
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emergency diagnosis made easy: matrix removal and analyte enrichment from raw saliva using paper-arrow mass spectrometry†
Yufeng
Zhou‡
a,
Tung-Ting
Sham‡
 a,
Cedric
Boisdon
a,
Barry L.
Smith
a,
Joanne C.
Blair
b,
Daniel B.
Hawcutt
cd and
Simon
Maher
*a
a,
Cedric
Boisdon
a,
Barry L.
Smith
a,
Joanne C.
Blair
b,
Daniel B.
Hawcutt
cd and
Simon
Maher
*a
aDepartment of Electrical Engineering and Electronics, University of Liverpool, Brownlow Hill, Liverpool, UK. E-mail: s.maher@liverpool.ac.uk
bDepartment of Endocrinology, Alder Hey Children's Hospital, Liverpool, UK
cNIHR Clinical Research Facility, Alder Hey Children's Hospital, Liverpool, UK
dDepartment of Women's and Children's Health, University of Liverpool, UK
First published on 4th September 2023
Abstract
Paracetamol overdose is a leading cause of acute liver failure that can prove fatal. Establishing paracetamol concentration accurately and quickly is critical. Current detection methods are invasive, time-consuming and/or expensive. Non-invasive, rapid and cost-effective techniques are urgently required. To address this challenge, a novel approach, called Paper-Arrow Mass Spectrometry (PA-MS) has been developed. This technique combines sample collection, extraction, enrichment, separation and ionisation onto a single paper strip, and the entire analysis process, from sample to result, can be carried out in less than 10 min requiring only 2 μL of raw human saliva. PA-MS achieved a LOQ of 185 ng mL−1, mean recovery of 107 ± 7%, mean accuracy of 11 ± 8% and precision ≤5% using four concentrations, and had excellent linearity (r2 = 0.9988) in the range of 0.2–200 μg mL−1 covering the treatment concentration range, surpassing the best-in-class methods currently available for paracetamol analysis. Furthermore, from a panel of human saliva samples, inter-individual variability was found to be <10% using this approach. This technique represents a promising tool for rapid and accurate emergency diagnosis.
Introduction
Paracetamol, also known as acetaminophen, is a commonly used medication for the management of fever and pain by both paediatric1 and adult patients2 globally. While it is generally well tolerated at therapeutic doses, overuse and overdose can lead to serious health consequences, including acute liver failure; paracetamol is the leading cause of liver failure in developed countries.3 In cases of frequent overdose, the medical outcomes can be severe, such as acute liver necrosis,4 which may require a liver transplant and can be fatal if left untreated.5 Additionally, a chronic overdose of paracetamol has been described as a “silent killer” due to the insidious nature of the liver damage it can cause over time.6Accurate measurement of paracetamol levels in blood is crucial for both therapeutic drug monitoring and diagnosis. Currently, several methods are available for this purpose, including immuno-enzymatic, spectrophotometric and chromatographic assays. While immuno-enzymatic assays are fast and automated,7–11 making them suitable for emergencies, they can also be expensive and prone to interference from various sources, such as exogenous pharmaceuticals like N-acetylcysteine (an antidote for paracetamol overdose) and endogenous metabolites like bilirubin.12 On the other hand, lower-cost spectrophotometric assays13–15 lack specificity, and the highly sensitive and specific liquid chromatography-mass spectrometry (LC-MS),16–22 which is considered to be the state of the art, is both time-consuming and expensive. Hence the need for a simpler, lower-cost and faster technique that can accurately measure paracetamol levels.
Ambient ionisation and, in particular, paper spray (PS) mass spectrometry (PS-MS) continues to gain popularity as an analytical measurement technique, in part due to its disposable nature and simplicity, amongst other benefits. It has shown promising results for a variety of applications such as diagnostic biomarkers,23 forensics,24 water analysis,25,26 drugs of abuse27–29 and therapeutic drug monitoring.30–32 One of the key advantages of PS-MS is its ability to provide fast and cost-effective analysis of mixtures directly while maintaining a sufficient level of specificity and sensitivity not far from that of LC-MS,33 although usually lower. This should make PS-MS an attractive choice for use with clinical samples since it enables rapid testing without the need for expensive and time-consuming separation procedures. It also offers the potential for point-of-care (POC) analysis. PS-MS has been successfully used to analyse a range of biological matrices such as blood,22,28,32,34–39 urine,17 saliva,40 tears and milk,41,42 demonstrating its potential for clinical research. Yet despite its promise, it has not been incorporated into routine clinical practice. Various challenges remain, particularly achieving adequate quantitation precision in accordance with strict clinical requirements.43
Saliva is a valuable biological matrix for assessing paracetamol overdose, offering several advantages over plasma sampling.44 Since 1988, paracetamol levels in saliva have been compared with human plasma/serum.14 Saliva sampling is non-invasive, easy to collect, and has lower costs and clinical burden, especially in the paediatric setting. Salivary paracetamol is a reliable indicator of drug bioavailability,45 pharmacokinetics44,46,47 and also self-poisoning.9,10 Several clinical studies have reported a strong correlation between paracetamol levels in saliva and those in plasma/serum.9–11,14,15,20
Combining salivary sampling with PS-MS could simplify sample collection and reduce analysis time for paracetamol quantification. However, despite several publications on paracetamol quantification, none to the best of our knowledge have used PS-MS. This is likely due to the significant signal suppression caused by the matrix effect during ionisation, which is a challenge for ambient ionisation in general, and especially when analysing complex biological matrices.37,40,42,48–51
Matrix interference occurs primarily during the ionisation of the analyte.52 For clinical samples, endogenous substances such as cells, proteins, lipids and salts are the most common interferences.52 When these substances co-elute with the target analyte, they can cause an ion suppression effect that can negatively impact quantitation. In our previous work, we found that direct analysis of paracetamol in saliva resulted in a low signal due to matrix interference, even after deproteination. Moreover, compared to a neat solution, the incomplete removal of the saliva matrix resulted in relatively poor spray stability.37
LC-MS is a popular technique for clinical analysis, but traditional methods also require extensive sample preparation to remove matrix interferences prior to MS analysis. Commonly used sample preparation methods include protein precipitation,37 liquid–liquid extraction, solid phase extraction (SPE), ultrafiltration and centrifugation.53 However, these methods have associated drawbacks. They usually lead to noticeable sample loss due to incomplete isolation of the analyte from the matrix.54 Also, the use of different solvent-based extraction methods can influence metabolite profiles, which impacts the biological interpretation of metabolomics data.55 Furthermore, such steps are often laborious, time-consuming and costly. Consumable costs might include an SPE column or ultrafiltration centrifuge tubes, which also impose limits on the minimum size (i.e., amount) of sample required.
Recently, some innovative approaches have been investigated to reduce the matrix effects that can interfere with the analysis of biomolecules in biological samples using PS-MS. These include supramolecular solvent microextraction from urine,56 membrane-based matrix removal from human biofluids,57 Nafion as a coating for cation exchange to enhance desalting,58 and chemically functionalised paper substrates to favour extraction of certain chemical classes.59,60 Although these promising approaches can improve the sensitivity and accuracy of biomolecule quantitation, they also require longer sample processing times (typically ≥∼20 min) as well as additional equipment and/or materials, which compromise the convenience and simplicity of PS.
This study proposes a novel solution to effectively eliminate matrix effects in biomedia analysis augmenting precise quantitation in keeping with stringent clinical analysis requirements, whilst maintaining a low-cost, rapid, simple and environmentally friendly method. When PS-MS was first introduced, the key achievement was an initial proof of concept demonstration of paper chromatography (PC) coupled with MS via paper-based electrospray ionisation.61 Yet this powerful hyphenation of PC and MS, with exceptional potential, has never been truly integrated nor properly harnessed. PS in its typical guise does not exploit any of the key benefits afforded by PC. Previous attempts to combine PC and PS relied on visual detection, making it challenging to identify the migration of invisible analytes,61–63 while analyte diffusion during PC separation led to a loss of signal intensity.
To overcome these issues, we developed a novel process harnessing the power of paper chromatography, via a bespoke paper geometry design, which effectively hyphenates PC and MS in a seamless manner facilitated by on-paper ionisation, without requiring visual indicators. The entire process of paper-arrow mass spectrometry (Fig. 1), from sample to result, is simple and can be readily completed in under 10 minutes requiring only 2 μL of raw sample, yielding superior analytical performance (LOD: 61 ng mL−1, LOQ: 185 ng mL−1, r2: 0.9988, mean recovery of 107 ± 7%, mean accuracy of 11 ± 8%, precision ≤ 5%), surpassing the current state-of-the-art.
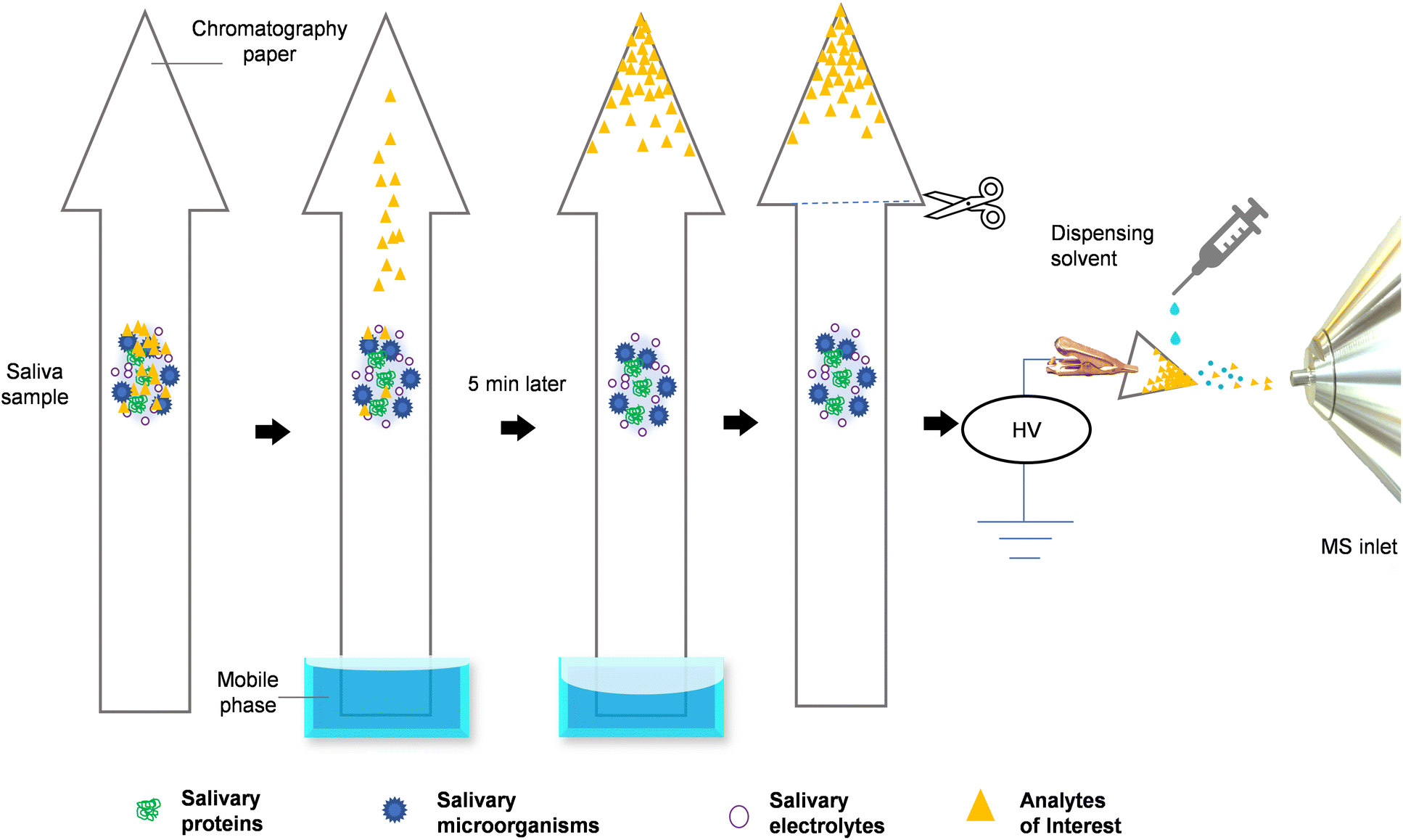 | ||
| Fig. 1 The figure depicts the whole process of paper-arrow MS analysis with a raw saliva sample. 2 μL of saliva is first applied to a paper arrow substrate, which is then introduced into the mobile phase via the base of the arrow's shaft. The analytes in the biofluid are separated and concentrated at the tip of the arrowhead through a ∼5 min process of PC. Finally, the arrowhead is cut off for PS-MS analysis. | ||
Experimental
Chemical and reagents
The details are described in the ESI (Method 1†).Standard solution preparation
The details are described in the ESI (Method 2†).Method development of PA-MS
The paper-arrow design and method development process consisted of four main steps (Fig. S1†), which are each covered in the following sub-sections (i–iv).In total, four different mobile phases were trialled: (1) pure ethyl acetate, (2) 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) of ethyl acetate:formic acid, (3) 10 mM ammonium formate in 9:1 (v/v) of ethyl acetate:formic acid, (4) 50 mM ammonium formate in 9:1 (v/v) of ethyl acetate:formic acid. In order to provide a reference point for the selection of the mobile phase, two additional experiments were conducted using PS-MS (Fig. S3b and d†). To do this, 100 μg mL−1 paracetamol samples were prepared in saliva or water as described earlier, and a 2 μL sample was applied to triangular paper (as shown in Fig. S2b†). The triangular paper pieces were then analysed without undergoing PC, using the same conditions as described in ESI Method 3.1.†
:1 (v/v) of ethyl acetate:formic acid, (3) 10 mM ammonium formate in 9:1 (v/v) of ethyl acetate:formic acid, (4) 50 mM ammonium formate in 9:1 (v/v) of ethyl acetate:formic acid. In order to provide a reference point for the selection of the mobile phase, two additional experiments were conducted using PS-MS (Fig. S3b and d†). To do this, 100 μg mL−1 paracetamol samples were prepared in saliva or water as described earlier, and a 2 μL sample was applied to triangular paper (as shown in Fig. S2b†). The triangular paper pieces were then analysed without undergoing PC, using the same conditions as described in ESI Method 3.1.†
Briefly, 2 μL of blank saliva was applied at the origin of the serrated paper strip and PC was carried out with the optimised mobile phase. After 12 min of PC, as described in the previous sub-section (i) (Selecting mobile phase for PC), the regions labelled 0–10 were cut apart, and 2 μL of 100 μg mL−1 paracetamol in water was applied onto each region. After drying in air at room temperature, full scan (m/z 50-180) MS data and single ion monitoring (SIM) of [M + H]+ (m/z 152.0706), [M + Na]+ (m/z 174.0525), and [M + K]+ (m/z 190.0265) of paracetamol were acquired (Fig. 2 and Fig. S4†).
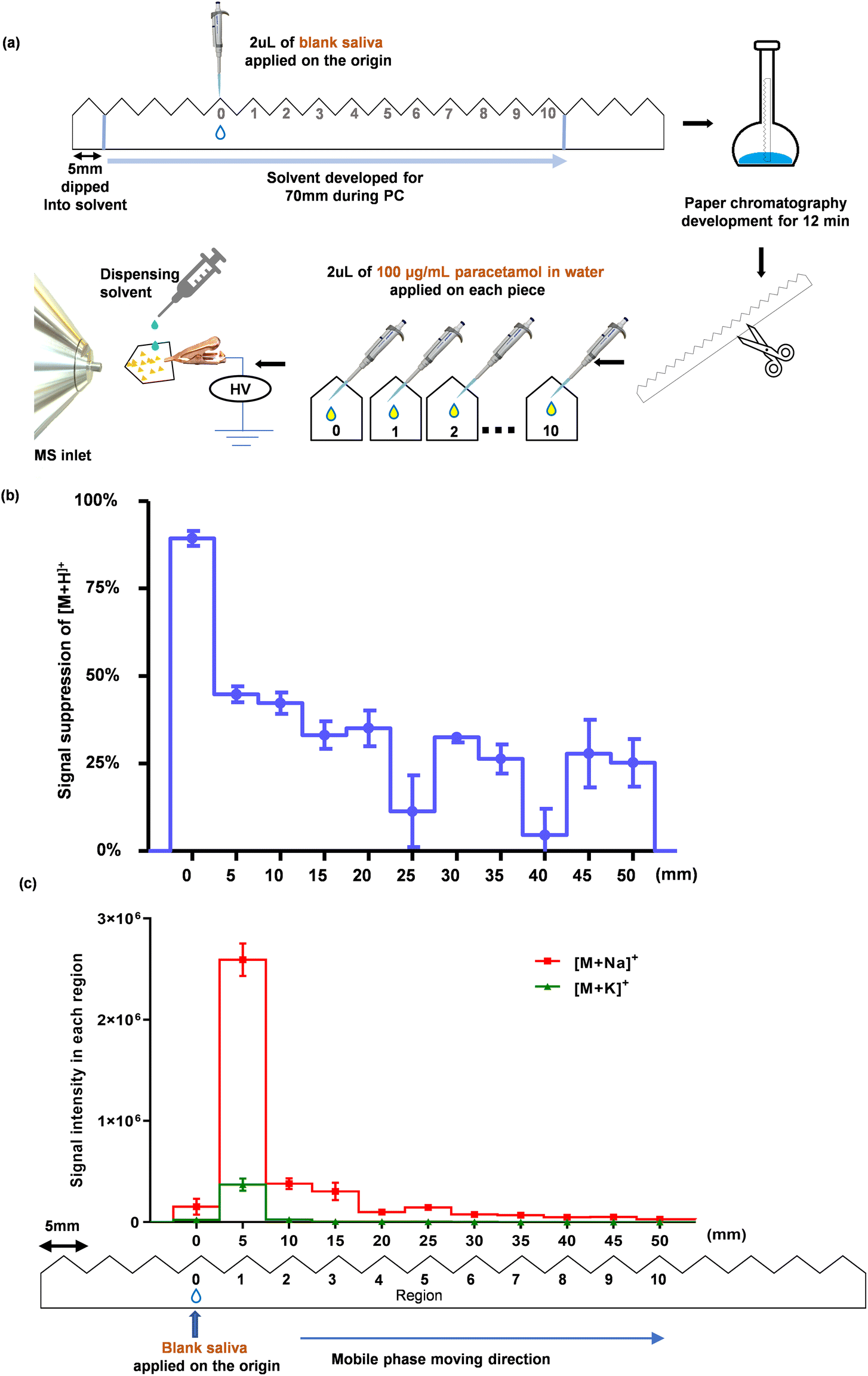 | ||
| Fig. 2 Estimation of the positions of saliva components after PC. (a) A schematic workflow illustrating the PC method, where 2 μL of blank saliva was treated with the mobile phase for 12 min. The paper strip was cut along the serrated regions (regions 0–10). 2 μL of water spiked with 100 μg mL−1 paracetamol was pipetted onto each piece of paper (i.e. each region). After they were air-dried, MS analysis was performed (further details are provided in ESI Method 3.1†). (b) Signal suppression extent of protonated paracetamol, [M + H]+, from a saliva sample for each region after PC was conducted; this was obtained by comparing the signals obtained from each region with that of blank water spiked with the same concentration of paracetamol i.e., standard dissolved in water added onto an independent piece of paper for direct analysis without PC (mean ± SD, n = 3). Ion suppression was calculated using Eqn (1). (c) SIM showing the intensity of [M + Na]+ and [M + K]+ ions of paracetamol in each region (mean ± SD, n = 3). | ||
To obtain a reference point to determine the extent of the signal suppression of [M + H]+, a serrated paper strip was cut into separate paper regions. Then, 2 μL of 100 μg mL−1 paracetamol in water was applied to each region. After drying, SIM of [M + H]+ was acquired. The signal suppression of [M + H]+ at each region was calculated using Eqn (1)
 | (1) |
:1 (v/v) of ethyl acetate:formic acid. SIM of [M + H]+, [M + Na]+, and [M + K]+ were acquired. The process of the experiment and corresponding data are shown in Fig. 3.
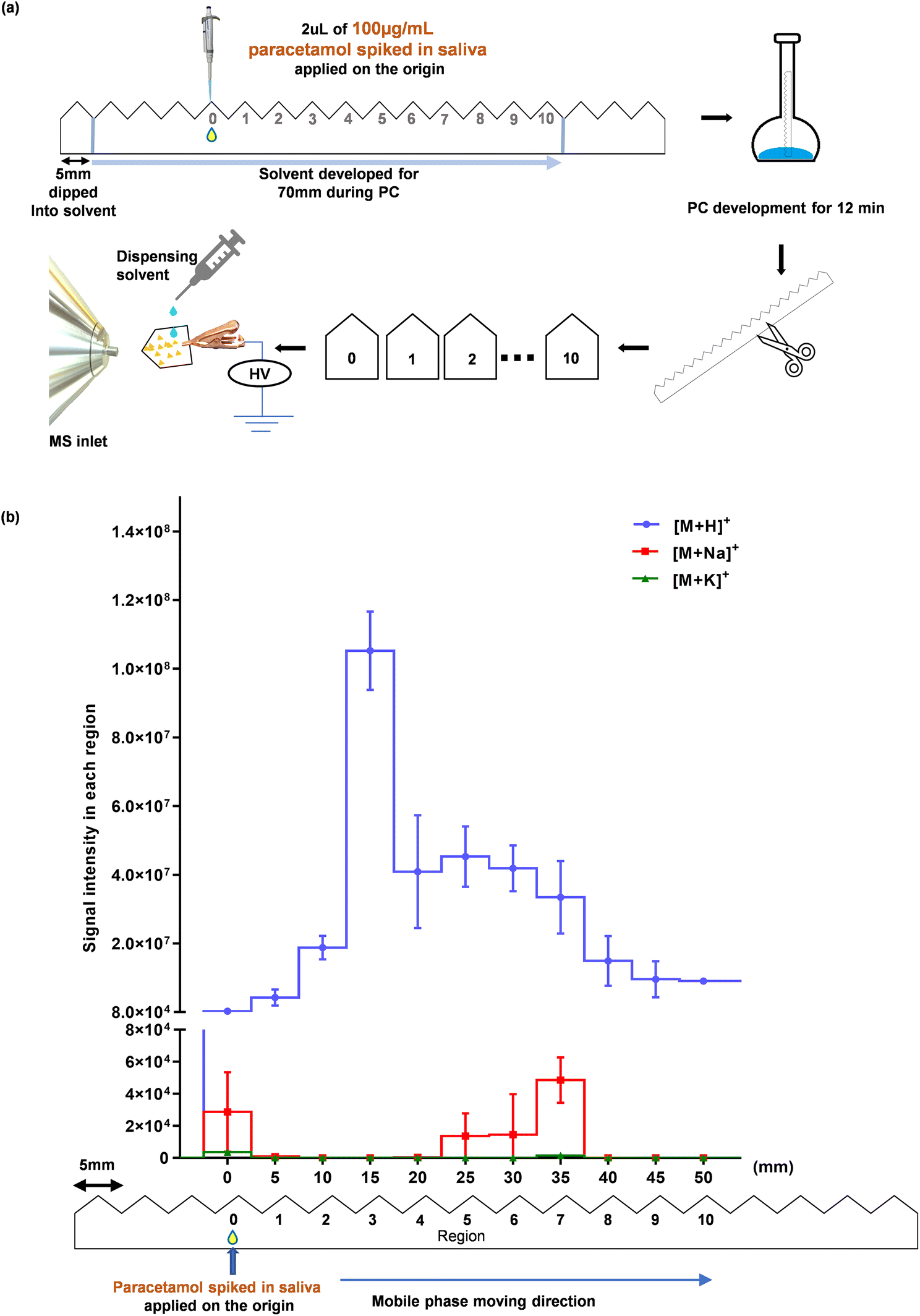 | ||
| Fig. 3 Identification of paracetamol's position after PC. (a) A schematic diagram of the workflow. 2 μL of saliva, spiked with 100 μg mL−1 of paracetamol was added at region 0. It was inserted into the mobile phase for 12 min of PC. After drying in the open air, the serrated paper strip was cut and each region was subjected to MS analysis (ESI Method 3.1†). (b) Ion intensities of [M + H]+, [M + Na]+ and [M + K]+ of paracetamol in each region (mean ± SD, n = 3). | ||
:1 (v/v) of ethyl acetate:formic acid. After drying in air, the arrowhead of the paper was manually cut off and the protonated molecular ion of paracetamol, [M + H]+, was monitored (Fig. 4).
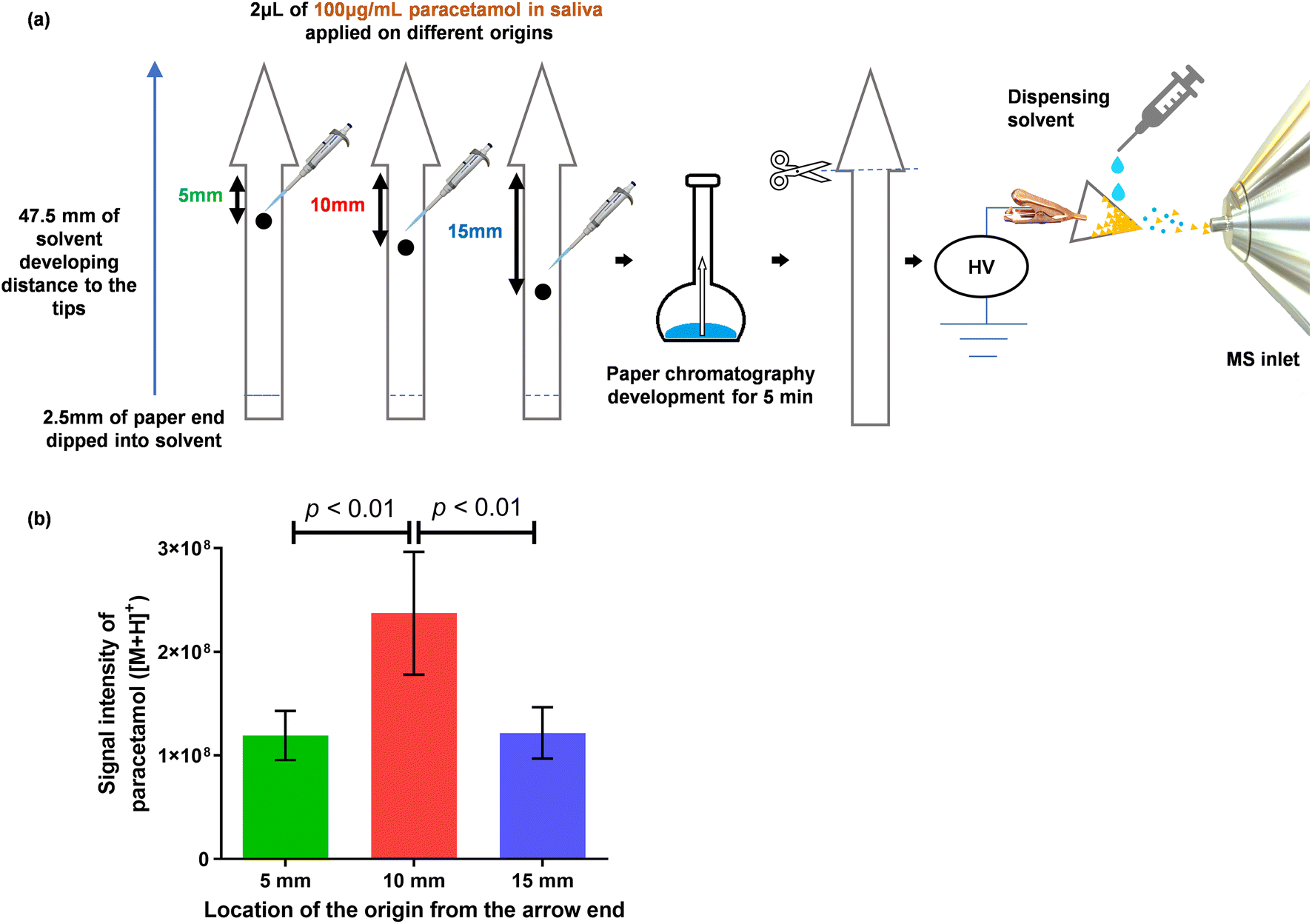 | ||
| Fig. 4 The figure presents a comparison of three positions of sample application on an arrow-shaped paper substrate. (a) Schematic presentation of the experiment in which 2 μL of 100 μg mL−1 paracetamol in saliva was spotted onto application points located at distances of 5 mm, 10 mm and 15 mm from the base of the arrowhead. After drying in air, the arrow-shaped paper strips were immersed in the mobile phase, and the front of the mobile phase reached the tip of the arrowhead within 5 min. (b) Intensities of [M + H]+ (mean ± SD, n = 4). One-way ANOVA reported p = 0.0032, Tukey's post hoc test was noted in the figure (p < 0.01). | ||
Evaluation of paper-arrow compared with paper spray
:1 (v/v) of ethyl acetate:formic acid as the mobile phase. The intensities of [M + H]+, [M + Na]+, and [M + K]+ were acquired by SIM (Fig. 5a and S5†).
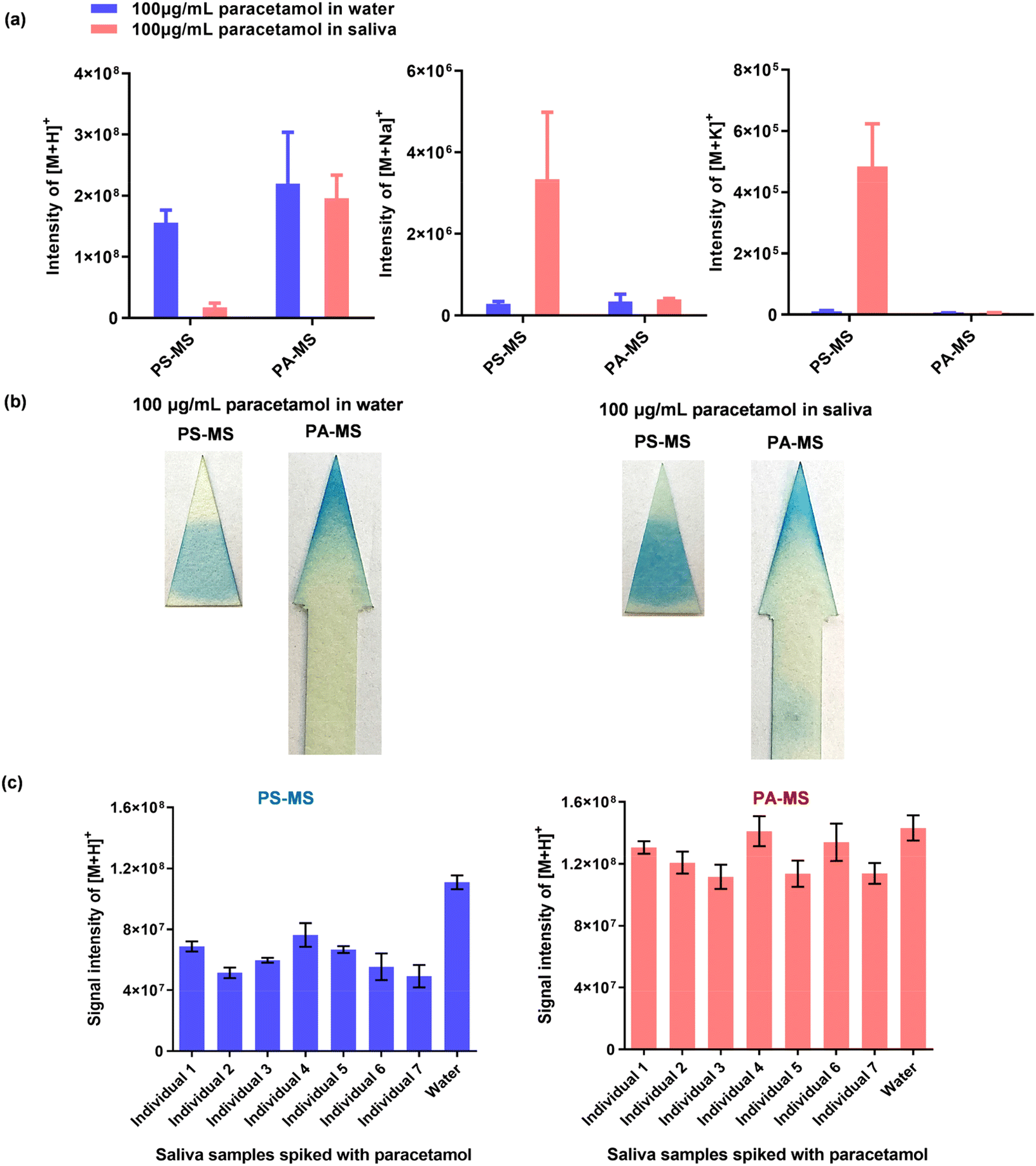 | ||
| Fig. 5 Comparison between PA-MS and PS-MS. (a) The intensities of [M + H]+, [M + Na]+ and [M + K]+ for 2 μL 100 μg mL−1 paracetamol in water or saliva, measured by PA-MS or PS-MS (mean ± SD, n = 3). (b) Staining results, illustrating the movement of paracetamol from water and saliva on triangular paper after PS-MS, and on arrow-shaped paper after PA-MS. For this analysis, 2 μL of 100 μg mL−1 paracetamol in water or saliva was applied and stained using a colourimetric method (ESI Method 6†). (c) Comparison of participants’ resting saliva samples and blank water, each spiked with 100 μg mL−1 paracetamol, and detected by PA-MS and PS-MS (mean ± SD, n = 3). | ||
| Detection method (n = 3) | Signal intensities of [M + H]+ from paracetamol | |||
|---|---|---|---|---|
| Mean | SD | CVa | Matrix effectb | |
| a CV = SD/mean. b Matrix effect = mean of the intensities of paracetamol from 7 participant's samples/mean of paracetamol intensities in water. | ||||
| PS-MS | 6.11 × 107 | 9.94 × 106 | 16.3% | 64.0% |
| PA-MS | 1.23 × 108 | 1.16 × 107 | 9.4% | 87.9% |
Method validation of paper arrow MS
:4, v/v), stood for 30 min at −20 °C, and then centrifuged for 20 min with 14000 rpm at 4 °C. The supernatant was diluted 4 times with water.
For the MS parameters of PA-MS/MS, the multiple reaction monitoring (MRM) transitions were: m/z 152.0706 → 110.0600 (quantifier) for paracetamol, and m/z 156.0957 → 114.0850 (quantifier) for paracetamol-D4. For UPLC-MS/MS, the MRM transitions were: m/z 151.94 → 109.95 as a quantifier for paracetamol and m/z 155.96 → 114.05 as quantifier for paracetamol-D4. Further details are given in ESI Method 3.†
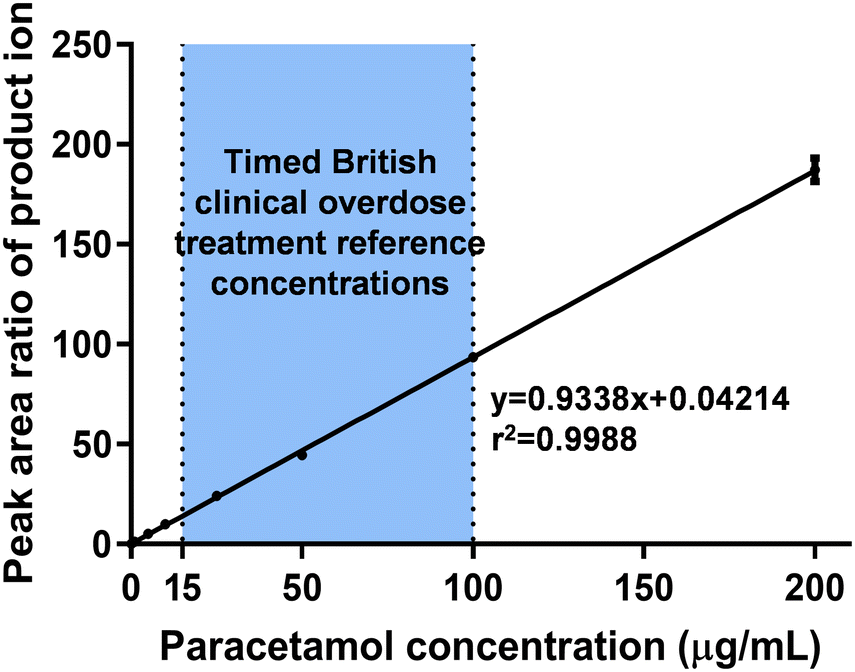 | ||
| Fig. 6 Calibration curve of salivary paracetamol detected by PA-MS/MS with a concentration range of 0.2–200 μg mL−1, covering the timed British clinical overdose treatment reference concentrations (as highlighted within the figure). Data expressed as mean ± SD, n = 5 and further details are provided in Table S7.† | ||
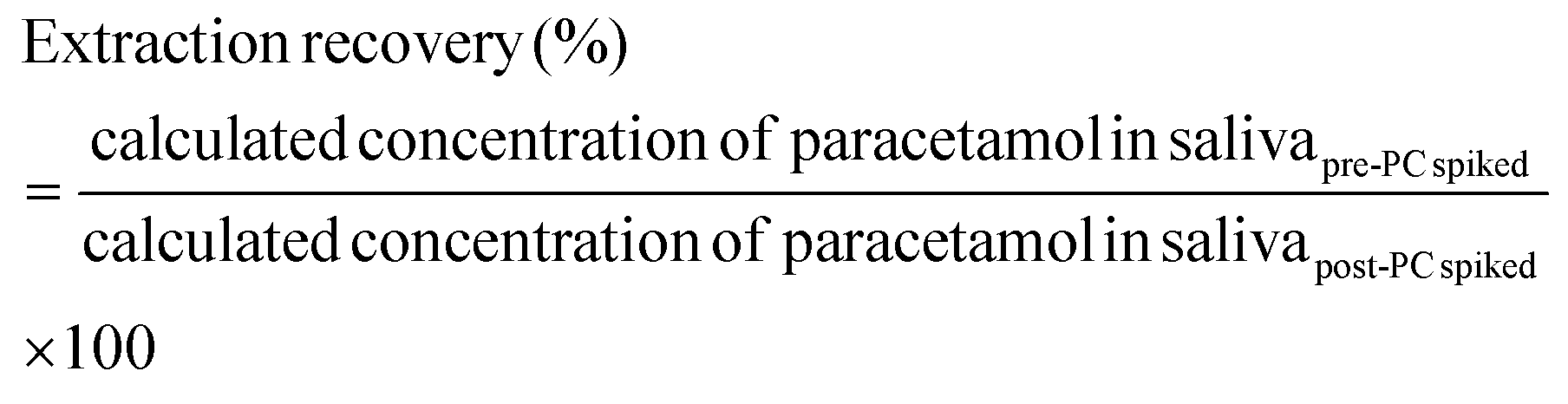 | (2) |
The matrix effects of PA-MS/MS were evaluated at four concentration levels of 0.2, 1, 25, and 100 μg mL−1 paracetamol with 1 μg mL−1 paracetamol-D4, in saliva and water. The samples were applied onto the origin of the arrow-shaped paper and detected after 5 min PC. The matrix effect was calculated with Eqn (3) and (4) following EMA guidelines (shown in Fig. 7).65
 | (3) |
 | (4) |
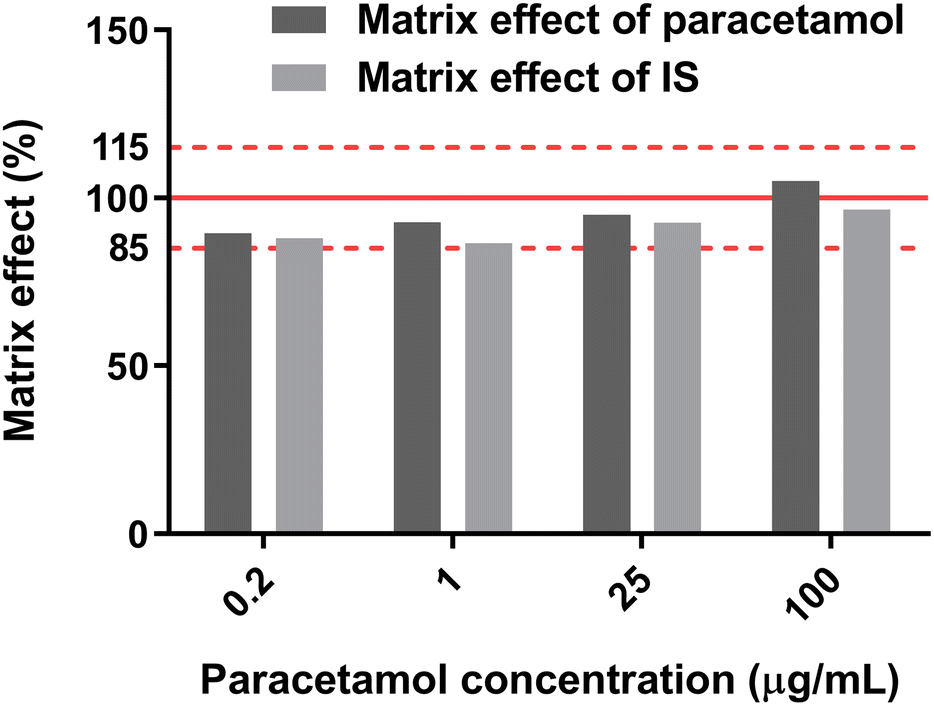 | ||
| Fig. 7 Matrix effects of paracetamol and paracetamol-D4 (IS) by PA-MS/MS. | ||
Statistical analysis
All above experiments were repeated at least 3 times. Average peak intensities of [M + H]+, [M + Na]+, and [M + K]+ were calculated for method development and verification of PA-MS/MS. Statistical analyses were performed and graphs were prepared by the software of GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, US). One-way ANOVA and Tukey's multiple comparison test were carried out for comparison among groups. A graph was created by plotting the average peak area ratio of the paracetamol quantifier over that of paracetamol-D4 versus the theoretical concentrations.Results and discussion
Design and method development of paper-arrow
In order to design and develop an effective paper arrow substrate, four main steps are carried out which are covered in the following:
Step 1. Selecting the mobile phase for PC
The development of PA-MS began by first selecting a suitable mobile phase, which is a key consideration for the success of PC.66 Guided by the literature,67,68 pure ethyl acetate was initially tested as the mobile phase but failed to sufficiently transport paracetamol away from the origin; this was evidenced by monitoring the protonated molecular ion peak of paracetamol, [M + H]+, which was most intense in the region closest to the origin, even after PC (Fig. S3b†). Upon further inspection, intense sodium adducts, [M + Na]+, and potassium adducts, [M + K]+, were also observed; the signal intensity of [M + Na]+ was similar to that of [M + H]+ at the origin when pure ethyl acetate was used as the mobile phase (Fig. S3c†). Metal ion adducts are likely formed due to the high sodium and potassium salt concentrations in raw saliva. The competition between protons and metal ions for paracetamol can affect the sensitivity and reproducibility of the analysis. The reproducibility of metal adduct ion formation is often poor and highly influenced by the sample preparation method and solution pH in conventional electrospray ionisation.69 Previous studies have shown that the addition of formic acid and volatile ammonium salts can suppress metal ion adduct formation and mainly produce [M + H]+ ions.70 Therefore, 10% formic acid and two concentrations of ammonium formate (NH4HCO2) in ethyl acetate were incorporated into the mobile phase to enhance the sensitivity of paracetamol in saliva.
The intensity of each serrated region after PC was compared with that of 2 μL of 100 μg mL−1 paracetamol in water or saliva using classic PS-MS/MS. Results showed that 50 mM NH4HCO2 in 9:1 ethyl acetate:formic acid (v/v) as the mobile phase produced the highest intensity of [M + H]+ at a distance of 15 mm from the origin, which was deemed sufficient (region 3, Fig. S3d†). Thus, the mobile phase used for PA-MS analysis in this study was confirmed to be 50 mM NH4HCO2 in 9:1 ethyl acetate:formic acid (v/v), which will be referred to as “the mobile phase” throughout this article.
Step 2. Identifying the location of saliva matrix after PC
The aim of this step is to assess the migration of the matrix (in our case saliva) in the mobile phase. The experimental process is illustrated in Fig. 2a. As human saliva is a complex biofluid comprising a vast array of organic and inorganic components,71 including over 770 prokaryotic species,72 0.9 ± 0.2 mg mL−1 protein (~3449 proteins),73 and 20–80 mmol L−1 sodium and 20 mmol L−1 potassium salts,71,74 it is impractical to identify the travelling distance of every single salivary component. Ultimately, our goal is to sufficiently separate paracetamol from those salivary components that will otherwise interfere with its MS analysis. Therefore, we adopted an approach in which the extent of ion suppression to paracetamol was used as an indicator to determine the location of the interfering saliva components after performing PC. As shown in Fig. 2b, the suppression extent of [M + H]+ at region 0 was high (89.3%), indicating that most of the interfering saliva components stayed close to the origin after performing the PC process. This made it possible to separate paracetamol from the saliva matrix. Although the other regions also showed some level of suppression of [M + H]+, a clear trend emerged: ion suppression reduced with increasing distance from the origin.
Interestingly, we observed the highest intensities of paracetamol adducts, [M + Na]+ and [M + K]+, at region 1 (5 mm from the origin), as shown in Fig. 2c. Their respective full scan MS spectra are shown in Fig. S4,† where the protonated molecular ion peak gradually intensified at subsequent regions. This suggests that sodium and potassium salts originating from the saliva did not travel much further than 5 mm from the sample application area. The results of this experiment enabled us to effectively determine the location of the salivary matrix. Based on these findings, it can be inferred that most salivary components, that would otherwise inhibit ionisation, are not effectively transported during PC, which provides a promising foundation for the extraction and separation of paracetamol from saliva.
Step 3. Identifying the Location of Paracetamol after PC Since it was deduced that most of the interfering saliva constituents remained close to the origin, the third step was to identify the location of paracetamol after being transported by PC. For this experiment, 2 μL of saliva mixed with 100 μg/mL paracetamol was spotted at the origin (Figure 3a). Unlike the previous experiment, no further spiking was conducted after PC. The intensities of [M + H]+, [M + Na]+, and [M + K]+ of each piece of paper at each region are plotted in Figure 3b. The intensity of [M + H]+ was lowest at the origin but increased by about 200-fold to its highest level at a distance of ~15 mm from the origin. This suggests that the majority of paracetamol was carried away from the origin by the mobile phase, enabling it to be effectively separated from the saliva matrix. It is worth noting that the peak intensities of [M + Na]+ and [M + K]+ were considerably lower across all regions, only slightly above the background signal (Fig. 3b). This significant decrease in the intensity at the origin, where paracetamol was added, is due to its transportation away from the sample application point. These findings clearly demonstrate that paracetamol can be effectively separated from salivary salts by PC.
Step 4. Optimising paper substrate design to concentrate paracetamol
After successfully separating paracetamol from the matrix using PC, the fourth and final step to complete the new method development was to integrate PC and MS into a seamless and efficient workflow. Previous attempts in the literature to combine these techniques were ad hoc and relied on visual detection to identify analyte regions for subsequent PS-MS analysis.61–63 Classic PC is limited to compounds that can be visibly identified (which also requires sufficient concentrations). Another major drawback is analyte diffusion during PC is not effectively controlled, resulting in a significant loss of analyte signal intensity. It is well known that chemicals inevitably diffuse in the mobile phase during chromatography,75 and this uncontrollable diffusion was also observed during our optimisation experiments using serrated paper strips. For instance, in Fig. 3b, we detected the protonated molecular ion of paracetamol, [M + H]+, with an appreciable signal intensity in region 2 and even as far as region 10, indicating that paracetamol had diffused over a large area, resulting in a significant loss of analyte signal intensity.
To address the issue of diffusion, we designed an arrow-shaped paper and modified the PC process using insights gained from the experiments already noted. The paper dimensions of the arrow-shaped paper are given in Fig. S1.† Contrary to the process of traditional PC, in which mobile phases are not allowed to reach the far end of the paper, we allowed the mobile phase front to reach the tip of the 47.5 mm long arrow-shaped paper within a timeframe of 5 min. Our aim was to rapidly transport paracetamol molecules to the tip of the arrowhead, thereby concentrating them, as depicted in Fig. 4a. Furthermore, three initial positions for sample spotting, at 5, 10 and 15 mm (measured from the base of the arrow head), were compared in relation to signal intensity. Fig. 4b shows that the highest signal intensity was achieved when the sample was applied at the 10 mm position.
Evaluation of paper-arrow compared with paper spray
So far, the PA-MS method for detecting paracetamol in saliva has been successfully developed, achieving a key goal of eliminating the matrix effect whilst providing analyte enrichment. With a simple, rapid and user-friendly process of PC, the intensity of the protonated paracetamol molecular ion, [M + H]+, was increased by more than an order of magnitude, significantly reducing the matrix effect, and yielding CV values that are within the required clinical range of ±15% across different individuals.
Method validation of PA-MS
| Matrix | Detection method | r 2 | LODa (ng mL−1) | LOQb (ng mL−1) |
|---|---|---|---|---|
| Calibration curves, precision and accuracy of each method are shown in Fig. S7 and Tables S2–S6,† respectively.a LOD = 3.3 × SD of response/slope.b LOQ = 10 × SD of response/slope.c Treatment of saliva samples: saliva samples were deproteinated with methanol (1:4, v/v), stood for 30 min under −20 °C, and then centrifuged for 20 min at 14000 rpm and 4 °C. The supernatant was diluted with water (1:4, v/v). |
||||
| Raw saliva | PA-MS/MS | 0.9969 | 61.10 | 185.15 |
| Pure water | PA-MS/MS | 0.9971 | 60.96 | 184.71 |
| Pre-treated salivac | UPLC-MS/MS | 0.9967 | 65.18 | 197.50 |
| Pre-treated salivac | PS-MS/MS | 0.9941 | 93.38 | 282.98 |
| Raw saliva | PS-MS/MS | 0.9864 | 135.91 | 411.83 |
Each calibration curve was meticulously constructed using seven concentration levels of paracetamol, which ranged from 5–1000 ng mL−1. Each concentration level was spiked with 500 ng mL−1 paracetamol-D4 to serve as the IS. The quantifier peak area ratios of paracetamol over paracetamol-D4 were measured and plotted against the theoretical concentrations to generate the calibration curves (Fig. S7†). The accuracy and precision of the calibration curves were evaluated using the correlation coefficient (r2), slope of the linear regression equation, limit of detection (LOD), and limit of quantitation (LOQ). Results from the five sets of experiments were compiled and presented in Table 2. Additional data relating to the evaluation of method precision and accuracy are provided in Tables S2–S6.†
The linearity of the paracetamol calibration curve in raw saliva using PA-MS/MS was found to be comparable to that obtained from pre-treated saliva using UPLC-MS/MS. Remarkably, the LOD and LOQ of paracetamol in raw saliva using PA-MS/MS were 61.10 ng mL−1 and 185.15 ng mL−1, respectively. Notably, these values were even lower than those achieved using UPLC-MS/MS and were found to be similar to the LOD and LOQ in pure water when analysed using the same method (PA-MS/MS).
Furthermore, the time and volume of solvent consumed by PA-MS/MS was 6.6 min and 65 μL per sample, much less than 62.5 min and 2700 μL per sample of UPLC-MS/MS (Table S8†). These findings suggest that the PA-MS/MS method is a better-performing, more cost-effective, efficient and sustainable approach for analysing saliva samples.
Compared to other studies for the direct detection of paracetamol in biofluids, PA-MS demonstrated significantly better sensitivity. For instance, a recent study reported a LOQ of 2.9 μg mL−1 for paracetamol in serum samples, which had been diluted five-fold and treated with an electrokinetic extraction syringe to remove proteins.77 In contrast, our PA-MS method allowed for the direct analysis of raw samples without the need for complex apparatus or dilution. The process of PC simultaneously achieved deproteination and desalting of the samples while concentrating the analyte of interest. These unique features of paper-arrow exhibit better sensitivity whilst making it a more practical solution, enabling analysis direct from raw saliva.
Lower LODs and LOQs for detecting paracetamol have been reported in some studies that treated biofluid samples with traditional methods like protein precipitation. For instance, R. K. Kam et al. reported a LOQ of 125 ng mL−1 for paracetamol in 20 μL of blood serum using LC-MS (LOQ defined as an S/N ratio higher than 10).78 However, if using the same quantification criterion, the LOQ in this study would be far superior, 5 ng mL−1 with a S/N of 135.93 (Table S2†). Other studies have used ±15% of CV and accuracy as the criteria to define the LOQ, and reported a value of 20 ng mL−1 for 50 μL of sample.79 In comparison, PA-MS/MS showed that the CV and accuracy for 50 ng mL−1 paracetamol in saliva were 3.4% and 3.2%, respectively, with only 2 μL of saliva sample (Table S2†). Furthermore, the sample pre-treatment methods used in those studies were more complicated and time-consuming than PA-MS/MS, our method achieved comparable sensitivity with lower sample volume and a much simpler treatment procedure.
In clinical practice, higher concentrations of paracetamol are typically detected in patients experiencing overdose.80 In response to new guidelines from the British government, which suggest treating paracetamol overdose with intravenous acetylcysteine, the recommended treatment threshold for timed plasma paracetamol concentration is now set at or above 100 μg mL−1 at 4 hours after ingestion and 15 μg mL−1 at 15 hours after ingestion.64 To facilitate this new guidance, a calibration curve was constructed covering a wider range of concentrations, from 0.2 μg mL−1 (LOQ) to 200 μg mL−1. The results demonstrated excellent linearity and accuracy, with the accuracy being less than 20% within the required guidelines (Fig. 6 and Table S7†).
| Spiked paracetamol concentration (μg mL−1) | Spiking time point | Calculated concentration (μg mL−1) (n = 3) | Recovery (Eqn (2)) | Accuracy (bias, pre-PC) | ||
|---|---|---|---|---|---|---|
| Mean | SD | CV | ||||
| 0.2 (LOQ) | Pre-PC | 0.240 | 0.004 | 1.7% | 106% | 19.9% |
| Post-PC | 0.226 | 0.04 | 18.1% | |||
| 1 | Pre-PC | 1.13 | 0.05 | 4.1% | 114% | 13.0% |
| Post-PC | 0.99 | 0.04 | 3.7% | |||
| 25 | Pre-PC | 25.18 | 0.31 | 1.2% | 98% | 0.7% |
| Post-PC | 25.77 | 0.11 | 0.4% | |||
| 100 | Pre-PC | 111.3 | 1.27 | 1.1% | 108% | 11.3% |
| Post-PC | 103.0 | 0.28 | 0.3% | |||
| Paracetamol concentration (μg mL−1) | Intra-daya | Inter-dayb | |||||
|---|---|---|---|---|---|---|---|
| Peak area ratios | Day | Peak area ratios | |||||
| Mean | SD | CV | Mean | SD | CV | ||
| a n = 10, mean accuracy ± SD = 9.0 ± 11.5%. b n = 5 per day (3 days), mean accuracy ± SD = 10.6 ± 7.7%. | |||||||
| 0.2 (LOQ) | 0.266 | 0.008 | 2.9% | Day 1 | 0.262 | 0.005 | 2.0% |
| Day 2 | 0.260 | 0.002 | 0.7% | ||||
| Day 3 | 0.264 | 0.003 | 1.2% | ||||
| 1 | 1.15 | 0.04 | 3.3% | Day 1 | 1.15 | 0.05 | 4.2% |
| Day 2 | 1.11 | 0.02 | 1.8% | ||||
| Day 3 | 1.14 | 0.03 | 2.4% | ||||
| 25 | 23.47 | 0.84 | 3.6% | Day 1 | 23.23 | 0.41 | 1.8% |
| Day 2 | 24.83 | 0.17 | 0.7% | ||||
| Day 3 | 24.37 | 0.34 | 1.4% | ||||
| 100 | 91.46 | 2.46 | 2.7% | Day 1 | 91.32 | 2.57 | 2.8% |
| Day 2 | 100.0 | 5.01 | 5.0% | ||||
| Day 3 | 102.2 | 2.88 | 2.8% | ||||
Conclusions
In conclusion, paper-arrow mass spectrometry combines sample collection, extraction, separation, enrichment and ionisation onto a single paper strip, achieving simple, fast, cost-effective and eco-friendly analysis – with superior analytical performance, consistent with clinical analysis requirements. The key technical advancement which underpins this achievement is the successful and seamless integration of paper chromatography with mass spectrometry. For PA-MS, we can say that ‘the whole is greater than the sum of its parts’. The process of PA-MS comprises three simple steps: direct sample application, paper chromatography and mass spectrometry analysis. It conforms with the principle of Ockham's razor, if one can ascribe this philosophical construct to competing analytical techniques rather than theoretical ideas. By combining sample collection, extraction, enrichment, separation and ionisation onto a single piece of paper, the entire process, from sample to result, can be completed in under 10 min, while achieving analytical performance that surpasses the current state of the art.The approach achieved a LOQ, for salivary paracetamol, as low as 185 ng mL−1, with excellent linearity across a wide concentration range, 0.2–200 μg mL−1, consistent with the clinical scenario of interest, using only 2 μL of saliva sample volume. Ongoing work includes an initial clinical validation study with Alder Hey Children's Hospital (Liverpool, UK), and future work will develop a point-of-care (POC) test format that can be performed in a clinical setting with a portable mass spectrometer.
Given the separation and enrichment performance of the paper-arrow, and the reduced inter-individual variation among human saliva samples, we envisage that this can significantly augment saliva sampling on a wider scale. A major constraint that has limited uptake of saliva sampling/analysis, in clinical practice, is the requirement that patients restrain from eating, drinking, smoking, chewing or using oral care products for a set time period (∼30 min–2 h) prior to providing a saliva sample. In this regard, ongoing work includes testing the performance of paper-arrow with saliva samples in which individuals have consumed various common food and drink types prior to sampling. Overcoming this challenge which besets saliva analysis in general, can yield significant patient benefits, particularly in a paediatric setting. Overall, the PA-MS method offers a simple, fast, convenient and non-invasive means to potentially risk stratify patients suspected of paracetamol overdose, and further research is underway to explore its potential in other medical scenarios.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are extremely grateful for the support received from the EPSRC (EP/V001019/1) and from Alder Hey Children's NHS Foundation Trust (Liverpool, UK).Notes and references
- D. J. Kanabar, Inflammopharmacology, 2017, 25, 1–9 CrossRef CAS PubMed.
- U. Klotz, Arzneimittelforschung, 2012, 62, 355–359 CrossRef CAS PubMed.
- R. T. Stravitz and W. M. Lee, Lancet, 2019, 394, 869–881 CrossRef CAS PubMed.
- D. G. Davidson and W. N. Eastham, Br. Med. J., 1966, 2, 497–499 CrossRef CAS PubMed.
- N. J. Thusius, M. Romanowicz and J. M. Bostwick, Psychosomatics, 2019, 60, 574–581 CrossRef PubMed.
- J. Pershad, M. Nichols and W. King, Pediatr. Emerg. Care, 1999, 15, 43–46 CrossRef CAS.
- F. Shihana, D. Dissanayake, P. Dargan and A. Dawson, Clin. Toxicol., 2010, 48, 42–46 CrossRef CAS PubMed.
- B.-Y. Chan, H.-M. Tsang, C. W.-Y. Ng, W. H.-W. Ling, D. C.-W. Leung, H. H.-C. Lee and C. M. Mak, J. Clin. Lab. Anal., 2019, 33, e22683 CrossRef PubMed.
- H. Wade, D. L. McCoubrie, D. M. Fatovich, J. Ryan, S. Vasikaran and F. F. Daly, Clin. Toxicol., 2008, 46, 534–538 CrossRef CAS PubMed.
- J. Ryan, C. Mandelt, H. Wade and S. D. Vasikaran, Ann. Clin. Biochem., 2009, 46, 149–151 CrossRef CAS PubMed.
- M. Smith, E. Whitehead, G. O'Sullivan and F. Reynolds, Br. J. Clin. Pharmacol., 1991, 31, 553–555 CrossRef CAS PubMed.
- J. Zeidler and P. A. Kavsak, Clin. Chem., 2011, 57, 1203–1204 CrossRef CAS PubMed.
- J. T. Afshari and T.-Z. Liu, Anal. Chim. Acta, 2001, 443, 165–169 CrossRef CAS.
- M. Nakano, N. I. Nakano, S. Funada and K. Iwasaka, Jpn. J. Clin. Pharmacol. Ther., 1988, 19, 549–553 CrossRef CAS.
- M. Sanaka, Y. Kuyama, S. Nishinakagawa and S. Mineshita, J. Gastroenterol., 2000, 35, 429–433 CrossRef CAS PubMed.
- T. Gicquel, J. Aubert, S. Lepage, B. Fromenty and I. Morel, J. Anal. Toxicol., 2013, 37, 110–116 CrossRef CAS PubMed.
- S. F. Cook, A. D. King, J. N. van den Anker and D. G. Wilkins, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 1007, 30–42 CrossRef CAS PubMed.
- R. B. Flint, P. Mian, B. van der Nagel, N. Slijkhuis and B. C. P. Koch, Ther. Drug Monit., 2017, 39, 164–171 CrossRef CAS PubMed.
- L. S. Jensen, J. Valentine, R. W. Milne and A. M. Evans, J. Pharm. Biomed. Anal., 2004, 34, 585–593 CrossRef CAS PubMed.
- N. M. Idkaidek, Saudi Pharm. J., 2017, 25, 671–675 CrossRef PubMed.
- M. Barfield, N. Spooner, R. Lad, S. Parry and S. Fowles, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 870, 32–37 CrossRef CAS PubMed.
- S. Maher, F. P. M. Jjunju and S. Taylor, Rev. Mod. Phys., 2015, 87, 113–135 CrossRef CAS.
- F. P. M. Jjunju, D. E. Damon, D. Romero-Perez, I. S. Young, R. J. Ward, A. Marshall, S. Maher and A. K. Badu-Tawiah, Sci. Rep., 2020, 10, 10698 CrossRef CAS PubMed.
- M. Jurisch and R. Augusti, Anal. Methods, 2016, 8, 4543–4546 RSC.
- S. Maher, F. P. M. Jjunju, D. E. Damon, H. Gorton, Y. S. Maher, S. U. Syed, R. M. A. Heeren, I. S. Young, S. Taylor and A. K. Badu-Tawiah, Sci. Rep., 2016, 6, 35643 CrossRef CAS PubMed.
- F. P. M. Jjunju, S. Maher, D. E. Damon, R. M. Barrett, S. U. Syed, R. M. A. Heeren, S. Taylor and A. K. Badu-Tawiah, Anal. Chem., 2016, 88, 1391–1400 CrossRef CAS PubMed.
- B. S. Frey, D. R. Heiss and A. K. Badu-Tawiah, Anal. Chem., 2022, 94, 4417–4425 CrossRef CAS PubMed.
- D. E. Damon, M. Yin, D. M. Allen, Y. S. Maher, C. J. Tanny, S. Oyola-Reynoso, B. L. Smith, S. Maher, M. M. Thuo and A. K. Badu-Tawiah, Anal. Chem., 2018, 90, 9353–9358 CrossRef CAS PubMed.
- B. S. Frey, D. E. Damon, D. M. Allen, J. Baker, S. Asamoah and A. K. Badu-Tawiah, Analyst, 2021, 146, 6780–6787 RSC.
- N. E. Manicke, P. Abu-Rabie, N. Spooner, Z. Ouyang and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2011, 22, 1501–1507 CrossRef CAS PubMed.
- R.-Z. Shi, E. T. M. El Gierari, N. E. Manicke and J. D. Faix, Clin. Chim. Acta, 2015, 441, 99–104 CrossRef CAS PubMed.
- R. D. Espy, N. E. Manicke, Z. Ouyang and R. G. Cooks, Analyst, 2012, 137, 2344–2349 RSC.
- T.-T. Sham, A. K. Badu-Tawiah, S. J. McWilliam and S. Maher, Sci. Rep., 2022, 12, 14308 CrossRef CAS PubMed.
- K. E. Yannell, K. R. Kesely, H. D. Chien, C. B. Kissinger and R. G. Cooks, Anal. Bioanal. Chem., 2017, 409, 121–131 CrossRef CAS PubMed.
- R. D. Espy, S. F. Teunissen, N. E. Manicke, Y. Ren, Z. Ouyang, A. van Asten and R. G. Cooks, Anal. Chem., 2014, 86, 7712–7718 CrossRef CAS PubMed.
- D. E. Damon, K. M. Davis, C. R. Moreira, P. Capone, R. Cruttenden and A. K. Badu-Tawiah, Anal. Chem., 2016, 88, 1878–1884 CrossRef CAS PubMed.
- B. L. Smith, C. Boisdon, D. Romero-Perez, T.-T. Sham, B. Bastani, Y. Zhou, S. McWilliam, A. K. Badu-Tawiah and S. Maher, Int. J. Mass Spectrom., 2022, 471, 116737 CrossRef CAS.
- D. E. Damon, Y. S. Maher, M. Yin, F. P. M. Jjunju, I. S. Young, S. Taylor, S. Maher and A. K. Badu-Tawiah, Analyst, 2016, 141, 3866–3873 RSC.
- S. Suraritdechachai, C. Charoenpakdee, I. Young, S. Maher, T. Vilaivan and T. Praneenararat, J. Agric. Food Chem., 2019, 67, 3055–3061 CrossRef CAS PubMed.
- H. Lee, C.-S. Jhang, J.-T. Liu and C.-H. Lin, J. Sep. Sci., 2012, 35, 2822–2825 CrossRef CAS PubMed.
- Y.-N. Yao, D. Di, Z.-C. Yuan, L. Wu and B. Hu, Anal. Chem., 2020, 92, 6207–6212 CrossRef CAS PubMed.
- Q. Wang, Y. Zheng, X. Zhang, X. Han, T. Wang and Z. Zhang, Analyst, 2015, 140, 8048–8056 RSC.
- W. Zhang, X. Wang, Y. Xia and Z. Ouyang, Theranostics, 2017, 7, 2968–2981 CrossRef CAS PubMed.
- N. Idkaidek and T. Arafat, Drug Res., 2014, 64, 559–562 CrossRef CAS PubMed.
- P. Retaco, M. González, M. T. Pizzorno and M. G. Volonté, Eur. J. Drug Metab. Pharmacokinet., 1996, 21, 295–300 CrossRef CAS PubMed.
- C. P. Babalola, F. A. Oladimeji and M. N. Femi-Oyewo, West Afr. J. Med., 2004, 23, 10–14 Search PubMed.
- A. M. Rittau and A. J. McLachlan, J. Pharm. Pharmacol., 2012, 64, 705–711 CrossRef CAS PubMed.
- C. Vega, C. Spence, C. Zhang, B. J. Bills and N. E. Manicke, J. Am. Soc. Mass Spectrom., 2016, 27, 726–734 CrossRef CAS PubMed.
- C. Zhang and N. E. Manicke, Anal. Chem., 2015, 87, 6212–6219 CrossRef CAS PubMed.
- N. M. Sarih, D. Romero-Perez, B. Bastani, M. Rauytanapanit, C. Boisdon, T. Praneenararat, H. A. Tajuddin, Z. Abdullah, A. K. Badu-Tawiah and S. Maher, Sci. Rep., 2020, 10, 21504 CrossRef CAS PubMed.
- D. E. Damon, Y. S. Maher, D. M. Allen, J. Baker, B. S. Chang, S. Maher, M. M. Thuo and A. K. Badu-Tawiah, Langmuir, 2019, 35, 13853–13859 CrossRef CAS PubMed.
- P. Panuwet, R. E. Hunter, Jr., P. E. D'Souza, X. Chen, S. A. Radford, J. R. Cohen, M. E. Marder, K. Kartavenka, P. B. Ryan and D. B. Barr, Crit. Rev. Anal. Chem., 2016, 46, 93–105 CrossRef CAS PubMed.
- W. Zhou, S. Yang and P. G. Wang, Bioanalysis, 2017, 9, 1839–1844 CrossRef CAS PubMed.
- A. D. Southam, L. D. Haglington, L. Najdekr, A. Jankevics, R. J. M. Weber and W. B. Dunn, Analyst, 2020, 145, 6511–6523 RSC.
- X. Duportet, R. B. M. Aggio, S. Carneiro and S. G. Villas-Bôas, Metabolomics, 2012, 8, 410–421 CrossRef CAS.
- F. M. de Oliveira, G. L. Scheel, R. Augusti, C. R. T. Tarley and C. C. Nascentes, Anal. Chim. Acta, 2020, 1106, 52–60 CrossRef PubMed.
- M. Zhang, F. Lin, J. Xu and W. Xu, Anal. Chem., 2015, 87, 3123–3128 CrossRef CAS PubMed.
- X. Song, M. Mofidfar and R. N. Zare, Front. Chem., 2021, 9, 807244 CrossRef CAS PubMed.
- W. Luo, T. A. van Beek, B. Chen, H. Zuilhof and G. I. J. Salentijn, Anal. Chim. Acta, 2023, 341673 CrossRef CAS.
- I. Pereira, J. Monaghan, L. R. Abruzzi and C. G. Gill, Anal. Chem., 2023, 95, 7134–7141 CrossRef CAS PubMed.
- H. Wang, J. Liu, R. G. Cooks and Z. Ouyang, Angew. Chem., Int. Ed., 2010, 49, 877–880 CrossRef CAS PubMed.
- M.-H. Cheng and C.-H. Lin, presented in part at the 2018 IEEE MEMS, Belfast, UK, 2018.
- Y.-C. Li, M.-H. Cheng and C.-H. Lin, presented in part at the 2020 IEEE 33rd International Conference on MEMS, Vancouver, BC, Canada, 2020.
- Medicines and Healthcare products Regulatory Agency, 2014.
- European Medicines Agency, ICH guideline 2022, M10.
- G. S. Shinde, P. Rao, R. Jadhav, P. Kolhe and D. Athare, Asian J. Pharm. Anal., 2021, 11, 45–48 CrossRef.
- H. Ibrahim, A. M. Hamdy, H. A. Merey and A. S. Saad, J. Chromatogr. Sci., 2020, 59, 140–147 Search PubMed.
- D. Mohamed, O. Hassan, N. Bahnasawy, A. S. Elnoby and S. Mowaka, Microchem. J., 2019, 150, 104146 CrossRef CAS.
- A. Leitner, J. Emmert, K. Boerner and W. Lindner, Chromatographia, 2007, 65, 649–653 CrossRef CAS.
- P. Marwah, A. Marwah and P. Zimba, J. Appl. Nat. Sci., 2020, 12, 180–192 CrossRef CAS.
- K. Ngamchuea, K. Chaisiwamongkhol, C. Batchelor-McAuley and R. G. Compton, Analyst, 2018, 143, 81–99 RSC.
- P. Sivadasan, M. K. Gupta, G. J. Sathe, L. Balakrishnan, P. Palit, H. Gowda, A. Suresh, M. A. Kuriakose and R. Sirdeshmukh, J. Proteomics, 2015, 127, 89–95 CrossRef CAS PubMed.
- C. Bacali, R. Vulturar, S. Buduru, A. Cozma, A. Fodor, A. Chiş, O. Lucaciu, L. Damian and M. L. Moldovan, Biomedicines, 2022, 10, 671 CrossRef CAS PubMed.
- B. Kumar, N. Kashyap, A. Avinash, R. Chevvuri, M. K. Sagar and K. Shrikant, Int. J. Contemp. Dent. Med. Rev., 2018, 011217 Search PubMed.
- V. A. Mirón-Mérida, M. Wu, Y. Y. Gong, Y. Guo, M. Holmes, R. Ettelaie and F. M. Goycoolea, Sens. Bio-Sens. Res., 2021, 32, 100421 CrossRef.
- N. Jehmlich, K. H. Dinh, M. Gesell-Salazar, E. Hammer, L. Steil, V. M. Dhople, C. Schurmann, B. Holtfreter, T. Kocher and U. Völker, J. Periodontal Res., 2013, 48, 392–403 CrossRef CAS PubMed.
- I. E. Mikhail, M. Tehranirokh, A. A. Gooley, R. M. Guijt and M. C. Breadmore, Angew. Chem., Int. Ed., 2020, 59, 23162–23168 CrossRef CAS PubMed.
- R. K. Kam, M. H. Chan, H. T. Wong, A. Ghose, A. M. Dondorp, K. Plewes and J. Tarning, Future Sci. OA, 2018, 4, FSO331 CrossRef PubMed.
- R. R. Taylor, K. L. Hoffman, B. Schniedewind, C. Clavijo, J. L. Galinkin and U. Christians, J. Pharm. Biomed. Anal., 2013, 83, 1–9 CrossRef CAS PubMed.
- H. H. Jeong, K. Cha, K. H. Choi and B. H. So, BMC Pharmacol. Toxicol., 2022, 23, 5 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00850a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |