown in Fig. S10 (ESI†). The left chamber was filled with the 1 M KI + 0.1 M I2 (dark brown); and the right, with K2SO4 electrolyte (colorless). In one cell, the chambers were separated by a separator comprised of glass fiber (GF), and in the other cell the separator was replaced by a composite of glass fiber and sPPB. For the cells separated by glass fiber, the color of the solution in the right chamber turns light brown after several minutes, becoming dark brown after 1 h. In contrast, the cell with the glass fiber/sPPB composite membranes exhibits much reduced crossover of triiodide ions even after 4 h. Quantitate analysis of the crossover of triiodide species from the H-cell was calculated using the starch titration method (described in the ESI†), and the result is shown in Fig. 3a. Employing a glass fiber separator, the triiodide crossover rate is 37.8 mol L−1 min−1; with the glass fiber/sPPB composite separator, the rate of triiodide crossover is decreased to 1.8 mol L−1 min−1.
 | ||
| Fig. 3 (a) Crossover of triiodide anions crossover through a glass fibre (GF) and glass fibre/sPPB composite) (GF + sPPB) separator with time. (b) Coulombic efficiency of the Zn//I2 battery with 2 h holding after each full charge state. (c) Coulombic efficiency of the Zn//I2 battery with 8 h holding after each full charge state. (d) Galvanostatic discharge–charge profiles of the bare Zn//I2 battery and Zn-sPPB//I2 battery obtained in the 2nd cycle at 1C. (e) Schematic illustration of the coordination environment in water and DFT-optimized geometry with sPPB. (f) Desolvation energy of Zn2+ in water and sPPB. | ||
Zn–I2 coin-cells were assembled using Zn as anode, I2/KI within active carbon as cathode, and ZnSO4 as electrolyte. Charge–discharge cycling was first conducted at 1C (211 mA g−1), holding for 2 h or 8 h after each full charge state. The test protocol is illustrated in Fig. S11 (Protocol A) and S12 (Protocol B) (ESI†). The ratio of discharge capacity to charge capacity is presented in Fig. 3b and c. For Protocol A, with a shorter holding time, Zn-sPPB//I2 cell has a little higher ratio (∼99%) in the whole process than that of bare Zn//I2 cell (95–97%); in Protocol B with a longer holding time (8 h) after each full charge, the discharge capacity/charge capacity between Zn-sPPB//I2 cell and bare Zn//I2 cell is increased (∼96% vs. <80%), indicating suppression of mobile of I3− species by the sPPB layer on the Zn anode. This is consistent with the visualized I3− crossover experiment in the H-type cell shown in Fig. S10 (ESI†) and Fig. 3a, in which the penetration of I3− is significantly hindered by sPPB. The galvanostatic discharge–charge profiles of the bare Zn//I2 cell and Zn-sPPB//I2 cell are displayed in Fig. 3d. By comparison, it can be observed that the discharge profiles of these two cells are almost in the same position (meaning they exhibit a similar discharge plateau, the grey circle), and the lower charging–discharging polarization gap of the Zn-sPPB//I2 cell originates from the charging process (green circle). Upon charging, the reaction on the anode corresponds to the reduction process Zn2+ to Zn, during which desolvation of Zn2+ is important.40 Thus, the energy to desolvate Zn2+ was calculated by density functional theory (DFT) as described in the ESI.† In ZnSO4 electrolyte, six water molecules surround the Zn2+ ion, leading to a high desolvation energy (19.1 eV, Fig. 3f). In the presence of sPPB layer, two –SO3− groups and four water molecules coordinate the Zn2+, leading to a decreased desolvation energy of 2.5 eV. Nafion also possesses a –SO3− head group, so we further compared the desolvation energies of Nafion and sPPB, the result of which is shown in Fig. S13 (ESI†), wherein a lower desolvation energy of Zn2+ in the presence of sPPB is observed.
Fig. 4a shows cyclic voltammograms of typical Zn-sPPB//I2 coin-cell recorded at different scan rates (0.2, 0.3, 0.5, 1.0, 2.0, 3.0 mV s−1). Anodic and cathodic peaks are observed at ∼1.2 V and ∼1.3 V, respectively. The peak current increases with increasing scan rate, following a power law relationship: i = avb.48 The b value typically falls between 0.5 and 1.0; with values of 0.5 representing a diffusion-controlled process as is observed for species dissolved in a medium; as opposed to values of 1.0, which are observed for surface bound-species or a capacitive dominant charge storage process. After a formula conversion, it can be expressed as:
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i = loga + blogv i = loga + blogv | (1) |
i versus logv, illustrated in Fig. 4b and found to be 0.837 and 0.820 (both are close to 1) for the anodic and cathodic redox processes, respectively. b value close to 1 indicate a high occurrence of the redox reactions on the surface or near-surface region of electrodes, in which case the faradaic process of the electrode in electrolyte is faster than a bulk phase transformation or ion-diffusion through the solid upon charging/discharging.49,50 This implies that the charge stored in the redox process of the Zn–I2 battery is simultaneously controlled by capacitive and diffusion contributions, but governed by the surface-induced capacitive behavior. The current i is therefore regarded as the combination of a capacitive contribution (k1) and a diffusion contribution (k2):| i = k1v + k2v1/2 | (2) |
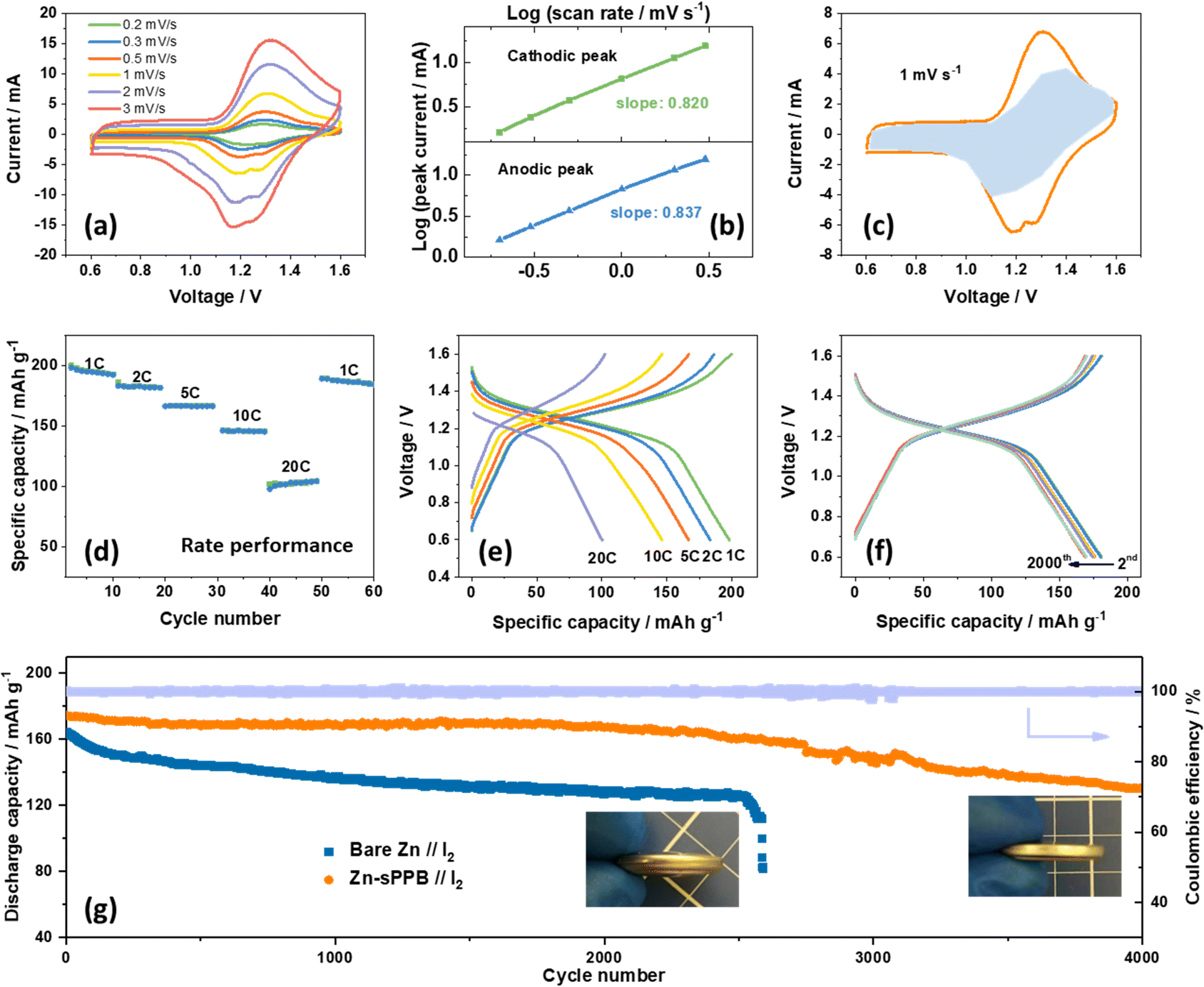 | ||
| Fig. 4 (a) CV curves of Zn-sPPB//I2 batteries at different scan rates. (b) logi and logv plots at anodic/cathodic peak currents. (c) CV curve with the pseudocapacitive (blue-shaded) contribution at 1 mV s−1. (d) Rate performance of the Zn-sPPB//I2 battery. (e) Galvanostatic discharge–charge profiles of the Zn-sPPB//I2 battery at different current densities. (f) Galvanostatic discharge–charge profiles of the Zn-sPPB//I2 obtained in the first 2000 cycles at 5C. (g) Comparison of long-term cycling performance at 5C (1055 mA g−1) with bare Zn and Zn-sPPB anodes (coulombic efficiency corresponds to the Zn-sPPB//I2 cell). | ||
To further understand the function of sPPB during charge/discharge cycles of the Zn-sPPB//I2 battery, XPS and ATR measurements were performed on the Zn-sPPB electrode during different charge/discharge states as marked in Fig. S15 (ESI†). XPS high-resolution spectra reveals the binding energy of the S 2s (Fig. S15b, ESI†) (S from the pendant SO3− group of sPPB) shifts to a higher energy (from 232.3 eV to 233.4 eV) during the charging (Zn2+/Zn reduction process) commensurate with increasing applied voltage, while the Zn 2p peaks simultaneously shifts to a lower binding energy (Fig. S15c, ESI†). Notably, the appearance of two split peaks in the Zn 2p spectra during charging is reasonably assigned to the coordination structure of the –SO3−–Zn2+–SO3−, which we interpret to indicate that the sulfonate groups on sPPB serve as a solvent sheath for Zn2+. In the following discharge process, the signals revert to their original binding energy, demonstrating a highly reversible process. The negligible shift of wavenumber in the range of ∼1000–1030 cm−1 (Fig. S15d, ESI†) indicates the stability of sPPB during charging/discharging.
Conclusion
In summary, electrochemical measurements, spectroscopic and imaging analyses, and DFT simulations support the hypothesis that sPPB facilitates the desolvation of Zn2+, playing a vital role in improving the Zn plating process by suppressing the dendrite growth, and serves to repel triiodide species against reaching the Zn anode – all of which enhance the cycle life of the Zn//I2 battery while maintaining the high specific capacity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) (Grant RGPIN-2018-03698) and made use of facilities in 4D LABS, SFU. The authors thank Dr Simon Cassegrain for the ATR measurements and fruitful discussions; and Dr Michael Adamski for synthesizing sPPB polymer.References
- A. Manthiram, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- B. Wu, X. Yang, X. Jiang, Y. Zhang, H. Shu, P. Gao, L. Liu and X. Wang, Adv. Funct. Mater., 2018, 28, 1803392 CrossRef.
- Y. Fu, Q. Wei, G. Zhang and S. Sun, Adv. Energy Mater., 2018, 8, 1703058 CrossRef.
- P. Lyu, X. Liu, J. Qu, J. Zhao, Y. Huo, Z. Qu and Z. Rao, Energy Storage Mater., 2020, 31, 195–220 CrossRef.
- C. Liu, X. Wang, W. Deng, C. Li, J. Chen, M. Xue, R. Li and F. Pan, Angew. Chem., Int. Ed., 2018, 130, 7164–7168 CrossRef.
- C. Yan, C. Lv, L. Wang, W. Cui, L. Zhang, K. N. Dinh, H. Tan, C. Wu, T. Wu and Y. Ren, J. Am. Chem. Soc., 2020, 142, 15295–15304 CrossRef CAS PubMed.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- Y. Li, Z. Wang, Y. Cai, M. E. Pam, Y. Yang, D. Zhang, Y. Wang and S. Huang, Energy Environ. Mater., 2022, 0, 1–29 Search PubMed.
- Z. Wang, J. Huang, Z. Guo, X. Dong, Y. Liu, Y. Wang and Y. Xia, Joule, 2019, 3, 1289–1300 CrossRef CAS.
- J. Hao, X. Li, S. Zhang, F. Yang, X. Zeng, S. Zhang, G. Bo, C. Wang and Z. Guo, Adv. Funct. Mater., 2020, 2001263 CrossRef CAS.
- Y. Yin, S. Wang, Q. Zhang, Y. Song, N. Chang, Y. Pan, H. Zhang and X. Li, Adv. Mater., 2020, 32, 1906803 CrossRef CAS PubMed.
- J. Ming, J. Guo, C. Xia, W. Wang and H. N. Alshareef, Mater. Sci. Eng., R, 2019, 135, 58–84 CrossRef.
- T. Xiong, Y. Zhang, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2020, 10, 2001769 CrossRef CAS.
- G. Liu, H. Huang, R. Bi, X. Xiao, T. Ma and L. Zhang, J. Mater. Chem. A, 2019, 7, 20806–20812 RSC.
- Y. Fu, Q. Wei, G. Zhang, X. Wang, J. Zhang, Y. Hu, D. Wang, L. Zuin, T. Zhou and Y. Wu, Adv. Energy Mater., 2018, 8, 1801445 CrossRef.
- F. Wan and Z. Niu, Angew. Chem., Int. Ed., 2019, 58, 16358–16367 CrossRef CAS PubMed.
- L. Wang, K.-W. Huang, J. Chen and J. Zheng, Sci. Adv., 2019, 5, eaax4279 CrossRef CAS PubMed.
- Z. Liu, P. Bertram and F. Endres, J. Solid State Electrochem., 2017, 21, 2021–2027 CrossRef CAS.
- L.-P. Wang, P.-F. Wang, T.-S. Wang, Y.-X. Yin, Y.-G. Guo and C.-R. Wang, J. Power Sources, 2017, 355, 18–22 CrossRef.
- G. Zampardi and F. La Mantia, Curr. Opin. Electrochem., 2020, 21, 84–92 CrossRef CAS.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed.
- C. Han, H. Li, R. Shi, T. Zhang, J. Tong, J. Li and B. Li, J. Mater. Chem. A, 2019, 7, 23378–23415 RSC.
- H. Y. Shi, Y. Song, Z. Qin, C. Li, D. Guo, X. X. Liu and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 16057–16061 CrossRef CAS PubMed.
- G. Li, Z. Yang, Y. Jiang, C. Jin, W. Huang, X. Ding and Y. Huang, Nano Energy, 2016, 25, 211–217 CrossRef CAS.
- B. Li, Z. Nie, M. Vijayakumar, G. Li, J. Liu, V. Sprenkle and W. Wang, Nat. Commun., 2015, 6, 6303 CrossRef CAS PubMed.
- K. Gong, X. Ma, K. M. Conforti, K. J. Kuttler, J. B. Grunewald, K. L. Yeager, M. Z. Bazant, S. Gu and Y. Yan, Energy Environ. Sci., 2015, 8, 2941–2945 RSC.
- Z. Yuan, Y. Yin, C. Xie, H. Zhang, Y. Yao and X. Li, Adv. Mater., 2019, 31, 1902025 CrossRef CAS PubMed.
- Y. Cheng, N. Zhang, Q. Wang, Y. Guo, S. Tao, Z. Liao, P. Jiang and Z. Xiang, Nano Energy, 2019, 63, 103822 CrossRef CAS.
- M. Xing, Z. Zhao, Y. Zhang, J. Zhao, G. Cui and J. Dai, Mater. Today Energy, 2020, 100534 CrossRef CAS.
- G.-M. Weng, Z. Li, G. Cong, Y. Zhou and Y.-C. Lu, Energy Environ. Sci., 2017, 10, 735–741 RSC.
- C. Xie, Y. Liu, W. Lu, H. Zhang and X. Li, Energy Environ. Sci., 2019, 12, 1834–1839 RSC.
- C. Xie, H. Zhang, W. Xu, W. Wang and X. Li, Angew. Chem., Int. Ed., 2018, 130, 11341–11346 CrossRef.
- J. Zhang, G. Jiang, P. Xu, A. G. Kashkooli, M. Mousavi, A. Yu and Z. Chen, Energy Environ. Sci., 2018, 11, 2010–2015 RSC.
- H. Yang, Y. Qiao, Z. Chang, H. Deng, P. He and H. Zhou, Adv. Mater., 2020, 32, 2004240 CrossRef CAS PubMed.
- N. Kang, Y. Lin, L. Yang, D. Lu, J. Xiao, Y. Qi and M. Cai, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- C. V. Pham, L. Liu, B. Britton, M. Walter, S. Holdcroft and S. Thiele, Sustainable Energy Fuels, 2020, 4, 1180–1190 RSC.
- C. Bai, F. Cai, L. Wang, S. Guo, X. Liu and Z. Yuan, Nano Res., 2018, 11, 3548–3554 CrossRef CAS.
- H. Pan, B. Li, D. Mei, Z. Nie, Y. Shao, G. Li, X. S. Li, K. S. Han, K. T. Mueller and V. Sprenkle, ACS Energy Lett., 2017, 2, 2674–2680 CrossRef CAS.
- Y. Li, L. Liu, H. Li, F. Cheng and J. Chen, Chem. Commun., 2018, 54, 6792–6795 RSC.
- Y. Cui, Q. Zhao, X. Wu, X. Chen, J. Yang, Y. Wang, R. Qin, S. Ding, Y. Song and J. Wu, Angew. Chem., Int. Ed., 2020, 132, 16737–16744 Search PubMed.
- T. J. Skalski, B. Britton, T. J. Peckham and S. Holdcroft, J. Am. Chem. Soc., 2015, 137, 12223–12226 CrossRef CAS PubMed.
- M. Adamski, T. J. G. Skalski, B. Britton, T. J. Peckham, L. Metzler and S. Holdcroft, Angew. Chem., Int. Ed., 2017, 56, 9058–9061 CrossRef CAS PubMed.
- T. J. G. Skalski, M. Adamski, B. Britton, E. M. Schibli, T. J. Peckham, T. Weissbach, T. Moshisuki, S. Lyonnard, B. J. Frisken and S. Holdcroft, ChemSusChem, 2018, 11, 4033–4043 CrossRef CAS PubMed.
- M. Adamski, T. J. G. Skalski, E. M. Schibli, M. Killer, Y. Wu, N. Peressin, B. J. Frisken and S. Holdcroft, J. Membr. Sci., 2020, 595, 117539 CrossRef CAS.
- D. MacKinnon, R. Morrison, J. Mouland and P. Warren, J. Appl. Electrochem., 1990, 20, 728–736 CrossRef CAS.
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC.
- Z. Cao, X. Zhu, D. Xu, P. Dong, M. O. L. Chee, X. Li, K. Zhu, M. Ye and J. Shen, Energy Storage Mater., 2020, 36, 132–138 CrossRef.
- T. Brezesinski, J. Wang, J. Polleux, B. Dunn and S. H. Tolbert, J. Am. Chem. Soc., 2009, 131, 1802–1809 CrossRef CAS PubMed.
- S. Liu, L. Kang, J. Zhang, E. Jung, S. Lee and S. C. Jun, Energy Storage Mater., 2020, 32, 167–177 CrossRef.
- S. Liu, L. Kang, J. Zhang, S. C. Jun and Y. Yamauchi, ACS Energy Lett., 2021, 6, 4127–4154 CrossRef CAS.
- J. Gamby, P. L. Taberna, P. Simon, J. F. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109–116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00171c |
| This journal is © The Royal Society of Chemistry 2022 |