
Mass spectrometry-based quantitation combined with time-dependent metabolomics to discover metabolic features in human neurogenesis using neural constructs generated from neural progenitor cells†
Xin
Wang
 *,
Zhenye
Gao
and
Wenxiu
Zhou
*,
Zhenye
Gao
and
Wenxiu
Zhou
School of Pharmacy, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, P. R. China. E-mail: xinwang2020@sjtu.edu.cn; Tel: +86-21-34204048
First published on 16th December 2022
Abstract
Direct studies focusing on the human brain are difficult to plan and conduct due to ethical and practical reasons. The advent of human pluripotent stem cell (hPSC)-derived neurons has revolutionized the research of the human brain and central nervous system, but relevant analytical techniques have been much less explored. Herein, we have designed a novel bioanalytical strategy to discover the characteristics of human neurogenesis using liquid chromatography-mass spectrometry-based quantitation and time-dependent metabolomics in combination with hPSC-derived neural constructs. To examine the growth of neurons in vitro, a quantitative method for the simultaneous measurement of N-acetylaspartic acid (NAA) and N-acetylglutamic acid (NAG) in a culture medium was established. The analysis of endogenous NAA and NAG concentrations over 28 days of neural cell culture not only illustrated the growth and maturation process of neural progenitors, but also confirmed the successful achievement of human neural constructs. Depending on the quantitative results, day 0, 10, 18, and 28 samples representing different growth phases were selected for further investigation of the global metabolic changes in developing human neurons. A versatile non-targeted, time-dependent metabolomics study identified 17 significantly changed metabolites and revealed the altered metabolic pathways including amino acid metabolism (tryptophan, phenylalanine, aspartate and beta-alanine metabolisms), pantothenate and coenzyme A biosynthesis, fatty acid metabolism, and purine and pyrimidine metabolism. The new metabolite profiles and overall metabolic pathways advance our understanding of human neurodevelopment. Additionally, the bioanalytical approach proposed in this study opens an interesting window for the capture and evaluation of the complex metabolic states of human neural cells, which would potentially be utilized in other in vitro models relevant to pathophysiology and treatment of neurological disorders, benefiting biomarker discovery and metabolic mechanism interpretation.
Introduction
Predictions of drug metabolism, drug–drug interactions and disease mechanisms have been mainly achieved in animal models and need to be evaluated and verified in human models.1,2 However, these efforts have been hampered by the limited access to and use of the rare and precious donated human tissues.3 Emergence of in vitro cellular models that accurately reflect human physiology would provide a reliable and cost-effective approach to address this problem.4 Among all the biological systems in our body, the human brain is particularly susceptible to toxic damage and environmental influences. Human pluripotent stem cells (hPSCs) can be successfully differentiated into neural progenitor cells (NPCs) and then into central nervous system (CNS)-related cell phenotypes including neurons and glial cells, which are invaluable for in vitro disease modeling, drug discovery, and regenerative therapy.5The hPSC-derived neurons represent a versatile tool for research in CNS physiology and pathology, and thus have dramatic potential to improve our understanding of the brain.6,7 However, culturing procedures must be carefully optimized and evaluated in order to generate reproducible models consisting of the same cell types.8 This challenge requires novel bioengineering and quality assurance techniques, for example, suitable biomarkers need to be identified and traced over extended culture periods.9 As a powerful quantitative instrument, liquid chromatography-mass spectrometry (LC-MS) could be promising for characterizing neural constructs.
Cell metabolism plays a crucial role in the survival, proliferation, and differentiation of NPCs.10 Metabolomics, the profiling of small molecules that are involved in metabolism in a biological system, is a reproducible, accurate and sensitive tool for analyzing metabolic changes.11,12 Metabolomics offers the closest direct measurement of a cell's physiological activity, and has advanced efforts to characterize cells’ fate, identify biomarkers, and investigate metabolic pathways.13 Recently, analyses of the metabolic changes accompanying maturation of several human in vitro models, such as the liver14,15 and heart,16 have been performed to better characterize and understand these organs. However, metabolomics study on human neurodevelopment is greatly lacking.
Based on the successful combination of hPSC-derived neural constructs, LC-MS quantitation, and metabolomics, we present here a novel bioanalytical approach for identifying the metabolic features linked to human neurogenesis. In order to examine the growth of neurons, a sensitive and selective LC-MS method has been established for the simultaneous quantitation of N-acetylaspartic acid (NAA)17 and N-acetylglutamic acid (NAG),18,19 which are important markers reflecting the function of the nervous system. NAA and NAG concentrations in medium samples were measured over 28 days of neural cell culture. Afterward, a non-targeted time-dependent metabolomics was conducted to demonstrate the significantly changed metabolites during neurodevelopment. The metabolic pathways altered in different growth phases were further discussed, focusing on the identified metabolites.
Experimental
Chemicals and reagents
Acetonitrile (ACN), methanol and formic acid (FA) of LC grade were from Sigma-Aldrich (MO, USA). Purified water was provided by the Hangzhou Wahaha Group (Zhejiang, China). NAA, NAG, 5-hydroxytryptophan, serotonin, phenylacetylglycine, tyrosine, spermidine, spermine, alpha-ketoisovaleric acid, butyrylcarnitine, guanine, inosine, uridine, and citric acid were all purchased from Sigma-Aldrich (MO, USA). N-Acetylaspartic acid-2,3,3-D3 (NAA-D3) was from CDN Isotopes Inc. (Quebec, Canada) and used as an internal standard for NAA and NAG quantitation.Formation of neural constructs
NPCs derived from the human H1 ES line were seeded at a density of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per PEG-transwell and cultured in a neural expansion medium for 28 days to undergo expansion and differentiation into neuronal and glial subpopulations (Fig. S1A†), characterized by increased immunostaining of the neural marker β3-tubulin and astrocyte marker GFAP (Fig. S1B†). The neural expansion medium consisted of DMEM/F12 HEPES (Life Technologies, 11330-032) supplemented with 220 µM L-ascorbic acid (Sigma-Aldrich A8960), 80 nM sodium selenite (Sigma-Aldrich S5261), 6.5 mM sodium bicarbonate (Sigma-Aldrich S5761), 5 ng mL−1 human FGF-2 (Fisher Scientific 507515502), and N-2 and B27 supplements at 1× final concentration (Life Technologies 17502-048 and 17504-044). The PEG-transwell was prepared using thiol–ene photopolymerization chemistry.20,21 The cells were allowed to attach overnight, and the medium was changed every 48 h with 1 mL in the basal compartment and 200 µL in the apical compartment. The apical medium was collected every two days for further quantitative analysis and metabolomics. Quality control (QC) samples were prepared by pooling aliquots (20 μL) of all the medium samples.
000 cells per PEG-transwell and cultured in a neural expansion medium for 28 days to undergo expansion and differentiation into neuronal and glial subpopulations (Fig. S1A†), characterized by increased immunostaining of the neural marker β3-tubulin and astrocyte marker GFAP (Fig. S1B†). The neural expansion medium consisted of DMEM/F12 HEPES (Life Technologies, 11330-032) supplemented with 220 µM L-ascorbic acid (Sigma-Aldrich A8960), 80 nM sodium selenite (Sigma-Aldrich S5261), 6.5 mM sodium bicarbonate (Sigma-Aldrich S5761), 5 ng mL−1 human FGF-2 (Fisher Scientific 507515502), and N-2 and B27 supplements at 1× final concentration (Life Technologies 17502-048 and 17504-044). The PEG-transwell was prepared using thiol–ene photopolymerization chemistry.20,21 The cells were allowed to attach overnight, and the medium was changed every 48 h with 1 mL in the basal compartment and 200 µL in the apical compartment. The apical medium was collected every two days for further quantitative analysis and metabolomics. Quality control (QC) samples were prepared by pooling aliquots (20 μL) of all the medium samples.
Sample preparation
000 rpm for 15 min. The supernatant of each sample was collected and dried by N2 reflux. Before analysis, the sample was redissolved in 20 μL of 98:2 water/ACN (v/v) for injection.
000 rpm. The supernatant was collected, dried by N2 reflux, and redissolved in 25 μL of 98:2 water/ACN (v/v) for LC-MS analysis.
Quantitation of NAA and NAG
The quantitation of NAA and NAG was performed on an Agilent 6430 Triple Quadrupole mass spectrometer (QqQ MS) connected to an Agilent 1200 LC system (Agilent Technologies, CA, USA). A Phenomenex C18 reversed-phase column (2.6 μm, 100 × 2.1 mm) was used for chromatography. The mobile phases comprised water (A) and ACN (B). A gradient elution program was employed during the separation (solution B: 2–16% in 3 min, 16–96% in 3 min, 96% for 1 min, 2% for 2 min). The flow rate of mobile phases was set at 0.2 mL min−1 with the column temperature being at room temperature and an injection volume of 4 μL.The multiple reaction monitoring (MRM) mode of QqQ MS was employed to establish a sensitive and selective quantitative method. MS was operated in the negative ion mode with a capillary voltage of 2800 V. The ion source parameters were as follows: gas temperature, 350 °C; gas flow, 10 L min−1; and nebulizer, 40 psi. Three transitions, 174.1 → 58.1 (NAA), 188.1 → 102.1 (NAG) and 177.1 → 58.1 (NAA-D3), were used for quantitation. Nitrogen was used as the collision gas with the optimized collision energies of 25 V for NAA quantitation and 20 V for NAG quantitation. LC-MS/MRM data were collected using MassHunter Data Acquisition for Triple Quadrupole B.06.00 (Agilent Technologies, CA, USA) and were analyzed using MassHunter Qualitative Analysis B.06.00 (Agilent Technologies, CA, USA).
Metabolomics analysis
An Agilent 6545 Accurate-Mass Quadrupole Time-of-Flight mass spectrometer (QTOF MS) coupled with an Agilent 1290 series LC system (Agilent Technologies, CA, USA) was used to detect and identify metabolites. Samples were analyzed in both positive and negative ion modes using an Agilent SB-C18 column (1.8 μm, 2.1 mm × 50 mm). The column was kept at 50 °C and the injection volume was 5 μL. Separation was performed by gradient elution using an aqueous phase (A) of 0.1% FA in water and an organic phase (B) of 0.1% FA in ACN, at a flow rate of 0.4 mL min−1. A gradient elution program was employed during the separation (solution B: 2–70% in 7 min, 70–100% in 5 min, 100% for 3 min, 100–2% in 1 min, post time for 4 min).An Agilent Dual AJS ESI source was operated and the ion source parameters were set as follows: gas temperature, 350 °C; dry gas flow, 12 L min−1; nebulizer, 50 psig; sheath gas temperature, 380 °C; sheath gas flow, 12 L min−1; capillary voltage, 3.5 kV; fragmentor, 120 V; and skimmer, 65 V. The MS scan range was 100–1100 m/z.
Data acquired by LC-MS were analyzed using the Molecular Feature Extraction (MFE) tool from MassHunter Qualitative Analysis (B.07.00) to obtain molecular features. The extraction algorithm “Small molecules (chromatographic)” was selected using the following parameters: ions ≥1000 counts; peak spacing tolerance = 0.0025 m/z, plus 7.0 ppm; isotope model = common organic molecules; and quality score ≥80. To identify different ion species coming from the same metabolite, H+, Na+, and K+ adducts were considered for positive ionization, while the H− and HCOO− adducts were considered for negative ionization. The extracted features were then analyzed with Mass Profiler Professional (MPP) software (Agilent Technologies, CA, USA). Molecular feature filtering was carried out using a minimum absolute abundance of 10000 counts. Data were aligned with a mass tolerance of 15 ppm and a retention-time window tolerance of 0.15 min, normalized by the percentile shift (75.0) method, followed by frequency filtering using the 80% rule.22 Next, principal component analysis (PCA) was applied to look for trends in the data and to determine if any of the injections was an outlier. PCA was performed based on all metabolites detectable in 80% of the subjects in at least one of the groups, using data that had been mean-centered and scaled to unit variance.23 The number of principal components was selected such that 85% of the total variance was explained. To find endogenous metabolites with significant differences among groups, metabolites with a fold change (FC) larger than 1.5 and a p-value (unpaired t-test) smaller than 0.05 were considered to be statistically significant. The exact masses of putative compounds with significant changes were searched against METLIN (version 3.7.1, https://metlin.scripps.edu), Human Metabolome Database (HMDB) (version 5.0, https://www.hmdb.ca), and MassBank (version 2.1.12, https://www.massbank.jp); the matched exact masses were stored as a list. To confirm the identity of metabolites, MS/MS analysis was carried out for the previously determined exact masses with collision energies of 20 and 40 V, and nitrogen was used as the collision gas. Commercially available metabolites were also verified by comparison of their MS/MS spectra with those of the corresponding chemical standards. Metabolites were reported based on the criteria set out by the Metabolomics Standard Initiative (MSI), in which four levels of metabolite identification were proposed, including the identified metabolite with the reference standard (level 1), putatively annotated metabolite (level 2), putatively annotated metabolite class (level 3), and unknown metabolite (level 4).24 Metabolite quantitation was performed with a single-point standard addition method using analytical standards of the individual compounds. For metabolites that are not commercially available, peak areas were employed to indicate the relative intensities.
Lastly, metabolic pathway analysis was conducted to sort the significantly changed metabolites into relevant biological pathways using MetaboAnalyst (version 5.0, https://www.metaboanalyst.ca), which is a web-based platform for comprehensive evaluation of metabolomics data.25,26 The pathway library of Homo sapiens (KEGG) was selected for the current study.
Results and discussion
Release of endogenous NAA and NAG during neurodevelopment
NAA is one of the most abundant amino acid derivatives in the human brain.27 NAA is predominantly located in the neurons, and has been shown to decrease with traumatic brain injury and stroke, returning to normal levels in accordance with cognitive recovery.28,29 Since brain NAA has been reported in experimental and clinical conditions to be associated with neuronal cell loss, NAA is used as a neuronal marker.17 Another N-acetylated amino acid, NAG, plays a vital role by acting as a precursor of peptide neurotransmitters in the CNS.18,19 In order to monitor the fate of neural constructs, we decided to trace the NAA and NAG secretion profiles in culture medium samples during neurodevelopment.An LC-MS/MRM method has been developed for the simultaneous quantitation of NAA and NAG. Fig. S2† shows the typical MRM chromatograms, and the peaks at 1.53 min and 1.92 min correspond to NAA and NAG, respectively. Even though the retention times of NAA and NAG are both less than 3 min, the MS scan experiment revealed that the whole method requires 9 min to allow all medium components to go through the column as well as column equilibration. The MRM parameters of the established LC-MS/MRM method are given in Table S1.† The deprotonated molecules of NAA (m/z 174.1) and NAG (m/z 188.1) were chosen as precursor ions, while the most intensive product ions of NAA (m/z 58.1) and NAG (m/z 102.1) were selected as quantitative ions. The selected quantitative ions and specific qualitative ions (m/z 88.1 for NAA and m/z 128.1 for NAG) were used for the confirmation. Structures of the above-mentioned quantitative and qualitative ions are shown in Fig. 1.
 | ||
| Fig. 1 Proposed structures of the quantitative and qualitative ions of NAA, NAG, and NAA-D3 in the negative ion mode. | ||
A series of standard mixtures of NAA and NAG were prepared for method validation, and 2 μM NAA-D3 was added as the isotope-labeled internal standard for compensating signal fluctuation and the unavoidable matrix effect. Mean peak area ratios of analytes to the internal standard were plotted against the concentrations of respective analytes to establish calibration equations. The results are presented in Table 1, and good linearities were obtained for NAA and NAG with the coefficients (R2) being no less than 0.999. The limit of detection (LOD) and the limit of quantitation (LOQ) were calculated as the concentrations corresponding to signal-to-noise ratios of 3 and 10, respectively. As a result, the LODs were found to be 2.6 nM for NAA and 0.5 nM for NAG; while the LOQs were 8.5 nM for NAA and 1.6 nM for NAG.
| Analyte | Linear range (nM) | Calibration curve | LOD (nM) | LOQ (nM) | ||
|---|---|---|---|---|---|---|
| Slope | Intercept | R 2 | ||||
| NAA | 10–2000 | 5.72 × 10−4 | 0.0088 | 0.9993 | 2.6 | 8.5 |
| NAG | 2–500 | 0.0023 | 0.0094 | 0.9990 | 0.5 | 1.6 |
The precisions and recoveries of the proposed method were measured with NAA and NAG spiked in blank media at three different concentrations (Table S2†). Intra- and inter-day RSDs were all below 13%. Recoveries were calculated using the following formula, %recovery = (detected concentration of the analyte with the calibration curve)/(actual concentration of the analyte spiked). The recoveries of NAA were in the range of 88.8%–101.0%, and the recoveries of NAG were between 95.6%–104.5%. Due to the matrix-free calibration curves constructed in the study, recoveries of this analytical method can be used to evaluate the matrix effect.30 The matrix effect is considered negligible when the recoveries are in the range of 85%–115%, showing that our method does not suffer from the negative matrix effect problems31,32 and is suitable for the analysis of NAA and NAG in complicated medium samples.
Lastly, the method was applied to determine endogenous NAA and NAG concentrations in medium samples over 28 days of neural cell culture. Consistent with previous publications,33,34 the levels of NAA in brain samples were much higher than those of NAG. As shown in Fig. 2, NAG concentrations increased along with the increase of culture time. As for NAA, the concentrations increased exponentially with the increase of culture time during day 2 to day 12, then increased linearly until day 18 and finally reached a balance, which tracked with the growth and maturation of NPCs. These results clearly demonstrated the maturation process of neural progenitors; on the other hand, they also proved that our human neural constructs were successfully achieved and could be utilized in future investigation.
 | ||
| Fig. 2 Profiling of NAA and NAG concentrations in culture medium samples over 28 days of the neurodevelopmental process using the LC-MS/MRM method. All data points represent the mean ± S.D. of three biological replicates and two technical replicates for each biological sample. | ||
Time-dependent metabolomics revealing potential indicators for neurodevelopment
In order to better understand the growth characteristics of NPCs, changes in global metabolite profiles in medium samples, based on different culture times, have been investigated. Representative time points were selected according to the quantitative results of endogenous NAA considering that NAA is an important brain osmolyte and a specific marker of neuronal health and viability. Fig. S3† shows the typical LC-MS total-ion chromatograms of day 18 and day 0 (control) samples acquired in both positive and negative ion modes. For quality assurance purposes, QC samples were prepared by pooling a small aliquot of all medium samples and were analyzed periodically together with the metabolomics samples. First, PCA was conducted on day 0, day 18, and QC samples to obtain a preliminary understanding of the metabolite profiling data. As shown in Fig. S4,† QC samples were clustered well, indicating that the instrument was operating stably throughout the testing process. Day 18 and day 0 samples were clearly classified into distinct groups under both positive and negative ion modes, which revealed the existence of metabolites with significant differences during human neurodevelopment. Next, a time-dependent metabolomics study was carried out to find metabolites contributing to the differences among day 0, day 10, day 18 and day 28 samples. The 3D PCA score plots demonstrated tight clustering of QC samples, as well as good separation of the four sample groups under positive and negative ion modes (Fig. 3). The metabolite profiling results are presented in Table S3.† | ||
| Fig. 3 PCA plots of day 0 (red), day 10 (grey), day 18 (blue), day 28 (brown), and QC (green) samples under both positive (A) and negative (B) ion modes, with the first three principal components accounting for 86.6% (A) and 82.1% (B) of the variance in analysis. Each dot represents a sample and each color represents the type of the sample. | ||
The metabolites were identified by database searches on exact masses, MS/MS analysis, and product ion interpretation.35,36 The commercially available metabolites were also confirmed by comparison of their MS/MS spectra with those of the corresponding chemical standards. In the end, we determined 17 MSI levels 1 and 2 metabolites that were significantly changed (FC > 1.5, p value <0.05) during neurodevelopment (Table 2). Changes in the intensities of these metabolites were visualized via a heat map and are shown in Fig. 4A. Analysis of the clean culture medium at each timepoint further demonstrated that tyrosine was a component of the culture medium and was consumed significantly during the growth of neural cells, whereas other metabolites were produced by the cells and released into the medium. Among these metabolites, the intensities of alpha-ketoisovaleric acid, spermidine, 4-phosphopantothenoylcysteine, spermine, and uridine showed a similar variation trend to the intensities of NAA, so they may also act as potential indicators for studying neural development (Fig. 4B).
 | ||
| Fig. 4 (A) Heat map of the 17 significantly changed metabolites identified by time-dependent metabolomics, and (B) 6 metabolites might serve as potential indicators for neurodevelopment. The heat map was drawn using R with gplots, and the red-green color represents the z-score transformed raw data of metabolites with significant differences among sample groups. Red or green colors indicate an increase or a decrease in metabolite intensities, respectively. | ||
| Metabolitea | HMDB | Exact mass | ESI+ | ESI− | Pathway | ||
|---|---|---|---|---|---|---|---|
| p value | Representative fragments (m/z) | p value | Representative fragments (m/z) | ||||
| a Metabolites labeled with * were verified with authentic standards (MSI level 1). | |||||||
| 5-Hydroxytryptophan* | 00472 | 220.0847 | 9.17 × 10−6 | 162.0542, 175.0837, 186.0626 | Tryptophan metabolism | ||
| Serotonin* | 00259 | 176.0949 | 2.61 × 10−5 | 132.0769, 160.0733 | Tryptophan metabolism | ||
| Acetyl-N-formyl-5-methoxykynurenamine | 04259 | 264.1097 | 7.14 × 10−5 | 136.0735, 150.0559, 247.1052 | Tryptophan metabolism | ||
| Phenylacetylglycine* | 00821 | 193.0732 | 3.64 × 10−5 | 65.0400, 76.0405, 91.0553 | 2.24 × 10−5 | 74.0252, 91.0553 | Phenylalanine metabolism |
| Tyrosine* | 00158 | 181.0739 | 1.94 × 10−6 | 93.0354, 107.0476, 119.0501 | Phenylalanine metabolism | ||
| Argininosuccinic acid | 00052 | 290.1234 | 4.25 × 10−4 | 70.0660, 116.0710, 176.0704 | Aspartate metabolism | ||
| N-Acetylaspartic acid* | 00812 | 175.0489 | 5.02 × 10−5 | 58.0301, 88.0401 | Aspartate metabolism | ||
| Spermidine* | 01257 | 145.1575 | 1.53 × 10−4 | 72.0820, 84.0820, 112.1125 | Beta-alanine metabolism | ||
| Spermine* | 01256 | 202.2157 | 3.86 × 10−6 | 72.0815, 84.0821, 112.1130, 129.1381 | Beta-alanine metabolism | ||
| 4-Phosphopantothenoylcysteine | 01117 | 402.0861 | 4.47 × 10−3 | 176.0395, 271.1250, 385.0849 | 3.83 × 10−3 | 78.9560, 96.9672 | Pantothenate and CoA biosynthesis |
| Alpha-ketoisovaleric acid* | 00019 | 116.0473 | 1.76 × 10−2 | 55.0191, 68.9965, 70.9997 | Pantothenate and CoA biosynthesis | ||
| Butyrylcarnitine* | 02013 | 231.1470 | 1.16 × 10−4 | 60.0824, 71.0503, 85.0295, 173.0817 | Fatty acid metabolism | ||
| N-Acryloylglycine | 01843 | 129.0425 | 2.36 × 10−5 | 84.0459, 130.0515 | 4.23 × 10−5 | 74.0238 | Fatty acid metabolism |
| Guanine* | 00132 | 151.0494 | 3.33 × 10−4 | 110.0337, 135.0302 | 2.47 × 10−4 | 65.0165, 108.0200, 133.0145 | Purine metabolism |
| Inosine* | 00195 | 268.0807 | 1.65 × 10−5 | 135.0345 | Purine metabolism | ||
| Uridine* | 00296 | 244.0695 | 1.93 × 10−8 | 66.0356, 110.0241, 140.3331 | Pyrimidine metabolism | ||
| Citric acid* | 00094 | 192.0270 | 4.94 × 10−5 | 85.0307, 87.0095, 111.0091 | TCA cycle | ||
Metabolic pathways associated with human neurogenesis
Based on the identified metabolites, a metabolic pathway analysis was conducted using MetaboAnalyst 5.0 and KEGG databases. Results suggested that the amino acid metabolism, pantothenate and coenzyme A (CoA) biosynthesis, fatty acid metabolism, and purine and pyrimidine metabolism were involved in human neurogenesis (Table 2 and Fig. 5). Perhaps the most interesting observations were the correlation between uptake and secretion of amino acids. In general, the aromatic amino acids (tyrosine, tryptophan, and phenylalanine) in the culture medium were transported into the neural cells as precursors for neurotransmitter synthesis, leading to decreased amounts in the medium with increasing culture time. The metabolomics analysis demonstrated that the intensity of tyrosine decreased in a time-dependent manner (Fig. 4A). Tryptophan and phenylalanine were not detected to be significantly changed, and this may be because their concentrations in the original culture medium were relatively high; so even though they were consumed, the FC data were still lower than 1.5. Nevertheless, the metabolites 5-hydroxytryptophan, serotonin, acetyl-N-formyl-5-methoxykynurenamine, and phenylacetylglycine were produced and released into the medium with significantly changed levels, indicating the activated tryptophan metabolism and phenylalanine metabolism during the neurodevelopmental process. Additionally, alterations of aspartate metabolism and beta-alanine metabolism were also observed, which could be related to the well-studied alanine, aspartate and glutamate metabolism in the human brain.37,38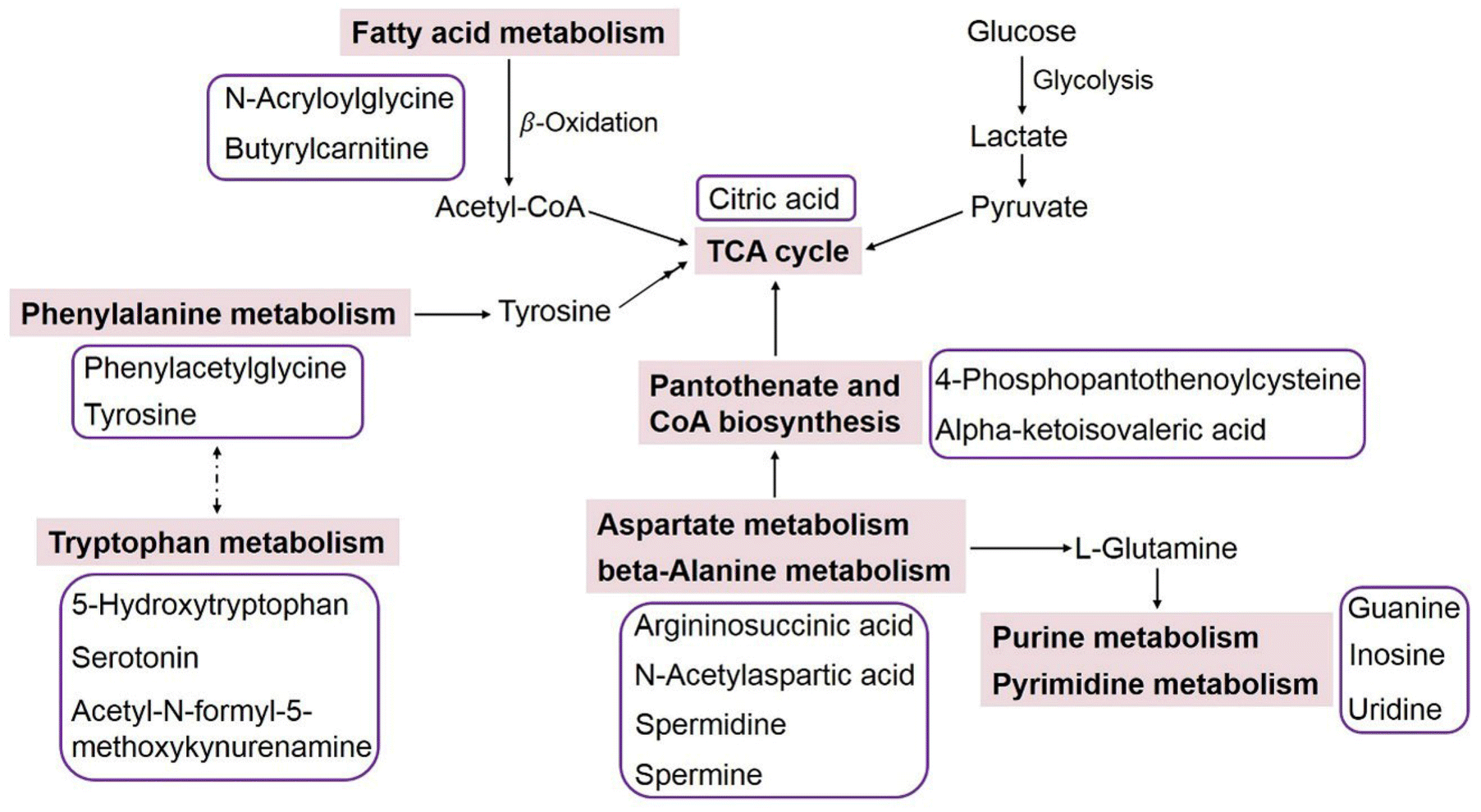 | ||
| Fig. 5 An overview of the integrated metabolic pathway network associated with human neurogenesis. Solid arrows represent direct metabolic reactions, and dashed arrows represent indirect connections between two pathways. The metabolites with significant changes during neurodevelopment are listed in purple frames and placed near the corresponding metabolic pathways. | ||
The intensities of 4-phosphopantothenoylcysteine and alpha-ketoisovaleric acid tracked with the growth and maturation of neural cells, indicating changed pantothenate and CoA biosynthesis in the developing human neurons. CoA is an essential cofactor for cell growth, and it functions as an acyl group carrier and carbonyl activating group in a multitude of biochemical transformations, including the tricarboxylic acid (TCA) cycle and fatty acid metabolism.39 The enhanced citric acid level during cell culture further demonstrated the activated TCA cycle. Significant increases in the levels of N-acryloylglycine and butyrylcarnitine were detected as well, and N-acryloylglycine is normally a minor metabolite of fatty acids, while the carnitine metabolite of butyrylcarnitine is associated with mitochondrial fatty acid beta-oxidation.40 Fatty acid beta-oxidation is responsible for generating ketone bodies for energy metabolism, and it has been shown that up to 20% of the total energy of the brain is provided by mitochondrial oxidation of fatty acids.41 Our metabolomics results have implied that fatty acid metabolism is involved in human neurogenesis.
Purine and pyrimidine metabolic pathways were also altered during neural cell culture. The levels of guanine, inosine and uridine in the culture medium were highly increased from day 0 to day 18, and then decreased from day 18 to day 28, which tracked with the growth status of the neural cells (Fig. 4). Purines and pyrimidines are known as building blocks for nucleic acid synthesis, so it is not surprising to observe fluctuations in purine and pyrimidine metabolism. In addition, purines are non-amino acid neurotransmitters of remarkable importance; pyrimidines are involved in polysaccharide and phospholipid synthesis and act via extracellular receptors to regulate a variety of physiological processes.42,43 Our results confirmed that purine and pyrimidine metabolism is sensitive to neural growth, and might play important roles in inducing or maintaining brain maturation. Considering that the metabolomics of the culture medium is concentrated on the metabolites released from the neural cells into the medium, an interesting area for future research will therefore be to perform metabolomics in the cells, which could capture the changes of those metabolites that could not be released into the medium.
Conclusions
This is the first example of successfully integrating human neural constructs, LC-MS quantitation, and time-dependent metabolomics to capture and evaluate key metabolic changes in human neurogenesis. First, endogenous NAA and NAG concentrations in medium samples were quantified over 28 days of neural cell culture. The quantitative results demonstrated the growth and maturation process of neural progenitors, and meanwhile showed the successful development of neural constructs. Based upon the maturation process, following time-dependent metabolomics was conducted for day 0, day 10, day 18 and day 28 samples. Novel metabolic features relevant to human neurodevelopment, including 17 significantly changed metabolites and 7 metabolic pathways, have been identified, indicating the alterations of amino acid metabolism, pantothenate and CoA biosynthesis, fatty acid metabolism, and purine and pyrimidine metabolism during neural growth. These metabolomics results allow enhanced understanding of human neurogenesis. Future research could focus on the validation of the proposed pathways using molecular and enzymatic tools.Author contributions
Xin Wang: conceptualization, methodology, investigation, supervision, writing – original draft, writing – review and editing, and funding acquisition. Zhenye Gao: formal analysis and investigation. Wenxiu Zhou: validation and investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 22104081) and the Shanghai Pujiang Program (Grant No. 20PJ1406300). We thank Prof. Steven R. Tannenbaum from Massachusetts Institute of Technology for offering very helpful advice on NAA and NAG detection.References
- F. Fontana, P. Figueiredo, J. P. Martins and H. A. Santos, Small, 2021, 17, 2004182 CrossRef CAS PubMed.
- D. E. Ingber, Adv. Sci., 2020, 7, 2002030 CrossRef CAS PubMed.
- J. P. Pirnay, E. Baudoux, O. Cornu, A. Delforge, C. Delloye, J. Guns, E. Heinen, E. Van den Abbeel, A. Vanderkelen, C. Van Geyt, I. Van Riet, G. Verbeken, P. De Sutter, M. Verlinden, I. Huys, J. Cockbain, C. Chabannon, K. Dierickx, P. Schotsmans, D. De Vos, T. Rose, S. Jennes and S. Sterckx, EMBO Rep., 2015, 16, 557–562 CrossRef CAS PubMed.
- G. Bassi, M. A. Grimaudo, S. Panseri and M. Montesi, Int. J. Mol. Sci., 2021, 22, 1195–1222 CrossRef CAS PubMed.
- K. W. Kelley and S. P. Pasca, Cell, 2022, 185, 42–61 CrossRef CAS PubMed.
- S. Velasco, B. Paulsen and P. Arlotta, Annu. Rev. Neurosci., 2020, 43, 375–389 CrossRef CAS PubMed.
- A. Tian, J. Muffat and Y. Li, J. Neurosci., 2020, 40, 1186–1193 CrossRef CAS PubMed.
- E. Fritsche, T. Haarmann-Stemmann, J. Kapr, S. Galanjuk, J. Hartmann, P. R. Mertens, A. A. M. Kaempfer, R. P. F. Schins, J. Tigges and K. Koch, Small, 2021, 17, 2006252 CrossRef CAS PubMed.
- D. Pamies, M. Leist, S. Coecke, G. Bowe, D. G. Allen, G. Gstraunthaler, A. Bal-Price, F. Pistollato, R. B. M. de Vries, H. T. Hogberg, T. Hartung and G. Stacey, ALTEX, 2022, 39, 30–70 Search PubMed.
- D. S. C. Bispo, C. S. H. Jesus, I. M. C. Marques, K. M. Romek, M. B. Oliveira, J. F. Mano and A. M. Gil, Stem Cell Rev. Rep., 2021, 17, 2003–2024 CrossRef PubMed.
- K. Sinclair and E. Dudley, in Advancements of Mass Spectrometry in Biomedical Research, Springer, Cham, 2019, pp. 613–633 Search PubMed.
- S. E. Murphy and J. V. Sweedler, Analyst, 2022, 147, 2918–2929 RSC.
- T. M. Domenick, E. L. Gill, V. Vedam-Mai and R. A. Yost, Anal. Chem., 2021, 93, 546–566 CrossRef CAS PubMed.
- R. Jellali, M. L. Bernier, Y. Tauran, F. Gilard, M. Danoy, T. Kido, A. Miyajima, Y. Sakai and E. Leclerc, Differentiation, 2020, 112, 17–26 CrossRef CAS PubMed.
- M. Danoy, Y. Tauran, S. Poulain, R. Jellali, J. Bruce, M. Leduc, M. Le Gall, F. Gilard, T. Kido, H. Arakawa, K. Araya, D. Mori, Y. Kato, H. Kusuhara, C. Plessy, A. Miyajima, Y. Sakai and E. Leclerc, Biotechnol. Bioeng., 2021, 118, 3716–3732 CrossRef CAS PubMed.
- V. J. Bhute, X. Bao, K. K. Dunn, K. R. Knutson, E. C. McCurry, G. Jin, W. H. Lee, S. Lewis, A. Ikeda and S. P. Palecek, Theranostics, 2017, 7, 2078–2091 CrossRef CAS PubMed.
- M. Warepam, K. Ahmad, S. Rahman, H. Rahaman, K. Kumari and L. R. Singh, Biomolecules, 2020, 10, 286–295 CrossRef CAS PubMed.
- E. Alonso, M. A. García-Pérez, J. Bueso and V. Rubio, Neurochem. Res., 1991, 16, 787–794 CrossRef CAS PubMed.
- L. Huang, X. Guo, P. Liu, Y. Zhao, C. Wu, C. Zhou, C. Huang, G. Li, Y. Zhuang, S. Cheng, H. Cao, C. Zhang, Z. Xu, X. Liu, G. Hu and P. Liu, J. Hazard. Mater., 2022, 422, 126849 CrossRef CAS PubMed.
- M. P. Schwartz, Z. G. Hou, N. E. Propson, J. Zhang, C. J. Engstrom, V. S. Costa, P. Jiang, B. K. Nguyen, J. M. Bolin, W. Daly, Y. Wang, R. Stewart, C. D. Page, W. L. Murphy and J. A. Thomson, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12516–12521 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- S. Bijlsma, L. Bobeldijk, E. R. Verheij, R. Ramaker, S. Kochhar, I. A. Macdonald, B. van Ommen and A. K. Smilde, Anal. Chem., 2006, 78, 567–574 CrossRef CAS PubMed.
- R. Goodacre, D. Broadhurst, A. K. Smilde, B. S. Kristal, J. D. Baker, R. Beger, C. Bessant, S. Connor, G. Capuani, A. Craig, T. Ebbels, D. B. Kell, C. Manetti, J. Newton, G. Paternostro, R. Somorjai, M. Sjostrom, J. Trygg and F. Wulfert, Metabolomics, 2007, 3, 231–241 CrossRef CAS.
- R. M. Salek, C. Steinbeck, M. R. Viant, R. Goodacre and W. B. Dunn, GigaScience, 2013, 2, 13–15 CrossRef PubMed.
- J. Chong and J. Xia, Bioinformatics, 2018, 34, 4313–4314 CrossRef CAS PubMed.
- Z. Pang, J. Chong, G. Zhou, D. A. D. L. Morais, L. Chang, M. Barrette, C. Gauthier, P. E. Jacques, S. Li and J. Xia, Nucleic Acids Res., 2021, 49, W388–W396 CrossRef CAS PubMed.
- E. V. Sullivan, E. Adalsteinsson, D. M. Spielman, R. E. Hurd and A. Pfefferbaum, Ann. Neurol., 2001, 50, 823–823 CrossRef CAS PubMed.
- R. J. Shannon, S. van der Heide, E. L. Carter, I. Jalloh, D. K. Menon, P. J. Hutchinson and K. L. H. Carpenter, J. Neurotrauma, 2016, 33, 319–329 CrossRef PubMed.
- N. Mazibuko, R. O'Gorman Tuura, L. Sztriha, O. O'Daly, G. J. Barker, S. C. R. Williams, M. O'Sullivan and L. Kalra, Diagnostics, 2020, 10, 482–493 CrossRef CAS PubMed.
- X. Wang, C. T. Garcia, G. Y. Gong, J. S. Wishnok and S. R. Tannenbaum, Anal. Chem., 2018, 90, 1967–1975 CrossRef CAS PubMed.
- A. Nasiri, R. Jahani, S. Mokhtari, H. Yazdanpanah, B. Daraei, M. Faizi and F. Kobarfard, Analyst, 2021, 146, 6049–6063 RSC.
- X. Wang, X. J. Li, Y. Bai and H. W. Liu, Chem. Commun., 2015, 5, 4615–4618 RSC.
- D. S. Ma, J. Y. Zhang, K. Sugahara, T. Ageta, K. Nakayama and H. Kodama, Anal. Biochem., 1999, 276, 124–128 CrossRef CAS PubMed.
- B. Tavazzi, R. Vagnozzi, D. Di Pierro, A. M. Amorini, G. Fazzina, S. Signoretti, A. Marmarou, I. Caruso and G. Lazzarino, Anal. Biochem., 2000, 277, 104–108 CrossRef CAS PubMed.
- X. Wang, M. Cirit, J. S. Wishnok, L. G. Griffith and S. R. Tannenbaum, Anal. Chem., 2019, 91, 8667–8675 CrossRef CAS PubMed.
- A. Verhoeven, M. Giera and O. A. Mayboroda, Analyst, 2020, 145, 3801–3808 RSC.
- S. Camandola and M. P. Mattson, EMBO J., 2017, 36, 1474–1492 CrossRef CAS PubMed.
- J. V. Andersen, K. H. Markussen, E. Jakobsen, A. Schousboe, H. S. Waagepetersen, P. A. Rosenberg and B. I. Aldana, Neuropharmacology, 2021, 196, 108719 CrossRef CAS PubMed.
- L. Mignani, B. Gnutti, D. Zizioli and D. Finazzi, Brain Sci., 2021, 11, 1031–1052 CrossRef CAS PubMed.
- M. M. Adeva-Andany, N. Carneiro-Freire, M. Seco-Filgueira, C. Fernandez-Fernandez and D. Mourino-Bayolo, Mitochondrion, 2019, 46, 73–90 CrossRef CAS PubMed.
- A. Panov, Z. Orynbayeva, V. Vavilin and V. Lyakhovich, Biomed Res. Int., 2014, 2014, 472459 Search PubMed.
- M. Fumagalli, D. Lecca, M. P. Abbracchio and S. Ceruti, Front. Pharmacol., 2017, 8, 941–958 CrossRef PubMed.
- D. Kundu and V. K. Dubey, Curr. Protein Pept. Sci., 2021, 22, 170–189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an01162j |
| This journal is © The Royal Society of Chemistry 2023 |