
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid sub-nanomolar protein determination in serum using electropolymerized molecularly imprinted polymers (E-MIPs)†
A. N.
Stephen
a,
S. R.
Dennison
 b,
M. A.
Holden
a and
S. M.
Reddy
*a
b,
M. A.
Holden
a and
S. M.
Reddy
*a
aDepartment of Chemistry, UCLan Centre for Smart Materials, School of Pharmacy and Biomedical Sciences, University of Central Lancashire, Preston, PR1 2HE, UK. E-mail: smreddy@uclan.ac.uk
bSchool of Pharmacy and Biomedical Sciences, University of Central Lancashire, Preston, PR1 2HE, UK
First published on 23rd September 2023
Abstract
Rapid detection of biologicals is important for a range of applications such as medical screening and diagnostics. Antibodies are typically employed for biosensing with high sensitivity and selectivity but can take months to prepare. Here, we investigate electropolymerized molecularly imprinted polymers (E-MIPs), which are produced in minutes as alternative-antibody rapid biosensors for the selective recognition of model proteins bovine haemoglobin (BHb) and bovine serum albumin (BSA). We evaluated two disposable screen-printed electrodes (SPE) designated AT-Au and BT-Au based on their different annealing temperatures. E-MIPs for BHb demonstrated an imprinting factor of 146![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 1 nM and 12:1 at 0.1 nM, showing high effectiveness of E-MIPs compared to their control non-imprinted polymers. The BHb imprinted E-MIP, when tested against BSA as a non-target protein, gave a selectivity factor of 6:1 for BHb. Sensor sensitivity directly depended on the nature of the SPE, with AT-Au SPE demonstrating limits of detection in the sub-micromolar range typically achieved for MIPs, while BT-Au SPE exhibited sensitivity in the sub-nanomolar range for target protein. We attribute this to differences in electrode surface area between AT-Au and BT-Au SPEs. The E-MIPs were also tested in calf serum as a model biological medium. The BT-Au SPE MIPs detected the presence of target protein in <10 min with an LOD of 50 pM and LOQ of 100 pM, suggesting their suitability for protein determination in serum with minimal sample preparation. Using electrochemical impedance spectroscopy, we determine equilibrium dissociation constants (KD) for E-MIPs using the Hill–Langmuir adsorption model. KD of BHb E-MIP was determined to be 0.86 ± 0.11 nM.
:1 at 1 nM and 12:1 at 0.1 nM, showing high effectiveness of E-MIPs compared to their control non-imprinted polymers. The BHb imprinted E-MIP, when tested against BSA as a non-target protein, gave a selectivity factor of 6:1 for BHb. Sensor sensitivity directly depended on the nature of the SPE, with AT-Au SPE demonstrating limits of detection in the sub-micromolar range typically achieved for MIPs, while BT-Au SPE exhibited sensitivity in the sub-nanomolar range for target protein. We attribute this to differences in electrode surface area between AT-Au and BT-Au SPEs. The E-MIPs were also tested in calf serum as a model biological medium. The BT-Au SPE MIPs detected the presence of target protein in <10 min with an LOD of 50 pM and LOQ of 100 pM, suggesting their suitability for protein determination in serum with minimal sample preparation. Using electrochemical impedance spectroscopy, we determine equilibrium dissociation constants (KD) for E-MIPs using the Hill–Langmuir adsorption model. KD of BHb E-MIP was determined to be 0.86 ± 0.11 nM.
1. Introduction
Biosensors capable of high-selectivity, low detection limits and rapid detection are desirable for applications in protein biomarker determination in clinical diagnostics.1–4 One of the most common ways to detect biomarkers is by immunoassay using monoclonal antibodies.5 Whilst these antibodies show high specificity and selectivity for their target molecules, there are distinct disadvantages to their use for target biorecognition in biosensors relating to their production times, which is important in applications such as medical screening and diagnostics.1–4 The production times for antibodies can be several months due to the time required for antibody–epitope pair identification and subsequent antibody production, and potential mutations can lead to further delays.6–8 Therefore, for rapid response and screening, an alternative means of biorecognition with quicker production times but similar selectivity and detection limits is desired.Molecularly imprinted polymers (MIPs)9 are synthetic receptors that are able to act as stable alternatives to natural receptors such as antibodies. They can be used in the same way as antibodies in immunoassay-type tests with the same level of specificity and selectivity, making them attractive for prospective biorecognition. MIPs are produced by the self-assembly of functional monomers around a template target molecule The self-assembly process is driven by intermolecular interactions, such as hydrogen bonding, van der Waals, and π–π interactions between the monomer (and crosslinker) and the target molecule.10–12 Free-radical polymerisation is then initiated either by chemical initiators,13 electrochemically14 or by using ultraviolet light.15 The target molecule is then removed from the polymer matrix.12 This removal is often achieved through chemical washing with a suitable solvent system. The cavity remaining within the polymer is, specific to the rebinding of target. MIPs can be prepared on much shorter timescales than antibodies, and do not require specific epitope pairs to be identified. MIPs can be formed around a biological target (template) typically proteins16,17 or viruses16,18via a self-assembly process in a one-pot chemical reaction in a matter of minutes.19,20 The target can then be selectively stripped from the polymer matrix, creating a cavity that will be specific to that target.
MIPs have been synthesised in several ways, requiring chemical13 or electrochemical initiation,16 resulting in micro and/or nanoparticle gel suspensions in solution or as thin films.21 The traditional method of MIP synthesis has been to make a monolith (bulk) MIP using acryloyl-based monomers such as acrylamide, acrylic acid and N-hydroxymethylacrylamide (NHMA) resulting in polymeric hydrogels. The polymer gel monolith is then broken down by manual sieving or grinding to produce micron-sized particles exposing target specific cavities on each particle surface. Due to the crude nature of the grinding process, there is limited control in the physical features of the final particles, resulting in the production of random nanoscale features in addition to the desired cavities. The MIPs produced in this way therefore have very little homogeneity and are prone to non-specific binding resulting in lower binding affinities for the target. More recent methods have looked at forming nanoscale MIPs (nanoMIPs), which make use of a bottom-up approach to form MIPs particles that are similar in dimension to the target. This results in a higher affinity MIP as binding sites are ‘one-to-one’ with the target protein. While offering superior affinities when compared with bulk MIP approach, both methods do not easily translate to integration with sensors. Layering of nanoMIPs on sensor surfaces such that the binding site is oriented correctly is a challenge requiring additional surface chemical modifications, for example, using 4-aminobenzoic acid, followed by a coupling procedure involving 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC)/N-hydroxysuccinimide (NHS) to connect the MIPs to the surface of sensor chips.22 This method has been reported using sensor systems such as the quartz crystal microbalance (QCM),23 surface plasmon resonance (SPR) chips,24 electrochemical electrodes, and screen-printed electrodes.16,25
Electrochemical thin film MIPs (EMIPs) can be synthesised directly on electrode sensor surfaces and have emerged as a promising biosensing technology. This approach makes use of free protein in the monomer solution. The monomer solution containing target template is exposed to the electrode surface and under controlled applied electrode potentials. The polymer is then directly formed on the surface of the sensor with the protein integrated into it, without the need to pre-modify the surface of the sensor chip. An elution step is required to remove the entrapped protein at the polymer surface. The resulting cavities (target binding sites) are located on the exposed surface of the thin film.16,25,26 Subsequent detection of target rebinding is facile for E-MIPs, and quantitative measurements of target binding can be obtained through cyclic voltammetry14 or electrochemical impedance spectroscopy (EIS).16
In this paper, we show the facile cyclic voltametric electrodeposition (in 3–4 min); electrochemical elution (in 1–2 min) and characterization of E-MIP thin films based on functionalised acrylamide monomer for protein biosensing. We demonstrate that the E-MIPs prepared on disposable electrodes can operate selectively in serum samples with minimum sample preparation, achieving nanomolar detection in under 10 mins. Further, we show that the EIS analysis of E-MIPs can be used to determine equilibrium dissociation constants (KD), which could provide a faster, cheaper, and more versatile way to obtain these measurements.
2. Experimental
2.1 Materials
N-Hydroxymethylacrylamide (NHMA, 48% w/v), N,N′-methylenebisacrylamide (MBAm), phosphate buffered saline tablets (PBS, 10 mM, pH 7.4 ± 0.2), potassium ferricyanide (K3[Fe(CN)6]), potassium chloride (KCl), sodium nitrate (NaNO3), potassium peroxydisulfate (KPS), haemoglobin from bovine blood (BHb), bovine serum albumin (BSA) and bovine calf serum (BCS) were used as received from Merck. Buffers were prepared in MilliQ water (resistivity 18.2 ± 0.2 MΩ cm). DropSens disposable screen-printed electrodes (SPEs) (Au-AT & Au-BT) comprising a gold working electrode (0.4 cm diameter), a platinum counter electrode and silver reference electrode were purchased from Metrohm (Runcorn, Cheshire, UK). AT-cut quartz crystal microbalance pieces (9.005 MHz nominal frequency) were kindly donated by Dr Aizawa and Dr Kurosawa (AIST, Tsukuba, Japan).2.2 E-MIP production using cyclic voltammetry
All electrochemical experiments were performed using a Metrohm Autolab PGSTAT204 potentiostat and NOVA2.1.4 software. Thin-film hydrogel layers were fabricated directly onto AT-Au and BT-Au screen-printed electrodes (SPEs; Metrohm) by electrochemical polymerization using cyclic voltammetry (CV). A 50 μL solution in PBS comprising 1.33 M NHMA as the functional monomer, 41.5 mM MBAm as the cross-linker, 0.29 M NaNO3, 48.15 mM KPS and 188 mM BHb was deposited onto the SPE. The potential was then cycled between −0.2 V and −1.4 V for 7 to 10 cycles at 50 mV s−1 (10 min, RT, 22 ± 2 °C). To remove template protein, elution was also carried out using CV between −0.5 V and 1.5 V for 5 cycles at 175 mV s−1 (≈5 min, RT, 22 ± 2 °C) in PBS (50 μL). A BSA MIP was prepared in an identical manner but with BSA replacing BHb as the template.Non-imprinted polymer controls (E-NIPs) were produced in a similar manner, but in the absence of the protein template, and eluted for consistency.
2.3 E-MIP mass determination using QCM
An approximation of mass of E-MIP deposited on the electrode was determined by repeating the E-MIP production process but substituting the gold working electrode of the BT-Au SPE disposable chip with the gold electrode (0.5 cm diameter) of a 9 MHz AT-cut quartz crystal microbalance piece. The counter and reference electrodes remained the same. Dry QCM frequency measurements were taken before (bare) and after E-MIP production (7 cycles), followed by protein elution and the mass of E-MIP deposited was determined using the Sauerbrey equation.27 Accounting for the differences in electrode area between SPE (0.1256 cm2) and QCM (0.1963 cm2), the mass of E-MIP was determined to be 7.9 ± 1.6 μg per BT-Au SPE chip.2.4 Electrochemical characterisation of E-MIP
Polymer deposition and elution was tracked via cyclic voltammetric scans (triplicate) of an external 5 mM potassium ferricyanide solution containing 0.5 M KCl as supporting electrolyte (50 mV s−1). Electrochemical impedance spectroscopy (EIS) measurements were conducted using the same redox couple, at a standard potential of 0.1 V (±0.01 V) with 10 scans of frequencies, and a sinusoidal potential peak-to-peak with amplitude 0.01 V in the 0.1–100000 Hz frequency range. An equivalent Randles circuit was fitted for all EIS experiments using the FRA32 module (see Fig. S1†). Template rebinding studies were performed by exposing E-MIP modified SPEs to 50 μL of target BHb or BSA (in the range of 0.1 nM–1 nM) for a period of 5 min. For matrix effect and biocompatibility testing, 15 μL of BHb (in the range of 0.1 nM–1 nM) was added to 150 μL of undiluted bovine calf serum and mixed by vortexing, after which a 50 μl aliquot was exposed to the E-MIP modified SPEs for 5 min. Subsequently, the excess and non-bound sample was removed by gentle washing with PBS. After each template rebind, the SPE chip was electrochemically interrogated in the presence of ferrocyanide redox marker (5 mM) using cyclic voltammetry and EIS.
2.5 AFM imaging
AFM images were recorded using a Bruker Dimension Icon® AFM with a NanoScope 6 controller. Images were obtained in fluid, using a phosphate buffer solution (PBS, 10 mM, pH 7.4 ± 0.2). Imaging was performed in Peak Force Tapping™ mode with silicon nitride cantilevers (SNL-10, nominal spring constant 0.35 N m−1 and SCANASYST-FLUID, nominal spring constant 0.7 N m−1). Representative images were collected to assess the surfaces of both bare and coated AT-Au and BT-Au electrodes. The coated electrodes were prepared through electrochemical polymerisation (see E MIP Production above), with both E-MIP and E-NIP coated electrodes imaged. Roughness measurements were collected for a minimum of three 20 μm × 20 μm scans for each surface, and the reported average roughness (Ra) and RMS roughness (Rq) data were obtained following a 1st order plane fit of the raw data.3. Results and discussion
Thin film MIPs were directly integrated with electrochemical electrodes through electropolymerization, resulting in electrochemically grown MIPs (E-MIPs). The polymer is directly formed upon the surface of the sensor with the protein integrated into it, without the need to modify the surface of the sensor chip. The controlled formation of thin layer E-MIP films upon an electrode surface was possible due to finely controlling the electrochemical generation of persulphate free radicals at the electrode surface in the presence of functional monomers such as acrylamide.8. The MIP is formed by interfacing a solution containing the monomer, crosslinker, target molecule, and an electroactive initiator at the surface of an electrode.Cyclic voltammetry was used for the reductive polymerisation of the NHMA monomer and the MBAm crosslinker at the electrode surface with each cycle contributing to surface polymerisation and growth of a thin film of E-MIP.15,16 This was done in the presence and absence of the BHb target, forming E-MIPs and E-NIP controls respectively. The cathodic potential sweeps allowed for the controlled production of persulphate radicals at the electrode, which in turn initiated the controlled layering of a poly(NHMA) E-MIP or E-NIP with each potential sweep. Fig. 1a shows a series of cyclic voltammograms for the progressive layering with each cycle of pNHMA E-MIP for BHb onto a BT-Au SPE. Typically, with each cycle, the cathodic peak current density increased due to cumulative attachment of polymer to the electrode surface. We investigated 1, 5, 7 and 10 cycles for electrodeposition of polymer and found that 7 cycles gave optimal desired differences between MIP and NIP after reloading of the target protein. Seven cycles were therefore used to deposit E-MIPs and E-NIPs in all subsequent studies.
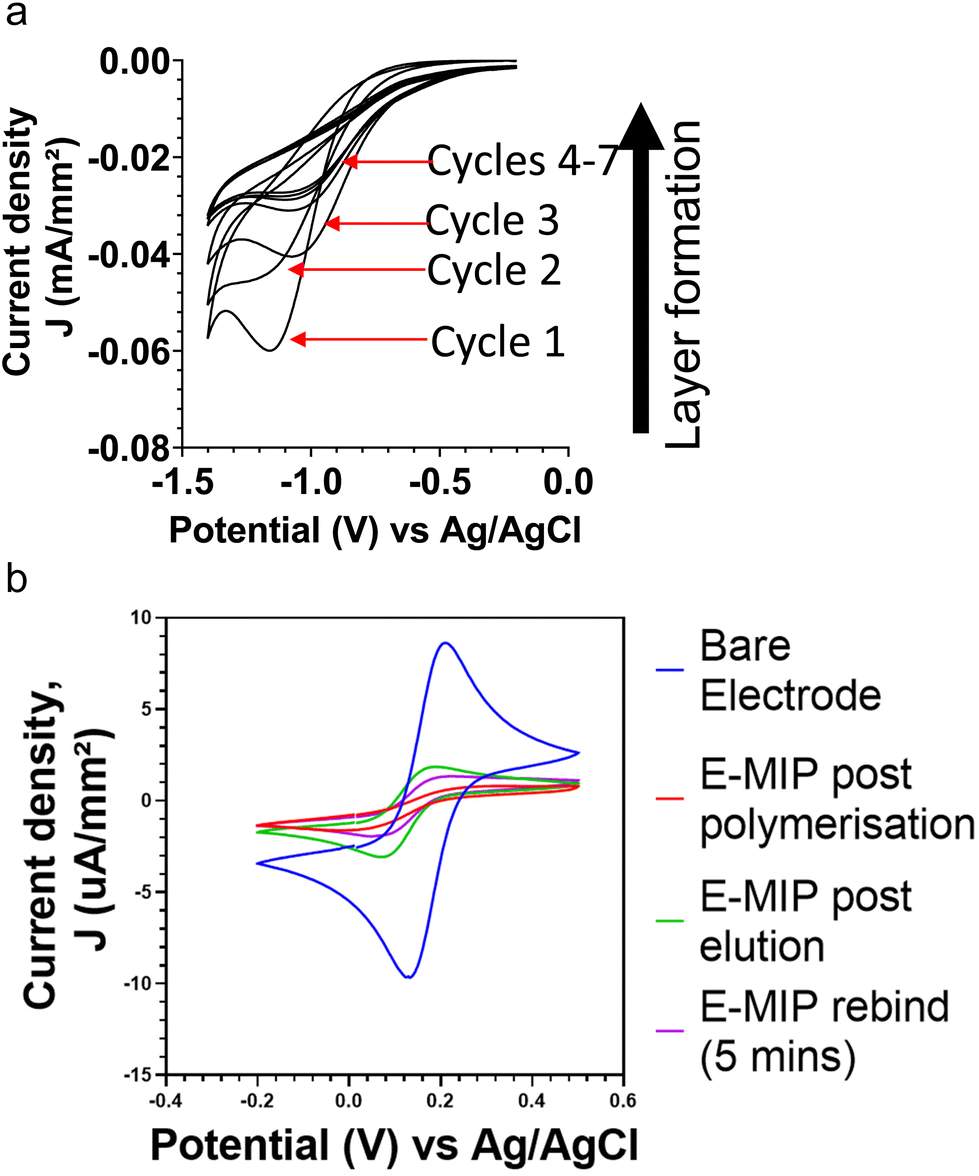 | ||
| Fig. 1 (a) Layer by layer deposition of pNHMA E-MIP on BT-Au SPE vs. Ag/AgCl reference electrode using cyclic voltammetry (−0.2 V and −1.4 V vs. Ag/AgCl reference; 7 cycles at 50 mV s−1). (b) CV of ferro/ferricyanide redox marker at different stages of electrode modification: bare (blue); post E-MIP polymerisation (red); post template, BHb elution (green); and after 5 min of template, BHb (1 nM) rebinding (purple). | ||
CV and EIS were used to investigate the protein binding to E-MIPs and E-NIPs. Fig. 1b compares cyclic voltammograms (CVs) for the model redox marker ferrocyanide at the bare electrode, after initial MIP deposition (7 cycles), after electrochemical elution of protein, and after protein reloading (5 min incubation period). The CVs follow the expected trend with the bare electrode displaying the largest anodic and cathodic peak current densities due to unimpeded access of redox marker to the electrode. The densities are significantly diminished following MIP and NIP polymer deposition. The template was then eluted, leaving surface-based cavities or binding sites specific to the selected target.7 After template elution, the MIP layer is at its most permeable and therefore has its lowest resistance to charge transfer. By using an appropriate redox marker, the E-MIP can be interrogated for presence of target template using either cyclic voltammetry, where a corresponding change in the reduction and oxidation peaks of the redox marker should be observed.17,18 Following the electrochemical protein elution cycle, only the E-MIP (and not the E-NIP) demonstrates a degree of recovery in peak currents for ferrocyanide marker. This is indicative of protein elution with E-MIP, resulting in the exposure of cavities (nanopores) on the MIP-layered electrode surface, and hence an increase in the diffusion of ferrocyanide to the electrode and subsequent detection. Many methods of MIP preparations and/or template elution still include the use of harsh solvents or acids which could be detrimental to the structure and conformation when biological species are imprinted. Our approach to elution using electrochemical methods avoids the use of such harsh chemicals, giving less possibilities for interferences in subsequent measurements. The null NIP effect is in agreement with our previous studies21,26 with the NIP signal remaining diminished since by design, they lack any protein selective cavities. Subsequent reloading of the target protein, BHb (1 nM) results in a decrease of redox marker signal for E-MIP due to selective binding of BHb within the cavities and a corresponding reduced access of ferrocyanide to the electrode surface. Again, the signal for E-NIP does not change. The E-NIP data is not shown as the cyclic voltammograms following E-NIP production, elution and protein rebinding all overlapped each other, with no discernible difference.
Electrochemical impedance spectroscopy-based interrogation can also be used, where a change in Nyquist plot parameters can be indicative of a protein binding event.19 As the E-MIP is exposed to the target, the cavities become filled. This decreases the permeability of the membrane and therefore, less of the redox marker can penetrate to the electrode surface, thus increasing the impedance. Therefore, the increase in impedance can be directly linked to the change in concentration of the target in the sample being analysed; the higher the target concentration, the more cavities that will be occupied and the lower the permeability of the film. This method allows for a direct measurement of the target binding event by the E-MIP. This is in contrast to for example spectroscopic methods that instead look at the residual amount of unbound target after rebinding to MIP has taken place.11,20,21 Therefore, with the E-MIP based electrochemical methods, one can directly track not only if the biomarker is present but also quantify the amount of target present.
Electrochemical impedance spectra (EIS) were obtained to further characterise and investigate E-MIP and E-NIP layers, including the binding and elution of protein. Fig. 2 compares EIS in the presence of ferro/ferricyanide at each stage of MIP and NIP production and characterisation. The Nyquist plots were truncated at 700 Ω in real impedance (ZRe) to aid with data interpretation. Each plot (except the bare electrode spectrum) is approaching the semi-circle arc, the diameter of which is indicative of the charge transfer resistance (RCT) value. RCT gives an indication of the ease with which the electrolyte and redox marker can be transported to the electrode surface. RCT values were extracted from the Randles circuit (see Fig. S1†) using the Nova 2.1.4 software. The bare electrode gives the lowest RCT as it has the least resistance to diffusion of the redox marker, and the RCT increases with polymer layer deposition on the electrode surface. Through investigation at 7 and 10 cycles, we determined that at 10 cycles, electrical interference was affecting the form of the Nyquist plots and hence the derived RCT values. This was confirmed in the Bode plots, where both the Bode modulus and phase dropped below 0.1 Hz, which resulted in this electrical interference. We therefore used 7 cycles for EIS studies.
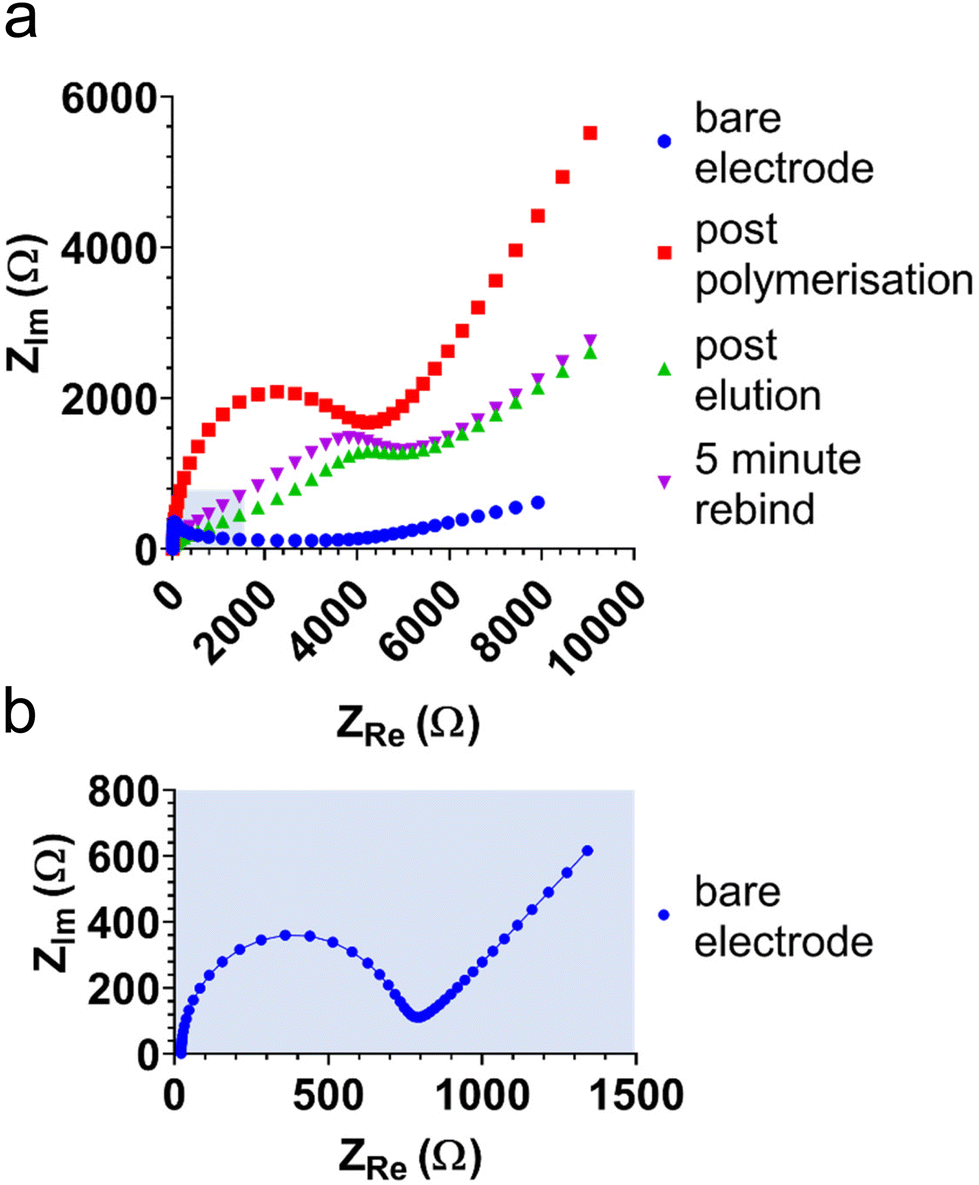 | ||
| Fig. 2 EIS Nyquist plots of ferro/ferricyanide redox marker at different stages of electrode modification (a): bare (see (b) for more detail); post E-MIP polymerisation; post template, BHb elution; and after 5 min of template, BHb rebinding. | ||
As well as polymer layer thickness, RCT is also related to the porosity of the polymer layer, with RCT decreasing as porosity (permeability to ferrocyanide) increases. As anticipated, the latter effect can be seen with the E-MIP (and not the E-NIP), which changes permeability between protein elution and subsequent protein reloading. Both CV and EIS therefore serve as useful tools to characterise and distinguish between E-MIP and E-NIP layers and investigate their interactions with proteins.
We investigated AT-Au and BT-Au screen printed electrodes (SPE) to determine the optimum electrode surface for protein detection and sensitivity following E-MIP layering. The BT-Au SPE has a lower annealing temperature (130 °C) compared with AT-Au SPE (800 °C).28 The annealing temperature has been shown to have a direct impact on the polycrystallinity of the gold electrodes with AT-Au SPE demonstrating lower crystallinity29 and surface roughness. Fig. 3 shows AFM images of (a) bare BT-Au SPE, (b) BT-Au SPE with E-MIP, (c) bare AT-AU SPE and (d) AT-Au SPE with E-MIP, with the underlying electrode morphology still visible in all cases. It is notable that the BT-Au SPE comprises sharper, more well-defined features with smaller individual clusters and a floret appearance. In comparison, the AT-Au electrodes are smoother. The BT-Au SPE samples also had a higher roughness both with and without the E-MIP or E-NIP layers (Table S1†). The average roughness Ra = 504 ± 45 nm for the BT-Au SPE, compared with Ra = 388 ± 105 nm for the AT-Au SPE. The BT-Au electrodes therefore present a larger surface area than the AT-Au electrodes, which in turn would directly influence the surface area of a subsequently electrodeposited MIP layer. Fresh E-MIPs using AT-Au and BT-Au SPEs were produced and their ability to electrochemically detect target protein over a concentration range was compared.
 | ||
| Fig. 3 AFM height images of (a) bare BT-Au SPE, (b) BT-Au SPE with E-MIP, (c) bare AT-Au SPE and (d) AT-Au SPE with E-MIP. | ||
Fig. 4a compares the change in ferro/ferricyanide peak currents from CV of AT-Au and BT-Au SPE modified with BHb E-MIPs at 1 nM of BHb rebinding. The large errors in Fig. 4a are due to a combination of the electrochemical method used (cyclic voltammetry) and the nature of the electrode used (AT-Au SPE). We are demonstrating that AT-Au SPE using CV is not fit for purpose and that the BT-Au SPE is superior for both CV and EIS application due to much lower errors. Fig. 4b shows the corresponding changes in RCT responses from EIS. Interestingly, the AT-Au SPE demonstrates a significant coefficient of variation when measuring peak current density at nM concentrations of protein compared with the BT-Au SPE demonstrating the AT-Au electrodes are less reliable than BT-electrodes at this concentration. When comparing relative RCT changes for a 1 nM addition of protein, only the MIP modified BT-Au SPE is sensitive to the addition of target protein. By contrast, the MIP modified AT-Au SPE showed a small and negative change in RCT. These results suggest that judicious selection of the electrodes, optimising for surface area, could be an important factor in improving E-MIP sensitivity.
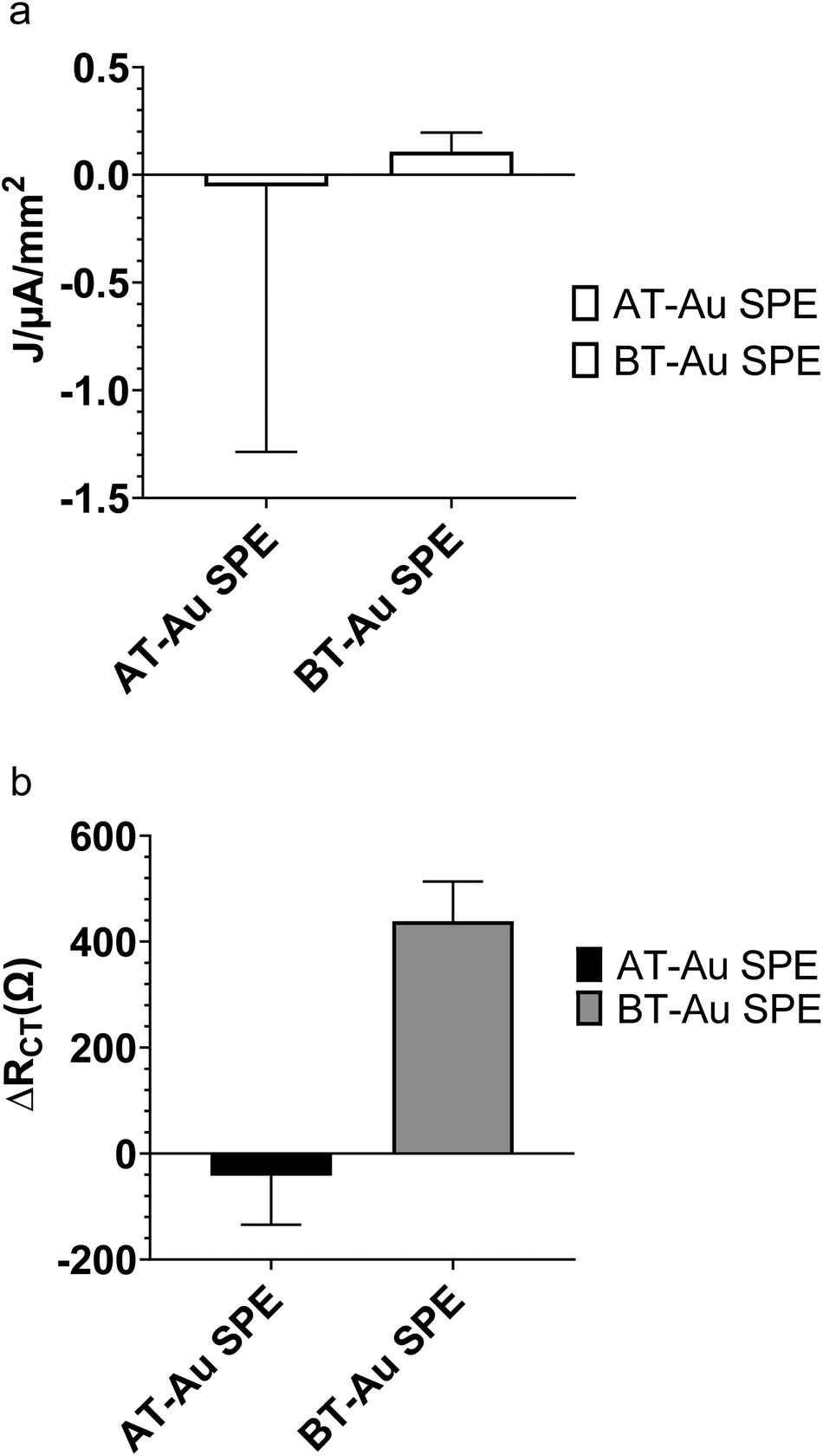 | ||
| Fig. 4 Comparison of performance of AT-Au and BT-Au SPEs, following MIP modification and protein (1 nM) rebinding. Effect on (a) CV peak anodic current change (J in μA mm−2) and (b) charge transfer resistance (RCT in Ω) change. Data represents mean ± S.E.M., n = 3. | ||
Due to its higher sensitivity, only the BHb MIP-modified BT-Au SPE electrode was studied further with EIS. The BHb sensor was able to measure down to 0.1 nM target protein with EIS (Fig. 5). A corresponding increase in the complex impedance (ZIm) is observed with increasing protein concentration.
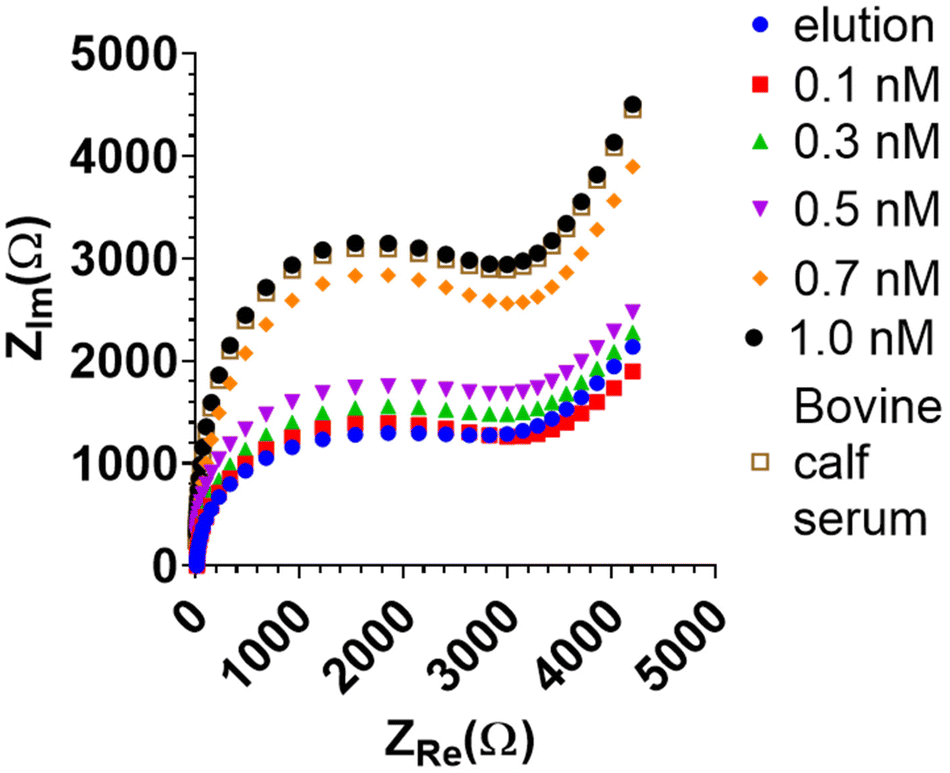 | ||
Fig. 5 EIS Nyquist plot of BHb MIP-modified BT-Au SPE electrode post-elution and after the addition of increasing concentrations of BHb (0.1 to 1.0 nM).  refers to determination of BHb (1 nM) spiked in 1/10 diluted bovine calf serum demonstrating 98.3% ± 1.5% recovery. refers to determination of BHb (1 nM) spiked in 1/10 diluted bovine calf serum demonstrating 98.3% ± 1.5% recovery. | ||
The extracted RCT values from EIS measurements were used to determine the dynamic linear range for the BHb MIP modified BT-Au SPE (Fig. 6). The large error bars in Fig. 6a, is only when the sensor becomes saturated at high protein loadings. Importantly, see Fig. 6b (which is an expansion of a section of Fig. 6a showing the linear range). Fig. 6b shows smaller (acceptable) errors at the lower concentrations in the analytically relevant linear dynamic range.
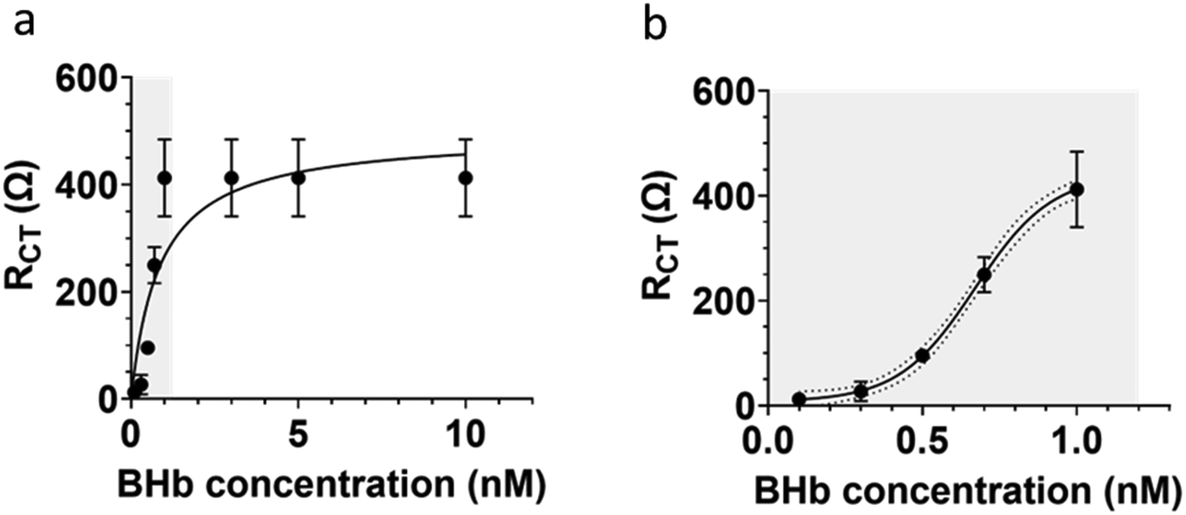 | ||
| Fig. 6 Standard curve for BHb determination using BHb MIP-modified BT-Au electrode (a). (b) Shows linear range in (a) expanded. Saturation occurs at 1 nM of target protein, indicating that all MIP cavities are occupied with protein. KD was determined to be 0.86 ± 0.11 nM using the Hill–Langmuir method. Data represents mean ± S.E.M., n = 3. | ||
Our narrow linear range (0.1 to 1 nM) is directly related to the capability and limitations of the electrochemical impedance analyser used and the protein (target) saturation limit of the E-MIP. Given the thin film nature of the E-MIP, imprinting and reloading is localised to being a surface-only phenomenon with therefore limited binding sites, and MIP saturation occurring at 1 nM protein. We investigated thinner and thicker layers of E-MIP (dictated by the number of CV cycles used to form the E-MIP) and identified 7 cycles to be the optimum for EIS signal generation. The sensor LOD was determined to be 50 pM, with an LOQ of 100 pM. Saturation of the signal occurred beyond 1 nM. Assuming the latter was the maximum protein binding capacity of the E-MIP thin film (Bmax), we can use the Hill–Langmuir method to determine the equilibrium dissociation constant KD for the E-MIP. We assumed the Hill coefficient is equal to 1, which is indicative of ligand (MIP) binding with no cooperativity to one site. The KD was then determined from the plot to be the protein concentration associated with 50% of binding sites being occupied (Bmax/2). The calculated KD was determined to be 0.86 ± 0.11 nM. From our QCM measurements, we determined the mass of E-MIP to be approximately 7.9 ± 1.6 μg per BT-Au SPE chip. The E-MIPs therefore demonstrate excellent sensitivity and superior affinity when compared with previously studied bulk microgel MIPs with KD = 4 μM30 and comparable to some nanoMIP formulations.31,32 For the latter nanoMIP formulations, it should be noted that surface plasmon resonance (SPR) was used to characterise rates of protein binding (kon) and unbinding (koff) to a pre-adsorbed monolayer of nanoMIP atop a SPR chip surface. KD was subsequently determined by taking a ratio of koff/kon. Recently, Bognár et al.33 used SPR to determine KD (2–5 nM) on electrosynthesised E-MIPs specific for the receptor binding domain (RBD) protein in SARS-CoV-2. Our KD determination, albeit comparable is a factor of 5 lower. Whereas EIS and SPR measurements can be made in comparable timescales (5–10 mins), the instrumentation required for EIS determinations in a mere 10% of the cost of that required for SPR at the time of writing this paper, and therefore EIS offers clear advantages for practical use. Moving forward, there needs to be some standardisation and harmonisation between EIS and SPR methods (e.g. using same proteins and MIPs) so that they can be directly compared in terms of equilibrium dissociation constants.
The BHb MIP modified BT-Au chip was compared against a BT-Au chip modified with a control non-imprinted polymer (E-NIP), prepared in the absence of a target protein. Since the E-NIP has not been exposed to a target template during preparation, one would expect the E-NIP to have no discernible binding affinity to BHb. As expected, the E-NIP showed only small RCT changes in the range 1–10 Ω (see ESI Fig. S2a†). This contrasts with the BHb-MIP returning signals in the 10–400 Ω range for 0.1 to 1 nM of protein. E-MIPs for BHb demonstrated a high imprinting factor of 146:1 at 1 nM and 12:1 at 0.1 nM. The imprinting factor gives a measure of performance MIP against a NIP and confirms that it is the imprinting that gives the superior binding affinity, and hence sensitivity, for the E-MIP biosensors.
As well as high sensitivity, it is important for biosensors to have a high selectivity too. The BHb MIP sensor was investigated for selectivity by testing against a non-target protein bovine serum albumin (BSA) in the range 0.1 to 1 nM (see ESI Fig. S2b†). The device returned a small response for BSA in this range, demonstrating a higher selectivity factor for BHb (BHb:BSA = 6:1). The protein chosen for these selectivity experiments was of similar size to the target protein (BHb 64.5 kDa, BSA 66.5 kDa), which means selectivity is achieved even under size compatible conditions. In other reports on E-MIP selectivity, the bovine albumin non-target was 20 kDa larger than their target protein A (in S. aureus), which could lead to size compatibility issues.34 This issue is further exacerbated by the acidic pH of the MES buffer solution used. At acidic pHs, bovine serum albumin is known to dimerize35–37 where protein-to-protein bonding occurs. This means the difference in size can range from 60 kDa to hundreds of kDa, potentially making it ineffective for testing cross-selectivity. We suggest that size compatibility be a key consideration for non-targets in selectivity studies. In this study, using size compatible proteins, we demonstrate that the E-MIPs are selective against a non-target protein.
The sensor was further assessed for selectivity by testing in a biologically relevant medium, bovine calf serum (BCS). This contains serum albumin, a common and the most prevalent protein present in blood and serum. The typical range of albumin in serum is 2.17–3.41 mM.38 Albumin concentrations even in this high range were not detected by the BHb MIP electrode when it was exposed to serum, demonstrating the selectivity and biocompatibility of the BHb MIP sensor. The BHb target (0.5 nM) was then spiked in neat calf serum (BCS) and gave a protein recovery of 88% ± 1%. At 1 in 10 dilution serum, the protein recovery was 98.3% ± 1.5% (Fig. 5), suggesting that some dilution is necessary to minimise matrix effects in determining the target.
A BSA E-MIP was then produced in a similar fashion to BHb E-MIP to determine the concentration of albumin in bovine calf serum (BCS). Given that the reported concentration of albumin in neat serum is 2.17–3.41 mM, the serum was diluted 1/106 so that the protein concentration could be determined within the nanomolar linear range of our highly sensitive E-MIP sensor. We determined the concentration to be 1.48 nM, equivalent to 1.48 mM ± 0.27 mM in the undiluted sample.
For MIPs to be effective in biosensors, it must be demonstrated that they can perform well in biological media such as serum or plasma. Poly-dopamine based MIPs for the NS1 protein from the Dengue fever virus showed a decrease in sensitivity from 1 ng mL−1–0.3 ng mL−1 to 5–200 ng mL−1 in a controlled buffer solution environment and serum, respectively,39 possibly due to biomatrix interference at the polymer/solution interface.40 With the improved selectivity and affinity in serum demonstrated by us, acrylamide-based E-MIPs are excellent candidates for rapid and reliable biosensing.
In a similar approach to us, Khan and co-workers34 also used electropolymerization to form E-MIPs around free protein in a solution to determine Protein A. Their MIP was applied to a single-walled carbon nanotube screen-printed electrode, and sensing was achieved using electrochemical impedance spectroscopy. However, our method offers distinct advantages compared to this work. In the first instance, we report higher sensitivity for our E-MIP approach, in the nanomolar range. Further, in the Khan et al.,34 methodology the protein elution step makes use of the enzyme proteinase K at 500 μg mL−1 in PBS, pH 7.4, for a 2.5-hour period. It is likely that template protein fragments remain within the cavities due to incomplete digestion during the enzyme-mediated template removal process.44 Their overall synthesis is also more time-consuming than the methodology in the work herein. In contrast, we have demonstrated a reagentless (electrochemical) method to elute the imprinted protein. Here, the elution step is enhanced taking less than five minutes. Therefore, the E-MIP sensor method developed here can be rapidly fabricated on demand and has higher sensitivity compared to previous MIP biosensors.
Table 1 summarizes a review of other pertinent reports on electropolymerization and chemical polymerization of protein MIPs followed by EIS interrogation, specifically in serum. There are few studies which investigate the use of a real serum sample. We highlight in the table the major significant advances of our method compared with these other reports. In particular, we are reporting a unique combination of (1) a very fast time to MIP fabrication and conditioning (10 min); (2) speedy rebinding (5 min) and (3) near 100% recovery of protein from spiked serum samples. This contrasts with the many hours taken for fabrication and conditioning methods reported by other researchers with significant under or overestimations in protein recovery. We attribute this to the superior biocompatibility of the base polymer (polyacrylamide) of NHMA that we are using compared with polymers used by others (e.g. polythiophene and poly-aminophenol) which are apparently prone to non-specific binding issues. Our recoveries are in agreement with Cieplak et al.41 who also used an acrylamide-based MIP. However, the latte group used a chemical method for MIP fabrication on sensor chip taking up to 120 h for fabrication and conditioning in contrast to the 10 mins of our electrochemical method. Where researchers have demonstrated sub-nanomolar recoveries like us, at best they are able to achieve only 92% recovery of target in spiked serum samples,42 even after 1/1000 dilution of serum to remove any matrix effects. Therefore, our additional significant advance is reliable excellent protein recovery from serum at sub-nanomolar determinations with minimum (1/10) dilution.
| Biomarker | Monomer | MIP fabrication and conditioning time | Rebinding time | Linear range and sensitivity | Rebinding efficiency | Ref. |
|---|---|---|---|---|---|---|
| Haemoglobin | NHMA | 10 minutes | 5 minutes at 22 °C | 0.1–1 nM | 98.3% ± 1.5% recovery of haemoglobin from bovine calf serum (1:10 diluted) |
Our study |
| LOD = 50 pM | ||||||
| Prostate specific antigen and myoglobin | Acrylamide | 120 hours | 90 minutes at 37 °C | 0.01–100 ng mL−1 (ca. 0.03–3 nM) | 98–102% recovery of proteins from human serum (dilution not given) | 43 |
| LOD = 5.4 pg mL−1 | ||||||
| Tau Protein | 3-Aminophenol | 2.5 hours | 30 minutes | 2.18 pM–2.18 nM | 92% recovery of Tau-441 from human serum (1:1000 diluted) |
42 |
| LOD = 0.024 pM | ||||||
| Albumin | 5,5′,5′′-Methanetriyltris(2,2′-bithiophene) | 50 minutes | 60 minutes | 12 to 300 pM | 96–117% recovery of human serum albumin from NORTROL control serum (dilution not given) | 41 |
| LOD = 0.25 pM |
4. Conclusions
We have developed a method for rapid biosensing based on electrochemically produced MIPs (E-MIPs). The polyacrylamide-based hydrogel E-MIPs can be synthesised within 10 minutes with rebinding and analysis achieved within 5 minutes. We produced highly selective E-MIPs for BHb and BSA, demonstrating excellent sensitivity using EIS. The E-MIP sensors demonstrated a linear range between 0.1 to 1 nM protein with a LOQ of 100 pM and LOD of 50 pM. This sensitivity was achieved in part through increasing the surface area of the electrode on which the E-MIP was deposited. The E-MIP sensor also performed well in neat serum, with near 100% recovery requiring only a 1/10 dilution of the serum. We have demonstrated that electrochemical impedance spectroscopy (EIS) is a viable technique to characterise and interrogate E-MIPs. We have also demonstrated that EIS interrogation of E-MIPs, in the presence of the model redox marker ferrocyanide, can be used to determine equilibrium dissociation constants using the Hill–Langmuir plot method. The KD for BHb E-MIP was determined to be 0.86 ± 0.11 nM, showing high sensitivity. Our approach makes use of a hydrogel-based E-MIP system as opposed to other MIP systems that use harsh solvents or acids that could impact biomolecules. This means that biomarkers can be detected in biologically relevant conditions. These results demonstrate the potential for E-MIPs as highly sensitive and rapid biosensors for protein biomarkers in disease diagnosis.Author contributions
SMR and ANS contributed to conception and design of the study. ANS performed the study and SMR and ANS performed the analysis. MAH and ANS performed AFM measurements and analysis. SMR and ANS wrote the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
The authors are grateful to the Royal Society of Chemistry COVID-19 Action fund (H20-188), the Daiwa Anglo-Japanese Foundation (13094/13916) and The Royal Society (IES\R3\193093) for funding this work.References
- T. J. Durkin, B. Barua and S. Savagatrup, ACS Omega, 2021, 6, 31390–31395 CrossRef CAS PubMed.
- S. S. Kordestani, F. S. Mohammadi, M. Noordadi, F. Rezaee and F. Fayyazbakhsh, Adv. Skin Wound Care, 2023, 36, 35–40 CrossRef PubMed.
- L. Lu, J. Yu, X. Liu, X. Yang, Z. Zhou, Q. Jin, R. Xiao and C. Wang, RSC Adv., 2020, 10, 271–281 RSC.
- J. J. S. Rickard, V. Di-Pietro, D. J. Smith, D. J. Davies, A. Belli and P. G. Oppenheimer, Nat. Biomed. Eng., 2020, 4, 610–623 CrossRef CAS PubMed.
- S. Singh, K. N. Tank, P. Dwiwedi, J. Charan, R. Kaur, P. Sidhu and K. V. Chugh, Curr. Clin. Pharmacol., 2018, 13, 85–99 CrossRef PubMed.
- K. Sakashita, K. Tsumoto and M. Tomita, J. Immunol. Methods, 2022, 511, 113384 CrossRef CAS PubMed.
- R. C. Ladner, Biotechnol. Genet. Eng. Rev., 2007, 24, 1–30 CrossRef CAS PubMed.
- S. Mitra and P. C. Tomar, J. Genet. Eng. Biotechnol., 2021, 19, 159 CrossRef PubMed.
- T. Sajini and B. Mathew, Talanta Open, 2021, 4, 100072 CrossRef.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812–1832 CrossRef CAS.
- J. J. BelBruno, Chem. Rev., 2019, 119, 94–119 CrossRef CAS PubMed.
- T. Takeuchi and J. Haginaka, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 728, 1–20 CrossRef CAS PubMed.
- A. M. Mostafa, S. J. Barton, S. P. Wren and J. Barker, TrAC, Trends Anal. Chem., 2021, 144, 116431 CrossRef CAS.
- H. El Sharif, S. Patel, E. Ndunda and S. Reddy, Anal. Chim. Acta, 2022, 1196, 339547 CrossRef CAS PubMed.
- S. A. Piletsky, I. Mijangos, A. Guerreiro, E. V. Piletska, I. Chianella, K. Karim and A. P. F. Turner, Macromolecules, 2005, 38, 1410–1414 CrossRef CAS.
- H. F. El Sharif, S. R. Dennison, M. Tully, S. Crossley, W. Mwangi, D. Bailey, S. P. Graham and S. M. Reddy, Anal. Chim. Acta, 2022, 1206, 339777 CrossRef CAS PubMed.
- M. Sullivan, W. Stockburn, P. Hawes, T. Mercer and S. Reddy, Nanotechnology, 2020, 32, 095502 CrossRef PubMed.
- S. P. Graham, H. F. El-Sharif, S. Hussain, R. Fruengel, R. K. McLean, P. C. Hawes, M. V. Sullivan and S. M. Reddy, Front. Bioeng. Biotechnol., 2019, 7 DOI:10.3389/fbioe.2019.00115.
- R. Schirhagl, Anal. Chem., 2014, 86, 250–261 CrossRef CAS PubMed.
- G. Vasapollo, R. D. Sole, L. Mergola, M. R. Lazzoi, A. Scardino, S. Scorrano and G. Mele, Int. J. Mol. Sci., 2011, 12, 5908–5945 CrossRef CAS PubMed.
- H. El Sharif, D. Stevenson and S. Reddy, Sens. Actuators, B, 2017, 241, 33–39 CrossRef CAS.
- J. McClements, L. Bar, P. Singla, F. Canfarotta, A. Thomson, J. Czulak, R. E. Johnson, R. D. Crapnell, C. E. Banks, B. Payne, S. Seyedin, P. Losada-Pérez and M. Peeters, ACS Sens., 2022, 7, 1122–1131 CrossRef CAS PubMed.
- A. Alassi, M. Benammar and D. Brett, Sensors, 2017, 17(12), 2799 CrossRef PubMed.
- X. Zhu and T. Gao, in Nano-Inspired Biosensors for Protein Assay with Clinical Applications, ed. G. Li, Elsevier, 2019, pp. 237–264 Search PubMed.
- H. F. El-Sharif, S. Patel, E. N. Ndunda and S. M. Reddy, Anal. Chim. Acta, 2022, 1196, 339547 CrossRef CAS PubMed.
- L. Bueno, H. El Sharif, M. Salles, R. Boehm, J. Narayan, T. Paixão and S. Reddy, Sens. Actuators, B, 2014, 204, 88–95 CrossRef CAS.
- K. K. Kanazawa and J. G. Gordon, Anal. Chem., 1985, 57, 1770–1771 CrossRef CAS.
- R. García-González, M. T. Fernández-Abedul, A. Pernía and A. Costa-García, Electrochim. Acta, 2008, 53, 3242–3249 CrossRef.
- A. Butterworth, E. Blues, P. Williamson, M. Cardona, L. Gray and D. Corrigan, Biosensors, 2019, 9, 22 CrossRef CAS PubMed.
- H. F. El-Sharif, D. M. Hawkins, D. Stevenson and S. M. Reddy, Phys. Chem. Chem. Phys., 2014, 16, 15483–15489 RSC.
- P. X. Medina Rangel, S. Laclef, J. Xu, M. Panagiotopoulou, J. Kovensky, B. Tse Sum Bui and K. Haupt, Sci. Rep., 2019, 9, 3923 CrossRef PubMed.
- Z. Altintas, A. Guerreiro, S. A. Piletsky and I. E. Tothill, Sens. Actuators, B, 2015, 213, 305–313 CrossRef CAS.
- Z. Bognár, E. Supala, A. Yarman, X. Zhang, F. F. Bier, F. W. Scheller and R. E. Gyurcsányi, Chem. Sci., 2022, 13, 1263–1269 RSC.
- M. A. R. Khan, F. T. C. Moreira, J. Riu and M. G. F. Sales, Sens. Actuators, B, 2016, 233, 697–704 CrossRef CAS.
- A. Brahma, C. Mandal and D. Bhattacharyya, Biochim. Biophys. Acta, 2005, 1751, 159–169 CrossRef CAS PubMed.
- V. Levi and F. L. González Flecha, Biochim. Biophys. Acta, 2002, 1599, 141–148 CrossRef CAS PubMed.
- G. Scatchard, A. C. Batchelder, A. Brown and M. Zosa, J. Am. Chem. Soc., 1946, 68, 2610–2612 CrossRef CAS PubMed.
- Henry's Clinical Diagnosis and Management by Laboratory Methods, ed. R. A. McPherson and M. R. Pincus, Saunders Elsevier, Philadelphia, 21st edn, 2007 Search PubMed.
- R. Arshad, A. Rhouati, A. Hayat, M. H. Nawaz, M. A. Yameen, A. Mujahid and U. Latif, Appl. Biochem. Biotechnol., 2020, 191, 1384–1394 CrossRef CAS PubMed.
- S. M. Reddy and P. M. Vagama, Anal. Chim. Acta, 1997, 350, 77–89 CrossRef CAS.
- M. Cieplak, K. Szwabinska, M. Sosnowska, B. K. C. Chandra, P. Borowicz, K. Noworyta, F. D'Souza and W. Kutner, Biosens. Bioelectron., 2015, 74, 960–966 CrossRef CAS PubMed.
- A. Ben Hassine, N. Raouafi and F. T. C. Moreira, Chemosensors, 2021, 9, 238 CrossRef CAS.
- P. Karami, H. Bagheri, M. Johari-Ahar, H. Khoshsafar, F. Arduini and A. Afkhami, Talanta, 2019, 202, 111–122 CrossRef CAS PubMed.
- D. Hawkins, D. Stevenson and S. Reddy, Anal. Chim. Acta, 2005, 542, 61–65 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an01498c |
| This journal is © The Royal Society of Chemistry 2023 |