
In-source fragmentation of nucleosides in electrospray ionization towards more sensitive and accurate nucleoside analysis†
Yu-Nan
Chen‡
a,
Xu-Yang
Shen‡
a,
Yue
Yu
b,
Chen-Yu
Xue
c,
Ying-Lin
Zhou
 *a and
Xin-Xiang
Zhang
a
*a and
Xin-Xiang
Zhang
a
aBeijing National Laboratory for Molecular Sciences (BNLMS), MOE Key Laboratory of Bioorganic Chemistry and Molecular Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing, China. Tel: +86-10-62754112; Fax: +86-10-62754112; E-mail: zhouyl@pku.edu.cn
bInstitute of Biotechnology Development, Qilu Pharmaceutical, Jinan, China
cKey Laboratory of Forensic Toxicology, Ministry of Public Security, Beijing, China
First published on 21st February 2023
Abstract
Nucleosides have been found to suffer in-source fragmentation (ISF) in electrospray ionization mass spectrometry, which leads to reduced sensitivity and ambiguous identification. In this work, a combination of theoretical calculations and nuclear magnetic resonance analysis revealed the key role of protonation at N3 near the glycosidic bond during ISF. Therefore, an ultrasensitive liquid chromatography-tandem mass spectrometry system for 5-formylcytosine detection was developed with 300 fold signal enhancement. Also, we established a MS1-only platform for nucleoside profiling and successfully identified sixteen nucleosides in the total RNA of MCF-7 cells. Taking ISF into account, we can realize analysis with higher sensitivity and less ambiguity, not only for nucleosides, but for other molecules with similar protonation and fragmentation behaviors.
1 Introduction
Mass spectrometry (MS) is a powerful and versatile tool for identification, quantification and mapping.1 Among the ion sources for MS, electrospray ionization (ESI) is generally considered “soft” and leads to few changes in the target analytes. However, in-source fragmentation (ISF) occurs naturally in ESI,2 wherein the pressure decreases sharply from the source to the analyzer, and the accelerating voltage increases the internal energy of the ions.3 Therefore, the ions become fragile, with the fragmentation ratios (FRs) varying for different structures and different instrumental designs.4 So far, there have been several works showing that peptides,5 sugar phosphates,2,6 lipids,3,4 phenethylamines,7etc. can fragment in the ESI process. For omics research, especially metabolomics2 and lipidomics,3 unintentional ISF leads to misannotation of analytes or even loss of their signals. Likewise, ISF hampers MS-based quantification of some analytes, reducing the sensitivity of detection.Nucleosides are endogenous structural components of nucleic acids. They have a large variety of potential structural modifications, which have drawn wide attention in the past decades.8 Among the modified nucleosides, epigenetically modified deoxycytosine (dC) derivatives, including 5-hydroxymethylcytosine (5mdC),9 5-hydroxymethylcytosine (5hmdC),10 5-formylcytosine (5fdC), and 5-carboxylcytosine (5cadC) are of particular interest.11 These nucleosides are involved in the active DNA demethylation in mammals, in which 5mdC is oxidized to the other three in the presence of ten-eleven translocation (TET) proteins.10–12 For detection of these modified deoxycytosines, sensitivity is a major concern due to their extremely low abundance in biological samples. For example, 5fdC constitutes merely 1–20 ppm of the DNA of mammal cells,11 making it necessary to develop detection methods with excellent sensitivity and selectivity. Currently, MS coupled with separation techniques such as high performance liquid chromatography (HPLC) is the most popular and recognized platform for quantification and qualification of nucleosides.10,11,13–16 Plenty of research has been done on methods to improve the performance of MS, including introducing additives to the mobile phase of HPLC17 and chemical derivatization. Chemical derivatization is an important post-column treatment that improves the ionization efficiency of analytes by introducing easily ionized and highly hydrophobic groups, thereby improving the MS response.18–21 Our group has shown that after fast labeling with a hydrazinotriazine reagent, the limit of detection (LOD) of 5hmdC, 5fdC and 5cadC can be significantly decreased by nearly 100 fold using the multiple reaction monitoring (MRM) mode of triple quadrupole MS.22,23
Here, we examined the MS1 spectra of nucleosides and found the existence of fragment ions. This suggested that ISF also took place in MS analysis of nucleosides, which has previously been neglected and lacked detailed investigation. In this work, several major nucleosides and their derivatives were analyzed to identify their ISF pattern. Confirming that ISF resulted from cleavage of the glycosidic bond, the separation method and MS parameters were changed, respectively, to determine the main factors affecting ISF. Furthermore, theoretical calculations and nuclear magnetic resonance (NMR) analysis were performed to determine the possible mechanism. According to the mechanism, we finally mitigated ISF by chemical derivatization and parameter optimization, and achieved highly-sensitive quantitation of 5fdC. Based on ISF, nucleosides of total RNA from MCF-7 were profiled within Q1 spectra.
2 Experimental section
2.1 Chemicals and reagents
LC-MS grade methanol (MeOH) and acetonitrile (ACN) were obtained from J. T. Baker®. Formic acid (FA, LC-MS grade) was purchased from Tokyo Chemical Industry (Tokyo, Japan). Acetic acid (HAc, 99.9985%) was purchased from Alfa Aesar (Ward Hill, MA, USA). Rhodamine B hydrazide (RBH) was purchased from J&K Scientific Ltd (Beijing, China). The ultrapure water used in this work was produced using a MilliQ system (Merk Millipore, Darmstadt, Germany).Adenosine (rA), cytidine (rC), 5-methylcytidine (mrC), 2′-deoxyadenosine (dA), 2′-deoxycytosine (dC), 2′-deoxyguanosine (dG), thymidine (dT) and 5mdC standards were purchased from J&K Scientific Ltd (Shanghai, China). N6-Methyladenosine (m6rA) was purchased from Macklin (Shanghai, China). Pseudouridine (ψ) was purchased from Toronto Research Chemicals (Ontario, Canada). 2′-O-Methylguanosine (rGm) and 2′-O-methylcytidine (rCm) were purchased from Aladdin (Shanghai, China). 2′-O-Methyladenosine (rAm), guanosine (rG) and uridine (rU) standards were purchased from Shanghai Yuanye Bio-Technology Co., Ltd (Shanghai, China). The 2′-O-methyluridine (rUm) standard was purchased from Meilunbio® (Dalian, China). N1-Methyladenosine (m1rA), 5hmdC, 5fdC, and 5cadC were purchased from Berry & Associates (Dexter, MI, USA). The three labeling reagents Me2N, Et2N, and i-Pr2N were synthesized according to our previous work.24 Their structures are shown in Fig. S1.†
2.2 Chemical labeling
5fdC has a carbonyl group that can easily react with the hydrazine group of our labeling reagents to form a Schiff base. For 5fdC, the derivatization reactions were performed by mixing a 10 μL aliquot of 100 nM (or less) 5fdC and 10 μL of 5 mM labeling reagent in MeOH, where 0.2 μL (1%) HAc was added for catalysis. After vortexing for several seconds, the mixture was dried and reconstituted in 40 μL of water containing 50% MeOH (v/v).2.3 Analysis of unlabeled and labeled nucleosides by LC-MS
A 1 μM mixed standard solution containing eight types of deoxyribonucleoside (dN), dA, dT, dC, dG, 5mdC, 5fdC, 5cadC and 5hmdC, and another containing twelve types of ribonucleoside, rA, rU, rC, rG, m1rA, m6rA, mrC, rAm, rUm, rCm, rGm and ψ, were prepared. Analysis of the nucleosides was performed on an LC-MS system consisting of a Thermo Scientific Q Exactive MS system with an ESI source (Thermo, USA) and a Thermo Scientific Dionex Ultimate 3000 HPLC (Thermo, USA). Data acquisition and processing were performed using Xcalibur (Thermo, USA). The HPLC separation was performed using a Zorbax StableBond Analytical SB-C18 column (2.1 mm × 100 mm, 3.5 μm, Agilent Technologies, USA) at 35 °C. Water containing 0.0085% FA (v/v, solvent A) and MeOH containing 0.0085% FA (v/v, solvent B) were employed as the mobile phase. A gradient of 0–5% B for 6 min, 5–80% B for 0.5 min, 80% B for 5 min, and 100% B for 5 min was used. The flow rate of the mobile phase was set at 0.3 mL min−1. For each run, the injection volume was 5 μL. The MS detection was performed in positive ESI mode. The nucleosides and labeled products were monitored using the full-scan mode within the m/z range of 100–700. The sheath gas and auxiliary gas flows were set as 35 and 10 arb. units, according to the recommendations of the instrument manual. The values of the spray voltage and capillary temperature were adjusted in the experiments.2.4 Synthesis of mg-level 5fdC-Et2N for NMR
20 μmol 5fdC and 20 μmol Et2N were mixed together in 1.8 mL methanol with 0.2 mL acetic acid, then heated at 60 °C for 3 h. The solvent was removed using a vacuum concentrator and the residue was reconstituted with 100 μL methanol. Thin layer chromatography was applied for purification of the product with 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) CH2Cl2:MeOH (Rf = 0.5). 2 mg white solid was obtained in a yield of 20%.
:1 (v/v) CH2Cl2:MeOH (Rf = 0.5). 2 mg white solid was obtained in a yield of 20%.
2.5 Theoretical calculations
The structure optimization and frequency calculation of the nucleosides were carried out using r2scan-3c.25 The single-point energy was calculated via density functional theory (DFT) using the B3LYP functional26 with D3 correction,27 the def2-TZVP basis set and the def2/J auxiliary basis set,28,29 accelerated by the RIJCOSX approximation.30 All calculations were carried out using ORCA 4.2.1,31,32 and the input files were generated with Multiwfn.332.6 Extraction and enzymatic digestion of total RNA
The total RNA of MCF-7 was extracted using a TransZol Up Plus RNA Kit (TransGen Biotech, Beijing, China) according to the manufacture's recommended procedure. 10× Nucleoside Digestion Mix Reaction Buffer and 1 μL of the Nucleoside Digestion Mix (New England Biolabs, USA) were added to extracted RNA for enzymatic digestion. The reaction mixture was then incubated at 37 °C for 3 h. The prepared sample could be analyzed using the HPLC-MS system directly.2.7 Profiling of nucleosides within Q1 spectra using Python
Q1 spectra (.raw) from HRMS (Q Exactive, Thermo) were first converted to .ms1 format using msConvert (ProteoWizard34). Then Python was applied for data transposition, alignment, extraction and plotting.3 Results and discussion
3.1 In-source fragmentation of nucleosides
The two mixed solutions of deoxyribonucleosides (dNs) and ribonucleosides (rNs) were analyzed by HPLC-MS, respectively. All nucleosides except pseudouridine (ψ) lost their ribose or deoxyribose ring to varying degrees in the ESI source during HPLC-MS, leaving the nitrogenous bases as the fragment ions (Fig. 1a and b, S1a†). The FR of each nucleoside was calculated according to the following formula: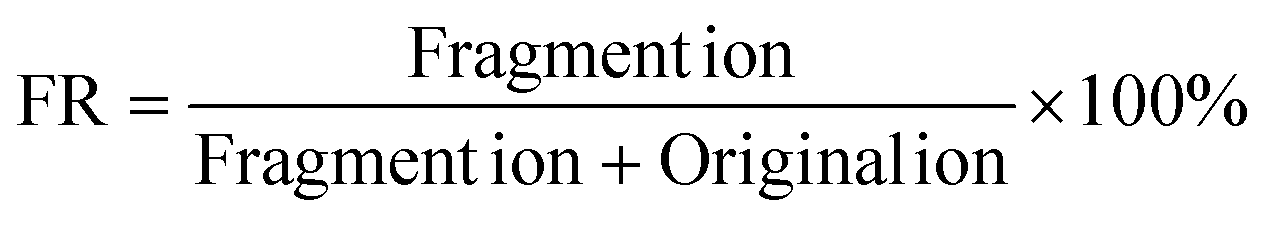 | (1) |
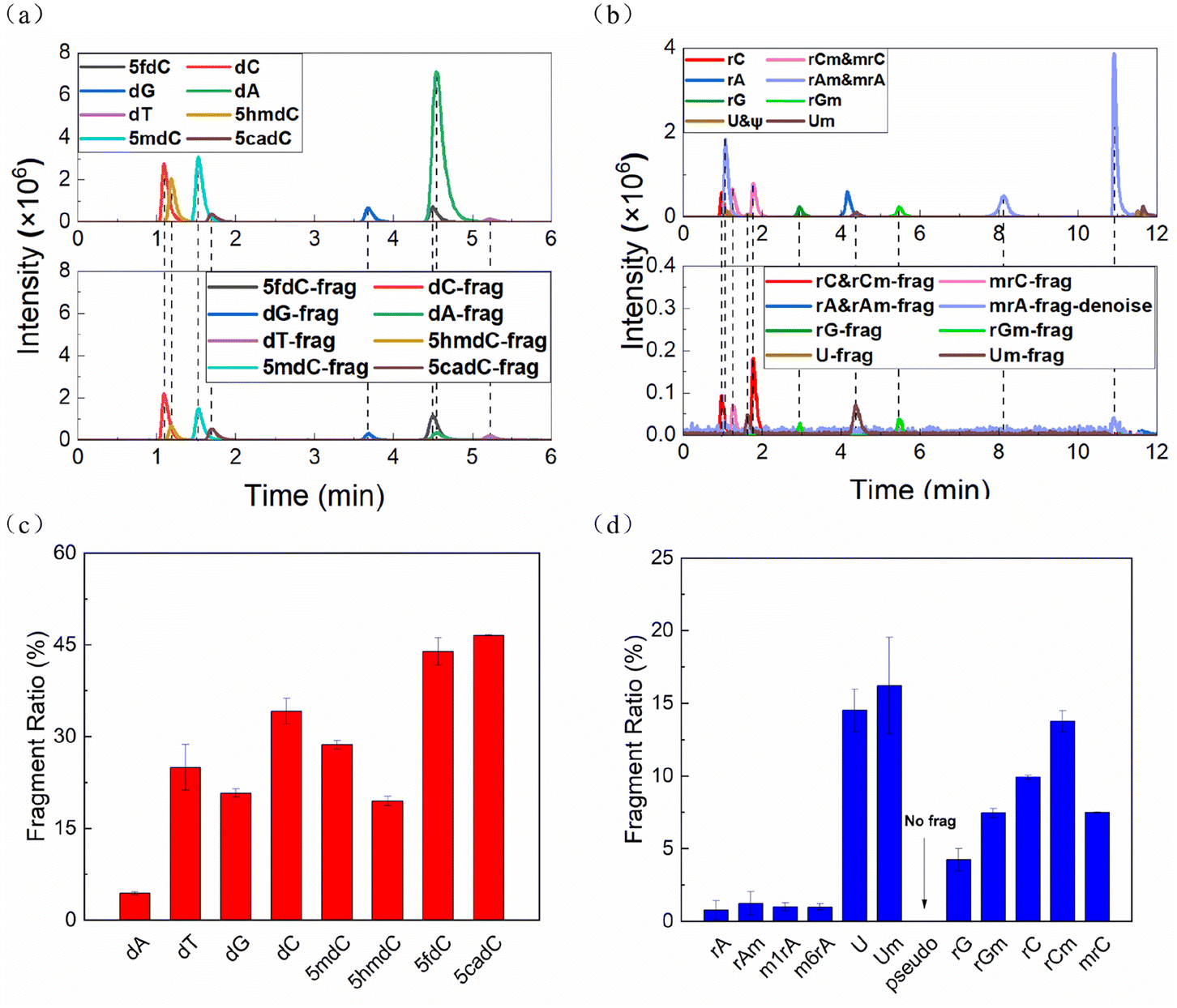 | ||
| Fig. 1 Extracted ion chromatograms (EICs) of the (a) deoxyribonucleoside mixture and (b) ribonucleoside mixture with HPLC-MS detection; FR of (c) deoxyribonucleosides and (d) ribonucleosides in the ESI ion source during HPLC-MS. | ||
The FRs of cytidines, uridines and their derivatives were particularly high, while the adenines and modified adenines were barely cleaved (Fig. 1c and d). Also, modifications affected the FR. For the same bases, dNs were more vulnerable than rNs. Methylation, taking rC as an example, on the C5 position of the base (mrC) stabilized the nucleoside, while methylation on the C2′ position of the ribose ring (rCm) had the opposite effect.
3.2 Labeling with hydrazine-based reagents and its influence on fragmentation
Based on previous work,22,23 we studied the effect of chemical derivatization on the fragmentation of 5fdC by using hydrazine-based labeling reagents including Me2N, Et2N, i-Pr2N and rhodamine B hydrazine (RBH) (Fig. S2†). The introduction of the hydrazinotriazine labeling reagents dramatically reduced the FRs of 5fdC from about 50% to about 10%. But it was more noteworthy that the outcome of RBH labeling was quite abnormal (Fig. S1 and S3†). RBH was used first for 5fdC labeling, having the same active hydrazine group but lower polarity and higher molecular weight, thus being favourable for improving ionization efficiency and MS responses. In the MS, however, it gave two kinds of species: singly charged and doubly charged forms. Both of these species could undergo ISF, but there was a huge difference between their FRs. The FR of the doubly charged ions (m/z: 347.671 → 289.647, 66%) was similar to that of 5fdC, while singly charged ions (m/z: 694.335 → 578.287, 17%) showed similar results to those of the other hydrazinotriazine labeling reagents. Therefore, we speculated that the nucleoside fragmentation was closely related to the difference in protonation between these two forms.3.3 Theoretical calculations and nuclear magnetic resonance evidence
To examine the mechanism of the ISF, we studied the relationship between the FR and molecular structure of cytosine, deoxycytosine and their derivatives. We calculated the free energy of formation (Gf) in a vacuum for these nucleosides, protonated at different positions including O2 and N3. As shown in Table S1,† the Gf of the molecules protonated at N3 was lower, indicating that all these nucleosides were more stable in a vacuum with protonation at N3. Likewise, the Gf of protonation of 5fdC-Et2N and 5fdC-RBH at different positions was calculated. Protonation at N2 resulted in the lowest Gf for 5fdC-Et2N (Table S1, Fig. S4†). We also prepared mg-level 5fdC-Et2N for NMR analysis. 13C spectra of 5fdC-Et2N in D-methanol and in D-methanol mixed with 1 mol L−1 HCl solution (3:1, v/v) were obtained, respectively. Distortionless enhancement by polarization transfer (DEPT) was also performed for peak assignment (Fig. S5†). Fig. 2a reveals that carbon atoms C6, C7 and C8 of 5fdC-Et2N experienced changes in chemical shift to lower field, indicating the occurrence of protonation at N2 of 5fdC-Et2N, which was in accordance with theoretical calculations.
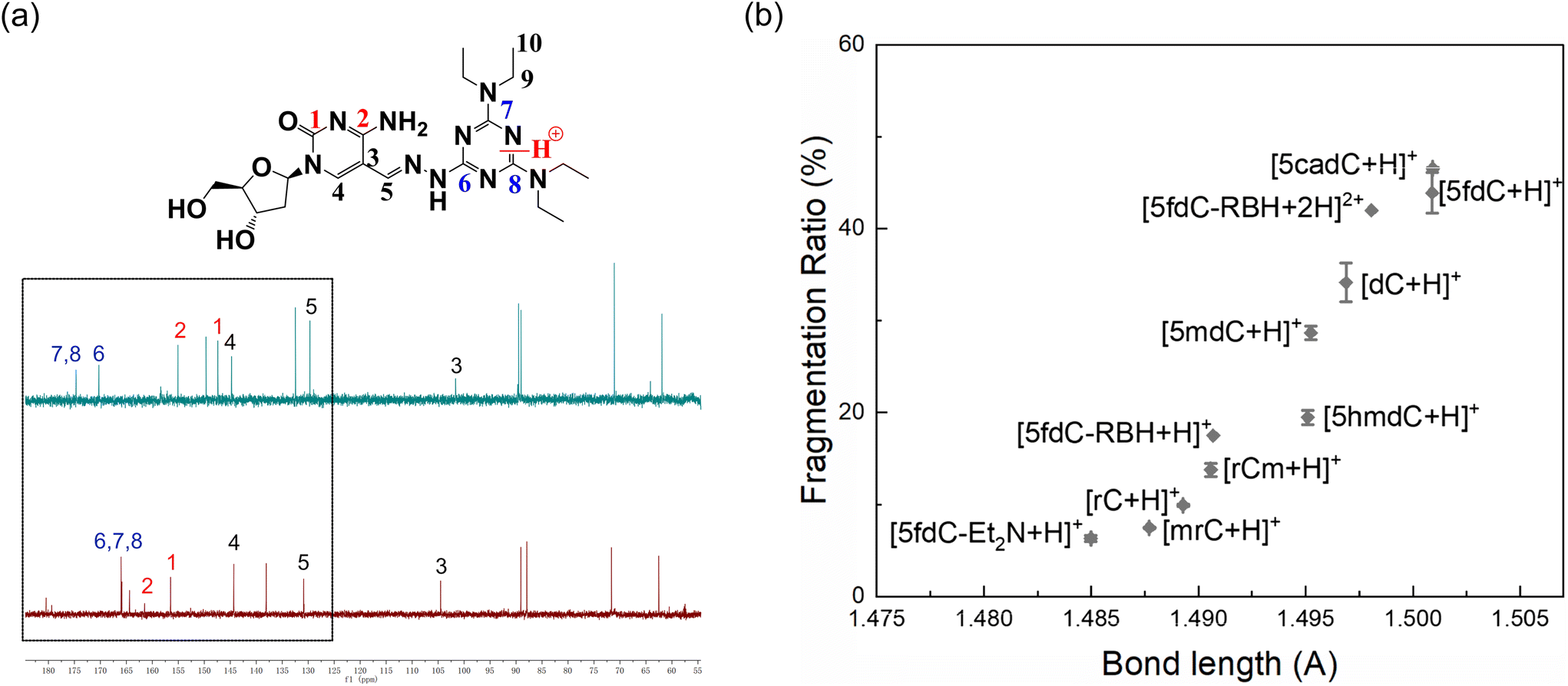 | ||
| Fig. 2 (a) 13C spectra of 5fdC-Et2N in D-methanol mixed with HCl solution (top) and D-methanol (bottom); (b) relationship between calculated bond lengths of protonated nucleosides and their FRs. | ||
For 5fdC-RBH (Table S1, Fig. S4†), molecules protonated at N1 were the most stable. But interestingly, the Gf of 5fdC-RBH gaining a proton at both N and N1 was even lower, indicating that 5fdC-RBH had a strong tendency to be doubly charged, which was consistent with experimental evidence.
The C–N bond lengths between the pyrimidine ring and the ribose were calculated for the optimized structures with the lowest Gf after protonation. The FR obtained in experiments and the C–N bond length given by calculations had a good positive correlation for the cytidines and their derivatives (Fig. 2b). Therefore, in the process of nucleoside ISF, the molecule becomes protonated and experiences charge transfer. Then the C–N bond is weakened and undergoes homolytic or heterolytic cleavage, followed by the migration of hydrogen radicals or protons between the two fragments, producing the fragment ions detected by MS (Fig. 3).
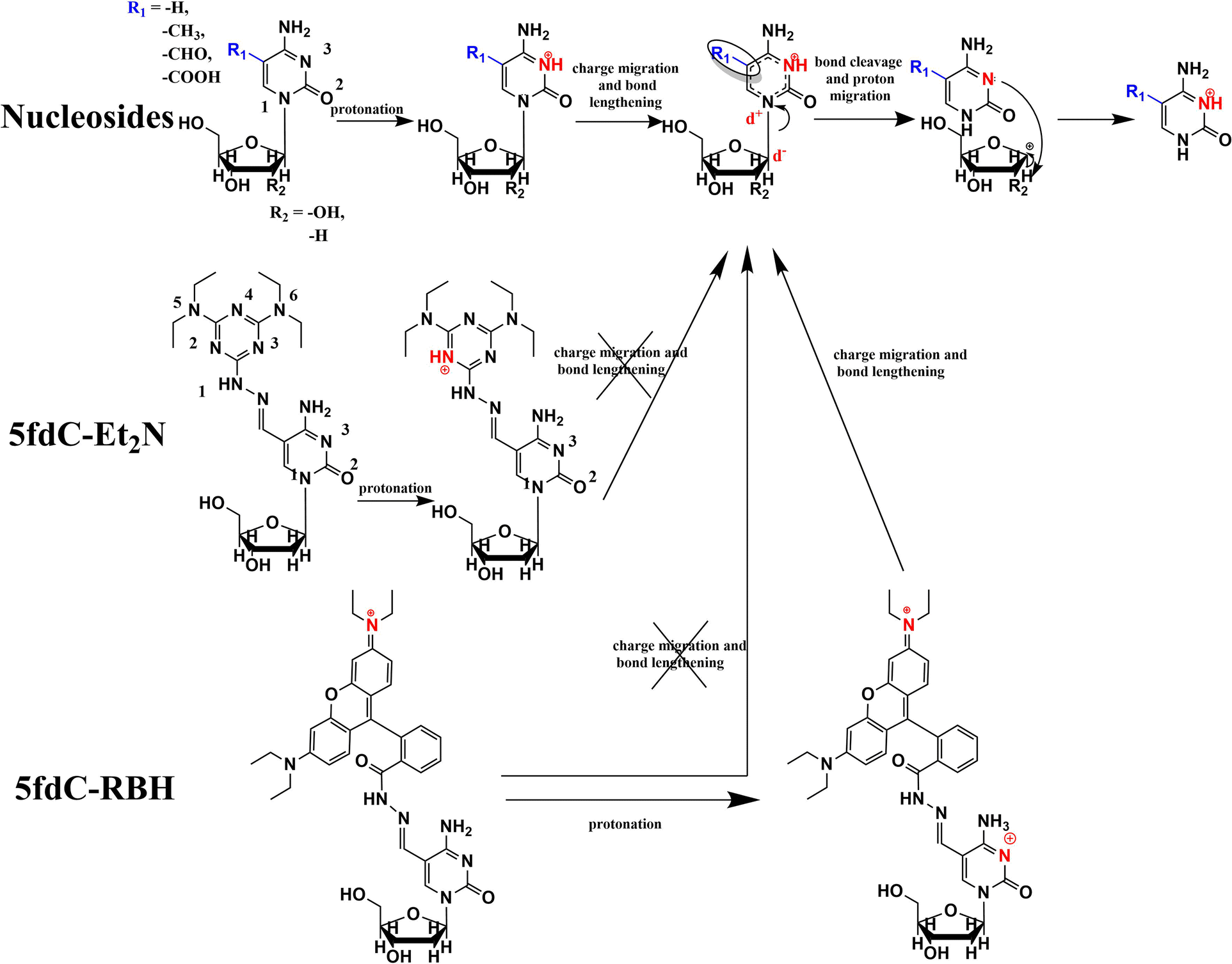 | ||
| Fig. 3 Proposed ISF mechanism of different modified cytidines after protonation. | ||
This assumed mechanism was also in accordance with experimental results. With the increase in the source temperature and proportion of water in the eluent, the nucleoside molecules could more easily bind protons and become fragmented, while the concentration and spray voltage did not have a significant role in the process (Fig. S6†). Also, different modifications influence the charge distribution and length of the C–N bond. Electron-donating groups (e.g. –CH3) on the nitrogenous base would make N1 less positively charged and decrease the FR, while the same on the ribose ring would make the carbon atoms more negative and lead to the opposite result.
3.4 Application of ISF towards more sensitive detection of the rare modified cytosine 5fdC and more accurate nucleoside profiling
As described above, under the same conditions as i-Pr2N labeling analysis, 5fdC-RBH was mainly doubly-charged and suffered severe fragmentation, with an FR of up to 70%. From the mechanism proposed above, we speculated that ISF could be mitigated by promoting the formation of singly-charged ions, whose FR was only 10%. Noticing that RBH and its derivatives undergo ring-opening and gain an additional proton under acidic conditions, we substituted the mobile phase of water and methanol with 0.0085% formic acid in water and non-protic acetonitrile. After careful adjustment of the MS conditions and Q1/Q3 ion pair (Fig. S7†), the signal to noise ratio (S/N) of the RBH labeling product was enhanced by 10 fold. Compared with our previous work,23 RBH showed better performance than i-Pr2N (Table S2†) and gave a LOD of 3 amol, which was 300 times lower than that of direct analysis of 5fdC (Fig. 4). As the most sensitive method for 5fdC detection, the introduction of RBH could further reduce the cost due to easier access to RBH and use of less sample.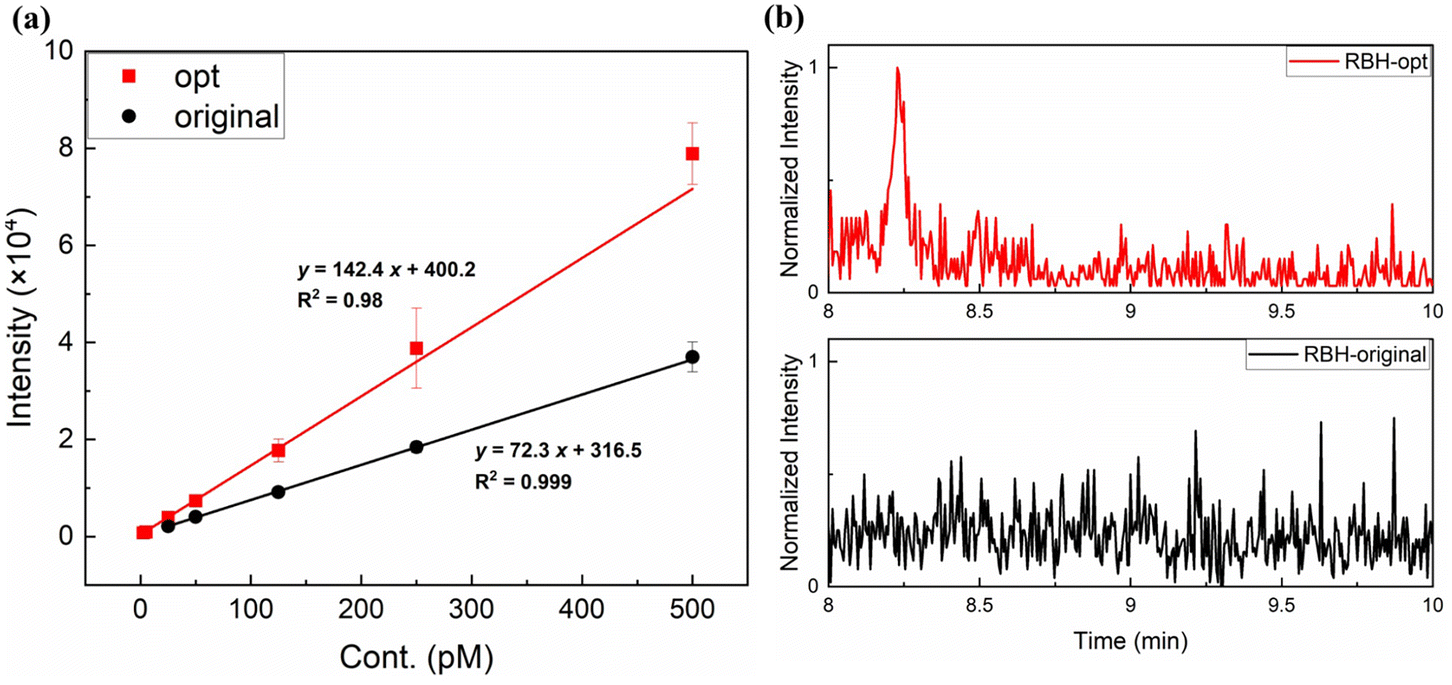 | ||
| Fig. 4 (a) The work curves of 5fdC-RBH under optimized (opt) and original conditions. (b) The MS responses of 3 amol 5fdC labeled with RBH, under original and optimized conditions. | ||
In addition to better sensitivity, with ISF we could also achieve the extraction and identification of nucleosides in complex unknown systems. Within the Q1 spectrum from high-resolution MS (HRMS), simultaneous peaks with a mass difference corresponding to a deoxyribose ring (116.048), ribose ring (132.043) or methylated ribose ring (144.059) indicate the presence of nucleosides or modified nucleosides. In the case of rCm and mrC, for example, they are identical in mass and difficult to differentiate without standards. However, using the ISF features, mrC (258.108 → 112.049) and rCm (258.108 → 126.066) could be easily identified (Fig. S8†). Finally, we extracted the rNs of MCF-7 cells and analyzed them directly by MS1 results of LC-HRMS. With Python, we configured all possible original and fragment ion pairs with a mass difference corresponding to ribose or methylated ribose, and plotted and calculated the EICs for peak detection (Fig. S9†). The time difference in their peaks represented the asynchronicity of the ion pairs. With a time difference threshold of <8 scans (0.05 min), sixteen nucleosides and modified nucleosides (Table S3, Fig S9†) were extracted, and further confirmed with standards or the database provided by MODOMICS.35–39
4 Conclusions
For nucleosides analyzed in positive mode MS, ESI becomes no longer “soft”, and a certain ratio of protonated nucleosides will fragment and lose their ribose ring. Combining theoretical calculations with NMR experiments, we speculated that the fragmentation was related to N3 protonation and the charge distribution near the glycosidic bond. Thus, the glycosidic bond was weakened and susceptible to cleavage in the mild environment. This was consistent with the factors that influence FRs, such as temperature, background buffer and chemical modifications. On this basis, we improved the quantification of 5fdC by RBH derivatization and fragmentation suppression. By adjusting the MS parameters, the FR of 5dfC–RBH was decreased from about 70% to 10%. Along with the labeling effect, the LOD of 5fdC was decreased by 300 times and reached 3 amol. Furthermore, our results showed that ISF should occur for almost all nucleosides with a glycosidic bond. Thus, we could achieve nucleosideomics by monitoring pairs of co-occurring peaks with certain mass differences. Using only Q1 spectra, we identified 16 nucleoside species in the total RNA of MCF-7 cells. Resolution and sensitivity permitting, we could even explore new epigenetic modifications.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22076003, 22174002 and 21775006). The theoretical calculation part of this work was supported by the High-Performance Computing Platform of Peking University.References
- B. Chen, B.-F. Yuan and Y.-Q. Feng, Anal. Chem., 2019, 91, 743–756 CrossRef CAS PubMed.
- Y.-F. Xu, W. Lu and J. D. Rabinowitz, Anal. Chem., 2015, 87, 2273–2281 CrossRef CAS PubMed.
- R. M. Gathungu, P. Larrea, M. J. Sniatynski, V. R. Marur, J. A. Bowden, J. P. Koelmel, P. Starke-Reed, V. S. Hubbard and B. S. Kristal, Anal. Chem., 2018, 90, 13523–13532 CrossRef CAS PubMed.
- A. Criscuolo, M. Zeller and M. Fedorova, J. Am. Soc. Mass Spectrom., 2020, 31, 463–466 CrossRef CAS PubMed.
- J.-S. Kim, M. E. Monroe, D. G. Camp, R. D. Smith and W.-J. Qian, J. Proteome Res., 2013, 12, 910–916 CrossRef CAS PubMed.
- P. M. Seitzer and B. C. Searle, J. Proteome Res., 2019, 18, 791–796 CrossRef CAS PubMed.
- D. Asakawa, H. Mizuno, E. Sugiyama and K. Todoroki, Anal. Chem., 2020, 92, 12033–12039 CrossRef CAS PubMed.
- Y. Fu and C. He, Curr. Opin. Chem. Biol., 2012, 16, 516–524 CrossRef CAS PubMed.
- J. A. Law and S. E. Jacobsen, Nat. Rev. Genet., 2010, 11, 204–220 CrossRef CAS PubMed.
- S. Kriaucionis and N. Heintz, Science, 2009, 324, 929–930 CrossRef CAS PubMed.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 2011, 333, 1300–1303 CrossRef CAS PubMed.
- S. Ito, A. C. D'Alessio, O. V. Taranova, K. Hong, L. C. Sowers and Y. Zhang, Nature, 2010, 466, 1129–1133 CrossRef CAS PubMed.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CrossRef CAS PubMed.
- Y.-F. He, B.-Z. Li, Z. Li, P. Liu, Y. Wang, Q. Tang, J. Ding, Y. Jia, Z. Chen, L. Li, Y. Sun, X. Li, Q. Dai, C.-X. Song, K. Zhang, C. He and G.-L. Xu, Science, 2011, 333, 1303–1307 CrossRef CAS PubMed.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 2011, 333, 1300–1303 CrossRef CAS PubMed.
- L. Hu, Z. Li, J. Cheng, Q. Rao, W. Gong, M. Liu, Y. G. Shi, J. Zhu, P. Wang and Y. Xu, Cell, 2013, 155, 1545–1555 CrossRef CAS PubMed.
- R. Yin, J. Mo, M. Lu and H. Wang, Anal. Chem., 2015, 87, 1846–1852 CrossRef CAS PubMed.
- Y. Dai, C.-B. Qi, Y. Feng, Q.-Y. Cheng, F.-L. Liu, M.-Y. Cheng, B.-F. Yuan and Y.-Q. Feng, Anal. Chem., 2021, 93, 6938–6946 CrossRef CAS PubMed.
- Y. Tang, J. Xiong, H. P. Jiang, S. J. Zheng, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2014, 86, 7764–7772 CrossRef CAS PubMed.
- H. P. Jiang, T. Liu, N. Guo, L. Yu, B. F. Yuan and Y. Q. Feng, Anal. Chim. Acta, 2017, 981, 1–10 CrossRef CAS PubMed.
- Y. Tang, S. J. Zheng, C. B. Qi, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2015, 87, 3445–3452 CrossRef CAS PubMed.
- F. Yuan, Y. Yu, Y.-L. Zhou and X.-X. Zhang, Anal. Chem., 2020, 92, 1605–1610 CrossRef CAS PubMed.
- Y. Yu, F. Yuan, X.-H. Zhang, M.-Z. Zhao, Y.-L. Zhou and X.-X. Zhang, Anal. Chem., 2019, 91, 13047–13053 CrossRef CAS PubMed.
- M.-Z. Zhao, Y.-W. Zhang, F. Yuan, Y. Deng, J.-X. Liu, Y.-L. Zhou and X.-X. Zhang, Talanta, 2015, 144, 992–997 CrossRef CAS PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Kossmann and F. Neese, J. Chem. Theory Comput., 2010, 6, 2325–2338 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. C. Chambers, B. Maclean, R. Burke, D. Amodei, D. L. Ruderman, S. Neumann, L. Gatto, B. Fischer, B. Pratt, J. Egertson, K. Hoff, D. Kessner, N. Tasman, N. Shulman, B. Frewen, T. A. Baker, M.-Y. Brusniak, C. Paulse, D. Creasy, L. Flashner, K. Kani, C. Moulding, S. L. Seymour, L. M. Nuwaysir, B. Lefebvre, F. Kuhlmann, J. Roark, P. Rainer, S. Detlev, T. Hemenway, A. Huhmer, J. Langridge, B. Connolly, T. Chadick, K. Holly, J. Eckels, E. W. Deutsch, R. L. Moritz, J. E. Katz, D. B. Agus, M. MacCoss, D. L. Tabb and P. Mallick, Nat. Biotechnol., 2012, 30, 918–920 CrossRef CAS PubMed.
- P. Boccaletto, M. A. Machnicka, E. Purta, P. Piątkowski, B. Bagiński, T. K. Wirecki, V. de Crécy Lagard, R. Ross, P. A. Limbach, A. Kotter, M. Helm and J. M. Bujnicki, Nucleic Acids Res., 2018, 46, D303–D307 CrossRef CAS PubMed.
- P. Boccaletto, F. Stefaniak, A. Ray, A. Cappannini, S. Mukherjee, E. Purta, M. Kurkowska, N. Shirvanizadeh, E. Destefanis, P. Groza, G. Avşar, A. Romitelli, P. Pir, E. Dassi, S. G. Conticello, F. Aguilo and J. M. Bujnicki, Nucleic Acids Res., 2022, 50, D231–D235 CrossRef CAS PubMed.
- S. Dunin-Horkawicz, A. Czerwoniec, M. J. Gajda, M. Feder, H. Grosjean and J. M. Bujnicki, Nucleic Acids Res., 2006, 34, D145–D149 CrossRef CAS PubMed.
- A. Czerwoniec, S. Dunin-Horkawicz, E. Purta, K. H. Kaminska, J. M. Kasprzak, J. M. Bujnicki, H. Grosjean and K. Rother, Nucleic Acids Res., 2009, 37, D118–D121 CrossRef CAS PubMed.
- M. A. Machnicka, K. Milanowska, O. O. Oglou, E. Purta, M. Kurkowska, A. Olchowik, W. Januszewski, S. Kalinowski, S. Dunin-Horkawicz, K. M. Rother, M. Helm, J. M. Bujnicki and H. Grosjean, Nucleic Acids Res., 2012, 41, D262–D267 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00047h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |