
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In vitro functional models for human liver diseases and drug screening: beyond animal testing
Alessia
Paradiso†
 a,
Marina
Volpi†
a,
Chiara
Rinoldi
b,
Nehar
Celikkin
c,
Nicola
Contessi Negrini
d,
Muge
Bilgen
c,
Giorgio
Dallera
d,
Filippo
Pierini
b,
Marco
Costantini
c,
Wojciech
Święszkowski
*a and
Silvia
Farè
*de
a,
Marina
Volpi†
a,
Chiara
Rinoldi
b,
Nehar
Celikkin
c,
Nicola
Contessi Negrini
d,
Muge
Bilgen
c,
Giorgio
Dallera
d,
Filippo
Pierini
b,
Marco
Costantini
c,
Wojciech
Święszkowski
*a and
Silvia
Farè
*de
aFaculty of Materials Science and Engineering, Warsaw University of Technology, Warsaw, Poland. E-mail: wojciech.swieszkowski@pw.edu.pl
bInstitute of Fundamental Technological Research, Polish Academy of Sciences, Warsaw, Poland
cInstitute of Physical Chemistry, Polish Academy of Sciences, Warsaw, Poland
dDepartment of Chemistry, Materials and Chemical Engineering “G. Natta”, Politecnico di Milano, Milan, Italy. E-mail: silvia.fare@polimi.it
eINSTM, National Consortium of Materials Science and Technology, Local Unit Politecnico di Milano, Milan, Italy
First published on 29th November 2022
Abstract
Liver is one of the most important and complex organs in the human body, being characterized by a sophisticated microarchitecture and responsible for key physiological functions. Despite its remarkable ability to regenerate, acute liver failure and chronic liver diseases are major causes of morbidity and mortality worldwide. Therefore, understanding the molecular mechanisms underlying such liver disorders is critical for the successful development of novel therapeutics. In this frame, preclinical animal models have been portrayed as the most commonly used tool to address such issues. However, due to significant species differences in liver architecture, regenerative capacity, disease progression, inflammatory markers, metabolism rates, and drug response, animal models cannot fully recapitulate the complexity of human liver metabolism. As a result, translational research to model human liver diseases and drug screening platforms may yield limited results, leading to failure scenarios. To overcome this impasse, over the last decade, 3D human liver in vitro models have been proposed as an alternative to pre-clinical animal models. These systems have been successfully employed for the investigation of the etiology and dynamics of liver diseases, for drug screening, and – more recently – to design patient-tailored therapies, resulting in potentially higher efficacy and reduced costs compared to other methods. Here, we review the most recent advances in this rapidly evolving field with particular attention to organoid cultures, liver-on-a-chip platforms, and engineered scaffold-based approaches.
1. Introduction
Three-dimensional (3D) in vitro models have become essential tools for understanding fundamental biophysical and molecular mechanisms of human biology. These systems have been recently proposed to recapitulate several tissues and organs, including kidney,1,2 lung,3,4 heart,5,6 and skeletal muscles,7,8 covering a broad spectrum of biomedical applications.To date, in vitro liver models have gained popularity due to the liver's critical role in various physiological processes – such as the regulation of fat metabolism, long-term mineral and vitamin storage, detoxification, and monitoring of innate and adaptive immunity – and increased mortality rates in the event of liver failure. Recent statistics, in fact, have shown that acute liver failure and chronic liver diseases (CLD) represent major causes of morbidity and death worldwide, causing approximately 2 million deaths (3.5% of total deaths) annually. Unfortunately, these figures are expected to continue rising due to the increasing obesity rate and alcohol consumption.9 As a result, understanding the molecular mechanisms underlying liver function and failure is critical for developing novel therapeutic strategies.
From a cellular and architectural point of view, the liver is shaped in a unique micro- and macro-architecture containing a variety of specialized cell types. Hepatocytes constitute the primary cell population fulfilling most of the liver functions. Other cell types include Kupffer cells, stellate cells, endothelial cells, and lymphocytes.10,11 Regarding its native architecture, the liver is microscopically arranged into hexagonal spatial units called lobules where hepatocytes line up in radial cords, separated by sinusoid branches of microvascular channels (Fig. 1). Such unique spatial and heterogeneous cellular organization generate a graded microenvironment enabling various metabolic functions to simultaneously occur in localized zones of the lobule.12 Another notable feature of the liver is its extraordinary regenerative capacity which allows vulnerable liver tissue to be fully replaced upon re-organization of the heterogeneous cell population – in particular hepatocytes and epithelial cells – restoring the hepatic functions required for body homeostasis.13–15
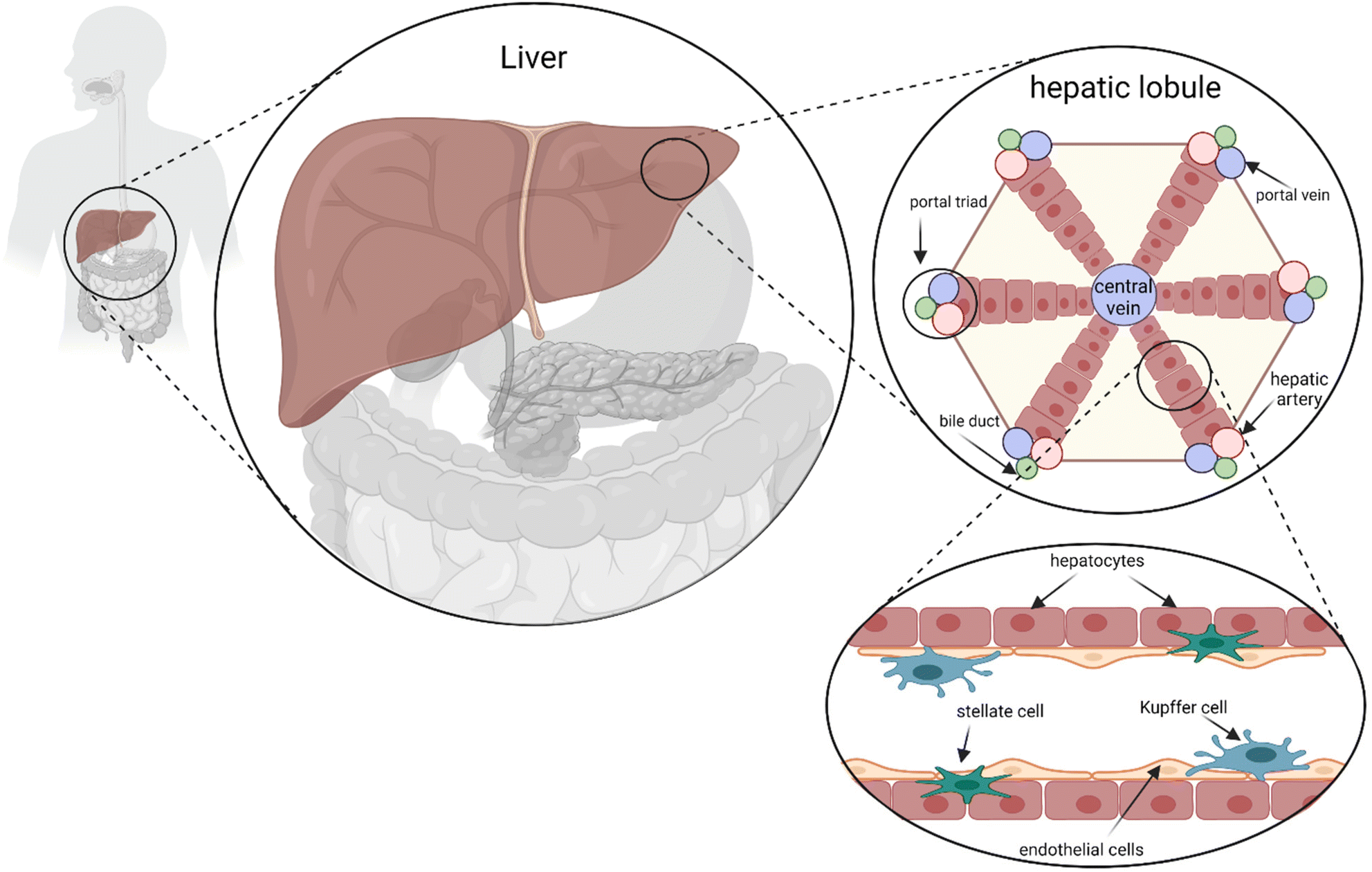 | ||
| Fig. 1 Human liver architecture and signaling. The hexagonal hepatic lobules are composed of sheets of hepatocytes radiating from a central vein towards six portal triads. Each portal triad is composed of a portal vein, a hepatic artery, and a bile duct. The lobule layers are mainly formed of hepatocytes along with endothelial cells, stellate cells, and Kupffer cells. | ||
Without a doubt, thoroughly recapitulating such complex biological processes within an in vitro liver model is currently out of our possibilities. Nevertheless, several in vitro assays and pre-clinical animal models have been developed to study further the liver's physiological functions, biomolecular mechanisms, and pathology development.16–20 Among in vitro models, 2D monolayer cultures (2DMCs) have been the most commonly used strategies due to their cost-efficacy, reproducibility, ease-to-perform, and easy-to-scale protocols.21 Despite the significant advantages of 2DMC, they also exhibit several limitations. For instance, the evaluation of specific hepato-functionality is possible only in short-term studies (e.g., 24–48 h) as cells progressively tend to lose their polarity and dedifferentiate into fibroblast-like cells.22,23 Along with 2DMCs, animal and humanized animal models are also relevant tools for modeling liver disease and accessing drug discovery processes. In this frame, rodents have been conventionally used for research and development purposes. However, such models cannot thoroughly recapitulate human liver architecture and functions, being thus poorly reliable for many diseases and hepatotoxicity studies.24,25 Herein, the development of 3D biomimetic human liver models for in vitro research is considered a promising bridge between 2DMCs and pre-clinical animal testing for drug screening, as well as for the investigation of CLD and the efficient design of patient-tailored therapies in liver diseases.26
In this review, we present an overview of the current state-of-the-art on main strategies for 3D liver model biofabrication. Specifically, engineered liver tissue constructs such as organoids,27 liver-on-a-chip platforms,28,29 and 3D scaffold-based constructs are thoroughly described (Fig. 2).30–32 Key advantages together with ongoing challenges and future outlooks of the current liver models are highlighted, especially from 3D manufacturing and biomaterials perspectives.
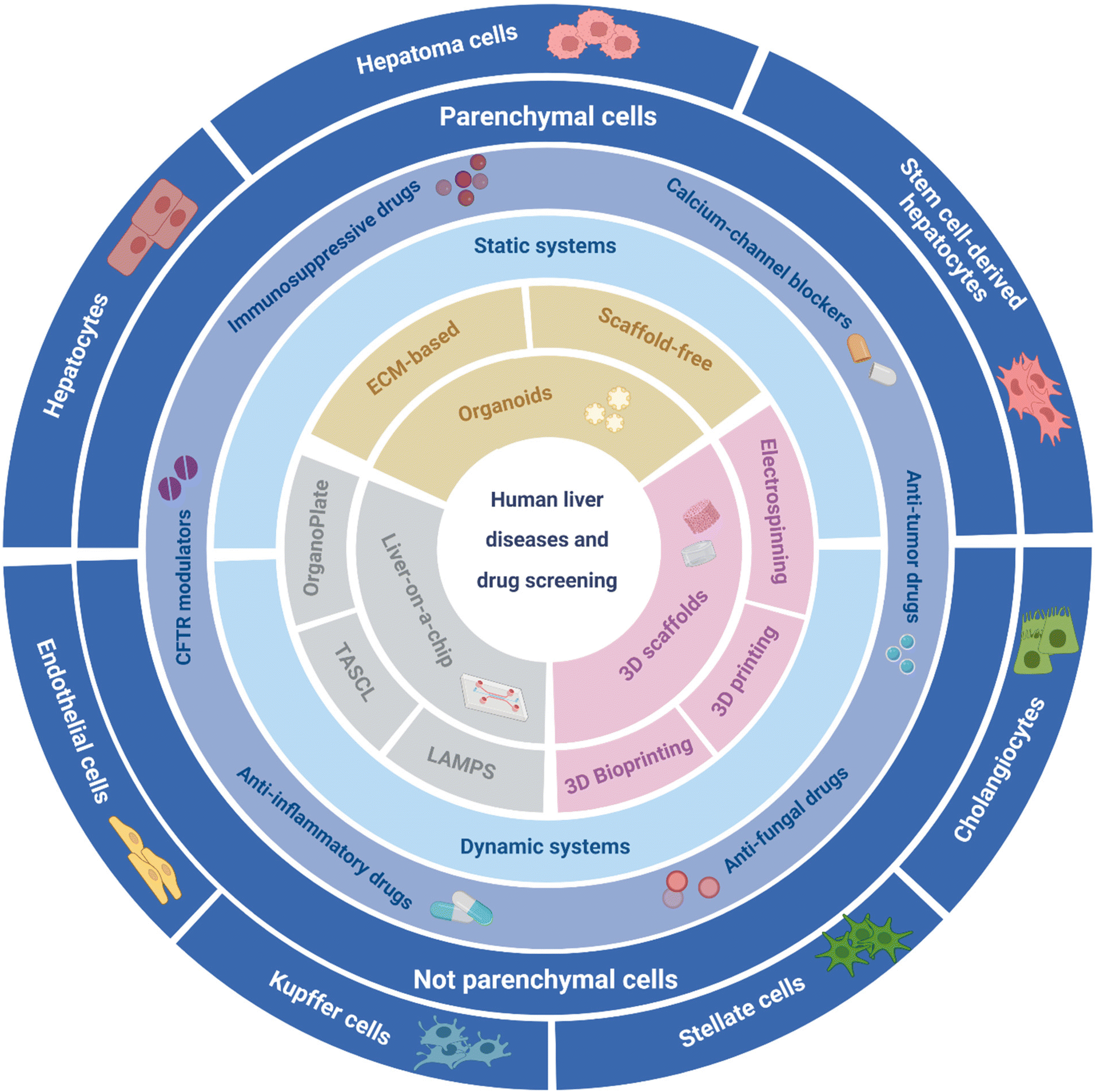 | ||
| Fig. 2 Representative scheme of relevant elements for developing in vitro functional human liver disease and drug screening models. The schematic represents the different strategies reported in this review to implement liver models, including Organoids (ECM-based and scaffold-free structures), liver-on-a-chip (OrganoPlate, TASCL, and LAMPS), and 3D scaffolds (3D printing, 3D bioprinting, and electrospinning techniques). The possibility of applying the models as static and dynamic systems is displayed. Additionally, various alternatives related to possible bioactive molecules (e.g., anti-tumor drugs, anti-fungal drugs, anti-inflammatory drugs, immunosuppressive drugs, mutant cystic fibrosis transmembrane conductance regulator (CFTR), and calcium-channel blockers) as well as parenchymal (e.g., hepatocytes, hepatoma cells, stem cell-derived hepatocytes) and not parenchymal (e.g., Kupffer cells, stellate cells, endothelial cells, cholangiocytes) cell types have been explored in the reported liver models. The design, optimization, and combination of the components have made possible the successful modeling of various liver disease models and liver drug screening platforms described in this review. | ||
2. Liver organoid models
Organoids are self-assembling 3D cell culture systems that recapitulate tissue-specific architectures, functionality, and spatial arrangement,33,34 promoting organ-specific cellular self-organization. Organoids are obtained from either tissue-specific progenitor cells or stem cells that are able to self-assemble in organ-specific cell types upon cell sorting and spatially restricted lineage commitment as in the in vivo organ.33,35 Moreover, they can be formed either in an extracellular environment or without using supportive biomaterials (i.e., spheroids).36,37 To date, the gold standard biomaterials used for organoid cultures is Matrigel.38,39 However, alternative materials such as collagen, laminin, fibrin, and cellulose nanofibril as supporting hydrogel-based scaffolds40–42 have gained significant attention due to their prominent features promoting the organoid formation and survival.43–45 In liver tissue engineering (LTE), 3D liver organoids are considered to mimic the miniaturized in vivo-like complex functionalities of healthy liver tissue, but also to model pathological liver conditions for the investigation of human diseases. Herein, stem cells, including induced pluripotent stem cells (iPSCs) and primary liver cells (e.g., primary human hepatocytes (PHH)), can be used to target functional patterns where cells are encouraged to self-assemble into functional 3D aggregates (Fig. 3A).46–52 Indeed, the self-organization ability of stem cells is crucial to boost the organ-specific mimicking, thus allowing for the generation of 3D constructs more stable than 2D cultures upon maturation of selected cell sources (e.g., embryonic stem cells (ESC), iPSCs and organ-specific cells as adult stem cells (ASCs))50,53–56 that can be obtained from tissue biopsies.57 Besides stem cell-derived cells, also cancer cell lines (e.g., HepG2, HepaRG, HUH7, primary liver cancer cells (PLC)) are generally used as cell sources for the development of liver organoids.52,58–61 | ||
| Fig. 3 Liver organoid-based models. (A) Schematic of organoid formation: in the case of healthy donors, the final organoids can be formed from stem cells, hepatocytes, cholangiocytes, endothelial cells, or their combination; in the case of pathologic cells (affected by tumor, hepatitis, etc.), the organoids represent a liver disease model formed from pathological or cancer liver cells. (B) Bright-field images of hepatic organoid growth at different culture passages (P). Reproduced with permission.72 Copyright 2019, The Authors, published by Elsevier. (C) Representative tissue biopsies of tumor and healthy liver used to form organoids. (D) Culture of organoids. Bright-field images of organoids derived from tumor and healthy livers: tumor organoids appear as compact spheroids, while healthy liver organoids show cystic structures. P = passage number; scale bar: 500 μm. (C and D). Reproduced with permission.75 Copyright 2018, The Authors, published by Elsevier. | ||
Organoid formation protocols have been mainly developed from principles in organogenesis. To achieve self-organization, it is essential that organoids should be treated with appropriate biochemical (in liver organoids, e.g., growth factors/glucose addition) and biophysical (e.g., matrix stiffness, mechanical properties) stimuli to promote maturation, tissue-specific or disease-specific differentiation. More specifically, organoid culture media must contain growth factor such as R-spondins (Rspo1) to promote organoid formation as well as Noggin (NOG) and bone morphogenetic protein (BMP) signaling antagonists to either inhibit or prevent other tissue-specific differentiations cues (e.g., mineralization).62,63 Parallelly, ascertained medium glucose levels have to be utilized to control cellular proliferation, investigate drug, and hepatic metabolism outcomes, as well as glucose production.64–66 Moreover, specific interactions among cell populations and biophysical environment should also be strictly controlled to recapitulate heterogeneous structures like the liver tissue.67,68 Organoids can be formed via 3D suspension culture systems (i.e., using ultra-low attachment plates), spinning bioreactors, air–liquid interface methods, and extracellular matrix (ECM)-based embedding matrices (e.g., Matrigel). Also, combinations of different methods may enhance the organoid formation and boost tissue-specific functions.69 For instance, ECM embedded organoids can be used as a scaffolding material, and bioreactors may be employed to improve nutrient absorption.70
One of the first attempts that demonstrated the successful generation of a functional human liver organoid from pluripotent stem cells was reported by Takebe et al., who obtained a vascularized functional human liver from iPSCs upon transplantation of liver buds.71 More recently, Akbari et al. obtained in vitro differentiation of endoderm-derived hepatic organoids into functional hepatocytes using a human-derived iPSC organoid culture system, producing both healthy and disease models from healthy human donors and citrullinemia patients, respectively.72 These liver organoid models were then further investigated to model the urea cycle disorder referred to as citrullinemia type 1 (CTLN1), a disease caused by a mutation in the argininosuccinate synthetase 1 (ASS1) gene causing ammonia accumulation in the blood. The authors modeled CTLN1, using EpCAM+ (Epithelial Cell Adhesion Molecule positive) endodermal cells as an intermediate, thus generating functional pathologic hepatic organoids (eHEPOs) that exhibited epithelial morphology and a pseudostratified structure (Fig. 3B).72 Moreover, this study indicated that eHEPOs were suitable to be expanded for more than six months without any significant loss in their phenotypic characteristics and proliferation rate, confirming the pluripotency of citrullinemia patient-derived iPSCs.72 Disease-related ammonia accumulation and ASS1-enzyme related overexpression were detected in CTLN1 patient organoids compared to healthy eHEPOs, thus recapitulating the urea cycle-related disease phenotype in the hepatic organoids. Human iPSCs have also been manipulated to generate heterogeneous functional liver organoids for modeling infectious diseases such as hepatitis B virus (HBV), which may cause life-threatening liver infections.73,74 Nie et al., investigated the host–HBV interactions causing hepatic dysfunction. The outcomes of the study were correlated to the patient-specific genetic background to develop a promising tool for personalized hepatitis treatment.73
Also, organoid platforms have been used to model alcoholic liver diseases (ALD), a CLD caused by an excess of alcohol in the liver. An interesting ALD-model was obtained using hESC-derived expandable hepatic organoids, which in turn incorporated human fetal liver mesenchymal cells (hFLMCs) to mimic ALD-related pathogenesis (e.g., liver inflammation).50 An ethanol (EtOH) treatment was assessed to recapitulate the ALD mechanism successfully. Upon optimization, the EtOH-treated hepatic organoids showed increased pro-inflammatory signaling of interleukins-1 (IL-1) and interleukins-17 (IL-17) compared to untreated control organoids, as well as fibrosis and ECM accumulation. Recently, ALD-related fatty liver diseases such as steatohepatitis have also been modeled with organoid cultures to recapitulate the pathology progression and potentially use of the developed platform for drug screening purposes.47 Multi-cellular human liver organoids (HLOs) were generated using 11 different types of healthy and diseased patient-derived iPSCs and hESCs. A free fatty acid exposure was assessed on HLOs via oleic acid treatment which significantly induced the progression of the steatohepatitis-like pathology over time (i.e., steatosis, inflammation, and fibrosis), as well as increased organoid stiffness due to the extensive liver fibrosis. Surely, the successful development of such multi-cellular in vitro organoid models should be considered relevant for further screening of human liver disease treatments.
Besides iPSCs and hESCs, primary cell-derived organoids have been reported as 3D models that may represent the donor tissue closely.75–77 In light of this, Gómez-Mariano et al. obtained adult human liver organoids from liver biopsies to model liver disease originating from different mutations of the alpha-1 antitrypsin (AAT) protein, an inhibitor produced by PHH that protects organs from infections and irritation effects.76 Such engineered organoids successfully recapitulated the typical features of deficient AAT liver cells. Indeed, results showed typical hypoalbuminemia and lower AAT secretion caused by AAT deficiency, providing a preclinical model for AAT-related liver diseases. Similarly, Nuciforo et al. generated long-term human liver tumoroids able to maintain histological features of the originating tumor from hepatocellular carcinoma (HCC) patient needle biopsies, allowing for the in vitro recapitulation of the cancer growth pattern up to 1 year (Fig. 3C and D).75 In the same study, patient-specific sensitivity to sorafenib – a kinase inhibitor drug used to treat HCC – was tested on tumoroids, leading to potentially reduced differences in the efficacy of current clinical HCC treatments among patients and ultimately suggested as a valid tool for tailored therapies.
Over the last decade, liver organoids have been widely investigated also for drug development and screening platforms, toxicity testing tools, as well as for the design of patient-specific therapies.78–80 For instance, patient-derived organoids have been described to validate drug studies for specific groups of patients to predict the effectiveness of therapies against drug-induced liver injuries (DILI).81 Relevant outcomes were shown by Skardal et al., who tested the cytotoxic effect of the chemotherapy medication 5-fluorouracil (5-FU) at different concentrations (i.e., from 0 up to 100 mM) on liver tumoroids for drug screening purposes.82 Organoids consisted of crosslinked dextran (Sephadex®) microbeads that were coated with thiol-functionalized hyaluronic acid (HA)/thiol-functionalized gelatin hydrogel blend by using a reduced pressure method, thus obtaining hyaluronic acid-coated microcarriers (HAMs). Subsequently, human colon carcinoma cells HCT-116 Tumor Foci and HepG2 cells were seeded on the HAMs surface to finally generate liver organoids with a rotating wall vessel bioreactor culture system. A 5-FU dose-dependent decrease in the organoid metabolism was revealed within the range 0–10 mM.82 Similar to 5-FU, patient-derived organoids treated with sorafenib has also showed a dose-dependent trend on decreased growth of hepatocellular carcinoma cells directly obtained from human tumor needle biopsies.75 An interesting study on patient-specific cholangiocyte organoid-laden collagen type-1/Matrigel culture revealed the potential cancer medication effect of VX-770 (i.e., Ivacaftor) on cystic fibrosis (CF) by restoring the CFTR gene, which causes such genetic disorder.49 In addition, octreotide- a synthetic analog of somatostatin- was also tested for polycystic liver disease reducing the organoid size, thus revealing its role in reducing the cyst size.48
Besides CF and liver tumors, organoids have also been employed to investigate in vitro the treatment of DILI. Indeed, DILI is one of the major causes of liver damage worldwide, and it can be classified as direct- and idiosyncratic-drug hepatoxicity.83–85 Direct hepatoxicity is mostly dose-dependent; on the other side, drug sensitivity reactions may lead to idiosyncratic reactions. Herein, various studies have been designed to assess the hepatic toxicity of medical drugs.86–89 Standard evaluation methods include the measurement of hepatic enzyme activity and structural changes in hepatocytes. An interesting study by Au et al. proposed a microfluidic drug screening platform for collagen type 1-based liver organoids treated with dexamethasone (i.e., CYP3A4 inducer) and ketoconazole (i.e., CYP3A4 inhibitor) to assess the model's drug metabolism.88 Liver organoids were formed in the custom-made device by co-culturing HepG2 and NIH 3T3 fibroblast cells embedded in the hydrogel matrix. Given that the organoid volume has decreased enabling cell–cell interaction and tissue functionalities due to higher and native-like cell densities in a reduced volume, a contractility test was performed.90 Results showed that the presence of the fibroblasts may reduce the volume of the organoid over 4 days, compared to sole HepG2-organoids. Likewise, albumin secretion after 4 days was significantly higher in co-culture organoids than in HepG2 mono-culture 3D structures, thus showing the role of fibroblasts in the improved functional activity of HepG2 hepatocytes and organoid densification. Additionally, the 3T3-HepG2 co-culture organoids treated with dexamethasone exhibited higher CYP activity and metabolism, while ketoconazole-treated organoids revealed a lower metabolism as expected from the chemical inducer and inhibitor treatment, respectively. Similar studies on CYP activity and organoid microstructure have been modeled using common drugs such as acetaminophen (APAP) and troglitazone on Matrigel-based 3D cultures.54,86,87 For instance, Ramli et al. developed hepatic organoids that closely mimicked the 3D interaction of two different human liver cell types (i.e., hepatocytes and cholangiocytes) to engineer an organized functional bile canaliculi system.54 A drug-induced cholestasis was modeled by incubating the organoids in troglitazone for different time periods, showing their role in the loss of the bile canaliculi system. Both hepatocyte and cholangiocyte functionalities were confirmed from the increased CYP450 activity expressed over the differentiation time and the alkaline phosphatase (biliary) activity, respectively; thus, suggesting that the model can be used to explore and further study liver cholestasis.
Alongside disease models, liver organoids in microfluidic systems have also gained attention for the possibility of developing large-scale high-throughput toxicity- and drug testing platforms.91 In a study proposed by Shinozawa et al., a robust protocol to form human iPSC (hiPSC)-based liver organoids for the preclinical identification of DILI has been developed and tested for 238 active components of commercially available drugs including antibiotics (erythromycin), chemotherapy agents (floxuridine), antivirals (Ritonavir), and anti-inflammatory drugs (nimesulide).81 Similarly, on a smaller scale, vascularized ECM-based human pluripotent stem cell-derived liver organoids have also been utilized for high-fidelity screening of different marketed drugs based on cholestatic and mitochondrial toxicity.80
Ultimately, the most significant advantage of the organoids is that they are the only 3D in vitro models that can be cryopreserved, presenting promising opportunities for specifically biobanking applications.92–95 Especially, cryopreservation of human ESC (hESC)-derived organoids hold great attention of biotech companies for pharmaceutical purposes. Despite ethical concerns on both research and therapeutic use of hESC organoids, their unlimited capability for self-renewal as well as the potential to differentiate into different tissues make them unique tools for personalized therapies to model liver diseases such as CF,49 ALD,47,50 and injuries,50 together with toxicity prediction purposes.86,92–95
3. Liver-on-a-chip platforms
Organ-on-a-chip platforms have been developed to scale single organ or tissue-specific functions down to the chip format.96 Typically, such platforms range from millimeters to centimeters in size and are characterized by microfluidic channels that guarantee a versatile manipulation of small amounts of fluids at the micro-scale. Thus, these tools provide a precise control over the flow regimes and an accurate timing-control of chemical reactions that the user may trigger.97 Herein, microchannels can reproduce in vivo tissue-specific cell densities by manipulating media-to-cell volume ratio.98,99 Moreover, the number of cells required to develop these models is significantly lower than that of macroscale systems (e.g., 3D scaffold-based models). Such a feature may be a critical advantage considering the limited availability and low proliferative character of primary hepatocyte cultures. However, the number of cells required is still a critical parameter which has to be carefully selected considering the dimension of the microfluidic platform.Among others, cell patterning is one of the most pioneering techniques in fabricating liver-on-a-chip platforms, as it enables appropriate positioning of the different cell populations in complex designed systems, including Liver Acinus MicroPhysiology System (LAMPS), Tapered Stencil for Cluster Culture (TASCL), and OrganoPlate® (Fig. 4A). In one work by Ho et al., hepatic-like lobule arrays have been designed to recreate the lobule pattern, allowing for the precise positioning of two different cell lines (i.e., human liver cancer cell line (e.g., HepG2) and human umbilical vein endothelial cells (HUVEC)).100 In such a system, an organized cell distribution with appropriate morphology was obtained by increasing liver-specific enzymatic activity.
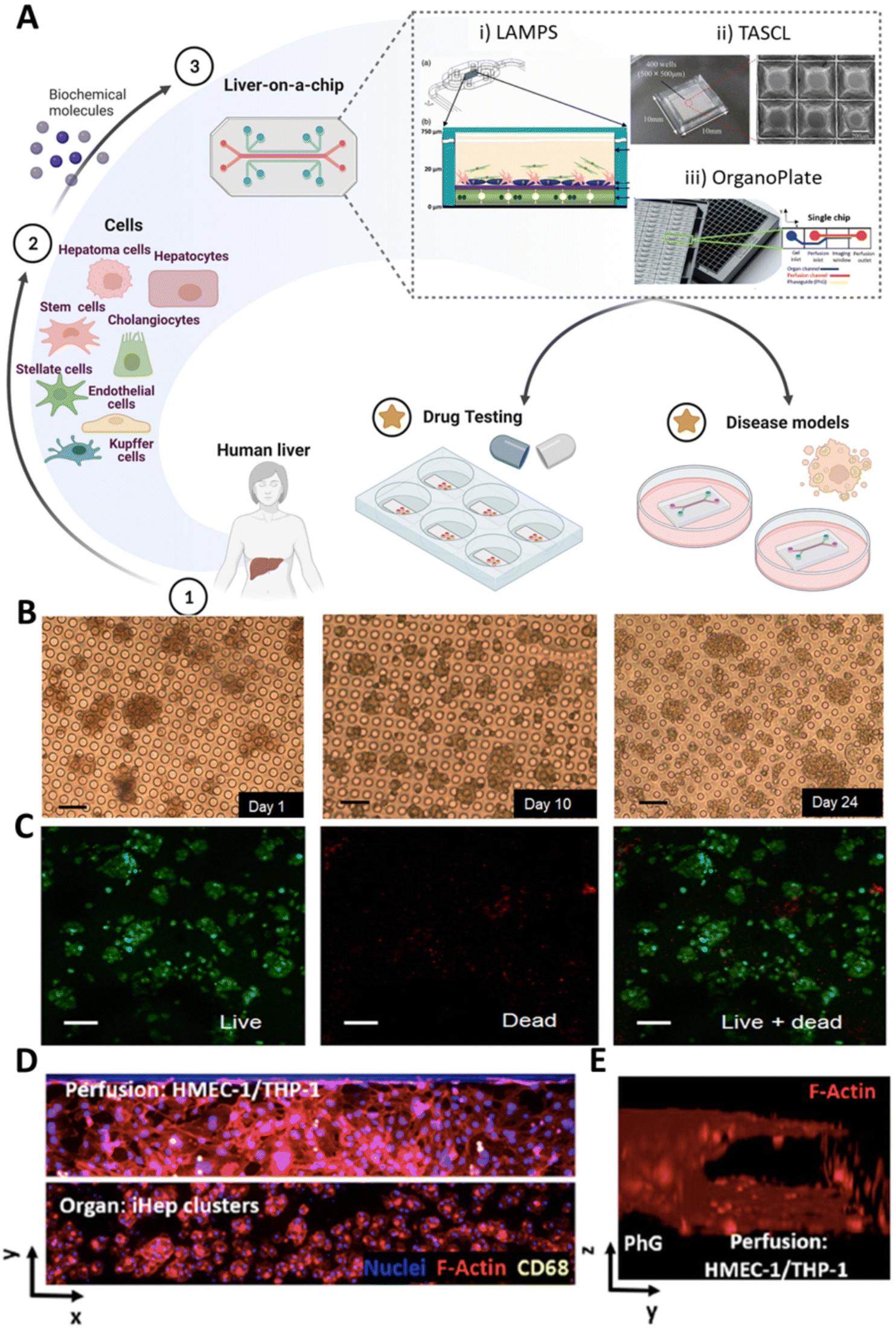 | ||
| Fig. 4 Liver-on-a-chip platforms. (A) Different cell sources, such as stem cells, endothelial cells, and liver-derived cells (e.g., healthy and pathological parenchymal and non-parenchymal cells) can be introduced into a microfluidic chip to obtain liver-on-a-chip models. The microfluidic devices, including LAMPS (i), TASCL (ii), and OrganoPlate® (iii), can be employed for disease modeling or drug testing purposes. (i) Reproduced with permission.115 Copyright 2017, SAGE. (ii) Reproduced under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).118 Copyright 2015, published by SAGE. (iii) Reproduced under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).120 Copyright 2021, The Authors, published by Elsevier. (B and C) Evaluation of hepatocyte spheroid response into the chip: (B) cell morphology observed in phase contrast micrographs up to 24 days of culture; (C) cell viability evaluated via live/dead staining at day 24 of culture. Scale bar: 100 μm. Reproduced under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).102 Copyright 2017, The Authors, published by Springer Nature. (D) Fluorescent images of 3-cell type liver-on-a-chip: organ and perfusion channels are seeded with pluripotent stem cell-derived hepatocytes (iHep) clusters and endothelial (HMEC-1) cells/Kupffer (THP-1) cells, respectively. Cells are stained with nuclei/F-actin/CD68 in blue/red/yellow colors. (E) 3D reconstruction of the perfusion channel. (D and E) Reproduced under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).120 Copyright 2021, The Authors, published by Elsevier. | ||
Similarly to other 3D in vitro models, liver-on-a-chip platforms provide a suitable microenvironment allowing for mimicking cell–cell interaction and in turn maintaining of cell morphology and polarization.101 Moreover, liver-on-a-chip platforms enable the study of both static and dynamic cell culture conditions. Although static cell cultures have been successfully employed in pre-clinical disease modeling and drug development studies, in vitro perfused cell culture systems are characterized by a more biomimetic condition (Fig. 4B and C).102 In fact, static culture systems may limit the nutrient supply and accumulate waste, likely inducing hepatocyte dedifferentiation and decreasing liver-specific functions.103 On the other hand, perfusable systems can provide continuous transfer of nutrients and metabolites to finally mimic the multiple cell–cell interactions of the liver sinusoidal structure.104,105 Furthermore, dynamic culture conditions allow for the creation of a stable oxygen gradient, thus replicating the peculiar metabolic zonation of the liver.28,106–109 For these attractive features, research on liver-on-a-chip platforms has gained enormous attention in recent years, and various strategies have been established.
For instance, Lee et al. designed a biologically inspired artificial liver sinusoid in a microfluidic chip that mimics the liver endothelial barrier layer. A collar region of parallel microchannels (2 μm × 1 μm, width × height) was designed to reproduce the endothelial-like barrier, which in turn surrounds a main cell inlet channel. A sinusoid-like flow channel was patterned at the frame of the chip, thus allowing for hepatic microcirculation. Herein, different mass transport parameters (e.g., channel length, height and width, fluidic resistance, volumetric flow rate, Reynolds number) were set.110 In the middle of the device, a cell culture area was placed. Primary rat and human hepatocytes (PRH and PHH, respectively) were loaded into the cell culture area. At the same time, the microfluidic platform was continuously perfused with culture medium in a standard incubator. Since the barrier channels had a cross-section much smaller than the cell diameter, hepatocyte packing was enabled without causing membrane damage. Moreover, due to the small scale of the device, continuous nutrient exchange was possible through simple diffusion, and extensive cell–cell interactions were allowed. In light of this, such culture conditions promoted high hepatocyte viability (>90%) for over 7 days of culture.
Another advanced perfusable 3D in vitro liver chip was developed to model Non-Alcoholic Fatty Liver Disease (NAFLD) by encapsulating sacrificial thermoresponsive poly(N-isopropyl acrylamide) (pNIPAAm) fibers into enzyme-crosslinked spheroid-laden gelatin hydrogels.111 The fibers were then dissolved away from the hydrogel with medium perfusion below the gel–sol transition temperature (i.e., 32 °C) to obtain hollow channels mimicking the liver microvasculature, thus generating an interconnected network of microchannels with physiologically relevant capillary-like diameters (i.e., 17.78 ± 8.73 μm on average). Liver cell spheroids were prepared using a monoculture of normal mouse liver hepatocytes and a hepatocytes/endothelial cells/HSC tri-culture. Here, the perfused system promoted the lipid metabolic activity of hepatocytes. Also, hollow gelatin-based channels with perfusion revealed higher albumin and CYP3A4 expression over 10 days of culture than non-perfused groups, confirming the role of micro-vascularization in liver maintenance. Afterward, NAFLD was modeled in its early inflammatory and late fibrosis stages (i.e., nonalcoholic steatohepatitis (NASH)) on the liver-on-a-chip platform upon palmitic acid treatment, a fatty acid that may induce reactive oxygen species (ROS)-mediated apoptosis in a dose-dependent manner. Results showed that a 10-day treatment led to intracellular ROS accumulation and increased inflammatory markers expression (interleukins-6 (IL-6) and Tumour Necrosis Factor α (TNFα)), thus successfully replicating inflammation, lipid accumulation, and fibrosis occurring during the progressive processes of NAFLD.111 Another study successfully developed a microfluidic chip to model NAFLD progression using different free fatty acid gradients to perfuse primary hepatocytes. Moreover, such a microfluidic platform was used to create oxygen-driven steatosis zonation to investigate the role of oxygen deprivation in hepatocyte lipid accumulation. The model successfully mimicked the sinusoidal lipid distribution on a single continuous tissue and ultimately showed that such fat zonation disappears under progressed steatosis.112
To boost the mimicking of in vitro hepatic microenvironment, liver-on-a-chip platforms can also be employed to real-time monitor parameters such as oxygen gradients and the expression of metabolic enzymes.108 Sato et al. monitored the cellular oxygen consumption rate of hepatocytes in a microfluidic device that can simultaneously expose cell layers to areas with different oxygen concentrations (i.e., area 1 – hyperoxia, areas 2/3 – physiological conditions, area 4 – hypoxia) and automatic medium exchange for long-term culture.113 The measurement of partial oxygen pressure was performed via a laser-assisted phosphorescence quenching method. The in vitro zone-specific mRNA expression of metabolic genes such as phosphoenolpyruvate carboxykinase (PEPCK) and glucokinase (GK) increased in the periportal and pericentral regions (i.e., areas 2/3) as in in vivo condition. Moya et al. used a microfluidic layer-by-layer device integrated with oxygen sensors to attain the real-time metabolic activity of the liver system.114 Similarly, Lee-Montiel et al. developed a microfluidic platform (i.e., LAMPS, Fig. 4A(i)).115,116 The LAMPS model was constructed in a microfluidic chamber (50 μl volume) to recapitulate the liver acinus structure and multiple zone-specific functions by recreating liver Zone 1 and Zone 3 oxygen microenvironments. The microfluidic chamber incorporated sequential cell layers composed of primary human endothelial cells, hepatocytes, and HUVECs, as well as human monocyte cells (Kupffer-like immune cells), LX-2, and immortalized human hepatic stellate cells (HSCs), where the structural organization of the model was improved by depositing a thin layer of porcine liver ECM between the hepatocytes and the endothelial cells to mimic the space of Disse.116 Finally, specific in vitro liver oxygen zonation was achieved by perfusing LAMPS at both 15 μl h−1 and 5 μl h−1 to recreate Zone 1 and Zone 3, respectively. These values were calculated via a computational model of oxygen flows directly installed into the device, thus considering media transport, material permeability, and cellular consumption. As a result, zonation-dependent lipogenesis (i.e., steatosis) showed that LAMPS could be used to model and investigate zone-specific liver metabolism and diseases, as Zone 3 was found to consistently possess higher lipid-filled cells than those located in Zone 1.115
Microfluidic platform features can also be exploited to perform liver drug screening and hepatotoxicity studies. For instance, Skardal et al. realized a microfluidic platform with four parallel culture chambers to produce multiple and identical liver structures for toxicology testing.117 A HA/gelatin-based hydrogel formulation containing liver cancer cells (HepG2) was mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with a Glycosyl/Gelin S/polyethylene glycol diacrylate (PEGDA) pre-polymer formulation (2:1:1 ratio) to obtain a hydrogel precursor cell-mixture with the addition of a photo-crosslinkable precursor. The solution was inoculated through separate channels for a localized in situ crosslinking. Once the chambers were filled, a photomask was positioned above the microfluidic device to selectively crosslink the structures via thiolene reaction. After 7 days of culture, high cell viability was quantified in the liver constructs (>75%). Samples were also tested upon EtOH treatment at different concentrations (0 mM–500 mM) to evaluate the constructs’ functionality and toxicity effects. A dose-dependent behavior was found, as albumin and urea secretion significantly decreased with the increase of EtOH concentration and exposure, suggesting the platform as a potential tool for drug development and toxicology screening.117
:1 ratio with a Glycosyl/Gelin S/polyethylene glycol diacrylate (PEGDA) pre-polymer formulation (2:1:1 ratio) to obtain a hydrogel precursor cell-mixture with the addition of a photo-crosslinkable precursor. The solution was inoculated through separate channels for a localized in situ crosslinking. Once the chambers were filled, a photomask was positioned above the microfluidic device to selectively crosslink the structures via thiolene reaction. After 7 days of culture, high cell viability was quantified in the liver constructs (>75%). Samples were also tested upon EtOH treatment at different concentrations (0 mM–500 mM) to evaluate the constructs’ functionality and toxicity effects. A dose-dependent behavior was found, as albumin and urea secretion significantly decreased with the increase of EtOH concentration and exposure, suggesting the platform as a potential tool for drug development and toxicology screening.117
Similarly, a patterned toxicity evaluation system was developed to simultaneously assess different drug screenings in the same microfluidic device (i.e., TASCL, Fig. 4A(ii)).118 The TASCL device was fabricated with an overall size of 10 mm × 10 mm, in which 400 microwells were designed and patterned with a top and a bottom aperture, measuring 500 μm × 500 μm (square) and 300 μm in diameter (circular) each, respectively. TASCL was seeded at different cell densities to investigate the formation of HepG2 spheroids. As a result, the microfluidic platform ensured suitable spherical aggregation, high viability, and albumin secretion upon highly precise positioning.
Also, commercial microfluidic platforms can be used for liver drug screening. Among them, OrganoPlate® platforms have been employed for hepatotoxicity studies (Fig. 4A(iii)).119,120 Such a device contains a specific number of microfluidic tissue culture chambers (e.g., 40, 64, or 96, according to the model) designed and embedded on the bottom of a commercial 364-well plate. Each culture chamber consists of one culture channel containing an ECM-based hydrogel of choice and up to two adjacent perfusion channels separated by specific phase guides to prevent the patterned hydrogel from flowing into the adjacent channels. Moreover, OrganoPlate® platforms are stimulated by a gravity-driven leveling technology that enables the induction of a continuous passive perfusion flow without using an external pump or tubing line. Thus, such microfluidic platforms allow for the separation of the cell culture area and the perfusion flow without any physical barrier, as well as indirect contact between the cells and the flow due to the polymerization of the hydrogel matrix. In particular, Bircsak et al. developed a liver-on-a-chip platform for high throughput hepatotoxicity screening, using an OrganoPlate® model with 96 culture chambers with two separate channels (i.e., one perfusable and one containing an ECM-based hydrogel).120 Specifically, hiPSC-derived hepatocytes (iHep) were seeded in the ECM channel and co-cultured with endothelial cells and THP-1 monoblasts differentiated to macrophages seeded in the perfusable channel. Such multicellular microfluidic structure allowed the formation of iHep clusters in the ECM-channel and a 3D tubular endothelial layer structure in the perfusable channel (Fig. 4D and E). After 15 days of culture, cells were viable and exhibited stable albumin and urea secretion. Moreover, an increase in the CYP3A4 activity and the decreased alpha-fetoprotein (AFP) secretion suggested further maturation of the iHeps. In addition, troglitazone and a small library of 159 compounds with known liver effects have been employed to successfully validate the device as a platform for liver drug testing.
Furthermore, different biomaterials such as ECM components and naturally derived hydrogels can be integrated into liver-on-a-chip platforms to enhance their biomimetic potential.119 The addition of a 3D in vivo-like tissue microenvironment can be beneficial for cell maturation. Hence, a 3D architecture can provide suitable physicochemical properties to mimic physiologically relevant conditions (i.e., cell–cell interaction and metabolic pathways). Although ECM-derived materials possess fast degradation rates and lack robust mechanical properties, components such as Matrigel, collagen, and fibronectin (FN) have been widely employed,115,121–123 as well as decellularized liver matrices,115,124 gelatin-based hydrogels,111 and hybrid formulations.125 For instance, Toh et al. performed a collagen-coating on a multiplexed microfluidic hepatocyte culture system, allowing for the investigation of the metabolic functions of hepatocytes.126 Furthermore, the liver chip was used to predict in vivo hepatotoxicity testing for five different drugs in an in vitro dose-dependent manner (i.e., acetaminophen, diclofenac, quinidine, rifampin, and ketoconazole). Also, the integration of liver decellularized ECM (dECM) and gelatin methacryloyl (GelMA) in a dynamic microfluidic-based 3D cell culture system displayed a linear dose-dependent drug response to the toxicity of Acetaminophen and sorafenib.125
In conclusion, coupling liver-on-a-chip technologies with perfusion-based systems can be beneficial for the development of 3D in vitro models that aim to functionally recapitulate the liver microarchitecture and realize robust platforms for high-throughput screening of hepatic diseases and drug compounds.
4. 3D liver scaffolds
Scaffolds are 3D structures designed to recapitulate the ECM role and provide a proper architecture for cell adhesion and maturation. 3D scaffolding approaches for engineering 3D hepatic tissues rely on cultivating cells within biodegradable scaffolds, either natural, synthetic, or hybrid. Given that scaffolding techniques allow superior control over cell morphology and spatial arrangement compared to organoids and 2D systems, such biofabrication methods can be easily used to model diseases, quantitatively evaluate cytotoxicity, and in turn, assess drug treatments. However, large-scale production and flow control of these constructs to mimic liver vascularization is still challenging. Biofabrication approaches such as electrospinning, micropatterning, casting, and 3D (bio)printing have been strategically exploited as 3D scaffolding approaches for LTE.127,128 Among others, biodegradable polymers such as polyethylene glycol (PEG), polycaprolactone (PCL), polylactic acid (PLA), poly-L-lactic acid (PLLA), poly-DL-lactide-co-glycolide (PLGA), polyvinyl alcohol (PVA), and thermoplastic biodegradable polyurethane (PU) polymers have been employed in the frame of LTE.129–134 These synthetic polymer-based scaffolds can provide structural support and allow the diffusion of nutrients, oxygen, and cell growth factors without being affected by possible animal-derived pathogenicity and batch-to-batch variability characterizing natural-based polymers.135 However, biocompatibility and ECM complexity still remain challenging to recapitulate. To address this issue, hybrid and/or natural polymer-based biomaterials can be used to fabricate liver scaffolds.136 Essential ECM components (e.g., hyaluronic acid, collagen, and FN) can be included in the hydrogel-based scaffolds to better replicate in vivo conditions, thus improving the cell–biomaterial interactions. The most common approach to fabricate in vitro liver models is based on the use of hydrogels that can mimic and resemble the complexity of the ECM microenvironment, induce and support cell adhesion, proliferation, and differentiation by providing chemical and physical signals. Among others, naturally derived hydrogels such as chitosan, alginate,137,138 gelatin,139–141 collagen,140,141 as well as blend formulations as gelatin/alginate/fibrinogen,142 and gelatin/alginate143 have been extensively explored.144 Natural materials are overall considered promising candidates for developing engineered liver tissues for selecting and fastening drug candidates. GelMA- and fibrin-based hydrogels have been used not only for the hierarchical vascularization of liver constructs (e.g., lobule-like microstructures, spheroids, fibers), but also for hepatocyte differentiation145–149 and drug screening.109,150 Given the soft nature and rapid degradation of most hydrogels, a crosslinking process (e.g., photo-mediated for GelMA, ionic for alginate) or a supporting framework made of synthetic biomaterials (e.g., PCL) is required to reach the structural integrity of the constructs for in vitro testing.151–154 Beyond these widely explored hydrogel-based formulations, the combination of different techniques and hybrid hydrogel biomaterials allowed to highlight the power of LTE multidisciplinary area to mimic the native complexity of the liver and investigate its pathogenic microarchitecture. Also, unpopular cell-based applications can be functionalized by coating the material surface with hydrogel-based formulations. For example, it was shown that paper may enhance the hepatic function of in vitro 3D liver models in the presence of human-induced hepatocytes (hiHEPs) and HUVECs co-culture upon Type I collagen.155In this section, a discussion of the most thought-provoking advances in 3D liver models obtained by 3D scaffolding strategies is provided. In light of this, biofabrication techniques such as 3D printing, 3D bioprinting, and electrospinning for in vitro disease modeling and drug testing are thoroughly described. Due to the limited possibility of developing controllable and complex architectures for physiologically relevant structures, more conventional methods like molding, extrusion, and micropatterning are out of the scope of this review.
4.1. 3D printed scaffolds
3D printing technology has been considered an emerging strategy to fabricate tailored structures for tissue engineering relying on the layer-by-layer deposition of biomaterial inks. A 3D CAD design that reproduces the scaffold complex architecture is subsequently converted into a code to produce a 3D construct with the desired geometry.156 Reproducibility, high control over the process and the scaffold size are considered the main advantages of this 3D deposition processing method.157,158 In LTE, the extrusion-based 3D printing approach allows for shaping constructs that can recapitulate the characteristic repetitive liver microstructure (e.g., lobule architecture). However, biomaterial characterization and scaffold geometry are currently gaining more attention over liver functionality outcomes, as LTE is a new trend in the field.159 In addition, costs, resolution, and lack of high throughput are potential limitations in the frame of liver disease modeling and drug testing. Typically, 3D printed engineered constructs with potential applications in human disease modeling have been investigated focusing on different geometries, pore size, and gene expression, revealing increased specific functions in more interconnected 3D printed scaffolds able to mimic the complex liver architecture.105,160–163 Based on the pore tortuosity influence on seeding efficiency,164 Lewis et al. showed that different pore geometry could positively modulate hepatocyte function and gene expression. They found that HUH-7 cells might improve CYP activity and bile transport upon seeding on 3D-printed gelatin scaffolds.161 Two different gelatin-based geometries were explored, with a strut orientation between subsequent layers of 90° or 60° of square boxes. Changes in the scaffold geometry were found to be responsible for gene expression over cell numbers. Indeed, scaffold architecture may significantly affect hepatocyte function, as shown by the higher activity of CYP3A4 and CYP2C9 in 60° geometry scaffolds, which also confirmed the higher formation of canalicular spaces. Different strategies to fabricate 3D perfused constructs have lately focused on highly porous structures. Indeed, interconnected pores allow cell growth and mass transport.30,165,1663D printed architectures have also been developed to evaluate the influence of 3D porous structures and dynamic mass transport on drug testing platforms.74,105 Vinci et al. proposed an interesting scalable in vitro liver model investigated in terms of 3D topology and convective flow, two primary cues influencing hepatocytes’ function.105 Synthetic materials such as PLGA and PLLA were used to prepare liver constructs employing a pressure-assisted microsyringe (PAM). The 3D scaffolds mimicked the characteristic size of hepatic lobules with three different layers, composed of 70 hexagonal unit cell structures each, thus creating an overall construct with high porosity suitable for cell penetration and adhesion. The authors showed that the 3D porous architecture increased cell density and promoted the formation of aggregates compared to 2D films, thus maximizing cell–cell interactions. Another novel bio-inspired 3D printed construct based on a PEG hydrogel was embedded with polydiacetylene (PDA) nanoparticles to fabricate a detoxification model that may provide an alternative strategy to drug intoxication.167 Herein, PDA nanoparticles showed their potential in neutralizing melittin toxins in vitro. Mimicking the liver lobule-like configuration, the platform was able to attract, catch and sense the melittin toxins, a harmful poisonous substance to humans that can result from animal bites and bacteria.
4.2. 3D bioprinted scaffolds
Similar to 3D printing technology, 3D bioprinting methods rely on computer-assisted layer-by-layer deposition strategies for the fabrication of clinically relevant constructs.168,169 However, 3D bioprinting offers the potential to encapsulate cells inside biomaterial inks, thus enhancing the biomimetic features towards the reproduction of a more realistic physiological and pathological liver microenvironment.18,170,171 Such cell-laden solutions (i.e., bioinks) are mainly based on hydrogels. Contrarily to other biomaterials, hydrogels possess unique properties, such as high water content and suitability for cell encapsulation.144 In addition to printability properties (i.e., viscosity, shear-thinning behavior), the choice of hydrogels is also determined by the ability to support cell viability and guarantee the performance of their physiological function.32 Specifically, hydrogels enriched with cell-binding sites are preferred to recreate a more cell-friendly environment. To this aim, protein hydrogels modified with arginine–glycine–aspartic acid (RGD) peptides (e.g., sodium alginate) or ECM-like hydrogels (e.g., collagen, gelatin, GelMA) are preferably selected.172–175 To further enhance the biomimicry potential, the liver ECM can also be employed to develop tissue-specific bioinks with the biochemical complexity of the native tissue. Moreover, hydrogel mechanical properties should be tuned to recreate the desired microenvironment stiffness to favor cell migration and reorganization.176 Finally, scaffold porosity, which is mainly dependent on hydrogel concentration and degree of crosslinking, must guarantee efficient transport of oxygen, nutrients, metabolites, and drug compounds.177 Regarding the encapsulated cells, both parenchymal and non-parenchymal cells have been employed. Parenchymal cell sources included hepatoma cell lines (e.g., HepG2, HUH-7, and HepaRG).138,178 Alternatively, hiPSCs, hESCs, and human hepatocytes can also be used.179–181 On the other side, liver non-parenchymal cells can include Kupffer cells, liver ECs, HSCs, and HUVECs (Fig. 5A).179,182 Based on the deposition method, 3D bioprinting approaches can be divided into three main methods: (i) droplet-based bioprinting, (ii) extrusion-based bioprinting, and (iii) light-assisted bioprinting. (i) Droplet-based bioprinting involves the precise dispensing of droplets of bioink through thermal, piezoelectric, and electro-hydrodynamic methods.170,183 For such 3D bioprinting method, the bioink selection is oriented to cell-laden hydrogels formulation characterized by low viscosity properties. (ii) Extrusion-based bioprinting relies on the deposition of continuous filaments of bioinks to form a 3D construct.184 By optimizing 3D printing parameters (e.g., temperature, pressure, velocity), a wide range of bioinks can be employed to fabricate high-resolution constructs. (iii) Light-assisted (e.g., UV- or visible-light) 3D bioprinting approach employs the selective curing of photo-crosslinkable bioink contained in a printing reservoir.29 Such a method exhibits a high printing resolution that guarantees the fabrication of sophisticated and complex biomimicking architecture in a high-throughput way.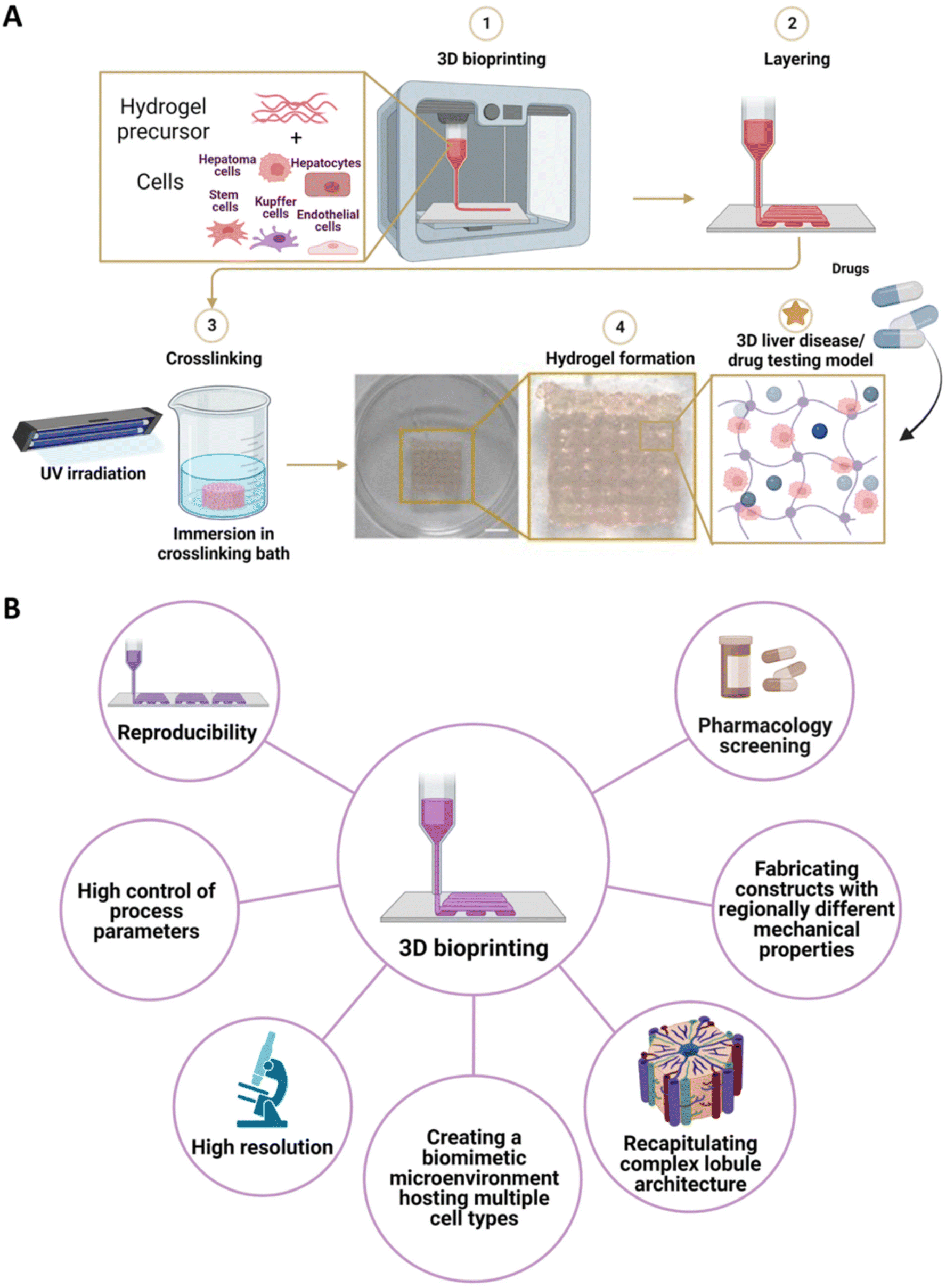 | ||
| Fig. 5 3D bioprinting methods for the production of 3D liver scaffolds. (A) 3D bioprinting of a hydrogel precursor solution is loaded with parenchymal, non-parenchymal liver-derived cells, cancer cells, stem cells, and/or cell lines. Afterward, the 3D printed construct is crosslinked via different methods (e.g., UV irradiation, crosslinking in a crosslinking bath) to allow the crosslinking of the hydrogel. (B) Advantages of the 3D bioprinting technique. | ||
3D bioprinting techniques have been widely used to develop highly accurate/precise/realistic disease liver models. A high geometrical complexity can be achieved by optimizing bioink formulations and 3D bioprinting parameters. Thus, a proper recapitulation of the liver structure similar to in vivo pathological conditions can be performed. For example, Wu et al. incorporated cellulose nanocrystals (CNCs) into an alginate/GelMA formulation to enhance shear-thinning behavior and shape fidelity upon bioink deposition.185,186 Indeed, CNCs were found to align along the printing direction, thus enhancing the bioink extrudability compared to pristine alginate/GelMA bioink. The bioink was extruded directly on a cover glass or in an alginate/calcium chloride (CaCl2) supporting bath and subsequently crosslinked by UV light exposure. A high-accuracy liver-biomimetic 3D honeycomb structure was successfully produced. Such structure was employed as a 3D platform for co-culturing two different cell types (i.e., NIH 3T3 and HepG2). Specifically, NIH 3T3/Alginate/GelMA/CNCs bioink was deposited to create the lobule-like structure, while HepG2/GelMA was deposited inside the hexagonal holes. The precise positioning of cells allowed for the alignment of NIH 3T3 fibroblasts and the formation of HepG2 spheroids, thus recapitulating the overall 3D biomimetic structure. Moreover, intracellular interactions enhanced albumin secretion compared to HepG2 culture used as a control. Pathological conditions can be reproduced by mimicking intracellular events and cross-talks. To this aim, 3D bioprinting enables the fabrication of biomimetic microenvironment hosting multiple cell types.187 For instance, Cuvellier et al. established a tri-culture model consisting of HepaRG/LX-2/HUVECs to recapitulate the progressive development of fibrosis in vivo.188 HepaRG/LX-2 were mixed with GelMA and 3D bioprinted in a square-shaped construct. Afterwards, scaffolds were crosslinked by UV-photopolymerization. HUVECs were seeded after one week of culture to recreate a pseudo-endothelial barrier following colonization of the surface structure. The 3D bioprinted structures exhibited long-term viability, proliferative ability, hepatocyte phenotype and functions. Moreover, the 3D bioprinted model was suitable for the interaction between parenchymal and non-parenchymal cells, which was modulated by the secretion of TGFβ-1 that in turn induced the activation of myofibroblastic genes (i.e., ACTA2 and COL1A1). As a result, a fibrillar collagen synthesis deposition was observed, which was not evidenced in monocultures of either HepaRG or LX-2.
Besides single-cell dispersion, 3D bioprinting was found to be suitable also for the encapsulation of 3D spheroids. Goulart et al. performed a study to compare 3D bioprinting of iPSC-derived hepatocyte-like spheroids and single cell dispersion, both in combination with iPSC-derived mesenchymal cells and ECs in alginate/Pluronic F-127 blend bioink formulation.180 3D hepatic spheroids were successfully 3D printed in a donut-shaped geometry construct and crosslinked with CaCl2 upon biofabrication. After 18 days of culture, the viability of the cultured spheroids was significantly higher than single cells. Moreover, 3D spheroids exhibited higher expression of liver-specific markers, including increased urea production and prolonged secretion of albumin. In addition, single cells revealed epithelial–mesenchymal transition, resulting in a rapid loss of hepatocyte phenotype. By tuning 3D printing parameters, constructs with tunable mechanical properties can be obtained. In particular, light-assisted 3D bioprinting technologies offer the possibility to modulate the photo-polymerization processes, thus allowing for the fabrication of constructs with regionally different mechanical properties. Such interesting feature provides a valuable tool for fabricating platforms that aim to replicate liver pathologic conditions (e.g., cirrhotic liver) characterized by higher mechanical properties than the native liver tissue in physiological conditions. Ma et al. used light-assisted 3D bioprinting to pattern a photo-crosslinkable GelMA/dECM formulation with tunable mechanical properties as a platform for studying the effects of stiffness on HCC progression.189 HepG2 cells were encapsulated in a lobule-like hexagonal structure with different mechanical properties to recapitulate both the native and cirrhotic liver tissue. For scaffolds with lower stiffness, cellular aggregation and spheroid formation were observed with increasing spheroid size during the culture period. Contrarily, reduced growth was observed for HepG2 cells cultured in scaffold with cirrhotic mechanical properties. Moreover, stiffer scaffolds showed an upregulation of invasion markers (i.e., insulin-like growth factor 2 (IGF-2)) compared to physiological native-like controls. To further investigate the HepG2 invasion potential, a biomimetic design consisting of three hexagonal lobules, each possessing different stiffness (e.g., soft, medium, and stiff scaffolds), was developed. Each hexagonal unit was interconnected with a collagen I-based scaffold to recapitulate the fibrous septa-like structure found in the native liver architecture. Such engineered cancer platform enabled the validation of the highest degree of invasion of HepG2 cultured in a cirrhotic mechanical environment into the surrounding stromal region.
The potential of 3D bioprinting in replicating highly biomimetic constructs has been used to produce liver platforms for drug testing purposes (Fig. 5B). Ma et al. employed a light-based 3D bioprinting system to produce a UV-crosslinked GelMA-based platform, aiming to embed hiPSC-derived hepatic cells and endothelial- and mesenchymal-originated supporting cells (i.e., HUVECs and adipose-derived stem cells) in a high-resolution hexagonal geometrical pattern that closely mimicked the in vivo hepatic lobule structure.190 The hydrogel-based triculture model promoted cell organization and alignment within the biomimetic architecture. Moreover, higher expression of hepatic markers (i.e., HNF4a, TTR, and albumin) and metabolic product secretion was observed, compared to the 2DMC and the 3D hepatic progenitor cells (HPC) monoculture model. In another study, Sun et al. used extrusion-based bioprinting to fabricate a 3D liver cancer model composed of HepG2 suspended in gelatin/alginate. Upon fabrication, the 3D-bioprinted scaffolds were crosslinked with CaCl2 solution, finalizing the 3D model for liver cancer.138 Compared to the 2D model counterpart, HepG2-laden 3D scaffolds showed higher expression of tumor related genes including ALB, AFP, CD133, EpCAM, and TGFβ-1 genes. To validate the cancer model, the anti-tumor response to several drug treatments (i.e., cisplatin, sorafenib, regorafenib) was evaluated. As a result, it was observed that the half-maximal inhibitory concentration (IC50) values of the tested drugs were higher in the HepG2-laden scaffold model compared to the 2D model. Such findings suggested a higher sensibility of the 2D model compared to the 3D scaffold. However, the IC50 values obtained in the 3D-HepG2 model were closer to the effective blood concentration of the drugs in the human body. These outcomes were explained by the higher levels of drug resistance autophagy-related genes (e.g., ABCB1, MDR-1, MRP1, and EGFR) expressed in the 3D-bioprinted model.
The employment of the same 3D printed model was furtherly extended to establish in vitro model for patient-specific drug screening for HCC.191 Cells were isolated from HCC-specimens collected from six patients and then combined with gelatin/alginate to produce the bioink. Patient-derived 3D bioprinted scaffolds were successfully fabricated, showing high viability and proliferation rate during long-term culture. Moreover, HCC-scaffolds allowed for retaining the features of the originating HCC tumors, including stable expression of the AFP biomarker, as well as the constant maintenance of genetic alterations and expression profiles. Furthermore, the efficacy of four commonly used empirical targeted drugs for HCC-affected patients was assessed. Most of the HCC-scaffold models derived from the six patients resulted to be insensitive to the four targeted drugs in treatments. However, four patient-specific HCC-derived 3D scaffolds were found to be positively responsive to one or more drugs. For such scaffolds, a dose-dependent manner was observed. For two patients, sorafenib and lenvatinib showed enhanced anti-tumor effects than other drugs, respectively. Such findings encouraged the classification of patient-derived HCC-laden scaffolds as a potential candidate in the field of personalized cancer treatments. 3D bioprinting was also found to be a suitable technology to preserve the differentiation futures of encapsulated cells. For instance, Faulkner-Jones et al. performed inkjet-based 3D bioprinting of hepatocyte-like cells (HLCs) differentiated from both hiPSCs and hESCs mixed with RGD/alginate-based hydrogel solution.172 A cell-laden hydrogel was printed by dispensing an array of droplets of the bioink, followed by an overprinting of droplets of CaCl2 solution to allow the crosslinking. The droplets were dispensed following a circular path, and a ring-shaped structure was finally obtained when adjacent droplets overlapped together and formed a single continuous layer. Upon fabrication, the cell-laden structures continued the differentiation process into hepatocyte-like cells (HLCs) over 24 days of culture. As a result, encapsulated cells were viable and preserved the hepatocyte marker expression validating such 3D printed model as a potential platform for drug testing studies.
4.3. Electrospun scaffolds
Electrospinning is a well-established fiber process employed to produce different interwoven nanofibrous scaffolds with high porosity, thus closely mimicking the native architecture of soft tissues. In a conventional electrospinning fabrication process, a high-voltage electric field is applied between the tip of a needle and the ground collector, allowing for the spinning solution to initially form a hemisphere drop at the tip of the needle. The surface charge on the polymer droplet increases, allowing for the formation of a polymer jet under an increasing high-voltage power towards the collector.192 By selecting the proper collector, nanofibers can be deposited to form a fibrous mat characterized by random or preferential fiber arrangement.192,193 Herein, electrospinning provides the possibility to tune high porosities and pore size distribution of the engineered constructs reaching a large surface area. Moreover, the features of electrospun fibers at the micro- and nano-scale and the surface roughness may also enable the mimicry of in vivo scenarios influencing the adhesion and proliferation rate of the cultured cells in the engineered microenvironment.193 Both natural and synthetic polymers can be used to obtain electrospun fibers. Among others, the application of synthetic polymers in this field involves PCL, PLA, and PLGA; while natural-based materials include chitosan and ECM-derived components. In this frame, a combination of different strategies may be applied to functionalize these materials (Fig. 6A). Hence, electrospun functionalized systems have been explored as liver disease models. For instance, Grant et al. proposed an electrospun system composed of PLA and human liver ECM (hLECM) components to create a liver disease model. The authors demonstrated that the combination of the synthetic polymer with the hLECM layer properly supports Transformed Human Liver Epithelial-3 (THLE-3) hepatocytes cultured on the surface, showing high cell viability and proper morphology.195 The possibility of designing drug testing models based on electrospun constructs has also been investigated. Fasolino et al. proposed an electrospun fibrous PCL model for hepatocarcinoma treatment by co-culturing two hepatic cell lines (i.e., HepG2, healthy human hepatocytes (HHH)).194 After 4 weeks, HepG2 cells showed lower proliferation compared to HHH cells. The anticancer effect of PCL on HepG2 was also confirmed by the reduced cell adhesion with respect to culture plates. Then, PCL fibers treated with doxorubicin (DOX) were cultured in the presence of HepG2-only and HepG2/HHH co-culture, respectively; thus, showing lower cell proliferation after 14 days compared to untreated PCL fibers and plate control. Considering that DNA damage induced by ROS production is one of the main mechanisms involved in both mutagenesis and carcinogenesis,196 the antioxidant effect of co-cultured fibers was also confirmed from the investigation of ROS generation induced by H2O2, a common genotoxic agent. Indeed, PCL fibers concurred to reduce ROS levels at day 1 of culture (i.e., HepG2, HepG2/HHH, healthy hepatocytes), thus validating the scaffold's ability to inhibit cancer development and progression. Drug treatment was similarly investigated by Grant et al. with a drug-induced electrospun PCL scaffold by introducing drugs that can inhibit histone deacetylases from enhancing the transcriptional activity, thus modulating the cellular response and influencing the ECM production.197 Electrospun PCL-scaffolds were seeded with 5637 human urinary bladder epithelial cells to produce ECM. To induce a drug treatment, culture medium was supplemented with either Valproic Acid (VA) or Sodium Butyrate (NaB). At the same time, controls were referred to as 3D scaffolds without an initial cell layer and no drug treatment. After 14 days, decellularization was assessed. Finally, the resulting scaffolds were further seeded with HepG2 to allow the constructs to form a functional liver cell layer in standard culture conditions and validate the platform. The drug-induced nanofibrous scaffolds showed a lower adhesion of HepG2 as well as positive effects on the expression of genes related to liver function (i.e., albumin) and metabolism (i.e., CYP450) when compared to controls. Additionally, the expression of key liver ECM proteins (e.g., collagen I, collagen IV, and fibronectin) outperformed in the case of PCL-based nanofibrous electrospun systems in comparison with tissue culture plates. On the other hand, PLGA was also employed to develop ECM-based 3D nanofibrous scaffolds using a wet-electrospinning technique.135 Brown et al. obtained by wet-electrospinning less dense and highly porous PLGA matrices compared to standard electrospinning technique,198,199 thus allowing to replicate the pore size of the acellular parenchyma of the liver and to accommodate the inclusion of human primary hepatocytes (hPHs) in the structures.200 Fibers were spun and collected using a mold soaked in an isopropyl alcohol/deionized water solution (7:3 (v/v)). Pluronic® F-108 was used as a surfactant. The scaffolds were then chemically modified with essential liver ECM proteins (type I collagen and FN) to closely mimic the liver microenvironment. Collagen was chosen because it is the major component of the physiological liver ECM, while FN is a critical binding ligand for integrin-mediated cell adhesion. hPHs cultured in both PLGA-ECM and PLGA scaffolds maintained high viability over two weeks. Specifically, hPHs embedded in PLGA-ECM constructs showed higher functionality and exhibited 10-fold greater albumin secretion, 4-fold higher urea synthesis, and elevated transcription of hepatocyte-specific CYP450 genes compared to unmodified PLGA scaffolds. The hepatocyte functionality was also higher comparing collagen to FN-bonded scaffolds. This evidence demonstrated that the incorporation of type I collagen into the biosynthetic wet-electrospun 3D scaffolds improved the hepatocyte functionality in vitro and suggested further in vitro investigations for pharmacology screening, as well as to preserve hepatocyte function for use in human cell therapy (Fig. 6B). Rajendran et al. designed a biomimetic chitosan nanofibrous scaffold fabricated by electrospinning with enhanced porosity, cellular adhesion, and spreading for long-term liver function by coating the surface of randomly oriented chitosan nanofibers with FN.201 The seeding of fibroblasts and primary rat hepatocytes revealed the beneficial impact of FN on focal adhesions formation and integrin-binding sites. Also, drug intake and metabolism were confirmed by high levels of CYP450 A1 enzyme activity.
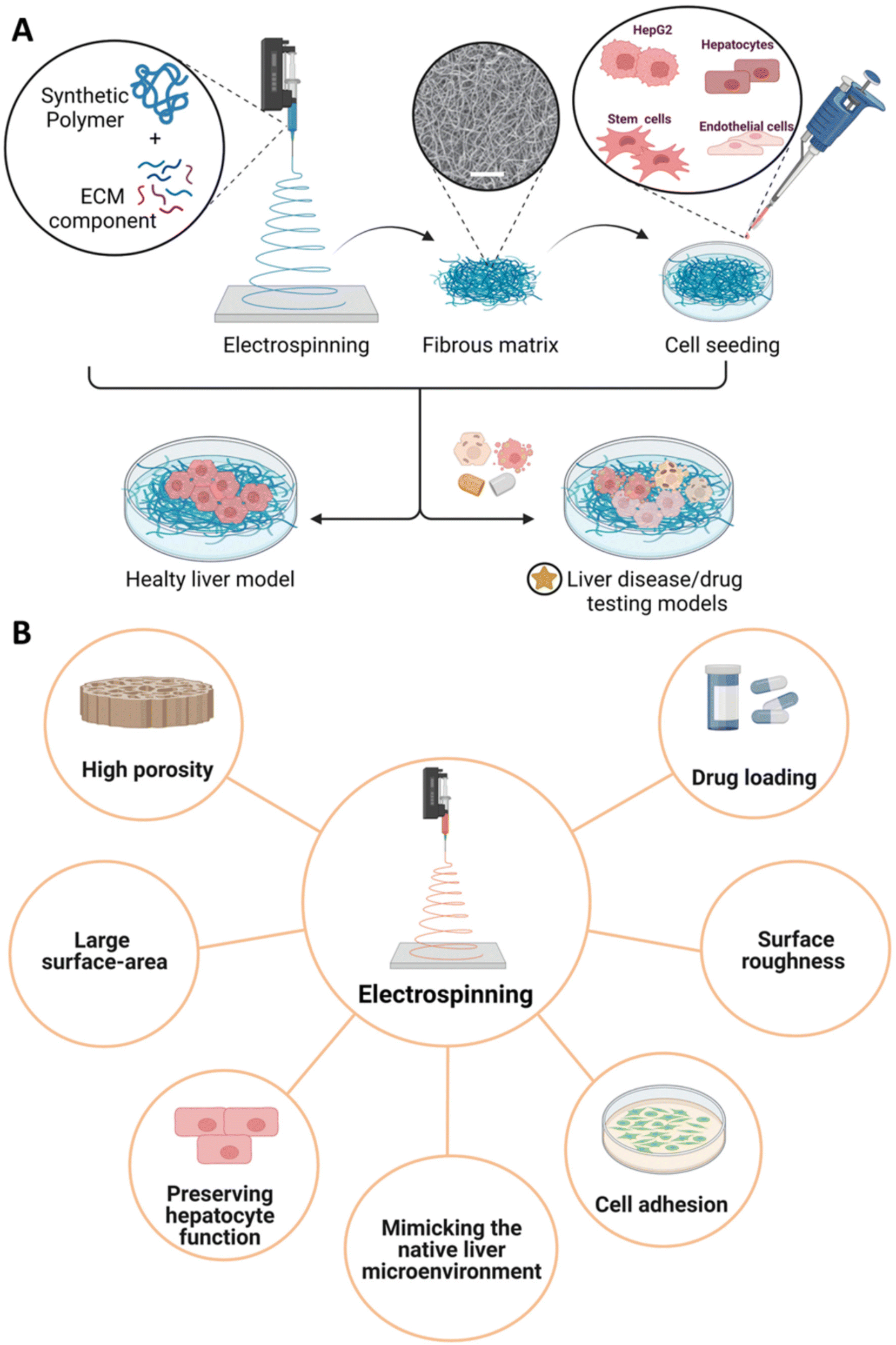 | ||
| Fig. 6 3D liver scaffolds fabricated by electrospinning technique. (A) Synthetic polymer(s) are mixed with ECM component(s) in a solution that is electrospun under electric field conditions. The resulting fibrous matrix is then seeded with liver-derived cells and/or cell lines to develop liver disease or drug testing models. Scale bar: 50 mm. (B) Advantages of the electrospinning technique. | ||
5. Ongoing challenges and future perspectives
To date, impactful advances in in vitro LTE have been made to accurately mimic liver-specific functions and predict drug responses (Table 1). Indeed, the recent literature reported a tendency to model liver diseases and assess drug testing by using 3D engineered constructs to closely mimic in vivo conditions. Despite such progress, there is an urgent need to extend the research to a large-scale human population to strongly validate in vivo the engineered systems. Parallelly, this stage could dramatically lower ethical concerns regarding the use of animal models, which may also impair human outcomes. Based on that, human co-culture-based LTE models that can recapitulate in vivo-like features would open up the possibility of bridging the gap between laboratory investigation and clinical applications. In Table 1, representative technological approaches and cell sources for in vitro 3D liver models are summarized. Moreover, relevant outcomes and major limitations of the proposed 3D liver models are also highlighted to identify the most effective strategy to develop the targeted system. In addition, the main advantages and drawbacks of the previously discussed liver models are summarized in Table 2. Due to their biocompatibility, most of the polymeric materials used are naturally derived. Thus, they are biodegradable and can properly support biological functions and cell growth, offering a suitable biomimetic environment to tailor specific liver architectures. Nevertheless, synthetic polymeric materials may be nondegradable, possess robust mechanical integrity, and maintain high-repeatability geometries. Also, they can be easily tuned to provide low immunogenicity. Although some of them are also used in the clinic, natural hydrogels suffer batch-to-batch consistency and poor mechanical properties, while synthetic polymers mainly lack cell-binding sites. Therefore, either the modification of single-origin materials or the combination of different materials is lately emerging as a new trend to get the most beneficial impact from in vitro 3D liver constructs.160,202–204 Additionally, to recreate the heterogeneous tissue microenvironment and enable physiological cell–cell interactions, both parenchymal (e.g., primary hepatocytes, hepatic-derived cell lines, stem cells) and non-parenchymal (e.g., Kupffer cells, ECs, HSCs, sinusoidal cells) cell types should be considered according to the final LTE purpose. However, multiple cell populations may require different material properties and network orientations to form characteristic microstructures, leading to a winding research path. For instance, primary hepatocytes are considered the gold standard for hepatotoxicity assessment as they can trigger specific liver functionalities and metabolism features. However, they have limited availability and are not advisable for long-term in vitro culture due to their rapid dedifferentiation time.205 Similarly, although liver cancer-derived cell lines (e.g., HepG2, HepaRG) have high proliferation capacity, they could lack metabolic activity such as urea formation, as a result of downregulated enzymatic activities of the ammonia detoxification cycle (e.g., Arginase I), and expression of phase I and phase II human enzymes.205,206 In addition, they have limited sensitivity for the prediction of human drug hepatotoxicity.207 In contrast, non-hepatic stem cells are usually suitable for investigating specific liver diseases and characteristic functions.208–210 Moreover, in vitro models and testing strategies aim to tackle the current challenge of properly mimicking the liver microenvironment, thus providing insights into the recreation of the in vivo physiology. Consequently, it is believed that a visionary road towards compliant materials for LTE relies on the manipulation and decellularization of ECM, which can appropriately preserve the hepatic microenvironment allowing for a comprehensive in vitro mimicry.124,211 Moreover, shortcomings in ECM availability may result in major drawbacks. On the other hand, in recent years this strategy has been gaining increased attention. Although 2D cultures are more convenient in terms of costs and ease of use, they do not reflect the in vivo conditions, due to low cell bioactivity and lacks in functional morphology.212–214 For this reason, 3D cultures allow for building more complex models to sustain the development of thorough toxicity studies, high-throughput drug screening platforms, and pathophysiology applications. However, in vitro research outcomes may be generally difficult to adapt thoroughly to in vivo human models. Such data adaptation is often performed by multiplying the maximum plasma concentration by a factor of 20× to 100×.215,216 On the other side, different pathways and mechanisms of toxicity of tested drug compounds may arise when using a wide range of concentrations.217 Similarly, drug concentrations relevant to the human in vivo situation should be strictly controlled;23 and effective dose calculation, blood volume, and cell number may diverge.218–220 To obtain these 3D in vitro LTE constructs, various approaches have been employed, and among others, there are organoids, liver-on-a-chip(s), and scaffolding techniques (e.g., electrospinning, 3D-bioprinting). In particular, liver-on-a-chip platforms can provide a dynamic microenvironment that allows for mimicking the perfusion flow system required for tissue vascularization.122,221 Such perfusable systems are more adequate for long-term studies to in-depth investigate liver tissue maturation under controllable fluid-dynamic stimuli (e.g., biochemical stimulation, signaling transduction) and for drug testing.105,222 Moreover, microfluidics allows for mimicking liver zonation and metabolism, which are still considered challenging when using 3D models such as organoids and scaffolds due to the complex liver physiology and heterogeneity.107 To our knowledge, a few studies successfully achieved liver zonation by using organoids223 and scaffold-based technologies.187 Nevertheless, such studies still rely on the use of microfluidic-based technologies.| In vitro model | Biomaterials | Cell source | Aim of the work | Main outcomes | Limitations | Ref. |
|---|---|---|---|---|---|---|
| Organoids | — | Patient-derived HCC-biopsies | Modeling of human liver cancer derived from tumor needle biopsies | • Long-term cultures | • Reduced number of testing patients | 75 |
| • Recapitulation of histological features and tumor markers expression comparable with the originating tumors | • Disadvantages related to liver biopsies (i.e., patient pain, bleeding, infection, accidental injury to nearby organs) | |||||
| • Tumor formation with histological features of the original biopsy upon injection of organoids into immunodeficient mice | ||||||
| • Dose-dependent trend in response to sorafenib treatment | ||||||
| — | HSC HepaRG | Modeling of drug-induced liver fibrosis | • Metabolically active hepatic organoids | • Absence of biomaterial providing physico-chemical cues to support cell-growth | 60 | |
| • Collagen secretion and HSC activation after exposure to pro-fibrotic compounds | • Replication of higher biomimetic fibrosis model that could be obtained including Kupffer cells, liver sinusoidal endothelial cells and PHH | |||||
| HA | HCT-116 | Modeling of liver-tumor organoids for tumor growth and drug response | • Enhanced mesenchymal phenotype for cells compared to 2D culture | • Use of cell seeding over hydrogel cell-encapsulation | 82 | |
| Gelatin PEGDA | HepG2 | • Dose-dependent behavior resulting from increased apoptosis and decreasing metabolism of HCT-116 with higher concentrations of 5-FU | ||||
| Liver-on-a-chip | Collagen | HepG2 NIH 3T3 | Microfluidic organoid platform for drug screening | • Automated platform to confine organoids and perform on-chip dilution for hepatotoxicity screening | • HepG2 (cancer cells) prevent the result translation to healthy hepatocytes | 88 |
| • Enhanced mimicry of fibroblasts contractile behavior and albumin secretion profiles compared to 2D systems | ||||||
| • CYP3A4 activities and necrotic/apoptosis consistent with the dose-dependent effect of treatment of chemical inducer/inhibitors on hepatoma cells | ||||||
| GelMA | HepG2 | Biomimetic liver tumor-on-a-chip model for toxicity testing | • Recapitulation of tumor microenvironment with biochemical factors and biophysical cues | • Use of animal-derived decellularized liver ECM instead of human-derived ECM | 125 | |
| Decellularized liver matrix | • Enhanced hepatocyte viability and functions under flow conditions | |||||
| • Linear dose-dependent drug response to the toxicity of APAP and sorafenib | ||||||
| Gelatin pNIPAAm | Mouse liver hepatocytes (AML12) | Implantable vascularized liver chip for cross-validation of disease treatment with animal model | • Successful implantation and maintenance of liver buds in perfusable 3D hydrogels | • Low reproducibility and control over the fabrication methods to produce hydrogel-based nanofibers for the generation of vessel-like networks | 111 | |
| Mouse endothelial cells | • Realistic replication of inflammation, lipid accumulation, and fibrosis due to the progression of NAFLD compared to animal model | |||||
| HSCs | • Similar response of liver-on-a-chip model upon the use of hepatic steatosis-reducing drug (i.e., restoration of mitochondrial activities, reduction of inflammation, oxidative stress, and lipid accumulation) compared to animal model | |||||
| 3D printed scaffolds | PLGA | HepG2 | Biomimetic 3D scaffold coupled with a modular bioreactor to investigate hepatocyte function | • Increased cell density of seeded culture compared to monolayer controls | • Low biochemical and biological potential of the 3D printed scaffold | 105 |
| PLLA | • Enhanced cell density, formation of aggregates, and gene expressions in 3D scaffolds compared with monolayer controls | |||||
| • 3D porous scaffold mimicking the characteristic geometry of hepatic lobules | ||||||
| • Increased cell metabolic in dynamic culture in comparison with static monolayer cultures | ||||||
| Gelatin | HUH-7 | Modulation of hepatocyte function and gene expression using different pore geometries | • Precise control over pore geometry and orientation | • Limited 3D scaffold design and architecture complexity | 161 | |
| • High viability and proliferation when cells are seeded on 3D-printed scaffolds with different geometries | ||||||
| • Enhanced hepatocyte functions (albumin secretion, CYP activity, bile transport) in the interconnected scaffold compared to less interconnected geometries and 2D controls | ||||||
| PEGDA | — | Bio-inspired 3D printed scaffold functionalized with nanocomposites hydrogel for detoxification studies | • Successful encapsulation of functional polydiacetylene nanoparticles with detoxification potential | • High expensive and no commercial 3D printing equipment | 167 | |
| • Highly biomimetic 3D scaffold structure enables access to toxins inside the hydrogel matrix | • Selection of inks limited to photo-crosslinkable hydrogels | |||||
| • Effective interaction of nanoparticles and toxins | ||||||
| • High functionality of nanoparticles enables the toxins capture | ||||||
| • Loss of virulence potential in toxin solution after treatment | ||||||
| 3D bioprinted scaffolds | Alginate | HepG2 | Bi-cellular liver biomimetic structure | • High printability of high-resolution hexagonal scaffolds without cell damage | • Potential cell damage risk due to shear stress induced by extrusion of highly viscous bioinks | 186 |
| Cellulose nanocrystal | NIH 3T3 | • Precise positioning of HepG2 and NIH 3T3 bioinks to mimic liver microenvironment | ||||
| GelMA | • NIH 3T3 alignment at the boundary of the hexagonal lobule | |||||
| • Enhancement of albumin secretion in the bicellular construct compared to NIH 3T3 and HepG2 singles cells culture | ||||||
| Liver decellularized ECM | HepG2 | Mimicking of cirrhotic liver environment to predict the HCC development and growth invasion | • Constructs with regionally varied mechanical properties | • Labor intensive 3D bioprinting procedure | 189 | |
| GelMA | • HepG2 reduced growth and high degree of stromal invasion from the nodules with cirrhotic-like stiffness compared to healthy controls | • Use of UV-photocrosslinking with potential cell damage risk | ||||
| Gelatin alginate | Patient-derived HCC | Platform for personalized medicine of human hepatocellular carcinoma | • Patient-derived scaffolds with high viability and proliferation rate | • Limited 3D scaffold design and geometry in terms of biomimicry and complexity | 138 | |
| • Good retainment of the features of the originating HCC tumors | ||||||
| • Effective response to four commonly used empirical targeted drugs showing a dose-dependent manner | ||||||
| Electrospun scaffolds | Liver decellularized ECM | THLE-3 hepatocytes | Nanofibrous hepatic-like scaffold for therapeutics research | • Bio-functional microenvironment | • Use of thermoplastic polymeric material with higher mechanical properties compared to the native liver tissue | 195 |
| PLLA | • Cell attachment and survival | |||||
| • Expression of key hepatic genes | ||||||
| • Hepatocyte growth and albumin production | ||||||
| Chitosan FN coating | Primary rat hepatocyte and 3T3-J2 fibroblasts | Biomimetic nanofibrous scaffolds and co-culture system for the long-term maintenance of liver functions | • Highly porous and randomly oriented nanofibrous structures | • Limited FN biomimetic potential compared to other biomaterials (e.g., decellularized ECM) | 201 | |
| • Enhanced cell attachment and spreading due to FN coating | ||||||
| • Formation of colonies, maintenance of cell morphology and function for prolonged periods of time | ||||||
| • High levels of CYP450 A1 enzyme activity and albumin secretion |
| In vitro model | Advantages | Disadvantages |
|---|---|---|
| Organoids | • Cost-effective procedure | • Labor-intensive procedures to develop suitable fabrication protocols |
| • Facile method | • Use of expensive cell sources (i.e., iPSC) | |
| • Ability to easily combine different cell types | • Relatively simple model with lack of flexibility enabling to reproduce complex tissue architecture and hierarchical structure | |
| • Cells self-organization properties | • Limited control on organoid size and uniformity | |
| • Cell–cell contact and interaction to reproduce in vivo conditions | • Lack of vascularization | |
| • Ability to mimic oxygen gradients and drug diffusion of the liver lobule | • Limited tissue availability in case of patient-derived tissue biopsies | |
| • Easy integration into 3D scaffold/microfluidic platform | ||
| • Long-term culture and maintenance of the overall metabolic configuration and liver-specific functionality | ||
| • Suitable for combination with soft biomaterials (i.e., hydrogels) | ||
| • Allow the development of personalized model by using patient-derived tissue biopsies or pluripotent stem cells | ||
| Liver-on-a-chip | • Cost-effective | • Expensive technology for the fabrication of the microfluidic platform |
| • Ability to recreate tissue-like microarchitecture by tailoring and adapting different layouts and design | • Low throughput | |
| • Low number of cells required and amount of tissue culture medium | • Multi-step fabrication process | |
| • Ability to recreate the in vivo liver natural and pathological physiology by generating dynamic mechanical and physicochemical stimuli | • Large amount of dead space | |
| • Ability to introduce fluidic channels to reproduce dynamic blood flow, wall shear stress, oxygen gradient, and metabolic zonation | • Not specific drug bindings to the chip biomaterial platform | |
| • Precise cell spatial distribution | • Expensive device (i.e., bioreactor, syringe pumps) coupled to the microfluidic chip to create in vivo condition | |
| • Ability to mimic the liver tissue functional unit (i.e., sinusoid, canicular system, acinus) | • Not suitable to reproduce physiological tissue-like structures in terms of complexity and size | |
| • Ability to mimic molecular mechanism of disease and drug action | • Limited selection of biomaterials to recreate a biomimetic microenvironment | |
| • Recapitulation of tissue/organ multi-cellular architectures and tissue–tissue interfaces to evaluate interorgan and intertissue interaction in drug metabolism. | ||
| 3D scaffolds | • High control over scaffold architecture and pore size | • Complex and expensive biofabrication apparatus Labor-intensive biofabrication procedures |
| • Suitable for the fabrication of highly biomimetic construct in terms of tissue architectures and biomaterials | • Challenging coupling of materials and biofabrication techniques suitable to create complex and high-resolution structure | |
| • Ability to precisely position different cell types and biomaterials to reproduce the complexity of the heterogeneous liver microenvironment in pathological and physiological state | • Not suitable to reproduce in vivo-like stimuli without perfusable systems | |
| • Tissue-like cell density and composition | • Material choice not always highly ECM biomimetic | |
| • Ability to load biomaterials with drug compounds | • Limited range of fabrication techniques allowing for functional cell encapsulation | |
| • Ability to fabricate constructs with different mechanical properties by easily changing fabrication parameter processes |
Although recent advances focus on more automated biofabrication approaches to avoid significant errors and bias, it is still strongly believed that a further step towards this research direction should be made. As a result, the data-driven repeatability of analytical methods for drug testing and toxicity studies would actively encourage predicting and identifying possible outcomes of these pioneering biomedical technologies.224 Moreover, bioinformatics would speed up the final process involving governmental agencies (e.g., FDA approval, EC regulations) to authorize and market such advanced LTE in vitro tools at a commercial scale. Among others, deep learning methods for DILI prediction and toxicogenomic have been lately realized.225–230 In this frame, machine learning algorithms have been applied to the chemical structure of small molecule drugs as a model to predict the DILI compound-specific risk at the preclinical stage and to identify patient-specific drug treatments.225 Similarly, an artificial intelligence-based approach was developed to forecast and rank gene expression features acquired from rodent livers exposed to drug compounds. A toxicogenomic open database231 was used to extract such features to predict liver toxicity, while the computational model was optimized to predict whether a drug compound can cause liver necrosis and identify target gene biomarkers as disease indicators.228 Here, the model identified predictor biomarkers that are involved in liver metabolism and detoxification (i.e., Car3, Crat, Cyp39a1, Dcd, Lbp, Scly, Slc23a1, and Tkfc), carcinogenesis as well as transcriptional regulation (i.e., Ablim3). In the future, similar methods could be used to boost the prediction of drug toxicity effects in humans. In fact, developing machine learning algorithms and bioinformatics methodologies are relatively cost-effective and could reduce the overall time to market safe drug products.
Moreover, the public debate should focus on emerging challenges such as biobanking frontiers, which would help long-term studies on patient-derived liver tissues for drug development. These biological samples, which can be collected in-hospital or during community screenings, can be maintained and used at a later stage for research purposes, at the border with healthcare outputs. Importantly, the engineered constructs should accomplish clinical-grade quality and good manufacturing practices (GMP).93 Herein, organoid models may closely recapitulate both the native and the diseased organs. Parallelly, they can be used to predict drug sensitivity and identify biomarkers for treatment response in different population groups. However, there is still an urgent need to fully validate the tissue heterogeneity and physiological relevance of 3D engineered in vitro models and the effectiveness of drug testing applications, respectively. Indeed, this would potentially allow for their clinical translation towards individualized treatments and therapy effects.232 Therefore, it is of foremost importance to clinically validate in vitro outcomes in humans.233–235
Similarly, somatic genome editing platforms (e.g., TALEN, ZFN, CRISPR/Cas9) could be also considered as an halfway point to design liver disease models for metabolic and acquired disorders with clinical therapy purposes, thus identifying responsible genes and mutational profile of liver diseases and hepatic cancer.93,211 For instance, such platforms were employed to model liver disease as HCC, inducing carcinogenesis in healthy liver 3D engineered constructs such as organoids.236
6. Conclusions
The production of small-scale 3D constructs that closely mimic the in vivo hepatic microenvironment is at the present a concrete option for disease modeling and drug testing. However, one of the primary challenges for in vitro liver models is the generation of reliable and accurate platforms to fully reflect the physiological function of the human liver. In particular, the main challenges are found in (i) validating in vitro scaled down LTE structures able to recapitulate in vivo human models, and (ii) perfusing such engineered models for a thorough characterization of the liver's functional complexity. Therefore, controlling the overall architecture of these platforms is essential for their mechanical and biological stability.151,237–239 Despite recent progress towards scalable 3D liver scaffolds for drug metabolism and toxicology studies, standardized testing platforms that aim to overcome animal models are still in their infancy and lack accurate approaches for the specific prediction and repeatability of data-driven results.240–243 However, it is strongly believed that the rapid advance of biofabrication technologies together with an improved clinical translation might extensively enhance the knowledge on disease pathogenesis and drug development for patient-specific applications.Abbreviations
| 2DMC | Two-dimensional monolayer culture |
| 3T3-J2 | Mouse embryonic fibroblasts |
| 5-FU | 5-Fluorouracil |
| ABCB1 | Atp binding cassette subfamily B member 1 |
| AAT | Alpha-1 antitrypsin |
| ACTA2 | Actin alpha 2 |
| AFP | Alpha-fetoprotein |
| ALD | Alcoholic liver disease |
| AML12 | Alpha mouse liver 12 hepatocytes |
| APAP | Acetaminophen |
| ASS1 | Argininosuccinate synthetase 1 |
| CF | Cystic fibrosis |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| CLD | Chronic liver disease |
| COL1A1 | Collagen type I alpha 1 chain |
| CNC | Cellulose nanocrystals |
| CTLN1 | Citrullinemia type 1 |
| CRISPR | Clustered regularly interspaced short palindromic repeat |
| CYP2C9 | Cytochrome P450 family 2 subfamily C member 9 |
| CYP3A4 | Cytochrome P450 3A4 |
| CYP450 | Cytochrome P450 |
| dECM | Decellularized extracellular matrix |
| DILI | Drug-induced liver injury |
| DNA | Deoxyribonucleic acid |
| DOX | Doxorubicin |
| ECM | Extracellular matrix |
| EGFR | Estimated glomerular filtration rate |
| EpCAM+ | Epithelial cell adhesion molecule positive |
| EtOH | Ethanol |
| FN | Fibronectin |
| GelMA | Gelatin methacryloyl |
| GK | Glucokinase |
| HA | Hyaluronic acid |
| HAM | Hyaluronic acid-coated microcarriers |
| HCC | Hepatocellular carcinoma |
| HCT-116 | Human colon carcinoma cells |
| HBV | Hepatitis B virus |
| HepG2 | Human hepatoma G2 cells |
| HepaRG | Human hepatic progenitor cells |
| hESC | Human embryonic stem cells |
| hFLMC | Human fetal liver mesenchymal cells |
| HHH | Healthy human hepatocytes |
| hiHEP | Human induced hepatocytes |
| hiPSC | Human induced pluripotent stem cells |
| HLC | Hepatocyte-like cells |
| hLECM | Human liver extracellular matrix |
| HMEC-1 | Human microvascular endothelial cells |
| HNF4A | Hepatocyte nuclear factor 4 alpha |
| HPC | Hepatic progenitor cells |
| hPH | Human primary hepatocytes |
| HSC | Hepatic stellate cells |
| HUH-7 | Human hepatoma cell line 7 |
| HUVEC | Human umbilical vein endothelial cells |
| IC50 | Half-maximal inhibitory concentration |
| IGF-2 | Insulin-like growth factor 2 |
| IPN | Interpenetrating network |
| iPSC | Induced pluripotent stem cells |
| LTE | Liver tissue engineering |
| LX-2 | Lieming Xu-2 human hepatic stellate cells |
| MDR-1 | Multidrug resistance protein 1 |
| MPS | Mucopolysaccharidosis |
| MRP1 | Multidrug resistance associated protein 1 |
| NaB | Sodium butyrate |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NIH 3T3 | Mouse embryonic fibroblasts |
| PCL | Polycaprolactone |
| PEG | Polyethylene glycol |
| PEGDA | Polyethylene glycol diacrylate |
| PHH | Primary human hepatocytes |
| PLA | Polylactide acid |
| PLC | Primary liver cancer cells |
| PLGA | Poly-DL-lactide-co-glycolide |
| PLLA | Poly-L-lactic acid |
| pNIPAAm | Poly(N-isopropyl acrylamide) |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PRH | Primary rat hepatocytes |
| PU | Polyurethane |
| PVA | Polyvinyl alcohol |
| RGD | Arginine–glycine–aspartic acid |
| RLC-18 | Rat liver cells |
| ROS | Reactive oxygen species |
| TALEN | Transcription activator-like effector nucleases |
| TGFβ-1 | Transforming growth factor beta 1 |
| THLE-3 | Transformed human liver epithelial-3 |
| THP-1 | Tamm-horsfall protein 1 kupffer cells |
| TNFα | Tumour necrosis factor A |
| TTR | Transthyretin |
| VA | Valproic acid |
| ZFN | Zinc finger nucleases |
Conflicts of interest
The authors declare no conflict of interest in this work.Acknowledgements
A. P., M. V. and W. S. acknowledge the financial support of Warsaw University of Technology (Subvention Funds), the National Centre for Research and Developments within the STRATEGMED program (project no. STRATEGMED3/305813/2/NCBR/2017) and in the framework of the project BIOMOTION (PL-TW/VI/3/2019). This work was partially supported by the First TEAM grant number POIR.04.04.00-00-5ED7/18-00, within the framework of the First TEAM programme of the Foundation for Polish Science (FNP), co-financed by the European Union under the European Regional Development Fund. C. R., F. P. and M. C. acknowledge the financial support from the Polish Ministry of Science and Higher Education through scholarships for outstanding young scientists. C. R. was partially supported by the Foundation for Polish Science (FNP). This study is also supported by the National Science Centre Poland (NCN) within PRELUDIUM 19 Project No. 2020/37/N/ST5/03272 to N. C. and SONATA 14 Project No. 2018/31/D/ST8/03647 to M. C.Fig. 1, 2, 3A, 4A, 5 and 6, and the graphical abstract are created with Biorender.com.
References
- S. Kishi, T. Matsumoto, T. Ichimura and C. R. Brooks, In Vitro Cell. Dev. Biol., 2021, 1–15 Search PubMed.
- A. Rizki-Safitri, T. Traitteur and R. Morizane, Function, 2021, 2, zqab026 CrossRef PubMed.
- I. Ziółkowska-Suchanek, Cells, 2021, 10, 141 CrossRef.
- B. Cunniff, J. E. Druso and J. L. van der Velden, Histochem. Cell Biol., 2021, 1–8 Search PubMed.
- I. Kulvinskiene, R. Aldonyte, R. Miksiunas, A. Mobasheri and D. Bironaite, Cell Biol. Transl. Med. Vol. 10 Stem Cells Tissue Regen, 2020, pp. 43–77 Search PubMed.
- C. Gardin, L. Ferroni, C. Latremouille, J. C. Chachques, D. Mitrečić and B. Zavan, Cells, 2020, 9, 742 CrossRef CAS PubMed.
- M. Costantini, S. Testa, E. Fornetti, C. Fuoco, C. Sanchez Riera, M. Nie, S. Bernardini, A. Rainer, J. Baldi and C. Zoccali, EMBO Mol. Med., 2021, 13, e12778 CAS.
- M. Costantini, S. Testa, P. Mozetic, A. Barbetta, C. Fuoco, E. Fornetti, F. Tamiro, S. Bernardini, J. Jaroszewicz, W. Swieszkowski, M. Trombetta, L. Castagnoli, D. Seliktar, P. Garstecki, G. Cesareni, S. Cannata, A. Rainer, C. Gargioli, W. Święszkowski, M. Trombetta, L. Castagnoli, D. Seliktar, P. Garstecki, G. Cesareni, S. Cannata, A. Rainer and C. Gargioli, Biomaterials, 2017, 131, 98–110 CrossRef CAS PubMed.
- S. Cheemerla and M. Balakrishnan, Clin. Liver Dis., 2021, 17, 365 CrossRef PubMed.
- R. P. Cunningham and N. Porat-Shliom, Front. Physiol., 2021, 12, 1–17 Search PubMed.
- J. Ozougwu and J. C. Ozougwu, Int. J. Res. Pharm. Biosci., 2017, 4, 13 Search PubMed.
- A. S. Serras, J. S. Rodrigues, M. Cipriano, A. V. Rodrigues, N. G. Oliveira and J. P. Miranda, Front. Cell Dev. Biol., 2021, 9, 1–30 Search PubMed.
- Y. Tao, M. Wang, E. Chen and H. Tang, Mediators Inflammation, 2017, 4256352 Search PubMed.
- G. K. Michalopoulos and B. Bhushan, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 40–55 CrossRef PubMed.
- W. Li, L. Li and L. Hui, Trends Cell Biol., 2020, 30, 329–338 CrossRef.
- H. Lee, S. Chae, J. Y. Kim, W. Han, J. Kim, Y. Choi and D. W. Cho, Biofabrication, 2019, 11(2), 025001 CrossRef CAS.
- M. Raasch, E. Fritsche, A. Kurtz, M. Bauer and A. S. Mosig, Adv. Drug Delivery Rev., 2019, 140, 51–67 CrossRef CAS PubMed.
- X. Ma, J. Liu, W. Zhu, M. Tang, N. Lawrence, C. Yu, M. Gou and S. Chen, Adv. Drug Delivery Rev., 2018, 132, 235–251 CrossRef CAS PubMed.
- Y. Parmentier, C. Pothier, N. Hewitt, L. Vincent, F. Caradec, J. Liu, F. Lin, M.-M. Trancart, F. Guillet and B. Bouaita, Xenobiotica, 2019, 49, 22–35 CrossRef CAS.
- Y. Kim, K. Kang, S. Yoon, J. S. Kim, S. A. Park, W. D. Kim, S. B. Lee, K.-Y. Ryu, J. Jeong and D. Choi, Organogenesis, 2018, 14, 1–12 CrossRef CAS PubMed.
- E. Milner, M. Ainsworth, M. McDonough, B. Stevens, J. Buehrer, R. Delzell, C. Wilson and J. Barnhill, Med. Drug Discovery, 2020, 8, 100060 CrossRef.
- C. M. Smith, C. K. Nolan, M. A. Edwards, J. B. Hatfield, T. W. Stewart, S. S. Ferguson, E. L. Lecluyse and J. Sahi, J. Pharm. Sci., 2012, 101, 3989–4002 CrossRef CAS.
- M. Vinken and J. G. Hengstler, Arch. Toxicol., 2018, 92, 2981–2986 CrossRef CAS.
- M. R. Mcgill and H. Jaeschke, Biochim. Biophys. Acta, Mol. Basis Dis., 2019, 1865, 1031–1039 CrossRef CAS PubMed.
- K. Yoshizato and C. Tateno, Expert Opin. Drug Metab. Toxicol., 2013, 9, 1419–1435 CrossRef CAS PubMed.
- Y. A. Nevzorova, Z. Boyer-Diaz, F. J. Cubero and J. Gracia-Sancho, J. Hepatol., 2020, 73, 423–440 CrossRef.
- K. Sakabe, T. Takebe and A. Asai, Clin. Liver Dis., 2020, 15, 3–8 CrossRef.
- C. H. Beckwitt, A. M. Clark, S. Wheeler, D. L. Taylor, D. B. Stolz, L. Griffith and A. Wells, Exp. Cell Res., 2018, 363, 15–25 CrossRef CAS.
- R. D. Pedde, B. Mirani, A. Navaei, T. Styan, S. Wong, M. Mehrali, A. Thakur, N. K. Mohtaram, A. Bayati, A. Dolatshahi-Pirouz, M. Nikkhah, S. M. Willerth and M. Akbari, Adv. Mater., 2017, 29, 1606061 CrossRef.
- J. S. Miller, K. R. Stevens, M. T. Yang, B. M. Baker, D.-H. T. Nguyen, D. M. Cohen, E. Toro, A. A. Chen, P. A. Galie and X. Yu, Nat. Mater., 2012, 11, 768–774 CrossRef CAS.
- L. R. Madden, T. V. Nguyen, S. Garcia-Mojica, V. Shah, A. V. Le, A. Peier, R. Visconti, E. M. Parker, S. C. Presnell and D. G. Nguyen, IScience, 2018, 2, 156–167 CrossRef CAS PubMed.
- J. Sarkar, S. C. Kamble and N. C. Kashikar, Chem, 2021, 3, 164–181 CAS.
- M. A. Lancaster and J. A. Knoblich, Science, 2014, 345, 1247125 CrossRef.
- M. Simian and M. J. Bissell, J. Cell Biol., 2017, 216, 31–40 CrossRef CAS.
- M. Hofer and M. P. Lutolf, Nat. Rev. Mater., 2021, 6, 402–420 CrossRef CAS PubMed.
- T. Agarwal, N. Celikkin, M. Costantini, T. K. Maiti and P. Makvandi, Bio-Des. Manuf., 2021, 4, 641–674 CrossRef.
- V. Magno, A. Meinhardt and C. Werner, Adv. Funct. Mater., 2020, 30, 2000097 CrossRef CAS.
- E. A. Aisenbrey and W. L. Murphy, Nat. Rev. Mater., 2020, 5, 539–551 CrossRef CAS PubMed.
- M. T. Kozlowski, C. J. Crook and H. T. Ku, Commun. Biol., 2021, 4, 1–15 CrossRef.
- Y. Wang, H. Liu, M. Zhang, H. Wang, W. Chen and J. Qin, Biomater. Sci., 2020, 8, 5476–5488 RSC.
- B. J. Klotz, L. A. Oosterhoff, L. Utomo, K. S. Lim, Q. Vallmajo-Martin, H. Clevers, T. B. F. Woodfield, A. J. W. P. Rosenberg, J. Malda and M. Ehrbar, Adv. Healthcare Mater., 2019, 8, 1900979 CrossRef CAS.
- M. Saheli, M. Sepantafar, B. Pournasr, Z. Farzaneh, M. Vosough, A. Piryaei and H. Baharvand, J. Cell. Biochem., 2018, 119, 4320–4333 CrossRef CAS.
- M. Krüger, L. A. Oosterhoff, M. E. van Wolferen, S. A. Schiele, A. Walther, N. Geijsen, L. De Laporte, L. J. W. van der Laan, L. M. Kock and B. Spee, Adv. Healthcare Mater., 2020, 9, 1901658 CrossRef.
- S. Ye, J. W. B. Boeter, M. Mihajlovic, F. G. van Steenbeek, M. E. van Wolferen, L. A. Oosterhoff, A. Marsee, M. Caiazzo, L. J. W. van der Laan and L. C. Penning, Adv. Funct. Mater., 2020, 30, 2000893 CrossRef CAS.
- N. Broguiere, L. Isenmann, C. Hirt, T. Ringel, S. Placzek, E. Cavalli, F. Ringnalda, L. Villiger, R. Züllig and R. Lehmann, Adv. Mater., 2018, 30, 1801621 CrossRef.
- T. Takebe, K. Sekine, M. Kimura, E. Yoshizawa, S. Ayano, M. Koido, S. Funayama, N. Nakanishi, T. Hisai, T. Kobayashi, T. Kasai, R. Kitada, A. Mori, H. Ayabe, Y. Ejiri, N. Amimoto, Y. Yamazaki, S. Ogawa, M. Ishikawa, Y. Kiyota, Y. Sato, K. Nozawa, S. Okamoto, Y. Ueno and H. Taniguchi, Cell Rep., 2017, 21, 2661–2670 CrossRef CAS.
- R. Ouchi, S. Togo, M. Kimura, T. Shinozawa, M. Koido, H. Koike, W. Thompson, R. A. Karns, C. N. Mayhew, P. S. McGrath, H. A. McCauley, R.-R. Zhang, K. Lewis, S. Hakozaki, A. Ferguson, N. Saiki, Y. Yoneyama, I. Takeuchi, Y. Mabuchi, C. Akazawa, H. Y. Yoshikawa, J. M. Wells and T. Takebe, Cell Metab., 2019, 30, 374–384 CrossRef CAS.
- F. Sampaziotis, M. Cardoso de Brito, P. Madrigal, A. Bertero, K. Saeb-Parsy, F. A. C. Soares, E. Schrumpf, E. Melum, T. H. Karlsen, J. A. Bradley, W. T. H. Gelson, S. Davies, A. Baker, A. Kaser, G. J. Alexander, N. R. F. Hannan and L. Vallier, Nat. Biotechnol., 2015, 33, 845–852 CrossRef CAS.
- M. Ogawa, S. Ogawa, C. E. Bear, S. Ahmadi, S. Chin, B. Li, M. Grompe, G. Keller, B. M. Kamath and A. Ghanekar, Nat. Biotechnol., 2015, 33, 853–861 CrossRef CAS PubMed.
- S. Wang, X. Wang, Z. Tan, Y. Su, J. Liu, M. Chang, F. Yan, J. Chen, T. Chen, C. Li, J. Hu and Y. Wang, Cell Res., 2019, 29, 1009–1026 CrossRef CAS PubMed.
- Y. Guan, D. Xu, P. M. Garfin, U. Ehmer, M. Hurwitz, G. Enns, S. Michie, M. Wu, M. Zheng, T. Nishimura, J. Sage and G. Peltz, JCI Insight, 2017, 2, e94954 CrossRef.
- M. Coll, L. Perea, R. Boon, S. B. Leite, J. Vallverdú, I. Mannaerts, A. Smout, A. El Taghdouini, D. Blaya, D. Rodrigo-Torres, I. Graupera, B. Aguilar-Bravo, C. Chesne, M. Najimi, E. Sokal, J. J. Lozano, L. A. van Grunsven, C. M. Verfaillie and P. Sancho-Bru, Cell Stem Cell, 2018, 23, 101–113 CrossRef CAS.
- S. S. Ng, K. Saeb-Parsy, S. J. I. Blackford, J. M. Segal, M. P. Serra, M. Horcas-Lopez, S. Mastoridis, W. Jassem, C. W. Frank and N. J. Cho, Biomaterials, 2018, 182, 299–311 CrossRef CAS.
- M. N. B. Ramli, Y. S. Lim, C. T. Koe, D. Demircioglu, W. Tng, K. A. U. Gonzales, C. P. Tan, I. Szczerbinska, H. Liang and E. L. Soe, Gastroenterology, 2020, 159, 1471–1486 CrossRef CAS.
- C. J. Hindley, L. Cordero-Espinoza and M. Huch, Dev. Biol., 2016, 420, 251–261 CrossRef CAS PubMed.
- S. J. Mun, Y.-H. Hong, H.-S. Ahn, J.-S. Ryu, K.-S. Chung and M. J. Son, Int. J. Stem Cells, 2020, 13, 279–286 CrossRef PubMed.
- J. Vives and L. Batlle-Morera, Stem Cell Res. Ther., 2020, 11, 1–4 CrossRef.
- J.-Y. Lee, H.-J. Han, S.-J. Lee, E.-H. Cho, H.-B. Lee, J.-H. Seok, H. S. Lim and W.-C. Son, Int. J. Mol. Sci., 2020, 21, 2982 CrossRef CAS PubMed.
- N. Dianat, H. Dubois-Pot-Schneider, C. Steichen, C. Desterke, P. Leclerc, A. Raveux, L. Combettes, A. Weber, A. Corlu and A. Dubart–Kupperschmitt, Hepatology, 2014, 60, 700–714 CrossRef CAS.
- S. B. Leite, T. Roosens, A. El Taghdouini, I. Mannaerts, A. J. Smout, M. Najimi, E. Sokal, F. Noor, C. Chesne and L. A. van Grunsven, Biomaterials, 2016, 78, 1–10 CrossRef CAS PubMed.
- B. Artegiani, L. van Voorthuijsen, R. G. H. Lindeboom, D. Seinstra, I. Heo, P. Tapia, C. López-Iglesias, D. Postrach, T. Dayton and R. Oka, Cell Stem Cell, 2019, 24, 927–943 CrossRef CAS.
- A. Correns, L.-M. A. Zimmermann, C. Baldock and G. Sengle, Matrix Biol. Plus, 2021, 11, 100071 CrossRef CAS.
- M. Urbischek, H. Rannikmae, T. Foets, K. Ravn, M. Hyvönen and M. de la Roche, Sci. Rep., 2019, 9, 1–11 CrossRef CAS.
- T. Tao, P. Deng, Y. Wang, X. Zhang, Y. Guo, W. Chen and J. Qin, Adv. Sci., 2022, 9, 2103495 CrossRef.
- Y. Lu, G. Zhang, C. Shen, K. Uygun, M. L. Yarmush and Q. Meng, Biotechnol. Bioeng., 2012, 109, 595–604 CrossRef CAS.
- C. M. Gamboa, Y. Wang, H. Xu, K. Kalemba, F. E. Wondisford and H. E. Sabaawy, Cells, 2021, 10, 3280 CrossRef CAS.
- G. Hong, J. Kim, H. Oh, S. Yun, C. M. Kim, Y. Jeong, W. Yun, J. Shim, I. Jang and C. Kim, Adv. Mater., 2021, 2102624 CrossRef CAS.
- T. Takebe and J. M. Wells, Science, 2019, 364, 956–959 CrossRef CAS PubMed.
- P. N. Bernal, M. Bouwmeester, J. Madrid-Wolff, M. Falandt, S. Florczak, N. G. Rodriguez, Y. Li, G. Größbacher, R. Samsom and M. van Wolferen, Adv. Mater., 2022, 2110054 CrossRef CAS PubMed.
- M. A. Lancaster, M. Renner, C.-A. Martin, D. Wenzel, L. S. Bicknell, M. E. Hurles, T. Homfray, J. M. Penninger, A. P. Jackson and J. A. Knoblich, Nature, 2013, 501, 373–379 CrossRef CAS.
- T. Takebe, K. Sekine, M. Enomura, H. Koike, M. Kimura, T. Ogaeri, R.-R. Zhang, Y. Ueno, Y.-W. Zheng, N. Koike, S. Aoyama, Y. Adachi and H. Taniguchi, Nature, 2013, 499, 481–484 CrossRef CAS PubMed.
- S. Akbari, G. G. Sevinç, N. Ersoy, O. Basak, K. Kaplan, K. Sevinç, E. Ozel, B. Sengun, E. Enustun, B. Ozcimen, A. Bagriyanik, N. Arslan, T. T. Önder and E. Erdal, Stem Cell Rep., 2019, 13, 627–641 CrossRef CAS.
- Y.-Z. Nie, Y.-W. Zheng, K. Miyakawa, S. Murata, R.-R. Zhang, K. Sekine, Y. Ueno, T. Takebe, T. Wakita and A. Ryo, EBioMedicine, 2018, 35, 114–123 CrossRef PubMed.
- Y. Xia, A. Carpentier, X. Cheng, P. D. Block, Y. Zhao, Z. Zhang, U. Protzer and T. J. Liang, J. Hepatol., 2017, 66, 494–503 CrossRef CAS PubMed.
- S. Nuciforo, I. Fofana, M. S. Matter, T. Blumer, D. Calabrese, T. Boldanova, S. Piscuoglio, S. Wieland, F. Ringnalda and G. Schwank, Cell Rep., 2018, 24, 1363–1376 CrossRef CAS.
- G. Gómez-Mariano, N. Matamala, S. Martínez, I. Justo, A. Marcacuzco, C. Jimenez, S. Monzón, I. Cuesta, C. Garfia and M. T. Martínez, Hepatol. Int., 2020, 14, 127–137 CrossRef.
- L. Broutier, G. Mastrogiovanni, M. M. A. Verstegen, H. E. Francies, L. M. Gavarró, C. R. Bradshaw, G. E. Allen, R. Arnes-Benito, O. Sidorova and M. P. Gaspersz, Nat. Med., 2017, 23, 1424–1435 CrossRef CAS.
- G. H. Underhill and S. R. Khetani, Cell. Mol. Gastroenterol. Hepatol., 2018, 5, 426–439 CrossRef PubMed.
- W. C. Peng, L. J. Kraaier and T. A. Kluiver, Exp. Mol. Med., 2021, 1–17 CAS.
- Y. Jin, J. Kim, J. S. Lee, S. Min, S. Kim, D.-H. Ahn, Y.-G. Kim and S.-W. Cho, Adv. Funct. Mater., 2018, 28, 1801954 CrossRef.
- T. Shinozawa, M. Kimura, Y. Cai, N. Saiki, Y. Yoneyama, R. Ouchi, H. Koike, M. Maezawa, R.-R. Zhang and A. Dunn, Gastroenterology, 2021, 160, 831–846 CrossRef CAS.
- A. Skardal, M. Devarasetty, C. Rodman, A. Atala and S. Soker, Ann. Biomed. Eng., 2015, 43, 2361–2373 CrossRef PubMed.
- N. Kaplowitz, Nat. Rev. Drug Discovery, 2005, 4, 489–499 CrossRef CAS PubMed.
- N. Prior, P. Inacio and M. Huch, Gut, 2019, 68, 2228–2237 CrossRef CAS PubMed.
- L. Meunier and D. Larrey, Front. Pharmacol., 2019, 10, 1482 CrossRef CAS PubMed.
- S. J. Mun, J.-S. Ryu, M.-O. Lee, Y. S. Son, S. J. Oh, H.-S. Cho, M.-Y. Son, D.-S. Kim, S. J. Kim, H. J. Yoo, H.-J. Lee, J. Kim, C.-R. Jung, K.-S. Chung and M. J. Son, J. Hepatol., 2019, 71, 970–985 CrossRef CAS PubMed.
- M. Sgodda, Z. Dai, R. Zweigerdt, A. D. Sharma, M. Ott and T. Cantz, Stem Cells Dev., 2017, 26, 1490–1504 CrossRef CAS PubMed.
- S. H. Au, M. D. Chamberlain, S. Mahesh, M. V. Sefton and A. R. Wheeler, Lab Chip, 2014, 14, 3290–3299 RSC.
- M. C. Bouwmeester, P. N. Bernal, L. A. Oosterhoff, M. van Wolferen, V. Lehmann, M. Vermaas, M. Buchholz, Q. Peiffer, J. Malda and L. J. W. van der Laan, Macromol. Biosci., 2021, 2100327 CrossRef CAS.
- T. Nakamura, K. Yoshimoto, Y. Nakayama, Y. Tomita and A. Ichihara, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 7229–7233 CrossRef CAS.
- T. Matsui and T. Shinozawa, Front. Genet., 2021, 2119 Search PubMed.
- S. Nuciforo and M. H. Heim, JHEP Rep., 2021, 3, 100198 CrossRef.
- S. N. Boers, J. J. M. van Delden, H. Clevers and A. L. Bredenoord, EMBO Rep., 2016, 17, 938–941 CrossRef CAS PubMed.
- S. N. Boers and A. L. Bredenoord, Nat. Cell Biol., 2018, 20, 642–645 CrossRef CAS.
- N. Sachs, J. de Ligt, O. Kopper, E. Gogola, G. Bounova, F. Weeber, A. V. Balgobind, K. Wind, A. Gracanin and H. Begthel, Cell, 2018, 172, 373–386 CrossRef CAS.
- L. R. Volpatti and A. K. Yetisen, Trends Biotechnol., 2014, 32, 347–350 CrossRef CAS PubMed.
- X. Hou, Y. S. Zhang, G. T. Santiago, M. M. Alvarez, J. Ribas, S. J. Jonas, P. S. Weiss, A. M. Andrews, J. Aizenberg and A. Khademhosseini, Nat. Rev. Mater., 2017, 2, 1–15 Search PubMed.
- S. Faley, K. Seale, J. Hughey, D. K. Schaffer, S. VanCompernolle, B. McKinney, F. Baudenbacher, D. Unutmaz and J. P. Wikswo, Lab Chip, 2008, 8, 1700–1712 RSC.
- D. Patel, A. Haque, Y. Gao and A. Revzin, Integr. Biol., 2015, 7, 815–824 CrossRef CAS PubMed.
- C.-T. Ho, R.-Z. Lin, R.-J. Chen, C.-K. Chin, S.-E. Gong, H.-Y. Chang, H.-L. Peng, L. Hsu, T.-R. Yew and S.-F. Chang, Lab Chip, 2013, 13, 3578–3587 RSC.
- P. Gissen and I. M. Arias, J. Hepatol., 2015, 63, 1023–1037 CrossRef PubMed.
- F. Yu, R. Deng, W. Hao Tong, L. Huan, N. Chan Way, A. IslamBadhan, C. Iliescu and H. Yu, Sci. Rep., 2017, 7, 14528 CrossRef.
- D. Huang, S. B. Gibeley, C. Xu, Y. Xiao, O. Celik, H. N. Ginsberg and K. W. Leong, Adv. Funct. Mater., 2020, 30, 1909553 CrossRef CAS.
- C. E. Suurmond, S. Lasli, F. W. van den Dolder, A. Ung, H. Kim, P. Bandaru, K. Lee, H. Cho, S. Ahadian and N. Ashammakhi, Adv. Healthcare Mater., 2019, 8, 1901379 CrossRef CAS PubMed.
- B. Vinci, D. Cavallone, G. Vozzi, D. Mazzei, C. Domenici, M. Brunetto and A. Ahluwalia, Biotechnol. J., 2010, 5, 232–241 CrossRef CAS PubMed.
- W. J. McCarty, O. B. Usta and M. L. Yarmush, Sci. Rep., 2016, 6, 1–10 CrossRef.
- R. Panday, C. P. Monckton and S. R. Khetani, in Seminars in Liver Disease, Thieme Medical Publishers, Inc., 2022, vol. 42, pp. 1–16 Search PubMed.
- J. Ahn, J.-H. Ahn, S. Yoon, Y. S. Nam, M.-Y. Son and J.-H. Oh, J. Biol. Eng., 2019, 13, 1–15 CrossRef CAS.
- J.-H. Lee, K.-L. Ho and S.-K. Fan, J. Biomed. Sci., 2019, 26, 88 CrossRef.
- P. J. Lee, P. J. Hung and L. P. Lee, Biotechnol. Bioeng., 2007, 97, 1340–1346 CrossRef CAS.
- J. B. Lee, J. S. Park, Y. M. Shin, D. H. Lee, J. Yoon, D. Kim, U. H. Ko, Y. Kim, S. H. Bae and H. Sung, Adv. Funct. Mater., 2019, 29, 1900075 CrossRef.
- B. Bulutoglu, C. Rey-Bedón, Y. B. A. Kang, S. Mert, M. L. Yarmush and O. B. Usta, Lab Chip, 2019, 19, 3022–3031 RSC.
- A. Sato, K. Kadokura, H. Uchida and K. Tsukada, Biochem. Biophys. Res. Commun., 2014, 453, 767–771 CrossRef CAS PubMed.
- A. Moya, M. Ortega-Ribera, X. Guimerà, E. Sowade, M. Zea, X. Illa, E. Ramon, R. Villa, J. Gracia-Sancho and G. Gabriel, Lab Chip, 2018, 18, 2023–2035 RSC.
- F. T. Lee-Montiel, S. M. George, A. H. Gough, A. D. Sharma, J. Wu, R. DeBiasio, L. A. Vernetti and D. L. Taylor, Exp. Biol. Med., 2017, 242, 1617–1632 CrossRef CAS PubMed.
- L. A. Vernetti, N. Senutovitch, R. Boltz, R. DeBiasio, T. Ying Shun, A. Gough and D. L. Taylor, Exp. Biol. Med., 2015, 241, 101–114 CrossRef.
- A. Skardal, M. Devarasetty, S. Soker and A. R. Hall, Biofabrication, 2015, 7, 31001 CrossRef PubMed.
- Y. Miyamoto, M. Ikeuchi, H. Noguchi, T. Yagi and S. Hayashi, Cell Med., 2015, 8, 47–56 CrossRef.
- M. Jang, P. Neuzil, T. Volk, A. Manz and A. Kleber, Biomicrofluidics, 2015, 9, 34113 CrossRef.
- K. M. Bircsak, R. DeBiasio, M. Miedel, A. Alsebahi, R. Reddinger, A. Saleh, T. Shun, L. A. Vernetti and A. Gough, Toxicology, 2021, 450, 152667 CrossRef CAS PubMed.
- M. S. Freag, B. Namgung, M. E. Reyna Fernandez, E. Gherardi, S. Sengupta and H. L. Jang, Hepatol. Commun., 2021, 5, 217–233 CrossRef CAS PubMed.
- K.-J. Jang, M. A. Otieno, J. Ronxhi, H.-K. Lim, L. Ewart, K. R. Kodella, D. B. Petropolis, G. Kulkarni, J. E. Rubins, D. Conegliano, J. Nawroth, D. Simic, W. Lam, M. Singer, E. Barale, B. Singh, M. Sonee, A. J. Streeter, C. Manthey, B. Jones, A. Srivastava, L. C. Andersson, D. Williams, H. Park, R. Barrile, J. Sliz, A. Herland, S. Haney, K. Karalis, D. E. Ingber and G. A. Hamilton, Sci. Transl. Med., 2019, 11, eaax5516 CrossRef CAS.
- J. C. Nawroth, D. B. Petropolis, D. V. Manatakis, T. I. Maulana, G. Burchett, K. Schlünder, A. Witt, A. Shukla, K. Kodella and J. Ronxhi, Cell Rep., 2021, 36, 109393 CrossRef CAS.
- H. Lee, W. Han, H. Kim, D.-H. Ha, J. Jang, B. S. Kim and D.-W. Cho, Biomacromolecules, 2017, 18, 1229–1237 CrossRef CAS.
- S. Lu, F. Cuzzucoli, J. Jiang, L.-G. Liang, Y. Wang, M. Kong, X. Zhao, W. Cui, J. Li and S. Wang, Lab Chip, 2018, 18, 3379–3392 RSC.
- Y.-C. Toh, T. C. Lim, D. Tai, G. Xiao, D. van Noort and H. Yu, Lab Chip, 2009, 9, 2026–2035 RSC.
- A. Carraro, W.-M. Hsu, K. M. Kulig, W. S. Cheung, M. L. Miller, E. J. Weinberg, E. F. Swart, M. Kaazempur-Mofrad, J. T. Borenstein, J. P. Vacanti and C. Neville, Biomed. Microdevices, 2008, 10, 795–805 CrossRef.
- T. Agarwal, B. Subramanian and T. K. Maiti, ACS Biomater. Sci. Eng., 2019, 5, 4167–4182 CrossRef CAS PubMed.
- S. Mohanty, L. B. Larsen, J. Trifol, P. Szabo, H. V. R. Burri, C. Canali, M. Dufva, J. Emnéus and A. Wolff, Mater. Sci. Eng., C, 2015, 55, 569–578 CrossRef CAS PubMed.
- Y. Liu, L. Zhang, J. Wei, S. Yan, J. Yu and X. Li, J. Mater. Chem. B, 2014, 2, 3029–3040 RSC.
- Y. Pang, Y. Horimoto, S. Sutoko, K. Montagne, M. Shinohara, D. Mathiue, K. Komori, M. Anzai, T. Niino and Y. Sakai, Biofabrication, 2016, 8, 35016 CrossRef CAS PubMed.
- W. Xu, X. Wang, Y. Yan and R. Zhang, J. Bioact. Compat. Polym., 2008, 23, 409–422 CrossRef CAS.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- K. R. Stevens, J. S. Miller, B. L. Blakely, C. S. Chen and S. N. Bhatia, J. Biomed. Mater. Res., Part A, 2015, 103, 3331–3338 CrossRef CAS.
- R. Grant, D. Hay and A. Callanan, Biomed. Phys. Eng. Express, 2018, 4, 65015 CrossRef.
- N. Contessi Negrini, A. Angelova Volponi, P. T. Sharpe and A. D. Celiz, ACS Biomater. Sci. Eng., 2021, 7, 4330–4346 CrossRef CAS.
- J. P. Miranda, A. Rodrigues, R. M. Tostões, S. Leite, H. Zimmerman, M. J. T. Carrondo and P. M. Alves, Tissue Eng., Part C, 2010, 16, 1223–1232 CrossRef CAS.
- L. Sun, H. Yang, Y. Wang, X. Zhang, B. Jin, F. Xie, Y. Jin, Y. Pang, H. Zhao, X. Lu, X. Sang, H. Zhang, F. Lin, W. Sun, P. Huang and Y. Mao, Front. Oncol., 2020, 10, 878 CrossRef.
- X. Wang, Y. Yan, Y. Pan, Z. Xiong, H. Liu, J. Cheng, F. Liu, F. Lin, R. Wu, R. Xhang, Q. Lu, R. Zhang and Q. Lu, Tissue Eng., 2006, 12, 83–91 CrossRef CAS.
- J. Cui, H. Wang, Q. Shi, T. Sun, Q. Huang and T. Fukuda, Molecules, 2019, 24, 1762 CrossRef CAS.
- B. H. Lee, H. Shirahama, M. H. Kim, J. H. Lee, N.-J. Cho and L. P. Tan, NPG Asia Mater., 2017, 9, e412 CrossRef CAS.
- S. Li, Z. Xiong, X. Wang, Y. Yan, H. Liu and R. Zhang, J. Bioact. Compat. Polym., 2009, 24, 249–265 CrossRef CAS.
- Y. Yan, X. Wang, Z. Xiong, H. Liu, F. Liu, F. Lin, R. Wu, R. Zhang and Q. Lu, J. Bioact. Compat. Polym., 2005, 20, 259–269 CrossRef CAS.
- S. Ye, J. W. B. Boeter, L. C. Penning, B. Spee and K. Schneeberger, Bioengineering, 2019, 6 Search PubMed.
- N. S. Bhise, V. Manoharan, S. Massa, A. Tamayol, M. Ghaderi, M. Miscuglio, Q. Lang, Y. Shrike Zhang, S. R. R. yo. Shin, G. Calzone, N. Annabi, T. D. Shupe, C. E. Bishop, A. Atala, M. R. Dokmeci and A. Khademhosseini, Biofabrication, 2016, 8, 014101 CrossRef.
- J. Cui, H. Wang, Q. Shi, P. Ferraro, T. Sun, P. Dario, Q. Huang and T. Fukuda, Acta Biomater., 2020, 113, 328–338 CrossRef CAS PubMed.
- R. D. Hickey and W. E. Naugler, Hepatology, 2018, 67, 2465–2467 CrossRef PubMed.
- X. Wang and C. Liu, Polymer, 2018, 10, 1048 Search PubMed.
- H. Bruns, U. Kneser, S. Holzhüter, B. Roth, J. Kluth, P. M. Kaufmann, D. Kluth and H. C. Fiegel, Tissue Eng., 2005, 11, 1718–1726 CrossRef CAS PubMed.
- Y. Li and E. Kumacheva, Sci. Adv., 2018, 4, eaas8998 CrossRef.
- F. J. O'Brien, Mater. Today, 2011, 14, 88–95 CrossRef.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- D. Mondal, M. Griffith and S. S. Venkatraman, Int. J. Polym. Mater. Polym. Biomater., 2016, 65, 255–265 CrossRef CAS.
- Y. Zhang, Q.-S. Wang, K. Yan, Y. Qi, G.-F. Wang and Y.-L. Cui, J. Biomed. Mater. Res., Part A, 2016, 104, 1863–1870 CrossRef CAS PubMed.
- Y. Wang, W. Su, L. Wang, L. Jiang, Y. Liu, L. Hui and J. Qin, Toxicol. Res., 2018, 7, 13–21 CrossRef CAS.
- S. V. Murphy and A. Atala, Nat. Biotechnol., 2014, 32, 773–785 CrossRef CAS PubMed.
- J. Malda, J. Visser, F. P. Melchels, T. Jüngst, W. E. Hennink, W. J. A. Dhert, J. Groll and D. W. Hutmacher, Adv. Mater., 2013, 25, 5011–5028 CrossRef CAS PubMed.
- A. Schwab, R. Levato, M. D'Este, S. Piluso, D. Eglin and J. Malda, Chem. Rev., 2020, 120, 11028–11055 CrossRef CAS.
- P. L. Lewis and R. N. Shah, Curr. Transplant. Rep., 2016, 3, 100–108 CrossRef.
- J. W. Lee, Y.-J. Choi, W.-J. Yong, F. Pati, J.-H. Shim, K. S. Kang, I.-H. Kang, J. Park and D.-W. Cho, Biofabrication, 2016, 8, 15007 CrossRef.
- P. L. Lewis, R. M. Green and R. N. Shah, Acta Biomater., 2018, 69, 63–70 CrossRef CAS.
- G. Mattei, C. Magliaro, A. Pirone and A. Ahluwalia, Organogenesis, 2018, 14, 129–146 CrossRef CAS.
- B. Wang, Q. Hu, T. Wan, F. Yang, L. Cui, S. Hu, B. Gong, M. Li and Q. C. Zheng, Int. J. Polym. Sci., 2016, 2862738 Search PubMed.
- J. M. Sobral, S. G. Caridade, R. A. Sousa, J. F. Mano and R. L. Reis, Acta Biomater., 2011, 7, 1009–1018 CrossRef CAS PubMed.
- F. Darus, R. M. Isa, N. Mamat and M. Jaafar, Ceram. Int., 2018, 44, 18400–18407 CrossRef CAS.
- T. Lu, Y. Li and T. Chen, Int. J. Nanomed., 2013, 8, 337 CrossRef PubMed.
- M. Gou, X. Qu, W. Zhu, M. Xiang, J. Yang, K. Zhang, Y. Wei and S. Chen, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- J. Idaszek, M. Volpi, A. Paradiso, M. N. Quoc, Ż. Górecka, M. Klak, G. Tymicki, A. Berman, M. Wierzbicki and S. Jaworski, Bioprinting, 2021, 24, e00163 CrossRef.
- M. Volpi, A. Paradiso, M. Costantini and W. Świȩszkowski, ACS Biomater. Sci. Eng., 2022, 8, 379–405 CrossRef CAS PubMed.
- W. Peng, D. Unutmaz and I. T. Ozbolat, Trends Biotechnol., 2016, 34, 722–732 CrossRef CAS PubMed.
- L. A. van Grunsven, Adv. Drug Delivery Rev., 2017, 121, 133–146 CrossRef CAS.
- A. Faulkner-Jones, C. Fyfe, D.-J. Cornelissen, J. Gardner, J. King, A. Courtney and W. Shu, Biofabrication, 2015, 7, 44102 CrossRef PubMed.
- C. Zhong, H.-Y. Xie, L. Zhou, X. Xu and S.-S. Zheng, Hepatobiliary Pancreatic Dis. Int., 2016, 15, 512–518 CrossRef.
- T. Hiller, J. Berg, L. Elomaa, V. Röhrs, I. Ullah, K. Schaar, A.-C. Dietrich, M. A. Al-Zeer, A. Kurtz, A. C. Hocke, S. Hippenstiel, H. Fechner, M. Weinhart and J. Kurreck, Int. J. Mol. Sci., 2018, 19, 3129 CrossRef.
- N. C. Negrini, N. Celikkin, P. Tarsini, S. Farè and W. Święszkowski, Biofabrication, 2020, 12, 025001 CrossRef CAS.
- Q. Mao, Y. Wang, Y. Li, S. Juengpanich, W. Li, M. Chen, J. Yin, J. Fu and X. Cai, Mater. Sci. Eng., C, 2020, 109, 110625 CrossRef CAS PubMed.
- M. Gori, S. M. Giannitelli, M. Torre, P. Mozetic, F. Abbruzzese, M. Trombetta, E. Traversa, L. Moroni and A. Rainer, Adv. Healthcare Mater., 2020, 2001163 CrossRef CAS.
- V. M. Lauschke, R. Z. Shafagh, D. F. G. Hendriks and M. Ingelman-Sundberg, Biotechnol. J., 2019, 14, 1800347 CrossRef.
- D. G. Nguyen, J. Funk, J. B. Robbins, C. Crogan-Grundy, S. C. Presnell, T. Singer and A. B. Roth, PLoS One, 2016, 11, e0158674 CrossRef.
- E. Goulart, L. C. de Caires-Junior, K. A. Telles-Silva, B. H. S. Araujo, S. A. Rocco, M. Sforca, I. L. de Sousa, G. S. Kobayashi, C. M. Musso, A. F. Assoni, D. Oliveira, E. Caldini, S. Raia, P. I. Lelkes and M. Zatz, Biofabrication, 2019, 12, 15010 CrossRef PubMed.
- H. Kizawa, E. Nagao, M. Shimamura, G. Zhang and H. Torii, Biochem. Biophys. Rep., 2017, 10, 186–191 Search PubMed.
- L. M. Norona, D. G. Nguyen, D. A. Gerber, S. C. Presnell, M. Mosedale and P. B. Watkins, PLoS One, 2019, 14, e0208958 CrossRef CAS PubMed.
- J. Nie, Q. Gao, J. Fu and Y. He, Adv. Healthcare Mater., 2020, 9, 1901773 CrossRef CAS PubMed.
- M. A. Heinrich, W. Liu, A. Jimenez, J. Yang, A. Akpek, X. Liu, Q. Pi, X. Mu, N. Hu, R. M. Schiffelers, J. Prakash, J. Xie and Y. S. Zhang, Small, 2019, 15, 1805510 CrossRef.
- Y. Wu, Z. Y. Lin, A. C. Wenger, K. C. Tam and X. Tang, Bioprinting, 2018, 9, 1–6 CrossRef.
- Y. Wu, A. Wenger, H. Golzar and X. Tang, Sci. Rep., 2020, 10, 20648 CrossRef CAS.
- G. Janani, S. Priya, S. Dey and B. B. Mandal, ACS Appl. Mater. Interfaces, 2022, 14, 10167–10186 CrossRef CAS.
- M. Cuvellier, F. Ezan, H. Oliveira, S. Rose, J.-C. Fricain, S. Langouët, V. Legagneux and G. Baffet, Biomaterials, 2021, 269, 120611 CrossRef CAS.
- X. Ma, C. Yu, P. Wang, W. Xu, X. Wan, C. S. E. Lai, J. Liu, A. Koroleva-Maharajh and S. Chen, Biomaterials, 2018, 185, 310–321 CrossRef CAS PubMed.
- X. Ma, X. Qu, W. Zhu, Y.-S. Li, S. Yuan, H. Zhang, J. Liu, P. Wang, C. S. E. Lai, F. Zanella, G.-S. Feng, F. Sheikh, S. Chien and S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2206–2211 CrossRef CAS PubMed.
- F. Xie, L. Sun, Y. Pang, G. Xu, B. Jin, H. Xu, X. Lu, Y. Xu, S. Du, Y. Wang, S. Feng, X. Sang, S. Zhong, X. Wang, W. Sun, H. Zhao, H. Zhang, H. Yang, P. Huang and Y. Mao, Biomaterials, 2021, 265, 120416 CrossRef CAS PubMed.
- M. Rahmati, D. K. Mills, A. M. Urbanska, M. R. Saeb, J. R. Venugopal, S. Ramakrishna and M. Mozafari, Prog. Mater. Sci., 2021, 117, 100721 CrossRef CAS.
- C. Rinoldi, E. Kijeńska, A. Chlanda, E. Choinska, N. Khenoussi, A. Tamayol, A. Khademhosseini and W. Swieszkowski, J. Mater. Chem. B, 2018, 6, 3116–3127 RSC.
- I. Fasolino, V. Guarino, M. Marrese, V. Cirillo, M. Vallifuoco, M. L. Tamma, V. Vassallo, A. Bracco, F. Calise and L. Ambrosio, Biomed. Mater., 2017, 13, 15017 CrossRef CAS.
- R. Grant, J. Hallett, S. Forbes, D. Hay and A. Callanan, Sci. Rep., 2019, 9, 6293 CrossRef PubMed.
- M. Mittal, M. R. Siddiqui, K. Tran, S. P. Reddy and A. B. Malik, Antioxid. Redox Signal., 2014, 20, 1126–1167 CrossRef CAS.
- R. Grant, D. C. Hay and A. Callanan, Tissue Eng., Part A, 2017, 23, 650–662 CrossRef CAS.
- Y. Yokoyama, S. Hattori, C. Yoshikawa, Y. Yasuda, H. Koyama, T. Takato and H. Kobayashi, Mater. Lett., 2009, 63, 754–756 CrossRef CAS.
- S. Khorshidi, A. Solouk, H. Mirzadeh, S. Mazinani, J. M. Lagaron, S. Sharifi and S. Ramakrishna, J. Tissue Eng. Regener Med., 2016, 10, 715–738 CrossRef CAS PubMed.
- J. H. Brown, P. Das, M. D. DiVito, D. Ivancic, L. P. Tan and J. A. Wertheim, Acta Biomater., 2018, 73, 217–227 CrossRef CAS PubMed.
- D. Rajendran, A. Hussain, D. Yip, A. Parekh, A. Shrirao and C. H. Cho, J. Biomed. Mater. Res., Part A, 2017, 105, 2119–2128 CrossRef CAS PubMed.
- M. M. Sk, P. Das, A. Panwar and L. P. Tan, Mater. Sci. Eng., C, 2021, 123, 111694 CrossRef CAS PubMed.
- F. Ghahremanzadeh, F. Alihosseini and D. Semnani, Int. J. Biol. Macromol., 2021, 174, 278–288 CrossRef CAS PubMed.
- Y.-H. Chen, Y.-H. Ku, K.-C. Wang, H.-C. Chiang, Y.-P. Hsu, M.-T. Cheng, C.-S. Wang and Y. Wee, Gels, 2022, 8, 149 CrossRef CAS PubMed.
- K. Zeilinger, N. Freyer, G. Damm, D. Seehofer and F. Knöspel, Exp. Biol. Med., 2016, 241, 1684–1698 CrossRef CAS PubMed.
- J. V. Castell, R. Jover, C. P. Martnez-Jimnez and M. J. Gmez-Lechn, Expert Opin. Drug Metab. Toxicol., 2006, 2, 183–212 CrossRef CAS PubMed.
- H. H. J. Gerets, K. Tilmant, B. Gerin, H. Chanteux, B. O. Depelchin, S. Dhalluin and F. A. Atienzar, Cell Biol. Toxicol., 2012, 28, 69–87 CrossRef CAS PubMed.
- X. Yang, Y. Meng, Z. Han, F. Ye, L. Wei and C. Zong, Cell Biosci., 2020, 10, 1–18 CrossRef.
- J. Deng, W. Wei, Z. Chen, B. Lin, W. Zhao, Y. Luo and X. Zhang, Micromachines, 2019, 10, 676 CrossRef.
- C. Luo, D. Lü, L. Zheng, F. Zhang, X. Zhang, S. Lü, C. Zhang, X. Jia, X. Shu and P. Li, Biomater. Sci., 2021, 9, 3776–3790 RSC.
- F. Jacob, R. D. Salinas, D. Y. Zhang, P. T. T. Nguyen, J. G. Schnoll, S. Z. H. Wong, R. Thokala, S. Sheikh, D. Saxena and S. Prokop, Cell, 2020, 180, 188–204 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS.
- A. Treyer and A. Müsch, Compr. Physiol., 2013, 3, 243 Search PubMed.
- C. Jensen and Y. Teng, Front. Mol. Biosci., 2020, 7, 33 CrossRef CAS PubMed.
- A. S. Serras, J. S. Rodrigues, M. Cipriano, A. V. Rodrigues, N. G. Oliveira and J. P. Miranda, Front. Cell Dev. Biol., 2021, 9, 626805 CrossRef.
- W. R. Proctor, A. J. Foster, J. Vogt, C. Summers, B. Middleton, M. A. Pilling, D. Shienson, M. Kijanska, S. Ströbel, J. M. Kelm, P. Morgan, S. Messner and D. Williams, Arch. Toxicol., 2017, 91, 2849–2863 CrossRef CAS PubMed.
- W. Albrecht, F. Kappenberg, T. Brecklinghaus, R. Stoeber, R. Marchan, M. Zhang, K. Ebbert, H. Kirschner, M. Grinberg, M. Leist, W. Moritz, C. Cadenas, A. Ghallab, J. Reinders, N. Vartak, C. van Thriel, K. Golka, L. Tolosa, J. V. Castell, G. Damm, D. Seehofer, A. Lampen, A. Braeuning, T. Buhrke, A.-C. Behr, A. Oberemm, X. Gu, N. Kittana, B. van de Water, R. Kreiling, S. Fayyaz, L. van Aerts, B. Smedsrød, H. Ellinger-Ziegelbauer, T. Steger-Hartmann, U. Gundert-Remy, A. Zeigerer, A. Ullrich, D. Runge, S. M. L. Lee, T. S. Schiergens, L. Kuepfer, A. Aguayo-Orozco, A. Sachinidis, K. Edlund, I. Gardner, J. Rahnenführer and J. G. Hengstler, Arch. Toxicol., 2019, 93, 1609–1637 CrossRef CAS.
- C. MoraesEqual contributions, J. M. Labuz, B. M. Leung, M. Inoue, T.-H. Chun and S. Takayama, Integr. Biol., 2013, 5, 1149–1161 CrossRef.
- P. Soltantabar, E. L. Calubaquib, E. Mostafavi, A. Ghazavi and M. C. Stefan, Organs-on-a-Chip, 2021, 3, 100008 CrossRef CAS.
- D. A. Tatosian and M. L. Shuler, Biotechnol. Bioeng., 2009, 103, 187–198 CrossRef CAS.
- K. Rennert, S. Steinborn, M. Gröger, B. Ungerböck, A.-M. Jank, J. Ehgartner, S. Nietzsche, J. Dinger, M. Kiehntopf and H. Funke, Biomaterials, 2015, 71, 119–131 CrossRef CAS PubMed.
- M. Darnell, M. Ulvestad, E. Ellis, L. Weidolf and T. B. Andersson, J. Pharmacol. Exp. Ther., 2012, 343, 134–144 CrossRef CAS PubMed.
- M. Yamada, R. Utoh, K. Ohashi, K. Tatsumi, M. Yamato, T. Okano and M. Seki, Biomaterials, 2012, 33, 8304–8315 CrossRef CAS PubMed.
- M. Puri, Assay Drug Dev. Technol., 2020, 18, 1–10 CrossRef CAS.
- T. Li, W. Tong, R. Roberts, Z. Liu and S. Thakkar, Chem. Res. Toxicol., 2020, 34, 550–565 Search PubMed.
- M. Chierici, M. Francescatto, N. Bussola, G. Jurman and C. Furlanello, Biol. Direct, 2020, 15, 1–10 Search PubMed.
- C. Feng, H. Chen, X. Yuan, M. Sun, K. Chu, H. Liu and M. Rui, J. Chem. Inf. Model., 2019, 59, 3240–3250 CrossRef CAS.
- B. P. Smith, L. S. Auvil, M. Welge, C. B. Bushell, R. Bhargava, N. Elango, K. Johnson and Z. Madak-Erdogan, Sci. Rep., 2020, 10, 1–27 CrossRef PubMed.
- V. M. Lauschke, Drug Metab. Rev., 2021, 1–8 Search PubMed.
- P. Kohonen, J. A. Parkkinen, E. L. Willighagen, R. Ceder, K. Wennerberg, S. Kaski and R. C. Grafström, Nat. Commun., 2017, 8, 1–15 CrossRef PubMed.
- Y. Igarashi, N. Nakatsu, T. Yamashita, A. Ono, Y. Ohno, T. Urushidani and H. Yamada, Nucleic Acids Res., 2015, 43, D921–D927 CrossRef CAS PubMed.
- L. Li, H. Knutsdottir, K. Hui, M. J. Weiss, J. He, B. Philosophe, A. M. Cameron, C. L. Wolfgang, T. M. Pawlik, G. Ghiaur, A. J. Ewald, E. Mezey, J. S. Bader and F. M. Selaru, JCI Insight, 2019, 4, e121490 CrossRef.
- E. Ho, S. Van Hees, S. Goethals, E. Smits, M. Huizing, S. Francque, B. De Winter, P. Michielsen and T. Vanwolleghem, Front. Med., 2019, 6, 183 CrossRef.
- J. F. Dekkers, C. K. van der Ent and J. M. Beekman, Rare Dis., 2013, 1, 939–945 Search PubMed.
- S. A. Dugger, A. Platt and D. B. Goldstein, Nat. Rev. Drug Discovery, 2018, 17, 183–196 CrossRef CAS.
- M. Matano, S. Date, M. Shimokawa, A. Takano, M. Fujii, Y. Ohta, T. Watanabe, T. Kanai and T. Sato, Nat. Med., 2015, 21, 256–262 CrossRef CAS.
- M. D. Sarker, S. Naghieh, N. K. Sharma and X. Chen, J. Pharm. Anal., 2018, 8, 277–296 CrossRef CAS.
- N. Annabi, J. W. Nichol, X. Zhong, C. Ji, S. Koshy, A. Khademhosseini and F. Dehghani, Tissue Eng., Part B, 2010, 16, 371–383 CrossRef CAS.
- M. Nomi, A. Atala, P. De Coppi and S. Soker, Mol. Aspects Med., 2002, 23, 463–483 CrossRef CAS.
- T. Takebe, R. Imai and S. Ono, Clin. Transl. Sci., 2018, 11, 597–606 CrossRef PubMed.
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah, M. Spitzer and S. Zhao, Nat. Rev. Drug Discovery, 2019, 18, 463–477 CrossRef CAS.
- P. Pound and M. Ritskes-Hoitinga, J. Transl. Med., 2018, 16, 1–8 CrossRef.
- S. J. Hollister, Nat. Mater., 2005, 4, 518–524 CrossRef CAS PubMed.
Footnote |
| † These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2023 |