
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection of coca alkaloids in oral fluid from coca leaf (tea) consumers: using solid phase extraction to improve validation parameters and widen the detection window
I.
Álvarez-Freire†
 b,
P.
Cabarcos-Fernández†
*b,
N. C.
Rubio
b,
A.
Moreda-Piñeiro
a,
M. J.
Tabernero-Duque
b,
I.
Sánchez-Sellero
b,
P.
Bermejo-Barrera
a and
A. M.
Bermejo-Barrera
b
b,
P.
Cabarcos-Fernández†
*b,
N. C.
Rubio
b,
A.
Moreda-Piñeiro
a,
M. J.
Tabernero-Duque
b,
I.
Sánchez-Sellero
b,
P.
Bermejo-Barrera
a and
A. M.
Bermejo-Barrera
b
aDepartamento de Química Analítica, Nutrición y Bromatología-Fac. de Química, Instituto de Materiales (iMATUS)-Universidad de Santiago de Compostela, Spain
bInstituto de Ciencias Forenses “Luís Concheiro” (INCIFOR), Fac. de Med., Universidad de Santiago de Compostela, Spain. E-mail: pamela.cabarcos@usc.es
First published on 18th October 2023
Abstract
Hygrine and cuscohygrine, two coca leaf alkaloids, have been previously proposed as markers to differentiate legal and illegal cocaine consumption. This is a very common problem in some countries of South America, where the consumption of coca leaves has a long tradition. Analytical methods focusing on the assessment of coca leaf alkaloids, such as cuscohygrine, hygrine, tropacocaine and t-cinnamoylcocaine, in oral fluid are virtually non-existent in forensic toxicology laboratories worldwide due to their lack of application. However, the problem of differentiating legal and illegal cocaine use in criminal justice, DUID (drug-impaired driving) and WDT (workplace drug testing) programs is growing. Therefore, researchers are obliged to develop methods to measure coca leaf alkaloids (cuscohygrine, hygrine and t-cinnamoylcocaine) in biological matrices for further validation for routine analyses in forensic toxicology laboratories. This work aims to optimize a previously published separation method by protein precipitation in oral fluid by using solid-phase extraction (SPE) coupled with liquid chromatography-tandem mass spectrometry (LC-MS/MS) operating in multiple reaction monitoring (MRM) mode. The use of SPE allowed the matrix effect and the background to be reduced in the chromatograms due to the obtained cleaner extracts. Consequently, improved detection and quantification limits were reached. Findings showed that the detection windows for coca leaf alkaloids were longer than three hours in real oral fluid samples from volunteers who drank a cup of coca tea. These detection windows are quite higher than those previously obtained when using the method based on separation by protein precipitation.
Introduction
Coca is a native South American plant with numerous alkaloid components, with cocaine being the psychoactive compound, the best alkaloid known.1 Coca has been used for millennia by indigenous people for numerous purposes, including ritual, social and physiological uses. Therefore, its uses have a long tradition in South America.2 In particular, the chewing of coca leaves, so-called “coqueo,” or drinking a coca leaf tea has been a long and revered practice since ancient times, besides in Argentina the consumption of coca leaves is legal due to an official Federal Law.3 This practice leads to positive test results for cocaine which makes it difficult to discriminate between legal consumption of coca (coca leaf chewing and coca tea drinking) and illegal cocaine use (sniffing, smoking or cocaine injection). Justice must frequently respond to the demands from victims regarding cocaine positive results in cocaine tests (urine and oral fluid) to elucidate a legal or an illegal practice of cocaine use.4 This also occurs in cases of driving tests under the influence of drugs (DUID), medical examinations at work (WDT), and anti-doping controls. For this reason, it is very important to be able to differentiate both practices from a toxicological point of view, which requires searching for suitable markers and developing analytical methods for their determination. Hygrine and cuscohygrine, two alkaloids present in the coca leaf, have been proposed as markers to differentiate both types of consumption in several biological samples since both substances are presumably lost during the cocaine preparation process.5–9Oral fluid (OF) is an alternative forensic sample for monitoring drugs of abuse. This biological sample has been used in clinical toxicology, criminal justice, for drugs of abuse testing in the workplace, and in DUID programs.10 The main advantages of OF are well documented in the scientific literature and include non-invasive and easy collection, difficult adulteration, and a better correlation with serum concentrations compared to urine. However, the results using this biological sample can be affected by factors such as sample pH, drug pKa, and drug plasma protein binding and volume of distribution. In addition, the device for OF collection is also quite important since it will condition sample dilution, drug stability, and drug adsorption.11
Despite the vast existing scientific literature on drugs of abuse in oral fluid, publications dealing with the analysis of coca leaf alkaloids, such as cuscohygrine (CUS), hygrine (HYG), t-cinnamoylcocaine (t-CIN), tropacocaine (TRO), anhydroecgonine methyl ester (AEME), and ecgonine methyl ester (EME), are scarce or almost non-existent. Only cocaine (COC) is widely mentioned.
The differentiation between the legal consumption of coca leaves and the illegal use of cocaine demands the development of methods capable of analyzing the existence of CUS, HYG, and also t-CIN in forensic matrices.
The objective of this work is to continue with the improvement of analytical methodologies for coca alkaloid assessment in oral fluid that can be used in criminal justice cases, DUID, and WDT for distinguishing between legal or illegal coca use. The development of new analytical strategies must enable detection of coca alkaloids (COC, CUS, HYG, t-CIN, and EME), meet international OF guideline requirements for COC (8 ng mL−1 or less), and allow the use of OF collecting devices. These purposes agree with Peters et al., who have stated that in addition to the purpose of the method development, a balance between the analytical problem and the expense and complexity of the method is needed.12
Our point of start has been the improvement of a previously published method for coca alkaloids in OF with the aim of decreasing the detection limit (LOD) and increasing the detection window of coca leaf alkaloids.13 It is intended to use 1 mL of oral fluid and SPE extraction (with the HLB Waters Oasis® extraction cartridge) in the same way as the routine method used in our laboratory for cocaine and benzoylecgonine (BE) and other analytes in forensic blood, meconium and hair samples.14,15 The applicability of the proposed method was demonstrated by analyzing three OF samples from volunteers after drinking coca tea.
Experimental
Reagents and chemicals
All standard solutions were prepared from stock standards (1 mg mL−1 dissolved in acetonitrile) of COC, BE, EME, t-CIN, AEME, and TRO supplied by LGC Standards, S.L.U. (Barcelona Spain). CUS (10 mg) was obtained from Toronto Research Chemicals Inc. (North York, Ontario, Canada). Deuterated analogous standard solutions (0.1 mg mL−1) of cocaine-d3 (COC-d3), benzoylecgonine-d3 (BE-d3), and ecgonine-d3 methyl ester (EME-d3) in acetonitrile were used to prepare 15 μg mL−1 internal standard mixtures. Deuterated analogous standards were supplied by LGC Standards, S.L.U. HYG was not commercially available at the time of the study, and it was obtained from coca leaves after extraction and further identification based on m/z (precursor ion) → m/z (product ion) transitions after coca leaf extract infusion and mass spectral recording. Acetonitrile and methanol (LC-MS grade) were purchased from Riedel-de Haën (Seelze, Germany), formic acid (98%) from Panreac (Barcelona, Spain) and ammonium formate (99%) from Fluka Analytical (Steinheim, Germany). Ultrapure water of 18 MΩ cm resistivity was from a water purification device from Millipore Co. (Bedford, MA, USA). The Waters Oasis® HLB extraction cartridge of 3 cm3/60 mg came from Waters, Spain.Working solutions
Three working standard calibrations were prepared in acetonitrile from stock solutions at concentrations of 10, 1 and 0.1 μg mL−1 for COC, BE, EME, t-CIN, TRO, AEME and CUS. Quality controls (QC) were prepared in acetonitrile at 10, 20 and 50 ng mL−1. A mix of deuterated analogues (COC-d3, BE-d3, EME-d3) was prepared in acetonitrile from stock solutions at a concentration of 1.5 μg mL−1. All working standards were stored at −20 °C when not in use.Calibrators and quality control (QC)
Seven drug-free OF samples spiked with drugs at concentrations of 1, 5, 10, 15, 20, 50, and 100 ng mL−1, and internal standards at 15 ng mL−1, and QC of 10, 20, and 50 ng mL−1 (also containing the internal standards at 15 ng mL−1) were prepared. Before being spiked, OF samples were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and 4 °C for 10 min. The spiked OF mixtures were subjected to the SPE procedure and the obtained extracts were further used for determining the working range, the detection limit (LOD) and the quantification limit (LOQ).
000 rpm and 4 °C for 10 min. The spiked OF mixtures were subjected to the SPE procedure and the obtained extracts were further used for determining the working range, the detection limit (LOD) and the quantification limit (LOQ).
Instrumental analysis and measurement (LC-MS/MS)
Determinations were carried out with a 3200 Q TRAP LC-MS/MS system (ABSciex, Concord, Canada), equipped with a Flexar FX-15 UHPLC binary chromatographic pump (PerkinElmer, Waltham, MA, USA), and a Flexar UHPLC autosampler (PerkinElmer). Analyst 1.6 software (ABSciex) was used for system control and data acquisition. MultiQuant 2.1 software (ABSciex) was used for data processing. Separations were performed with an Infinity LabPoroshell 120 Hilic (2.7 μm, 2.10 × 100 mm) from Agilent Technologies (Santa Clara, CA, USA). Temperature control of the column was performed with a GECKO 2000 column heater (temperature control from 30 to 80 °C) from Amchro GmbH (Hattersheim, Germany). Measurements were carried out under similar conditions as in our previous work.13 The gradient elution used was 20 mM ammonium formate in ultrapure water (pH 4.2) (A) and an acetonitrile/methanol (4:1) mixture (B) as a mobile phase for compound separation at a flow-rate of 300 μL min−1. The gradient was run as the following scheme: from sample injection until 0.1 min, gradient elution to 8% (A); from 0.1 to 2 min, a linear gradient to 9% (A); from 2 to 6 min, a linear gradient to 15% (A); from 6 to 11 min, a linear gradient to 60% (A); from 11 to 12 min, isocratic elution with 60% (A); from 12 to 13 min, a linear gradient to 40% (A); from 13 to 14 min, a linear gradient reduces to 8% (A); and from 14 to 20 min, isocratic elution back to 8% (A). Chromatographic separations were performed at 40 °C and the chromatographic time was 20 min.
The criteria that were used on the data acquisition parameters for each transition of the multireaction monitoring mode (MRM) are listed in Table 1. At least two precursor → ion product ion transitions were monitored for each analyte to ensure the specificity of the measurements (the presence of an analyte was confirmed when all qualifying MRM transitions in each chromatographic series were identified). The MRM transitions that offered the most sensitive MRM transitions were finally used for quantification. The optimized ion source parameters (positive ionization) were set at 400 °C for temperature, 5.000 V for voltage, 20 psi for curtain gas (N2), 15 psi for nebulizer gas (N2), and HIGH mode for collision gas (N2).
| Analyte name | Precursor ion, amu | Product ion, amu | ISTD | RT, min |
|---|---|---|---|---|
| a Amu: atomic mass unit; ISTD: internal standard; RT: retention time. | ||||
| BE | 290.1 | 168.2 | BE-d3 | 2.16 |
| 105.1 | ||||
| 77.0 | ||||
| COC | 304.1 | 182.0 | COC-d3 | 2.48 |
| 105.1 | ||||
| 82.0 | ||||
| 77.0 | ||||
| EME | 200.1 | 182.2 | EME-d3 | 4.20 |
| 82.2 | ||||
| 67.1 | ||||
| 41.1 | ||||
| t-CIN | 330.2 | 182.1 | COC-d3 | 2.28 |
| 103.1 | ||||
| 77.1 | ||||
| 51.1 | ||||
| TRO | 246.1 | 124.1 | COC-d3 | 3.20 |
| 77.1 | ||||
| 67.1 | ||||
| 51.1 | ||||
| CUS | 225.1 | 84.1 | COC-d3 | 11.38 |
| 42.1 | ||||
| HYG | 142.1 | 84.1 | — | 3.50 |
| 42.1 | ||||
| AEME | 182.0 | 122.0 | COC-d3 | 2.50 |
| 91.0 | ||||
| 65.0 | ||||
| BE-d3 | 293.1 | 171.2 | 2.20 | |
| 80.0 | ||||
| COC-d3 | 307.1 | 185.1 | 2.61 | |
| 85.1 | ||||
| EME-d3 | 203.1 | 185.1 | 4.30 | |
| 85.1 | ||||
Oral fluid and coca leaf samples
(b) Improvements on method development were checked by studying several parameters such as matrix effect (ME), LOD, and LOQ, using spiked OF samples. Therefore, OF samples taken from three laboratory volunteers after drinking a cup of coca tea were analyzed. Blank OF samples were taken from the three volunteers at −10 min (10 min before drinking a cup of coca tea). Then, OF samples were taken from volunteer 1 at 30, 60, 120 and 180 min after drinking, and from volunteers 2 and 3 at 30 and 180 min after drinking. The three volunteers drunk coca tea brewed from the same coca mate (brand and batch).
000 rpm and 4 °C for 10 min. One mL aliquots were transferred to a 10 mL glass tube and mixed with 10 μL of 1.5 μg mL−1 internal standard (IS) solution (mixture of COC-d3, BE-d3 and EME-d3) plus 1 mL of borate buffer pH 9.2. The mixtures were vortexed for 1 min before being loaded on the SPE cartridges. In addition, blank OF samples were processed in the same way, and after extraction they were spiked with several concentrations of standards and deuterated ISs. These samples were used as set B when assessing ME.
Coca leaves cut into small pieces (50 mg) were mixed with 5 mL of methanol/acetonitrile/2 mM ammonium formate (25:25:50, v/v/v), and the mixture was mechanically stirred for 15 min.7 Subsequently, the liquid phase was loaded into a SPE cartridge, and the extract was 1:50 and 1:100 diluted with acetonitrile/methanol (4:1). This extract was mainly used as a HYG control (commercial controls/standards for HYG are not available) and to confirm the presence of COC, t-CIN, EME, CUS, and AEME alkaloids (Fig. 1).
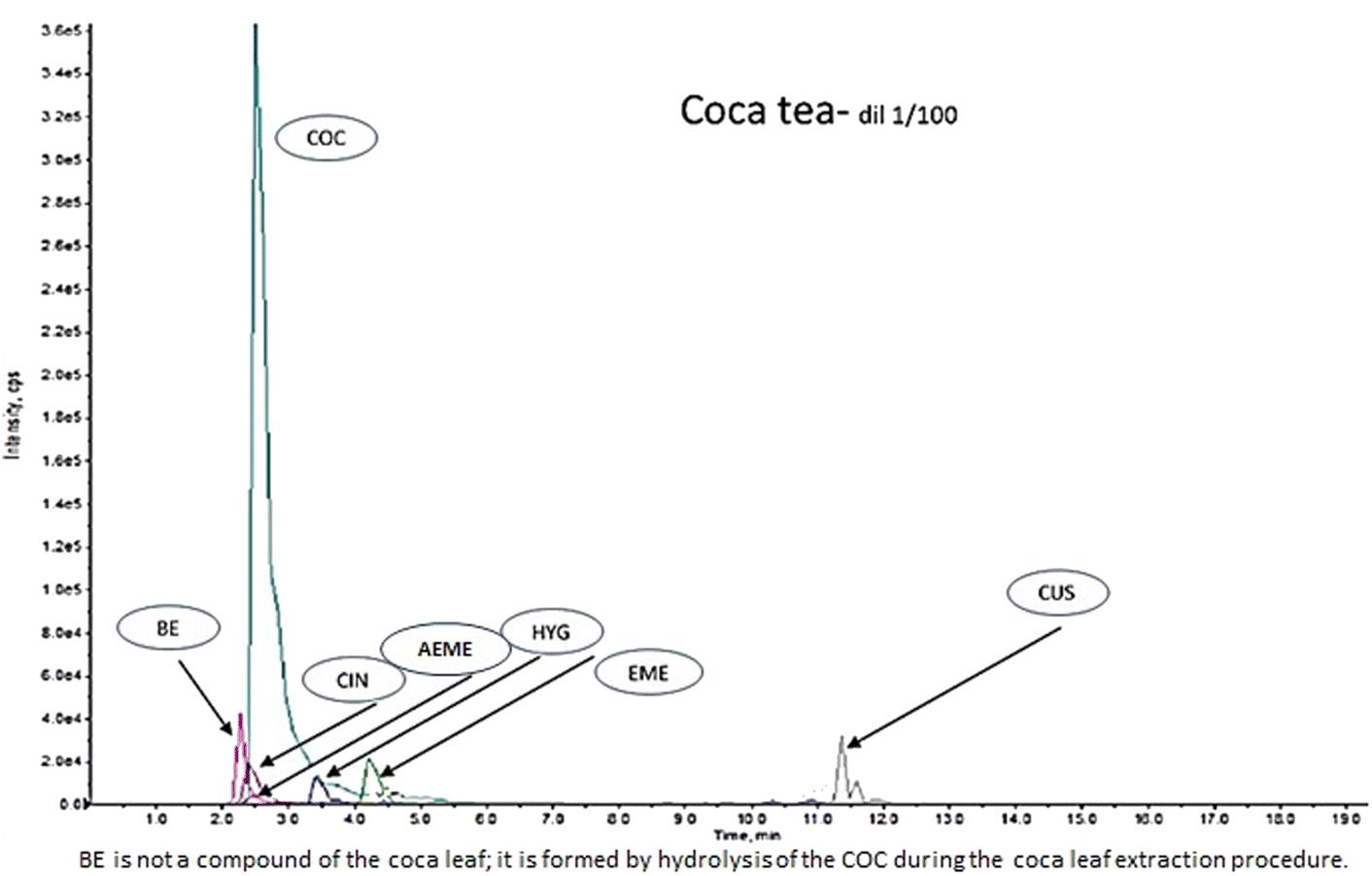 | ||
| Fig. 1 Coca tea (quantifier ion) LCMSMS chromatogram. (1) Liquid extraction of coca leaves. (2) Afterwards, extraction and cleaning by SPE. | ||
:1). The reconstituted samples were transferred to an auto-sampler, and 20 μL was injected in duplicate onto the LC-MS/MS.
Method development
Improvements on the performance of the previously published method were evaluated through the assessment of the LOD and LOQ, and the study of ionization suppression/enhancement (ME), interferences and carryover.13 These studies were performed following the guideline used in forensic toxicology formerly known as Scientific Working Group for Forensic Toxicology (SWFTOX).15The method development should comply with the recommendations for DUID and WDT, and therefore, the cut-off to confirm COC and BE in OF was set at 8 ng mL−1 for DUID and WDT since a cut-off of 20 ng mL−1 was established by DUID, and WDT recommends cut-off values of 15 ng mL−1 and 30 ng mL−1 to detect COC and BE in OF depending on the international guides.17–19 For the other analytes (EME, TRO, t-CIN, AEME, CUS), there are no recommendations/cut-off values, and the same criteria for COC and BE were used (cut-off of 8 ng mL−1 for each analyte).
The calibration range for COC, BE, EME, AEME, t-CIN, and TRO was set between 5 and 100 ng mL−1, whereas the range was varied from 10 to 100 ng mL−1 for CUS. Six calibration curves (each calibration at six concentration levels in addition to the blank sample) were prepared and run in duplicate on three different days (the OF from volunteers was analyzed each day to corroborate improvements on ME, LOD, and LOQ parameters, and also on the window of detection of leaf alkaloids in OF samples from coca tea drinkers). QC samples at 10, 20 and 50 ng mL−1 were analyzed in duplicate when performing each calibration curve. The variation between replicates in the calibration standards and the bias were lower than 20%. The choice of the concentration levels for calibration (5–100 ng mL−1) was done on the basis of the most frequent concentrations found in people who drink coca tea, including concentrations (higher and lower) close to the cut-off values given by DUID and WDT guidelines.
The LOD and LOQ values were established as 3 and 10 times the signal-to-noise ratio (S/N), respectively. The LOD was not determined for COC and BE because the LOQ was considered low enough for the objective of this research.
The ME (ionization suppression/enhancement) was evaluated following the strategy post extraction addition as follows:16 ME% = 100 − B/A × 100 where A is the average peak area of the analyte at a certain concentration in standard solutions, and B is the average peak area of the analyte (same concentration) but added to the SPE extracts after drug-free OF sample pre-treatment. ME was evaluated twice (two days) in triplicate at 20 and 50 ng mL−1 for COC, CUS, TRO, BE, t-CIN, AEME, and EME. Negative ME% values show ion suppression, whereas positive values mean ion enhancement.
The study of interferences (effect of the presence of common compounds in OF) was established by analyzing ten drug-free OF samples by the proposed method (without adding internal standards) and verifying that there were no chromatographic signals at the retention times and MRM transitions for each analyte. In addition, similar experiments were performed by adding ISs (15 ng mL−1) for evaluating interferences from stable isotope ISs.
Carryover was evaluated by monitoring two extracts obtained from two drug-free OF samples just after analyzing extracts from OF samples containing the analytes at several concentrations.
Results and discussion
The calibration model was the simple linear regression model using the least squares method. The R2 on the curve for all analytes was higher than 0.990. The carryover required a methanol blank injection between samples and, in addition, the autosampler was adjusted to clean the injector needle four times before injection and four times after sample injection (2-propanol was used to rinse the injector needle). No carryover was detected when using OF samples spiked until 500 ng mL−1 under this condition. No significant matrix components or IS interferences were observed when analyzing the ten drug-free OF samples.The LOD and LOQ and the ME were the parameters that had the best performance when compared with the previous method.13Table 2 shows the LOD and LOQ values and the ME for the method based on acetonitrile protein precipitation (0.2 mL OF) and after SPE extraction (1 mL of OF).13 The LOQs were reduced substantially for almost all analytes: from 50 ng mL−1 to 15 ng mL−1 for CUS, from 10 ng mL−1 to 5 ng mL−1 for EME and TRO, and from 5 ng mL−1 to 1 ng mL−1 for COC and BE. However, Table 2 shows that variations were not found for t-CIN and AEME. The LOD was found to be 1 ng mL−1 for EME, TRO and t-CIN, 5 ng mL−1 for AEME, and 10 ng mL−1 for CUS. The LOD for COC and BE was found to be lower than 1 ng mL−1. These findings show that the LOQ achieved for CUS does not fit with the requirement of 8 ng mL−1 as a cut-off established for COC and BE confirmation in OF.
| ng mL−1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| CUS | EME | TRO | t-CIN | COC | AEME | BE | ||
| a PP: protein precipitation extraction; SPE: solid-phase extraction; ne: not established. b Published in JAT 2019; 1–7. | ||||||||
| PPb | LOD | ne | 5 | ne | ne | ne | 5 | ne |
| LOQ | 50 | 10 | 10 | 5 | 5 | 10 | 5 | |
| ME | 13 | −67 | −59 | −74 | −61 | −59 | −34 | |
| SPE | LOD | 10 | 1 | 1 | 1 | ne | 5 | ne |
| LOQ | 15 | 5 | 5 | 5 | 1 | 10 | 1 | |
| ME | 19 | −37 | −17 | −16 | −17 | −12 | 10 | |
The ionization suppression has been found to be very important when using the published protein precipitation method with ME values higher than −55% for EME, TRO, t-CIN, COC, and AEME, and −34% for BE.13 However, ionization suppression was markedly reduced for EME, TRO, t-CIN, COC, AEME and BE when using SPE (Table 2). Even, ionization enhancement (+10) was found for BE, whereas CUS was the only analyte that retained the ionization enhancement around (+13, +19).
Table 3 lists the alkaloid concentrations in OF from volunteers whose samples were collected by spitting. It can be seen that the detection window for HYG, CUS, EME, BE, COC, and t-CIN was higher than 180 min (HYG was an exception in OF from volunteer 3), and we must take into account larger time windows in future OF sampling. Regarding AEME (a marker of smoked cocaine abuse), the detection window was up to 60 min in volunteer 1, whereas this alkaloid was not detected in volunteers 2 and 3.20,21 Similarly, TRO was also not detected in any OF samples. These results can be explained taking into account that AEME and TRO are alkaloids that are present in low concentration in coca leaves.22–24 As an example, Fig. 2 shows LC-MS/MS chromatograms of HYG and CUS in OF from volunteer 1 at 30, 60, 120 and 180 min after coca tea consumption.
| ng mL−1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| HYG | CUS | AEME | EME | TRO | BE | COC | t-CIN | |
| a OF: oral fluid taken by spit. nd: not detectable. HYG: It was considered positive when the signal meets the requirements quoted in LC-MS/MS conditions. Bold numbers: concentration below or above the calibration curve. | ||||||||
| Volunteer 1 | ||||||||
| OF_Blank | nd | nd | nd | nd | nd | nd | nd | nd |
| OF_30min | Positive | 765.9 | 323.1 | 167.7 | nd | 115.6 | 793.5 | 138.6 |
| OF_60 min | Positive | 73.6 | 6.9 | 125.2 | nd | 121.1 | 126.3 | 40.1 |
| OF_120 min | Positive | 30.7 | nd | 73.0 | nd | 31.4 | 4.4 | 1.8 |
| OF_180 min | Positive | 35.6 | nd | 69.7 | nd | 14.1 | 3.0 | 1.6 |
|
||||||||
| Volunteer 2 | ||||||||
| OF_30min | Positive | 99.8 | nd | 138.0 | nd | 76.7 | 330.1 | 46.0 |
| OF_180 min | Positive | 18.8 | nd | 110.4 | nd | 32.2 | 1.1 | 1.4 |
|
||||||||
| Volunteer 3 | ||||||||
| OF_30min | Positive | 204.7 | — | 178.0 | nd | 179.8 | 840.1 | 108.3 |
| OF_180 min | nd | 20.5 | nd | 56.0 | nd | 54.0 | 7.0 | 3.1 |
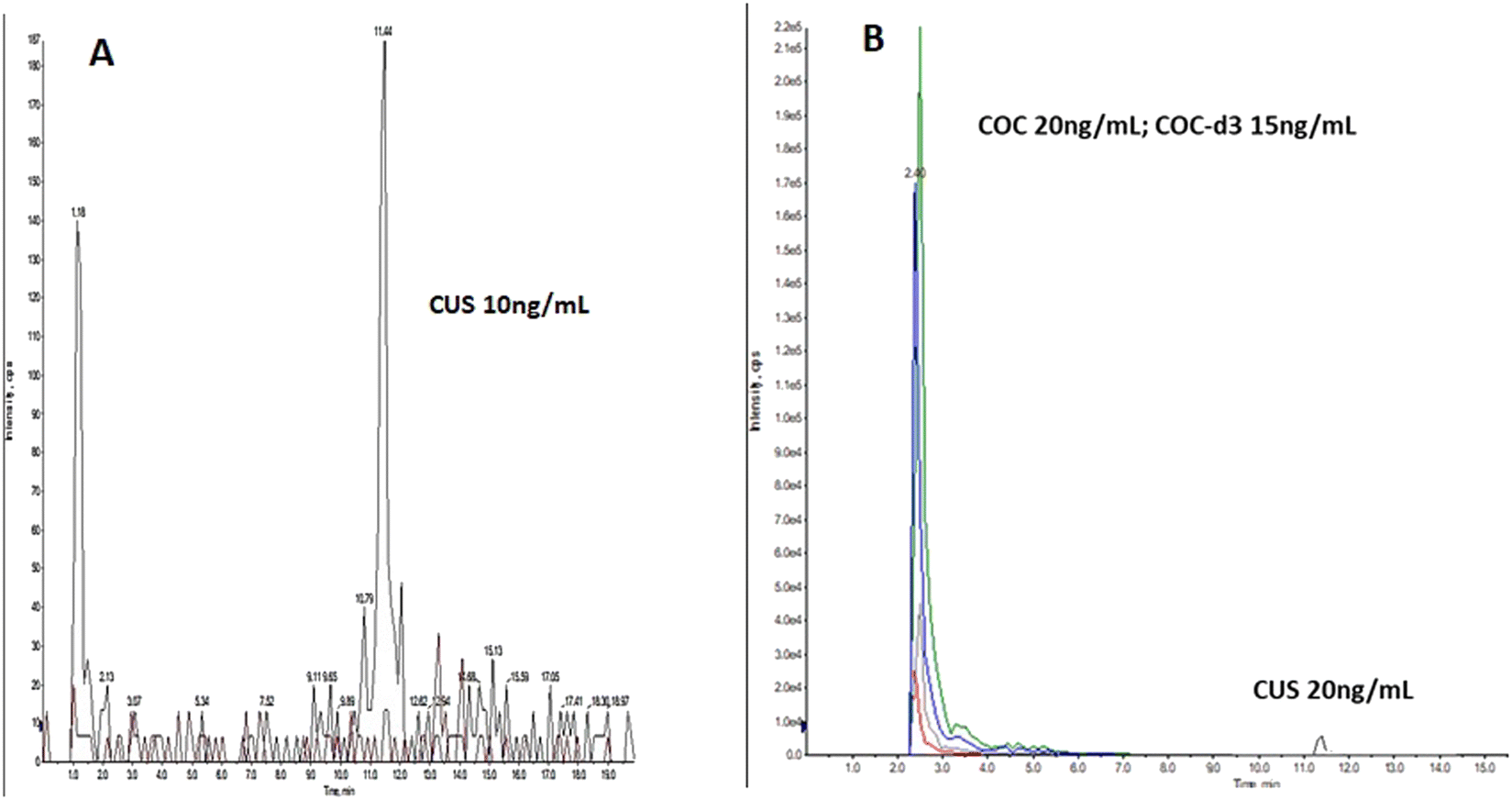 | ||
| Fig. 2 Oral fluid LCMSMS ion chromatogram, spiked with: (A) 10 ng mL−1 CUS, (B) 20 ng mL−1 COC, 15 ng mL−1 COC-d3 and 20 ng mL−1 CUS. | ||
Considering the cut-off values for COC and BE confirmation in OF (8 ng mL−1 in DUID and WLDT), BE concentration is above the international guideline requirement in all OF samples while COC is below the cut-off point at 120 min in volunteer 1, and at 180 min in volunteers 2 and 3 (COC was measured only at 30 min and 180 min in OFs from volunteers 2 and 3). These results show COC/BE ratios higher than 1 at times near to coca tea intake, and the ratios reverse to values lower than 1 at larger times (COC is metabolized mainly to BE and EME). Coca tea consumption markers (HYG and CUS) and coca leaf alkaloids such as EME and t-CIN are positive throughout the time window analyzed. We cannot rule out that the high concentrations of some of the analytes (CUS, COC, EME, t-CIN, and AEME) found at 30 min could be due to OF contamination by coca tea residues in the mouth (Fig. 3).
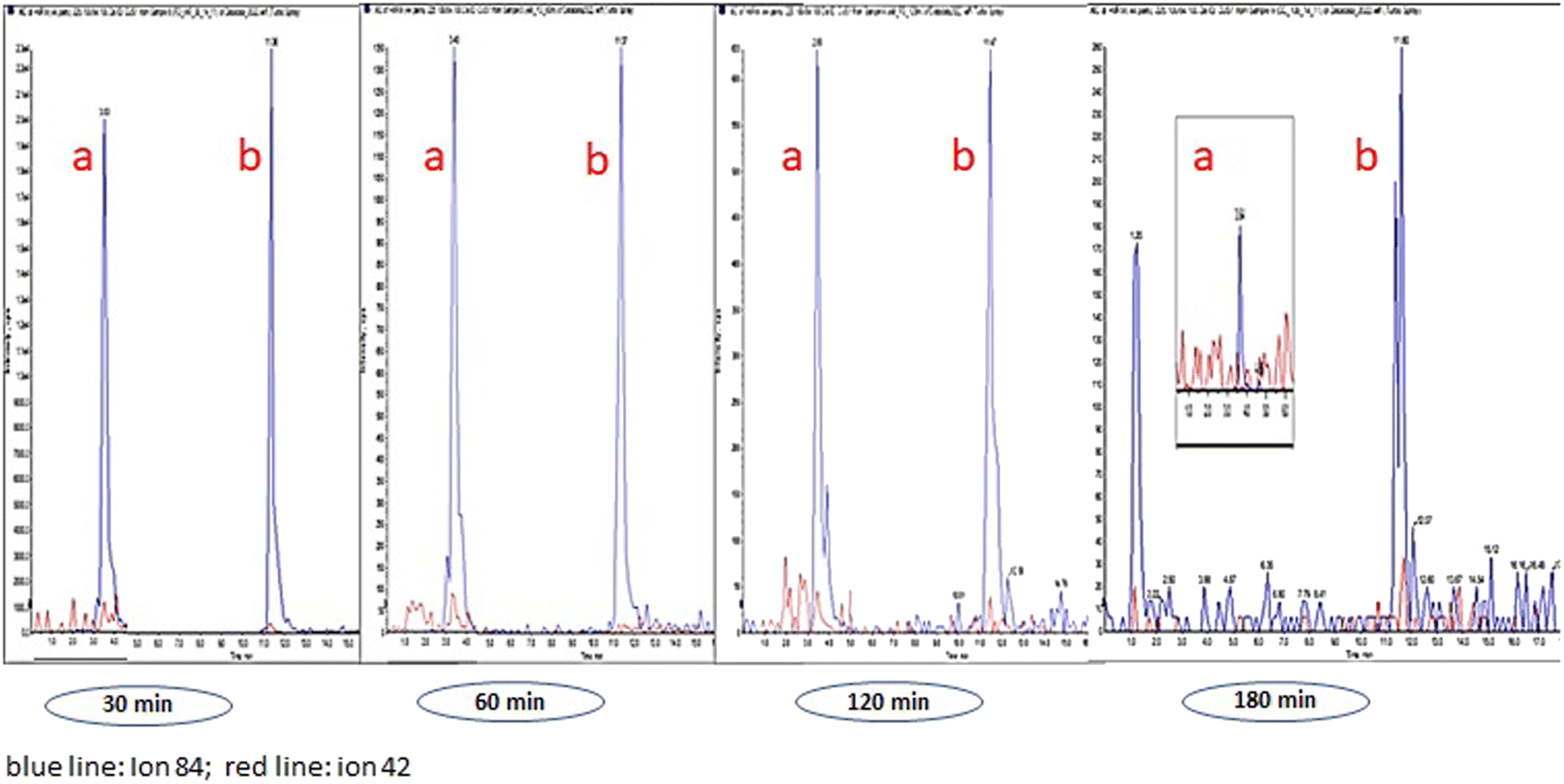 | ||
| Fig. 3 HYG (a) and CUS (b). LCMSMS chromatogram (volunteer 1). Sampling by spitting at 30, 60, 120 and 180 min. | ||
Conclusions
The use of SPE as an extraction technique has been found to improve the assessment of coca alkaloids in OF samples. The SPE extraction method (Waters Oasis® HLB extraction cartridges) and a large OF sample volume of 1 mL improve significantly the LOD of the previous method by minimizing the ionization suppression and background in the chromatographic spectrum. OF samples analyzed from volunteers after drinking a cup of coca tea seem to confirm this conclusion. The modifications introduced in the method not only extended the range of detection of coca leaf alkaloids but also the markers of coca leaf consumption (CUS, HYG, and t-CIN) were found to remain positive when COC and/or BE were above the cut-off of 8 ng mL−1. The developed SPE method fulfills an important requirement in the development of the method, i.e. detecting CUS and HYG (positive window) when COC and/or BE concentrations are still above the internationally established cut-off values. This first conclusion needs to be confirmed by analyzing OF samples from a larger number of volunteers. The next step in the optimization of this method before being used in the toxicology laboratory for routine analyses will be the selection of an OF collector device since OF samples taken by spitting are impractical for WDT or DUID control and unpleasant for the OF donor. Finally, results of this work contribute to solving a problem in some Latin American countries where coca leaf consumption is traditional, deeply rooted, and legal. Although the proposed coca leaf use markers (HYG, CUS, and t-CIN) are not conclusive to rule out joint cocaine use, they are useful to confirm that there was coca leaf use, and they can help judicial authorities (DIUD and WDT) to differentiate both types of consumption.Ethical statement
The authors declare therefore that all volunteers have signed informed consent for allowing the use of the provided oral fluid sample in this study.Conflicts of interest
There are no conflicts to declare.References
- A. S. Biondich and J. D. Joslin, Emerg. Med. Int., 2016, 4048764 Search PubMed.
- V. B. Stolberg, J. Ethn. Subst. Abuse, 2011, 10(2), 126–146 CrossRef PubMed.
- Argentine Federal Law 23737, art.15. O.B. October 10, 1989, Ley 23737/1989, https://www.argentina.gob.ar/, accessed January 2023.
- E. Feisthauer, A. Ameline, L. Gheddar, N. Arbouche, J. S. Raul and P. Kintz, J. Anal. Toxicol., 2022, 46(1), 108–113 CrossRef CAS PubMed.
- N. C. Rubio, S. Strano-Rossi, M. J. Tabernero, L. Anzillotti, M. Chiarotti and A. M. Bermejo, Forensic Sci. Int., 2013, 227, 60–63 CrossRef PubMed.
- N. C. Rubio, S. Strano-Rossi, M. J. Tabernero, J. L. González, L. Anzillotti, M. Chiarotti and A. M. Bermejo, Forensic Sci. Int., 2014, 243, 30–34 CrossRef CAS PubMed.
- N. C. Rubio, M. Hastedt, J. Gonzalez and F. Pragst, Int. J. Legal Med., 2014, 129(1), 69–84 CrossRef PubMed.
- N. C. Rubio, D. Thurmann, F. Krumbiegel and F. Pragst, Drug Test Anal., 2017, 9, 323–326 CrossRef CAS PubMed.
- N. C. Rubio, A. Moreda-Piñeiro, P. Bermejo-Barrera and A. M. Bermejo, Acta Toxicol. Argent., 2019, 27(2), 72–80 Search PubMed.
- W. M. Bosker and M. A. Huestis, Clin. Chem., 2009, 55(11), 1910–1931 CrossRef CAS PubMed.
- S. M. R. Wille, V. Di Fazio, S. W. Toennes, J. H. P. van Wel, J. G. Ramaekers and N. Samyn, Drug Test Anal., 2015, 7(3), 178–186 CrossRef CAS PubMed.
- F. T. Peters, D. K. Wissenbach, F. P. Busardò, E. Marchei and S. Pichini, Curr. Pharm. Des., 2017, 23(36), 5455–5467 CAS.
- N. C. Rubio, P. Bermejo-Barrera, A. Bermejo and A. Moreda-Piñeiro, J. Anal. Toxicol., 2019, 43(3), 196–202 CrossRef CAS PubMed.
- P. Cabarcos, M. J. Tabernero, I. Álvarez, P. López, P. Fernández and A. M. Bermejo, J. Anal. Toxicol., 2010, 34(9), 539–542 CrossRef CAS PubMed.
- P. López, A. M. Bermejo, M. J. Tabernero, P. Fernández and I. Alvarez, Anal. Lett., 2006, 39(11), 2307–2316 CrossRef.
- Scientific working groups for forensic toxicology (SWGTOX) standard practices for method validation in forensic toxicology, J. Anal. Toxicol., 2013, 37, 452–474 Search PubMed.
- B. K. Logan, A. L. D'Orazio, A. L. Mohr, J. F. Limoges, A. K. Miles, C. E. Scarneo, S. Kerrigan, L. J. Liddicoat, K. Scott and M. A. Huestis, J. Anal. Toxicol., 2018, 42(2), 63–68 CrossRef CAS PubMed.
- European Guidelines for Workplace Drug Testing in Oral Fluid 2022-11-01 Version 3.0 FINAL, EWDTS, http://www.ewdts.org/.
- Proposed Revisions to Mandatory Guidelines for Federal Workplace Drug Testing Programs-Oral Fluid, 2015, Federal Register, 80 FR 28053-2015 Version, Department of Health and Human Services, SAMHSA, https://www.federalregister.gov/documents/2015/05/15/2015-11523/mandatory-guidelines-for-federal-workplace-drug-testing-programs, accessed March 30, 2023.
- S. W. Toennes, A. S. Fandiño and G. Kauert, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 735(1), 127–132 CrossRef CAS PubMed.
- J. Takitane, V. Leyton, G. Andreuccetti, H. Gjerde, V. Vindenes and T. Berg, Forensic Sci. Int., 2018, 289, 165–174 CrossRef CAS PubMed.
- E. L. Johnson and S. D. Emche, Ann. Bot., 1994, 73(6), 645–650 CrossRef CAS.
- E. L. Johnson, Ann. Bot., 1995, 76(4), 331–335 CrossRef CAS.
- J. F. Casale, S. G. Toske and V. L. Colley, J. Forensic Sci., 2005, 50(6), 1402–1406 CAS.
Footnote |
| † Joint first authors. |
| This journal is © The Royal Society of Chemistry 2023 |