
A microfluidic chip-based multivalent DNA walker amplification biosensor for the simultaneous detection of multiple food-borne pathogens†
Zhenli
Xu
a,
Jiaqi
Wang
a,
Zhijian
Jia
b,
Yong-Xiang
Wu
 *a,
Ning
Gan
*a and
Shaoning
Yu
a
*a,
Ning
Gan
*a and
Shaoning
Yu
a
aKey Laboratory of Advanced Mass Spectrometry and Molecular Analysis of Zhejiang Province, Institute of Mass Spectrometry, School of Material Science and Chemical Engineering, Ningbo University, Ningbo, Zhejiang province 315211, China. E-mail: wuyongxiang@nbu.edu.cn; ganning@nbu.edu.cn
bSchool of Materials and Chemical Engineering, Ningbo University of Technology, Ningbo, Zhejiang province 315211, China
First published on 16th January 2023
Abstract
The rapid, simultaneous, sensitive detection of the targets has important application prospects for disease diagnosis and biomedical studies. However, in practical applications, the content of the targets is usually very low, and signal amplification strategies are often needed to improve the detection sensitivity. DNAzyme-driven DNA walkers are an excellent signal amplification strategy due to their outstanding specificity and sensitivity. Food-borne pathogens have always been a foremost threat to human health, and it is an urgent demand to develop a simple, rapid, sensitive, and portable detection method for food-borne pathogens. In addition, there are various species of pathogens, and it is difficult to simultaneously detect multiple pathogens by a single DNA walker. For this reason, a substrate strand with three rA cleavage sites was cleverly designed, and a multivalent DNA walker sensor combined with the microfluidic chip technology was proposed for the simultaneous, rapid, sensitive analysis of Vibrio parahaemolyticus, Salmonella typhimurium, and Staphylococcus aureus. The developed sensor could be used to detect pathogens simultaneously and efficiently with low detection limits and wide detection ranges. Moreover, the combination of gold stirring rod enrichment and DNA walker achieved double amplification, which greatly improved the detection sensitivity. More importantly, by changing the design of the substrate chain, the sensor was expected to be used to detect other targets, thus broadening the scope of practical applications. Therefore, the sensor can build novel detection tool platforms in the field of biosensing.
Introduction
In recent years, a variety of biosensing technologies have been developed using the unique biochemical recognition capabilities of organisms, which were widely applied in medical care, genetic analysis, environmental monitoring, food testing, etc.1 However, in practical application, the concentration of target biomolecules in living organisms is usually very low.2 To achieve efficient detection, the signal amplification strategy is often needed to improve the detection sensitivity.3 At present, some common signal amplified strategies, such as hybrid chain reaction (HCR), rolling circle amplification (RCA), strand displacement amplification (SDA), and DNA walker, can obtain significant signal amplification effects.4–7 Among these assays, DNA walker had become increasingly used owing to its simplicity and high sensitivity.8 DNA walker is mainly constructed by integrating three basic components: walking strands, walking tracks, and driving forces.9 It can cleave DNA substrates under the action of driving forces and move autonomously along the programmed nucleic acid trajectory for improving the detection sensitivity of trace biological samples.10 Usually, DNA walkers consist of strand displacement, protein enzymes, and DNAzymes that act as drivers to propel DNA walkers. However, in the process of strand displacement, double-stranded (ds) DNA strands as waste and the dilution of the system because of the necessity of externally adding single-stranded (ss) DNA strands for hybridization control limit the potential applicability of DNA walking devices controlled by fuel strands.11 Besides, the protein enzyme was susceptible to experimental conditions (temperature, pH, and ionic concentration), which easily led to the poor detection reproducibility and accuracy.12 Meanwhile, the high cost and easy inactivation of protein enzymes also greatly limit their application.13 In view of the above limitations, DNAzymes have aroused immense interest in various biomedical and biosensor applications due to their unique and excellent characteristics including high catalytic efficiency, good chemically and thermally stability, excellent specificity, and easy preparation.14–16 With the assistance of specific metal ions, DNAzymes enable quick and efficient catalysis of the continuous cleavage of a single RNA linkage embedded in their complementary DNA substrates.17 In addition, it was easy to control the DNAzyme activity by regulating the depletion or addition of the cofactor metal ions, which can switch the DNA walker off or on.18,19 Hence, combining DNA walker with DNAzyme cleavage reaction may possess an excellent prospect of being applied to various biological analyses. However, due to the single design and fixed length of the fuel chain (substrate chain) and swing arms (DNAzyme chains), the DNA walker is almost limited to the detection of a single target.20 Furthermore, DNA walker usually uses gold nanoparticles or magnetic nanoparticles as walking tracks.21,22 These nanoparticles have a strong adsorption effect on DNA and are likely to produce steric hindrance during the DNA walker process, which was not conducive to the walker.23 Therefore, it is necessary to improve the DNA walker system to address the above limitations.As a common public health security hazard, food-borne pathogens have always been a foremost threat to human health.24,25 According to the report, more than 90% food poisoning diseases are caused by pathogenic bacteria.26 Therefore, there is an urgent demand to develop a simple, rapid, and portable detection method for food-borne pathogens. According to China's food safety national standards,27 many food-borne pathogens are required to be undetectable in foods. The content of foodborne pathogenic bacteria in food is very low, which requires high sensitivity of bacterial detection methods. Therefore, using signal amplification or enrichment methods can achieve lower detection limit and better detection effect. In addition, the matrix in food is complex and diverse, manifold bacterial pathogens coexist in a single food sample in most instances, while most of the current investigations focus on single bacterial detection;28–30 thus, there is an intense necessity for detecting multiple bacterial pathogens simultaneously. Microfluidic chip (MC) technology can manipulate the fluid in the micrometer scale through a high voltage electric field and then integrate the process of sample injection, separation, and detection into a square centimetric level microchip analysis platform.31,32 Due to the excellent separation effect of MC, it also can achieve the simultaneous detection of multiple targets.33,34 Hence, MC plays an important role in the field of rapid analysis and simultaneous detection of multiple samples on account of its simple and fast operation, low sample consumption, favorable separation efficiency, and friendly environment.35
Herein, a multivalent DNA walker sensor combined with MC technology was developed to achieve simultaneous, rapid, and sensitive detection of multiple pathogens such as Vibrio parahaemolyticus, Salmonella typhimurium, and Staphylococcus aureus. Firstly, for improving the detection sensitivity, functionalized gold rod (labelled with 4-mercaptophenylboronic acid) was used to capture targets, resulting in the enrichment of the bacteria for the first signal amplification. Secondly, to solve the limitation of the low concentration and detection of single target, a multivalent DNA walker sensor was designed for the second signal amplification, including a set of signal probes (substrate chain labelled by phenylboronic acid) with three rA cleavage sites and three sets of capture probes (Zn2+-specific DNAzyme chains labelled with different antibodies). Thirdly, to solve the steric hindrance caused by common nanoparticles, the targets’ pathogenic bacteria were directly used as walking tracks. Thus, not only was the adsorption of DNA to the walking track reduced, but the higher specific surface area of bacteria can load more signal molecules, resulting in better signal amplification. When the pathogens existed, the signal probe and the capture probe could be respectively self-assembled on the surface of the pathogen through the reaction of cis-diols with polysaccharides and antigens with antibodies, respectively. It is worth noting that, benefiting from the specificity of the antibody, different capture probes would self-assembled onto the corresponding pathogenic bacteria, respectively. Then, under the action of Zn2+-driven DNAzyme (8–17 enzyme), the three capture probes can cleavage the specific rA site on the signal probe to trigger DNA walker based on the surface of the pathogenic bacteria (walking track), resulting in many DNA fragments of three different lengths. After that, the obtained DNA fragments of different lengths were separated and detected by MC, which can realize the simultaneous quantitative detection of three pathogenic bacteria. Importantly, by changing the design of the signal probe and capture probes, the DNA walker sensor was expected to be a versatile platform for the simultaneous detection of different targets.
Experimental section
Functionalization of the gold stirring rod
The gold stirring rod with the function of bacterial enrichment was made of 4-mercaptophenylboronic acid (4-MPBA) and a gold-plated stirring rod through the Au–S bond. Firstly, the gold stirring rod was cleaned by a heated piranha solution (the ratio of H2SO4 and 30% H2O2 was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and deionized water to remove organic impurities on the stirring rod. Secondly, the gold stirring rod was put into the MPTMS solution (100 μL ethanol, 33 μL 0.1 M HCl, 200 μL MPTMS, 667 μL water) and placed for 1 h. Then, the gold stirring rod was taken out and rinsed 3 times with PBS (pH 7.4). Finally, the stirring rod was put into 100 mL AuNPs solution for 1 h to obtain the gold stirring rod @ AuNPs. Afterward, the obtained gold stirring rod @AuNPs was immersed in a 10 mM 4-MPBA solution (diluted with ethanol) at 4 °C overnight. Then, the 4-MPBA-modified stirring rod was rinsed with deionized water and immersed in 1 mM 6-mercapto-1-hexanol (MCH) solution overnight to block the excess gold sites. The obtained gold stirring bar was stored at 4 °C until further experiment. The picture of the gold stirring rod is shown in Fig. S8B.†
:1) and deionized water to remove organic impurities on the stirring rod. Secondly, the gold stirring rod was put into the MPTMS solution (100 μL ethanol, 33 μL 0.1 M HCl, 200 μL MPTMS, 667 μL water) and placed for 1 h. Then, the gold stirring rod was taken out and rinsed 3 times with PBS (pH 7.4). Finally, the stirring rod was put into 100 mL AuNPs solution for 1 h to obtain the gold stirring rod @ AuNPs. Afterward, the obtained gold stirring rod @AuNPs was immersed in a 10 mM 4-MPBA solution (diluted with ethanol) at 4 °C overnight. Then, the 4-MPBA-modified stirring rod was rinsed with deionized water and immersed in 1 mM 6-mercapto-1-hexanol (MCH) solution overnight to block the excess gold sites. The obtained gold stirring bar was stored at 4 °C until further experiment. The picture of the gold stirring rod is shown in Fig. S8B.†
Preparation of signal probe and capture probes
The preparation of the signal probe was accomplished by a copper-free click chemistry strategy. First, E17 (10 μM) was mixed with excess acetylene ethyl phenylboronic acid (PBA, 100 μM, 2% DMSO), and then ascorbic acid (2.5 mM), copper sulfate (100 μM), and THPTA (500 μM) were added to the mixed solution to catalyze the click reaction (all of them were final concentrations). After 0.5 h, the product was separated by SDS-PAGE, and the product band was cut with a knife, and then soaked in TE buffer to redissolve the product to obtain the signal probe PBA@E17, which was stored at 4 °C.The preparation of the capture probes was referred from the literature,36 which was achieved through the coupling of antibody and DNAzymes (P-VP, P-ST, P-SA) with a little revision. The steps were as follows. Firstly, the antibody was incubated with a 30-fold molar excess of DBCO-NHS for 30 min at room temperature and then ultrafiltered (three times) with a 30 kDa MWCO centrifugal filter, obtaining the DBCO-antibody. Secondly, the azido-modified DNA in a 20-fold molar excess was incubated with the DBCO-antibody for 30 min. Finally, the above solution was washed with PBS and centrifuged by ultrafiltration to remove excess DNA. The resulting capture probes were stored at 4 °C.
Detection of the multivalent DNA walker biosensor
The functionalized gold stirring rod was taken into a mixture solution containing three target pathogenic bacteria together, and was stirred at 120 rpm for 40 min to achieve the capturing and enrichment of pathogenic bacteria. Then, the above stirring rod, capture probes, and signal probes were mixed simultaneously (molar ratio of the capture probes to the signal probe 1:10, pH = 8.0), and incubated at room temperature for 30 min. At this point, due to the specificity of the antibodies, the different capture probes were immobilized on the respective bacterial surfaces, while the signal probe was bound to the bacterial surface via PBA. The reacted stirring rod was then taken out, immersed in the solution containing Zn2+ (100 μM), and incubated for 30 min at room temperature. As a result, the different capture probes selectively cleave the specific rA sites on the signal probe, initiating DNA walking on the surface of the corresponding bacteria, obtaining a large number of DNA fragments of different lengths (three types of bacteria correspondingly produce three different lengths of DNA fragments) and enabling signal amplification. Next, the abovementioned fragments were detected by MC, the separation and detection of DNA were obtained by controlling the voltage change, and then the detection of the target pathogenic bacteria was realized.
Results and discussion
Design of the biosensor and its simultaneous detection mechanism
The development of simultaneous, rapid, and sensitive detection methods has always been a research focus in the field of bioanalysis. However, it is difficult to detect low-content targets directly using traditional detection methods. Therefore, it is necessary to use some strategies for signal amplification. DNA walker was often used as an effective signal amplification strategy because it can perform multiple motions in succession under a single trigger event, producing copious signalling molecules. In the meantime, MC was an excellent choice for the simultaneous detection of targets because of its outstanding separation results. Based on this, a multivalent DNA walker sensor combined with MC technology was developed to achieve simultaneous, rapid, and sensitive detection of multiple pathogens such as Vibrio parahaemolyticus, Salmonella typhimurium, and Staphylococcus aureus.The principle of this work was described in Scheme 1. First, a gold stir bar functionalized with 4-mercaptobenzeneboronic acid was used to enrich for the target pathogen due to the fact that the cis-diol on the phenylboronic acid can bind to the polysaccharide on the bacterial surface.37 Then, it was mixed and reacted with both the signal probe (phenylboronic acid modified one) and the capture probe (antibody modified one). At that moment, the signal probe and the capture probe can be respectively self-assembled on the surface of the pathogen through the reaction of cis-diols with polysaccharides and antigens with antibodies (Scheme 1A). In this case, both the signal and capture probes were confined to the surface of the pathogen, allowing the two to hybridize in close proximity to each other. It was worth noting that benefiting from the specificity of the antibody, the different capture probes would be self-assembled onto the corresponding pathogenic bacteria, respectively. Next, under the action of Zn2+, the three capture probes can cleave the specific rA site on the signal probe to trigger DNA walker based on the surface of the pathogenic bacteria (walking track), resulting in many DNA fragments of three different lengths (Scheme 1B). Therefore, a functionalized gold rod was used to enrich the targets and was integrated with the DNA walker strategy to achieve double signal amplification. Finally, the obtained DNA fragments of different lengths were separated and detected by MC. MC consisted of S (sample reservoir), B (buffer reservoir), SW (sample waste reservoir), BW (buffer waste reservoir), and microchannels. The sample was injected from S, and the voltages were adjusted as S = 280 V, SW = 510 V, B = 320 V, and BW = 0 V for 1 min. At this time, three DNA fragments of different lengths converged at the intersection. Then, the voltages changed to S = 250 V, SW = 250 V, B = 0 V, BW = 1000 V. In the meantime, the sample will flow from the intersection to the detection region. Due to the different lengths of the three DNA fragments, the longer DNA fragments had a slower movement electrophoresis speed during, resulting in the peaks of the three DNA fragments occurring at different times for separation purposes. The peak intensities of the three DNA fragments were corresponding to their corresponding bacterial concentrations, thus enabling the simultaneous quantitative detection of multiple target pathogenic bacteria (Scheme 1C). If the capture probes were not modified with the antibodies, it cannot be immobilized on the surface of the corresponding target bacteria; thus, the DNA walker cannot be formed and the corresponding signal cannot be detected. Meanwhile, the targets’ pathogenic bacteria were directly used as walking tracks, which effectively avoided the generation of false positive signals. If the pathogenic bacteria were absent, there was not only a lack of primer strands to initiate DNA walkers but also a walking track for DNA walkers. In that case, even if there is a small amount of signal leakage and the enzyme cleavage reaction occurs, DNA walker cannot be formed to amplify the catalytic signal. In addition, compared with traditional DNA walker tracks (such as gold nanoparticles and magnetic beads), the higher specific surface area of the bacteria can load more signal molecules, resulting in better signal amplification.
 | ||
| Scheme 1 The principle of the multivalent DNA walker sensor Au-Rods @ PBA @ Bacteria @ DNA-Walker to simultaneously detect three pathogenic bacteria. | ||
Characterization of probes and their combination with targets
To explore the process of probe synthesis, the principles of their preparation are briefly displayed in Fig. 1A. The signal probe (E17-PBA) was prepared by the click reaction of PBA (alkynyl modified) with the E17 (azido-modified) DNA strand. The capture probes were prepared by the click and amide reactions of the coupling agent DBCO-NHS (containing both alkynyl and ester groups) with the azido-modified DNAzyme strand and antibodies containing amino groups, respectively. Simultaneously, for the purpose of illustrating the successful preparation of the signal probe and the capture probes, they were verified by MC.38,39 As shown in Fig. 1B, lanes 1–8 were strips of E17, E17-PBA, P-VP, P-VP-Ab, P-SA, P-SA-Ab, P-ST, and P-ST-Ab, respectively. From the figure, when E17 was bound to phenylboronic acid and P-SA, P-ST and P-VP bound to their corresponding antibodies, the depth of the color of the DNA strands corresponding to lanes 2, 4, 6, 8 became shallow, and new electrophoresis bands appeared upper, which demonstrated the successful preparation of the signal probe (E17-PBA) and the capture probes (P-VP-Ab, P-SA-Ab, P-ST-Ab).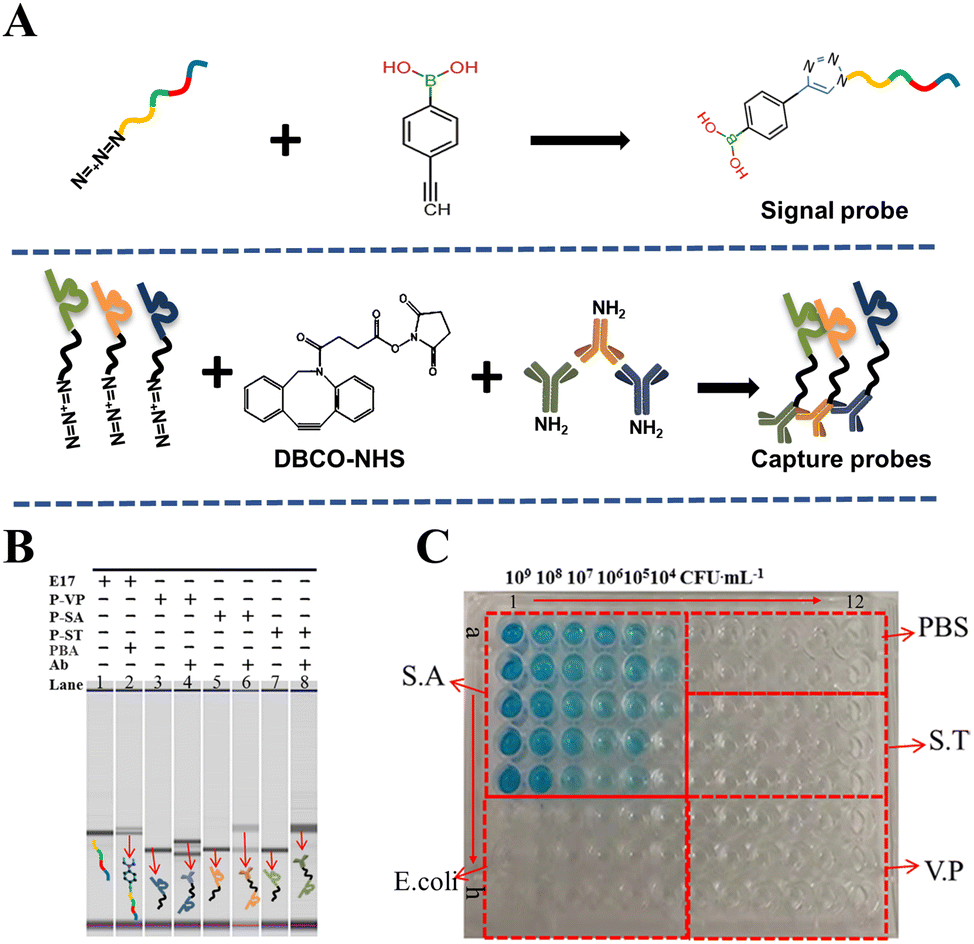 | ||
| Fig. 1 (A) The principle of signal probe and capture probes’ preparation. (B) Microfluidic chip proved the preparation of the signal probes and capture probes. (0.2 μM E17, P-SA, P-ST, P-VP, 2 μM PBA, 0.01 μM antibody, all final concentrations). (C) Verification of the binding of P-SA-Ab to S.A by ELISA. (All bacteria were cultured for 6 h, and the incubation time of P-SA-Ab with the bacteria was 1 h). | ||
With the further purpose of exploring whether the capture probe can bind to its corresponding bacteria, the enzyme-linked immunosorbent assay (ELISA) method was used for characterization. As displayed in Fig. 1C, a1–e6, f1–h6, f7–h12, c7–e12, and a7–b12 represent the ELISA results of Staphylococcus aureus (ATCC 25923), Escherichia coli (ATCC 25922), Vibrio parahaemolyticus (ATCC 17802), Salmonella typhimurium (ATCC 14028), and PBS (pH = 7.4), respectively. Columns 1–6 and 7–9 both showed the concentration gradients of different bacteria (109, 108, 107, 106, 105, and 104 CFU mL−1), and columns a–e and f–h displayed the concentration gradients of the capture probes (1:5, 1:10, 1:20, 1:40, and 1:80). It could be concluded from the figures that the blue color changes only occurred in the presence of S.A, while the other bacteria and the blank control group showed no color change, which meant that the prepared P-SA-Ab capture probe had excellent specificity binding to S.A.40 The conditions of the remaining two capture probes (P-ST and P-VP) binding with their corresponding bacteria are revealed in Fig. S1.† From the blue distributions in Fig. S1A and S1B,† it can be perceived that the binding of the P-ST-Ab and P-VP-Ab capture probes to their corresponding bacteria also had prominent specificity.
Characterization of the multivalent DNA walker sensor
To clearly observe the enrichment of the target pathogenic bacteria by the gold stirring rod, the individual bacteria, gold stirring rod, and gold stirring rod after the enrichment of bacteria were photographed by a scanning electron microscope (SEM) separately for directly observing the enrichment effect. Fig. 2A–C and D depicted the SEM images of the three pathogenic bacteria and gold stirring rod, respectively. It can be seen from the figures that V.P and S.T were both rod-shaped with smooth surfaces, while the V.P were slimmer than S.T, S.A was spherical and had distinct boundaries, and all three bacteria exhibit unique morphology.41 The surface finish of the gold stirring rod alone was inadequate, which may be attributed to the soaking time of the piranha solution being insufficient, resulting in incomplete cleaning. Fig. 2E and F respectively show the enrichment effect of unmodified and modified phenylboronic acid gold stirring rod on pathogenic bacteria. According to the figures, though the experimental conditions were identical, they were rare even when no pathogenic bacteria was captured on the gold stirring rod without the modification of phenylboronic acid, while the gold stirring rod modified with phenylboronic acid was loaded with different bacteria on a large area, which indicated that the gold rod-modified by phenylboronic acid not only greatly improved the capturing ability of pathogenic bacteria but can also capture a variety of pathogenic bacteria simultaneously.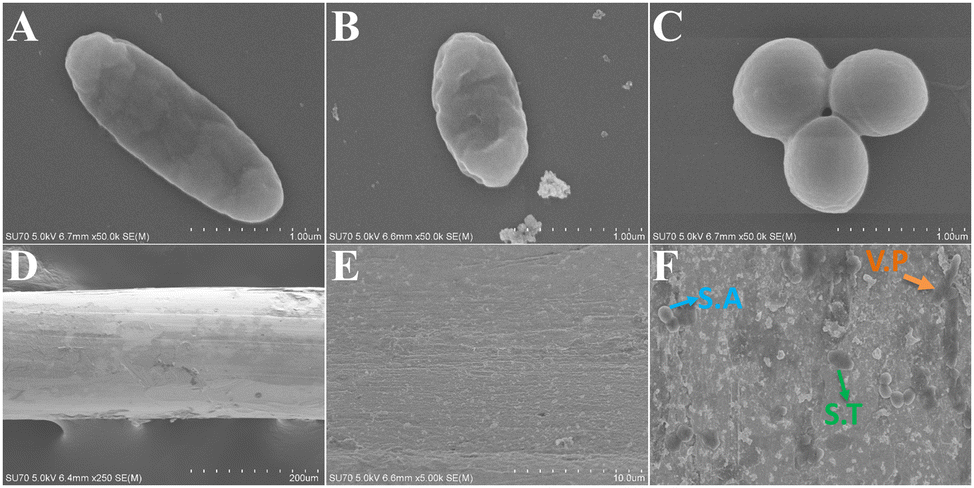 | ||
| Fig. 2 SEM of (A to C) single bacteria (from left to right: V.P, S.T, S.A), (D) single gold stirring rod and (E) its surface, (F) gold stirring rod simultaneously enriched three target pathogenic bacteria. | ||
To support the above point of view, a confocal microscope was used to prove the enrichment ability of the functionalized gold stirring rod for pathogenic bacteria. The pathogenic bacteria were stained with AO dye (the excitation wavelength at 488 nm, emission wavelength at 515 nm), and the confocal microscopy images of the pathogenic bacteria enriched by the gold stirring rod were obtained (Fig. 3). As can be clearly observed from the figures that all the subsequent gold stirring rods modified with phenylboronic acid (Fig. 3D–F) produced fluorescent signals and were evenly distributed in the field of view, however, the unmodified gold stirring rods had no fluorescence (Fig. 3A–C), which showed that the functionalized gold stirring rods were obtained from the enrichment of three target pathogenic bacteria indeed.
 | ||
| Fig. 3 Confocal microscopy images of bacteria enriched with gold stirring rod (A, B, C represents the images of V.P, S.T, and S.A enriched without PBA, respectively, and D, E, F represents the images of V.P, S.T, and S.A enriched with PBA, respectively). The concentration of pathogenic bacteria was 1 × 106 CFU mL−1 and PBA was 100 μM, the fluorescent dye was AO with excitation wavelength of 488 nm and emission wavelength of 515 nm, the enrichment time was 40 min. The scale bar is 50 μm. | ||
Feasibility of multivalent DNA walker amplification biosensor
The catalytic effect of Zn2+ on DNAzyme was demonstrated by the MC technology, and the specific cleavage relationship between the substrate chain and the DNAzyme chains was verified. From Fig. 4A, lanes 1–4 were the electrophoresis bands corresponding to the E17 substrate chain, P-VP, P-SA, and P-ST DNAzyme chains. Lanes 5–7 were the electrophoretic bands of the products obtained by mixing E17 with P-VP, P-SA, and P-ST respectively. When Zn2+ was presented, E17 hybridized with the corresponding DNAzyme chain, triggering the cleavage effect of DNAzyme. Compared with lane 1, the concentration of the band corresponding to E17 decreased, and two new bands were produced in lanes 5, 6, and 7. The new bands originated from the cleavage DNA fragments, which were obtained by combining P-VP, P-SA, and P-ST with E17 to form an 8–17 DNAzyme conformation for cutting the corresponding rA sites and liberating different length of DNA chains. These results clearly showed that the capture probes based on 8–17 DNAzyme designed by us can be utilized to cleave E17 and generate three characteristic DNA fragments that can be separated and detected by MC.42 | ||
| Fig. 4 Microfluidic chip technology to confirm the feasibility of the capture probes’ specific cleavage of the signal probe. (A) Without pathogenic bacteria. (The probes were dispersed in PBS buffer (pH 7.4), their final concentration was 0.5 μM, and the cleavage time were 0.5 h). (B) With pathogenic bacteria. (C) The curve of corresponding electrophoresis of (B). (The concentration of pathogenic bacteria was 1 × 106 CFU mL−1 and probes were 0.2 μM; other conditions were the same as in (A)). | ||
For the sake of testing the performance of the DNA walker whose walking track was our pathogenic bacteria, MC was applied to detect its feasibility and the mutual interference between them. As explained in Fig. 4B, lane 1 represented the presence of the E17-PBA signal probe, there was only one band that appeared in the electrophoresis image. When P-VP-Ab and 106 CFU mL−1 V.P were added to the solution, a new DNA strand (about 8 bp, 16 nt) was generated in the corresponding electrophoretic image, and the signal intensity of the corresponding E17-PBA probe strand was reduced (lane 2). When P-ST-Ab and 106 CFU mL−1 S.T were added to the solution, a DNA strand of approximately 12 bp (24 nt) was produced, and the signal intensity of the E17-PBA probe strand decreased (lane 3). Similarly, when P-SA-Ab and 106 CFU mL−1 S.A were presented, a DNA strand of approximately 18 bp (36 nt) emerged, and the E17-PBA probe concentration also declined (lane 4). These results manifested that the pathogenic bacterium can be successfully used as walking track for the DNA walker, and the characteristic DNA fragments can be obtained. Moreover, supposing that the solution contained two target pathogenic bacteria and their corresponding capture probes synchronously, such as lane 5, there were S.T as well as S.A, with the concentration of the E17-PBA probe decreased and new bands were generated at 18 bp and 12 bp. In the presence of both S.A and V.P, or both S.T and V.P and their corresponding capture probes, likewise, the concentration of E17-PBA probe decreased, and the new bands of 18 bp, 8 bp, and 12 bp, 8 bp were produced in lanes 6 and 7, separately. Noteworthy, when the three kinds of bacteria and their corresponding capture probes existed in the meantime, the E17-PBA probe concentration in lane 8 weakened, and 18 bp, 12 bp, and 8 bp bands appeared in lane 8 homochromously, which illustrated that when multiple pathogenic bacteria coexisted, different pathogenic bacteria will emancipate the corresponding signal chains through the DNA walker, and there was no internal interference between them. As a result, the detection strategy can realize the simultaneous detection of multiple targets. Fig. 4C displayed the peak images obtained by the separation and simultaneous detection of three target pathogenic bacteria of MC. The results are shown in Fig. 4B, and the concurrent quantification of the three pathogenic bacteria was successfully achieved.
Optimization of experimental parameters
To obtain the best performance of the biosensor, the Zn2+ concentration, cleavage time, pH, and stirring rod's enrichment time were optimized. The cleavage of the substrate chain by the DNAzyme was catalyzed by Zn2+; accordingly, the concentration of Zn2+ has a certain effect on the DNA walker. Hereby, the influence of Zn2+ concentration in the range of 10–250 μM was analyzed. As shown in Fig. S2A,† when the signal intensity was the highest, the Zn2+ concentration was 100 μM. Next, the impact of the cleavage time of DNA walker in the range of 10–60 min was evaluated. With the extension of the incubation time, the signal gradually increased and reached a plateau at 30 min (Fig. S2B†). The results showed that 30 min was enough to complete the DNA walker cleaving process. The pH also had a major influence on the binding of phenylboronic acid to pathogenic bacteria. Consequently, the pH of the reaction buffer was the basic parameter that needed to be optimized. The influence of pH on the experiment was analyzed in the pH range of 3.0–10.0. Fig. S2C† showed that when the pH was 8.0, the signal intensity reached the maximum value, and then, as the pH value further increased, the signal value began to decrease, indicating that 8.0 was the optimal pH value for the experiment. In addition, the enrichment time of the gold stirring rod for pathogenic bacteria was also an important factor affecting the detection performance of the sensor. When the enrichment time reached 40 min, the signal value did not change much with the extension of time (Fig. S2D†), which revealed that the enrichment of the pathogenic bacteria was basically achieved in 40 min. Moreover, since the ratio between the capture probes and the signal probe played an important role in DNA walker signal amplification, we compared the signal responses of the two at different molar ratios. Fig. S3† clearly showed that the signal intensity of the three pathogenic bacteria attained the maximum when the molar ratio was 1:10 (capture probe/signal probe), and this molar ratio was applied in subsequent experiments.
Specificity and stability of the multivalent DNA walker sensor
Since multiple bacterial pathogens and target bacteria coexist in most instances, the specificity of the sensor should be confirmed. E. coli, Listeria monocytogenes, and Edwardsiella were selected as interfering bacteria to evaluate the specificity of our sensor (Fig. 5A). As expected, under optimal conditions, if there were no target bacteria in the sample, extremely low signal values in the blank sample and other bacteria samples were observed. In contrast, once the target such as V.P or S.T or S.A was presented, significant signals were generated accordingly, and there was no mutual interference between the three target bacteria, which confirmed that our work had excellent selectivity. Besides, the stability of the sensor within 10 days was also evaluated (Fig. 5B). Compared with the initial signal value, after 10 days, the signal intensity values of V.P, S.T, and S.A (1 × 106 CFU mL−1) decreased very slightly; each experiment was performed 3 times in parallel, and the relative average deviation was less than 5.0%, demonstrating that the DNA walker amplification sensor possessed satisfactory stability and repeatability. | ||
| Fig. 5 (A) The selectivity performance of the sensor with the interference of different bacteria. (B) Stability of the biosensor. (The concentration of bacteria was 1 × 106 CFU mL−1, pH was 8.0, the reaction buffer was PBS buffer and time was 0.5 h). | ||
The sensing performance of multivalent DNA walker amplification biosensors for V.P, S.T, and S.A
The dynamic responses of a series of V.P, S.T, and S.A with different concentrations under optimal conditions were simultaneously measured by the MC method to understand the detection limit of the DNA walker amplification sensor for the target (Fig. 6). From Fig. 6A, as the concentration of the three target pathogenic bacteria was enhanced from 10 to 108 CFU mL−1, the corresponding signal intensity value also rose, proving that the concentration value of the target bacteria and the intensity of signal were proportional. The standard curves of the logarithm of the concentration of V.P, S.T, and S.A and their signal intensity were plotted. For Fig. 5B, C, and D, the linear equations were IV.P = 2.39 × log[V.P] − 1.97, IS.T = 10.68 × log[S.T] − 7.98, and IS.A = 14.31 × log[S.A] − 7.70. The detection range of V.P, S.T, and S.A was 1 × 102–1 × 108, 1 × 101–1 × 108, and 1 × 101–1 × 108 CFU mL−1, and the detection limit was 28, 10, and 9 CFU mL−1, respectively. In addition, some previous methods for the simultaneous detection of bacteria were compared with this method to explain our advantages (Table S4†). The detection limits of the three bacteria were obtained by this method respectively, indicating a wider detection range and lower target detection limits than most previously available methods. Apart from this, for the detection of three kinds of bacteria, first, the reason why our method greatly shortened (<1 h) the detection time compared with the fluorescence method of Wu's group (>2 h) was due to the labelling of the fluorescent probe that they modified on the DNA strand, which increased the complexity of probe preparation and thus prolonged the time.43 The probes did not require any fluorophore labelling and were simple to prepare. For another, the MC detection platform can realize the simultaneous injection and detection of multiple samples, further reducing the detection time. | ||
| Fig. 6 (A) Microfluidic chip technology determination of V.P, S.T, and S.A in the concentration range from 10 CFU mL−1 to 1 × 108 CFU mL−1. (B), (C), and (D) Correspond to the calibration plots for V.P, S.T, and S.A, respectively. (The signal probe was 0.2 μM and the capture probes were 0.02 μM, pH was 8.0, the reaction buffer was PBS buffer, and time was 0.5 h). | ||
Determination of the biosensor in different water samples
To further determine the applicability of the method in actual samples, different water samples and mixed spiked samples with target bacteria were used as actual samples were analyzed for their detection conditions. The results are summarized in Table S5.† The recovery rates of V.P, S.T, and S.A in the four aquatic samples were all above 90%, and the RSD values were all less than 10%. The above results indicated that the biosensor platform can be used to simultaneously detect three kinds of foodborne bacteria that may coexist in complex samples and has great potential for the simultaneous detection of V.P, S.T, and S.A in water samples.Discussion of the multivalent DNA walker amplification effect
The loading number of the probes on the targets bacteria over time was measured by UV spectrophotometry to verify the signal amplification effect of the designed sensor. As can be seen from Fig. S4,† the DNA loading on the surface of the pathogenic bacteria had basically reached saturation at 30 min, and the loading number of the probes remained almost unchanged with the further increase in time. According to Lambert–Beer law, the DNA concentrations on V.P, S.T, and S.A were calculated to be 3.16 × 10−7, 1.2 × 10−6, and 1.5 × 10−6 M; thus, the loading number of the probes on the above three pathogenic bacteria was further calculated to be 3.16 × 10−15, 1.2 × 10−14, and 1.5 × 10−14 mol CFU−1, respectively. In addition, to determine the enzymatic activity of the designed sensor, the kinetics of the enzymatic reaction was quantitatively described using the classic Michaelis equation (eqn (1)), | (1) |
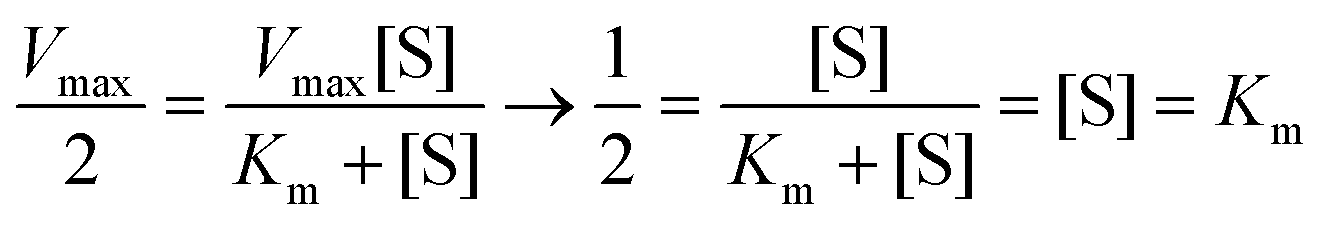 | (2) |
Thus, the Km value was the substrate concentration when the enzyme reaction rate reached half the maximum reaction rate (eqn (2)). Therefore, the changes in the reaction rate at different substrate concentrations were measured; the consequences were observed from Fig. S4,†Km (V.P) = 0.23 μM, Km (S.T) = 0.3 μM, and Km (S.A) = 0.34 μM, which indicated good affinity between the capture probes and the signal probe.
DNA walker was used to take gold nanoparticles as the walking tracks. Thereby, the signal amplification effects of the target pathogens and gold nanoparticles can be compared as walking tracks. Fig. S5† revealed that under the same conditions, the signal amplification effect of the DNA walker on the targets were better than that on gold nanoparticles. The possible reason was that gold nanoparticles had strong adsorption effect on DNA, which produced a certain steric hindrance effect and was not conducive to the progress of DNA walker. As the target, pathogenic bacteria not only effectively avoided the steric hindrance effect but, at the same time, its larger specific surface area can accommodate more probe strands; thus, its signal amplification effects were greater over gold nanoparticles. The key concept of the DNA walker was the high mobility of the DNA walker after activation. DNAzyme consists of a catalytic core with two binding domains on both sides. Once the DNA walker was activated, the DNAzyme hybridizes with the substrate strand through the two binding domains. For this reason, the influence of the binding length between the DNAzyme chain and the substrate chain on the experiment was studied. Fig. S6† showed that when the base binding numbers of the signal probe and corresponding capture probes on V.P, S.T, and S.A were 9/8, 10/8, and 10/8, respectively, the signal was the strongest. A longer binding domain can significantly increase the binding efficiency between the capture probes and the signal probe, thereby enhancing the probability of cleavage. However, a very long binding domain will prevent the capture probes from hybridizing with the next signal probe to reduce the mobility of the DNA walker, thus affecting the reaction rate. Thus, the base binding lengths of 9/8, 10/8, and 10/8 of the capture probes and the signal probe were selected as the probe chains for V.P, S.T, and S.A, respectively. Furthermore, through the bioluminescence method, it has been proved that the pathogenic bacteria used in the experiment were alive (Fig. S7†). Therefore, the DNA walker amplification sensor can be applied for the detection of live bacteria.
Conclusion
In summary, a multivalent DNA walker sensor combined with MC technology was developed to successfully realize the simultaneous, rapid, selective, and sensitive detection of V.P, S.T, and S.A. Antibody-functionalized DNAzyme chains endowed the sensor with excellent specificity. At the same time, the ingenious design of the substrate chain (with three rA cleavage sites) successfully achieved the simultaneous quantitative detection of the targets of the three pathogens. The multivalent DNA walker sensor applied the target pathogen directly as walking tracks, effectively avoided the generation of false positive signals, and reduced the steric hindrance effect caused by traditional nanoparticle tracks. In addition, the developed sensor integrated gold stirring rod enrichment and DNA walker to achieve dual signal amplification, which significantly improved the detection sensitivity. The detection time of V.P, S.T, and S.A was within 30 min. The detection range of V.P, S.T, and S.A was 1 × 102–1 × 108, 1 × 101–1 × 108, and 1 × 101–1 × 108 CFU mL−1, and the detection limit was 28, 10, and 9 CFU mL−1, respectively. It is worth mentioning that by changing the design of the signal probe and the capture probe, the DNA walker sensor is expected to become a general platform for the simultaneous detection of different targets.Author contributions
Zhenli Xu: Writing – original draft, investigation, data curation and analysis. Jiaqi Wang: Investigation and data curation. Zhijian Jia: Writing instructions. Yong-Xiang Wu, Ning Gan, and Shaoning Yu: Project administration, funding acquisition, directed the project and modified the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21974074), Zhejiang Province Public Welfare Technology Application Research Analysis Test Plan (No. LGC20B050006), Zhejiang Provincial and Ningbo Natural Science Foundation (Y23B050013, Y23B050014, Y23C200022, 2019A610194), Major Scientific and Technological Tasks of Ningbo (Grant no. 2021Z056, 2022Z170, 2022S011) and K. C. Wong Magna Fund in Ningbo University.References
- G. Maduraiveeran, M. Sasidharan and V. Ganesan, Biosens. Bioelectron., 2018, 103, 113–129 CrossRef CAS PubMed.
- X. Weng, C. Zhang and H. Jiang, LWT – Food Sci. Technol., 2021, 151, 112172 CrossRef CAS.
- Y. Ji, L. Zhang, L. Zhu, J. Lei, J. Wu and H. Ju, Biosens. Bioelectron., 2017, 96, 201–205 CrossRef CAS PubMed.
- X. Peng, J. Zhu, W. Wen, T. Bao, X. Zhang, H. He and S. Wang, Biosens. Bioelectron., 2018, 118, 174–180 CrossRef CAS PubMed.
- J. Chen, Y. R. Baker, A. Brown, A. H. El-Sagheer and T. Brown, Chem. Sci., 2018, 9, 8110–8120 RSC.
- S. Gong, S. Zhang, X. Wang, J. Li, W. Pan, N. Li and B. Tang, Anal. Chem., 2021, 93, 15216–15223 CrossRef CAS PubMed.
- Q. Kang, M. He, B. Chen and B. Hu, Sens. Actuators, B, 2021, 345, 130370 CrossRef CAS.
- J. Dong, D. Zhang, C. Li, T. Bai, H. Jin and Z. Suo, Bioelectrochemistry, 2022, 146, 108134 CrossRef CAS PubMed.
- S. D. Mason, Y. Tang, Y. Li, X. Xie and F. Li, Trends Anal. Chem., 2018, 107, 212–221 CrossRef CAS.
- D. Wang, C. Vietz, T. Schröder, G. Acuna, B. Lalkens and P. Tinnefeld, Nano Lett., 2017, 17, 5368–5374 CrossRef CAS PubMed.
- M. Skugor, J. Valero, K. Murayama, M. Centola, H. Asanuma and M. Famulok, Angew. Chem., Int. Ed., 2019, 58, 6948–6951 CrossRef CAS PubMed.
- E. Xiong, D. Zhen, L. Jiang and X. Zhou, Anal. Chem., 2019, 91, 15317–15324 CrossRef CAS PubMed.
- M. Xiao, K. Zou, L. Li, L. Wang, Y. Tian, C. Fan and H. Pei, Angew. Chem., Int. Ed., 2019, 58, 15448–15454 CrossRef CAS PubMed.
- J. Chen, Z. Luo, C. Sun, Z. Huang, C. Zhou, S. Yin, Y. Duan and Y. Li, Trends Anal. Chem., 2019, 120, 115626 CrossRef CAS.
- Y. Li, G. A. Wang, S. D. Mason, X. Yang, Z. Yu, Y. Tang and F. Li, Chem. Sci., 2018, 9, 6434–6439 RSC.
- P. Q. Ma, C. P. Liang, H. H. Zhang, B. C. Yin and B. C. Ye, Chem. Sci., 2018, 9, 3299–3304 RSC.
- F. Wang, J. Elbaz, C. Teller and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 295–299 CrossRef CAS PubMed.
- L. Song, Y. Zhuge, X. Zuo, M. Li and F. Wang, Adv. Sci., 2022, 9, 2200327 CrossRef CAS PubMed.
- Y. Takezawa, L. Hu, T. Nakama and M. Shionoya, Angew. Chem., Int. Ed., 2020, 59, 21488–21492 CrossRef CAS PubMed.
- Y. Yin, G. Chen, L. Gong, K. Ge, W. Pan, N. Li, J. O. a. Machuki, Y. Yu, D. Geng, H. Dong and F. Gao, Anal. Chem., 2020, 92, 9247–9256 CrossRef CAS PubMed.
- J. Huang, L. Zhu, H. Ju and J. Lei, Anal. Chem., 2019, 91, 6981–6985 CrossRef CAS PubMed.
- F. Luo, F. Chen, Y. Xiong, Z. Wu, X. Zhang, W. Wen and S. Wang, Anal. Chem., 2021, 93, 4506–4512 CrossRef CAS PubMed.
- H. Gan, J. Wu and H. Ju, Anal. Chim. Acta, 2019, 1074, 142–149 CrossRef CAS PubMed.
- L. Ji, L. Zhang, H. Yang, S. Liang, J. Pan, Y. Zou, S. Li, Q. Li and S. Zhao, J. Colloid Interface Sci., 2022, 621, 489–498 CrossRef CAS PubMed.
- F. Mi, M. Guan, C. Hu, F. Peng, S. Sun and X. Wang, Analyst, 2021, 146, 429–443 RSC.
- J. C. Jokerst, J. A. Adkins, B. Bisha, M. M. Mentele, L. D. Goodridge and C. S. Henry, Anal. Chem., 2012, 84, 2900–2907 CrossRef CAS PubMed.
- F. Huang, H. Zhang, L. Wang, W. Lai and J. Lin, Biosens. Bioelectron., 2018, 100, 583–590 CrossRef CAS PubMed.
- S. Yao, B. Pang, Y. Fu, X. Song, K. Xu, J. Li, J. Wang and C. Zhao, Sens. Actuators, B, 2022, 359, 131581 CrossRef CAS.
- J. Kampeera, P. Pasakon, C. Karuwan, N. Arunrut, A. Sappat, S. Sirithammajak, N. Dechokiattawan, T. Sumranwanich, P. Chaivisuthangkura, P. Ounjai, S. Chankhamhaengdecha, A. Wisitsoraat, A. Tuantranont and W. Kiatpathomchai, Biosens. Bioelectron., 2019, 132, 271–278 CrossRef CAS PubMed.
- J. E. Lee, H. Mun, S. R. Kim, M. G. Kim, J. Y. Chang and W. B. Shim, Biosens. Bioelectron., 2020, 151, 111968 CrossRef CAS PubMed.
- M. Wu, W. Chen, G. Wang, P. He and Q. Wang, Food Chem., 2016, 209, 154–161 CrossRef CAS PubMed.
- Y. Zhao, D. Zeng, C. Yan, W. Chen, J. Ren, Y. Jiang, L. Jiang, F. Xue, D. Ji, F. Tang, M. Zhou and J. Dai, Analyst, 2020, 145, 3106–3115 RSC.
- J. Yin, Z. Zou, Z. Hu, S. Zhang, F. Zhang, B. Wang, S. Lv and Y. Mu, Lab Chip, 2020, 20, 979–986 RSC.
- Y. Shang, X. Xiang, Q. Ye, Q. Wu, J. Zhang and J.-M. Lin, Trends Anal. Chem., 2022, 147, 116509 CrossRef CAS.
- W. Chen, T. Li, S. He, D. Liu, Z. Wang, W. Zhang and X. Jiang, Sci. China: Chem., 2011, 54, 1227–1232 CrossRef CAS.
- M. Du, J. Zheng, S. Tian, Y. Liu, Z. Zheng, H. Wang, J. Xia, X. Ji and Z. He, Anal. Chem., 2021, 93, 5606–5611 CrossRef CAS PubMed.
- Y. Tsuchido, R. Horiuchi, T. Hashimoto, K. Ishihara, N. Kanzawa and T. Hayashita, Anal. Chem., 2019, 91, 3929–3935 CrossRef CAS PubMed.
- N. Gan, L. Xie, K. Zhang, Y. Cao, F. Hu and T. Li, Sens. Actuators, B, 2018, 272, 526–533 CrossRef CAS.
- L. Xie, Y. Cao, F. Hu, T. Li, Q. Wang and N. Gan, Microchim. Acta, 2019, 186, 547 CrossRef PubMed.
- H. Xiang, F. Cao, D. Ming, Y. Zheng, X. Dong, X. Zhong, D. Mu, B. Li, L. Zhong, J. Cao, L. Wang, H. Ma, T. Wang and D. Wang, Appl. Microbiol. Biotechnol., 2017, 101, 6671–6681 CrossRef CAS PubMed.
- C. Xu, J. Li, L. Yang, F. Shi, L. Yang and M. Ye, Food Control, 2017, 73, 1445–1451 CrossRef CAS.
- Y. Chen, Y. Xiang, R. Yuan and Y. Chai, Nanoscale, 2015, 7, 981–986 RSC.
- S. Wu, N. Duan, Z. Shi, C. Fang and Z. Wang, Anal. Chem., 2014, 86, 3100–3107 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an01941h |
| This journal is © The Royal Society of Chemistry 2023 |