
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffectiveness of a novel gene nanotherapy based on putrescine for cancer treatment†
Saínza
Lores
 ab,
Manuel
Gámez-Chiachio
cd,
María
Cascallar
abd,
Carmen
Ramos-Nebot
cd,
Pablo
Hurtado
de,
Sandra
Alijas
ae,
Rafael
López López
abde,
Roberto
Piñeiro
de,
Gema
Moreno-Bueno
*cdf and
María
de la Fuente
*abdg
ab,
Manuel
Gámez-Chiachio
cd,
María
Cascallar
abd,
Carmen
Ramos-Nebot
cd,
Pablo
Hurtado
de,
Sandra
Alijas
ae,
Rafael
López López
abde,
Roberto
Piñeiro
de,
Gema
Moreno-Bueno
*cdf and
María
de la Fuente
*abdg
aNano-Oncology and Translational Therapeutics Unit, Health Research Institute of Santiago de Compostela (IDIS), Travesía da Choupana s/n, Santiago de Compostela, 15706, A Coruña, Spain. E-mail: sainza.lores@gmail.com; maria.cascallarc@gmail.com; maria.fuente.freire@sergas.es; Tel: +34-981-955-706
bUniversidade de Santiago de Compostela (USC), Praza do Obradoiro, s/n, Santiago de Compostela, 15782, A Coruña, Spain
cTranslational Cancer Research Laboratory, Department of Biochemistry, Autonomous University of Madrid, School of Medicine, “Alberto Sols” Biomedical Research Institute CSIC-UAM, IdiPaz, Arturo Duperier 4, 28029, Madrid, Spain. E-mail: mgamez@iib.uam.es; cramos@iib.uam.es; gmoreno@iib.uam.es
dBiomedical Cancer Research Network (CIBERONC), 28029 Madrid, Spain
eRoche-CHUS Join Unit. Translational Medical Oncology Group (ONCOMET), Health Research Institute of Santiago de Compostela (IDIS), Travesía da Choupana s/n, Santiago de Compostela, 15706, A Coruña, Spain. E-mail: felurh@gmail.com; sandraalijasperez@gmail.com; rafa.lopez.lopez@gmail.com; Roberto.Pineiro.Cid@sergas.es
fMD Anderson International Foundation, Gómez Hemans s/n, 28033 Madrid, Spain
gDIVERSA Technologies SL, Edificio Emprendia, Universidade de Santiago de Compostela, Campus Vida s/n, 15782 Santiago de Compostela, Spain
First published on 15th February 2023
Abstract
Gene therapy has long been proposed for cancer treatment. However, the use of therapeutic nucleic acids presents several limitations such as enzymatic degradation, rapid clearance, and poor cellular uptake and efficiency. In this work we propose the use of putrescine, a precursor for higher polyamine biosynthesis for the preparation of cationic nanosystems for cancer gene therapy. We have formulated and characterized putrescine-sphingomyelin nanosystems (PSN) and studied their endocytic pathway and intracellular trafficking in cancer cells. After loading a plasmid DNA (pDNA) encoding the apoptotic Fas Ligand (FasL), we proved their therapeutic activity by measuring the cell death rate after treatment of MDA-MB-231 cells. We have also used xenografted zebrafish embryos as a first in vivo approach to demonstrate the efficacy of the proposed PSN-pDNA formulation in a more complex model. Finally, intratumoral and intraperitoneal administration to mice-bearing MDA-MB-231 xenografts resulted in a significant decrease in tumour cell growth, highlighting the potential of the developed gene therapy nanoformulation for the treatment of triple negative breast cancer.
Introduction
The FDA defines gene therapy as a medicine that “administrates genetic material to modify or manipulate the expression of a gene product or to alter the biological properties of living cells for therapeutic use”.1 Successful gene therapy is based on two key points, transporting nucleic acids to the precise site of action, and delivering an adequate dose of genetic material. The transport of naked nucleic acids presents some obstacles such as a short half-life because of the rapid enzyme degradation, a fast clearance, as well as a low cellular uptake and nonspecific biodistribution.2–5 Viral vectors have been proposed for this purpose but present several risks such as immunogenicity, cytotoxicity and mutagenesis that limit their application and subsequent market approval.6 In contrast, non-viral vectors, although less efficient, produce low immune responses, do not have restrictions on the size of the carried genetic material, and are easy to manufacture with good yields. Besides, it is possible to associate a large amount of genetic material because of their high surface–volume ratio.5,7–10 Importantly, in 2018 the FDA approved for the first time the use of lipid nanoparticles with a short interfering RNA (siRNA) for the treatment of polyneuropathies.11 Recently, in 2020, two COVID-19 mRNA-based vaccines making use of nanotechnology to transport nucleic acids were successfully approved,12 opening a new era for gene nanomedicines.Cationic nanosystems are potential candidates for the development of an effective gene therapy due to their ability to associate nucleic acids by simply establishing electrostatic interactions. DOTAP and DOTMA have been widely used to develop cationic lipid nanosystems, most typically liposomes;5,7,13,14 however, it has been proved that the in vivo transfection with these lipids is poor.15 In addition to cationic lipids, cationic polymers have been extensively used as non-viral vectors, such as polyethyleneimine (PEI), considered one of the gold standards for gene transfection, poly(2-dimethylaminoethyl methacrylate) (PDMAEMA) or poly(amidoamine) (PAMAM).16–18 We describe here the development of biodegradable and biocompatible nanocarriers based on putrescine, an organic cation which is a precursor for higher polyamine biosynthesis, relevant for some biological processes such as cell proliferation, differentiation or chromatin remodelling.19,20 This natural polyamine presents several advantages in the development of non-viral vectors for gene therapy. Its cationic character provides the possibility to associate negative groups, in this case, nucleic acids. Indeed, putrescine has a “natural” ability to associate nucleic acids and stabilize them.21–24 Moreover, due to their cationic nature, polyamines are described to act as free radical scavengers and have therefore shown the ability to protect DNA against damage from free radicals.25 One of the key aspects to develop an efficient gene carrier is to achieve a good internalization by the target cells, which allows the subsequent delivery of its cargo. In this point, putrescine can provide a high uptake by the cancer cells and also a selective interaction, due to the described polyamine transport system (PTS);26,27 because of the cellular functions in which polyamines are involved, all eukaryotic cells need to maintain their intracellular levels. This fact is enhanced in cancer cells, which have to keep higher levels of polyamines to meet their metabolomic needs, as lately demonstrated.26 Nanocarriers based on polyamines such as spermidine and spermine have already been proposed for gene delivery,28–30 while putrescine has been used for providing functional groups for surface conjugations.31 Moreover, putrescine nano-crystalsomes have been recently developed for the treatment of metastatic breast cancer, improving the intracellular accumulation of doxorubicin.32 To the best of our knowledge, its use for gene delivery purposes has not been explored yet.
In this work, we propose the incorporation of a putrescine derivative, oleamide-modified putrescine, in sphingomyelin nanosystems (SNs), previously developed by our group,14,33–39 and composed by vitamin E, a GRAS-listed excipient,40 and sphingomyelin, one of the main components of cell membranes.41 We aim to prove the therapeutic potential of putrescine-SNs (PSNs) that associate a plasmid encoding the apoptotic Fas Ligand (FasL)42,43 for treating highly aggressive triple-negative breast cancer (TNBC). It accounts for 15–20% of all breast cancers, and has the worst mortality rate.44,45 Due to the loss of estrogen and progesterone receptors and the human epidermal growth factor 2 (HER2), and thus, lack of therapeutic targets, the treatment options are limited to surgery, radiotherapy and, mainly, anthracyclines and taxanes-based chemotherapy45,46 and therefore the development of new therapeutic strategies answer to a clinical demand. In addition, because of the high implication of genetic mutations in this type of cancer, gene therapy represents a promising therapeutic approach.47–49
Materials and methods
Materials
Vitamin E (DL-α-tocopherol), Mowiol®, culture media, phosphate-buffered saline (PBS), polyvinylpyrrolidone (PVP), ampicillin, puromycin, trypsin, paraformaldehyde (PFA) and poly-L-lysine and nuclease-free water were acquired from (Merck-Millipore, Madrid, Spain). Sphingomyelin (Lipoid E SM) was kindly provided by Lipoid GmbH (Ludwigshafen, Germany). The lipidic putrescine, (9Z)-N-(4-aminobutyl)-9-octadecenamide, was synthesized by Galchimia SA (Galicia, Spain). C11 TopFluor Sphingomyelin (N-[11-(dipyrrometheneboron difluoride)undecanoyl]-D-erythro-sphingosyl-phosphorylcholine was provided from Avanti Polar Lipids (Alabaster, AL, USA). Ethanol of analytical grade was acquired from VWR (Barcelona, Spain). Hoechst was purchased from Thermo Fisher Scientific (Massachusetts, USA). The plasmid pcDNA4TO-mito-mCherry-10xGCN4_v450 was acquired in AddGene (plasmid #60914; https://n2t.net/addgene:60914; RRID:Addgene_60914) (Massachusetts, USA). ORF expression clone for FasL (NM_000639.2) was acquired in Tebu-Bio. pReceiver-M61 control vector (EX-NEG-M61) was purchased in GeneCopoeia (Rockville, USA). MDA-MB-231 and HCC38 breast cancer cell lines were purchased from the American Type Culture Collection (ATCC).Putrescine derivative
Galchimia SA has synthesized a putrescine derivative to facilitate its incorporation into the nanosystems. Putrescine was covalently bound to oleic acid through an amide linker, between a primary amine from the polyamine and the carboxylic acid from the fatty acid. Then, it was characterized by 1H-NMR (proton resonance 300 MHz) in CDCl3 and HPLC-MS/UPLC-MS with a SunFire Column C18 (3.5 μm, 2.1 × 100 mm) (Waters, USA). Sample was dissolved in methanol and mobile phases used were: solvent A (acetonitrile 50%, methanol 50%), solvent B (water), and solvent C (100 mM ammonium acetate pH 7). Flow rate was set at 0.30 mL min−1. Separation was achieved using the following mobile phase gradient: t = 0 min, 10% A, 85% B, and 5% C; t = 5 min, 10% A, 85% B, and 5% C; t = 15 min, 95% A, 0% B, and 5% C; t = 25 min, 95% A, 0% B, and 5% C. Solid NMR spectra were measured at 298 K in a Bruker NEO-750 spectrometer (750 MHz proton frequency) equipped with a Varian T3 solid triple resonance probe 1H/X/Y that uses zirconia rotor of an outer diameter of 3.2 mm with an effective sample capacity of 22 μL which corresponds to approximately 30 mg of the powder sample. The probe was adapted to the Bruker spectrometer by a A2B conversion kit (Revolution NMR, LLC). The spectrometer control software was TopSpin 4.x. All the spectra were processed with MestreNova software v14.0 (Mestrelab Research Inc.). Carbon-13 chemical shifts were referenced externally to the methylene signal of crystalline α-form of glycine at 43.5 ppm. The spectra were measured under MAS of 20 kHz.Plasmid DNA extraction
To study the ability of the developed cationic nanosystems to incorporate nucleic acids in their surface, the plasmid mCherry (pcDNA4TO-mito-mCherry-10xGCN4_v4) was selected as a model for the whole characterization. Additionally, pReceiver-M90-FASLG or pReceiver-M61-control constructions were extracted to evaluate the therapeutic effect. First, bacteria were growth in 25 mL of previously autoclaved sLB agar medium (55 mg mL−1), with ampicillin (100 mg mL−1) at 37 °C overnight (O/N) with shaking. Then, when colonies were grown, one single colony was picked and seeded in 100 mL of sLB broth (buffered) (54.48 mg mL−1), again with 100 mg mL−1 of ampicillin, at 37 °C for one day in the orbital shaker. After that, plasmid extraction was performed with the Plasmid DNA MaxiPrep Kit (Norgen Biotek Corporation, Canada), following the steps of the protocol. Next, concentration of the obtained pDNAs was measured with the NanoDrop™ 2000 (Thermo Fisher Scientific, Massachusetts, USA). Part of the obtained mCherry plasmid was labelled with Cy5 with the Label IT® Tracker™ Intracellular Nucleic Acid Localization Kit (Madison, USA), following the protocol.Preparation of putrescine-sphingomyelin nanosystems (PSN)
Oil in water (O/W) nanosystems were prepared by ethanol injection. In this method, 100 μL of an ethanolic phase containing 5 mg of vitamin E (Vit E) and different amounts of sphingomyelin (SM) and the lipidic-putrescine (C18-Pt) (as disclosed in Table 1) were injected under magnetic stirring at 700 rpm in 1 mL of Molecular Grade Water. The suspension was kept under stirring at room temperature for 5 min.Association of the pDNA (PSN-pDNA)
Once the plasmids were extracted, 5 μg of pDNA were dissolved in 100 μL of H2O nuclease-free and added over 100 μL of preformed nanosystems, for 20 min under magnetic stirring at 500 rpm to achieve the association. After this time, obtained nanocarriers (PSN-pDNA) were physiochemically characterized by DLS, NTA, and STEM, as previously detailed. PSN and PSN-pDNA were also evaluated in terms of colloidal stability, by determining their size, PdI and ZP along the time under storage conditions (12 weeks at 4 °C), and upon incubation with supplemented cellular medium (DMEM) at 37 °C for 4 h.Physicochemical characterization
The nanosystems were characterized using a Zetasizer® (NanoZS Malvern Instruments, England) to determine the particle size and polydispersity index (PdI) by dynamic light scattering (DLS). All samples were analysed after dilution by 10-fold with Milli-Q water in disposable microcuvettes (ZEN0040), at room temperature, and with a detection angle of 173°. Moreover, the Z potential (ZP), indicative of the surface charge of the nanocarriers was determined using the same instrument, by Laser Doppler Anemometry (LDA) after dilution by 40-fold with Milli-Q water, in folded capillary cells cuvettes (DTS 1070, Malvern Instruments). Each sample is given by 3 measurements and the average of independent samples is provided (n = 3).Samples were also analysed by Nanoparticle Tracking Analysis (NTA) to determine the concentration of particles per millilitre and the size distribution. The measurement was carried out with a NanoSight NS3000 System (Malvern Instruments, Worcestershire, UK) equipped with a 488 nm laser and a sCMOS camera. The samples were diluted 1000-fold in ultrapure water and measured with a camera level of 14.
Scanning transmission electron microscope (STEM)
Morphological examination was performed by scanning transmission electron microscopy (STEM) using a Field Emission Scanning Electron Microscopy (FESEM) Ultra Plus (Zeiss, Germany) operating at 20 kV. Before that, 20 μL of the nanocarrier were mixed with 20 μL of phosphotungstic acid 2% (w/v) to stain. The mixture was placed in a copper grid for 6 hours, washed with 500 μL of water and dried overnight under vacuum.Efficiency of association of pDNA
The association efficiency was determined by agarose gel electrophoresis. A fixed amount of pDNA (0.2 μg) was loaded onto 1% agarose gel with Sybr Gold, loading buffer and tris-borate-EDTA (TBE) as buffer. Gel was run at 100 mV for 40 min and was analysed with the ChemiDoc™ MP Imaging System (Bio-Rad, California, USA). The efficiency of association was inversely proportional to the amount of pDNA that was displaced attached to the Sybr Gold.Twelve weeks after the association, the electrophoresis was repeated to monitor the bond between PSN and pDNA. The stability of the association was also tested in cellular medium (DMEM) supplement or not with 1% fetal bovine serum (FBS, Gibco) after the incubation for 4 h at 37 °C.
DNase protection assay
Degradation assay of PSN-pDNA against nucleases was carried out with PerfeCTa® DNase I kit (Quantabio, Massachusetts, USA). Briefly, equivalents of both naked pDNA and PSN-pDNA containing 2 μg of plasmid were incubated with 10 μL reaction buffer and 2 U of DNases at 37 °C for 5 and 30 min, in a reaction volume of 100 μL. After these time points, the reactions were stopped with 10 μL of stop buffer. After that, to extract the remaining pDNA, a mixture of phenol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :chloroform:isoamyl alcohol (25:24:1, v/v/v) (Thermo Fisher Scientific, Massachusetts, USA) was added to the samples at equal volume, mixed by inversion and centrifuged at 12000 RCF for 3 min. The supernatant (aqueous phase) was moved to a new tube and mixed by inversion with an equal volume of chloroform. Samples were centrifuged again at 12000 RCF for 3 min and the supernatants were placed in new tubes, mixed with 2 volumes of isopropanol, and centrifuged at 12000 RCF for 15 min. Supernatants were carefully removed and 500 μL of 70% ethanol were added. Samples were centrifuged at 12000 RCF for 15 min and the supernatant was removed again. Finally, pellets were dissolved in 100 μL of nuclease-free water and analysed by 1% agarose gel electrophoresis was carried out.
:chloroform:isoamyl alcohol (25:24:1, v/v/v) (Thermo Fisher Scientific, Massachusetts, USA) was added to the samples at equal volume, mixed by inversion and centrifuged at 12000 RCF for 3 min. The supernatant (aqueous phase) was moved to a new tube and mixed by inversion with an equal volume of chloroform. Samples were centrifuged again at 12000 RCF for 3 min and the supernatants were placed in new tubes, mixed with 2 volumes of isopropanol, and centrifuged at 12000 RCF for 15 min. Supernatants were carefully removed and 500 μL of 70% ethanol were added. Samples were centrifuged at 12000 RCF for 15 min and the supernatant was removed again. Finally, pellets were dissolved in 100 μL of nuclease-free water and analysed by 1% agarose gel electrophoresis was carried out.
Cell culture
MDA-MB-231 and HCC38 TNBC cell lines were obtained from the American Type Cell Culture (ATCC). Cells were cultured following the supplier conditions. Briefly, cells were cultured in DMEM medium or RPMI-1640 medium (Gibco), respectively, supplemented with 10% fetal bovine serum (Gibco), 10 mM glutamine (Life Technologies) and 1% penicillin/streptomycin (Invitrogen). Cells were authenticated by STR-profiling according to ATCC.Transfection assay
Transfection efficiency was studied in MDA-MB-231 cells. Eighty thousand cells per well were seeded on a 24-well plate. After 24 h, cells were washed and 200 μl of medium without supplement were added. Then, 1 μg of PSN-pDNA-mCherry were incubated for 4 h, when cells were washed with PBS and 500 μl of DMEM were added. As a negative control, 40 μL of water were incubated with the cells in the same conditions. While, as a positive control, Lipofectamine™ 2000 Transfection Reagent was used according to manufacturer indications (Thermo Fisher Scientific, Massachusetts, USA). Expression of the reported protein, mCherry, was observed under the fluorescent microscope (Leica DMi8), 24 hours post-transfection.Immunofluorescence staining
Cells were grown to 50–70% confluency on coverslips in 24-well plates and subjected then to different experimental immunofluorescence analyses. To assess the intracellular uptake and trafficking of nanosystems, cells were treated with 0.44 mg mL−1 PSN-pDNA (TopFluor-labelled PSN/Cy5-labelled pDNA) for different time points. Specifically, to test the intracellular trafficking, cells were treated with PSN-pDNA for 4 h then medium was replaced, and cells were cultured on complete medium for 2 and 4 h. After, cells were fixed with 4% paraformaldehyde for 20 min at RT followed by 5 min permeabilization with 0.1% Triton™ X-100. Then, coverslips were incubated with corresponding primary antibodies, caveolin-1 (3267, Cell Signalling Technology, Massachusetts, USA), clathrin heavy chain (ab21679, Abcam, Cambridge, UK), EEA1 (3288, Cell Signaling Technology), LAMP1 (9091, Cell Signaling Technology), calreticulin (PA3-900, Invitrogen) or GM130 (610822, BD Biosciences). After, incubation with secondary antibody was carried out using Alexa Fluor 546-labelled (A-11035, Invitrogen) for 1 h at RT. Cell nuclei were stained with DAPI (D1306, Invitrogen). Images were captured on a LSM710 confocal microscope (ZEISS, Oberkochen, Germany). The analysis and quantification were done using ImageJ software.Cellular viability assays
The cytotoxic effect of PSN-pDNA encoding for the Fas Ligand protein (PSN-pDNA-FasL), compared to the empty vector (PSN-pDNA-control), were checked on MDA-MB-231 and HCC38 using the AlamarBlue™ reagent (Bio-Rad, Madrid, Spain) according to the manufacturer's instructions. Briefly, 10000 cells per well were seeded on 96-well plates in triplicates. After 24 h, cells were treated with different concentrations of both formulations (from 0.25 to 1 mg mL−1 in medium without supplement) for 4 h. Then, cells were washed once with PBS and cultured with complete medium for 72 h. Spectrophotometric analysis was performed at wavelengths of 570 and 600 nm with a Synergy HT reader (BioTek Instruments).
Cell apoptosis analysis
Cell death rate was quantified by flow cytometry using the Annexin V-FITC stained apoptosis detection kit (Immunostep, Salamanca Spain) and propidium iodide (PI, Sigma-Aldrich) according to the manufacturer's instructions. Briefly, MDA-MB-231 cells were transfected with different concentrations of PSN-pDNA-FasL or PSN-pDNA-control constructions. Cells were treated for 4 h and incubated for 72 h with fresh medium allowing the expression of the plasmid. Annexin V-FITC positive cells alone (A+/PI−) and Annexin V-FITC and PI doubled stained (A+/PI+) were defined as apoptotic cells. The apoptosis analysis was conducted using an FC500 flow cytometer (Beckman Coulter, California, USA).In vivo studies in zebrafish embryos
In vivo assays were performed in zebrafish (Danio rerio) embryos by microinjection of PSN-pDNA in xenografted embryos with cancer cells. For the manipulation of the embryos, these were kept in E3 medium with 1× PTU and 5% tricaine metasulfonate, an anaesthetic compound used for zebrafish. The microinjection was carried out with a binocular loupe (SMZ745, Nikon, Japan), the IM 300 Microinjector (Narishige, Japan) and needles made with the PC-10 Puller (Narishige) from glass capillaries (Harvard Apparatus, Massachusetts, USA). GFP-labelled MDA-MB-231 cells were resuspended in PVP 2% and 200–300 cells were injected into the yolk sac. After 24 hours, PSN-pDNA-control and PSN-pDNA-FasL, previously concentrated 10 times by the SpeedVac Concentrator (Savant SPD111V-120, Cambridge Scientific, Massachusetts, USA), were microinjected into the yolk sac. Zebrafish embryos were photographed by fluorescence microscopy (Leica DMi8) each 24 h for 72 h. After that, pictures were analysed by ImageJ and quantified by QuantiFish (version 2.1.1).51Zebrafish embryos used in the study (120 hpf) are not considered animals, based on the current EU legislation (Directive 2010/63/EU of the European Parliament and the Council of 22 September 2010) as at this stage of development they are not independently feeding larval forms.
In vivo studies in mice
All the experimental procedures with mice were approved by the internal ethical research and animal welfare committee (IIB, UAM), and by the Local Authorities (Comunidad de Madrid, PROEX 235.6/20). They complied with the European Union (Directive 2010/63/UE) and Spanish Government guidelines (Real Decreto 53/20133). Orthotropic breast tumour xenografts were performed in female nu/nu mice (Charles River) following standard procedures.52 MDA-MB-231 (1 × 106) cells were injected into the right fifth mammary fat pad (mfp) of 7-week-old female NMRI-nu immunodeficient mice (Janvier Labs) and tumour growth was assessed as described previously.52,53 When tumour diameter reached approximately 0.5 cm, the animals were randomized into four groups of treatment: PSN-pDNA-control and PSN-pDNA-FasL which were intratumorally administrated, and PSN-pDNA-control and PSN-pDNA-FasL intraperitoneally injected. PSN-pDNA-control and PSN-pDNA-FasL (TopFluor-labelled) were concentrated 6 times by the SpeedVac Concentrator Jouan RC1010 until achieving 15.68 mg mL−1 of PSN and 150 μg mL−1 of pDNA. From the concentrated PSN, 100 L were intratumorally or intraperitoneally injected twice a week for 3 weeks, obtaining the final treatment concentration of 78.30 mg kg−1 of PSN and 0.75 mg kg−1 of pDNA. Tumour growth was monitored twice a week and measured using a calliper. The tumour volume was calculated using the formula L × W2 × (π/6), where L is the length and W, the width of the tumour xenografts. At the end of the experiment, mice were sacrificed after 24 h of the last treatment, and subsequently, PSN-pDNA biodistribution in the tumours and in the different organs was quantified by ex vivo fluorescence using an IVIS-Lumina XR In Vivo Imaging System (PerkinElmer). To reduce the tissue autofluorescence background, the fluorescence emitted by the same organs and tumours of untreated mice was subtracted.Results and discussion
Development of cationic putrescine nanosystems
The development of novel targeted therapeutic innovations continues to be one of the main challenges in current oncology. For the particular case of TNBC there is still an urgent need to develop efficient therapies.54,55 Gene therapies have a real potential for the development of targeted anticancer therapeutics.56,57 Previous results from our group highlight the potential of SNs for the development of new cancer therapeutics.34–38 For gene therapy applications, they have already been tested for delivery of microRNA (miRNA) and pDNA upon modification with the cationic lipids DOTAP and stearylamine.14,58 Here, we have selected putrescine, a natural polyamine presented in eukaryotic cells for which cancer cells are ravenous due to their increased metabolic activity,26,59 as the cationic material for the development of a new type of cationic SNs14,33,34 for cancer gene therapy applications. Despite the many advantages of putrescine, its hydrophilic character hampers its use. To successfully incorporate the putrescine into SNs, we used a lipid-putrescine derivative, (9Z)-N-(4-aminobutyl)-9-octadecenamide (Fig. S1†), which was commercially synthesized and characterized by Galchimia SA (1H-NMR, 13C-NMR, and HPLC-MS/UPLC-MS, Fig. S2†). Peaks were assigned as follows: 1H NMR (300 MHz, CDCl3) δ = 5.87 (s, 1H, NH), 5.42–5.25 (m, 2H, 2CH), 3.24 (q, J = 6.3, 2H, CH2), 2.71 (t, J = 6.5, 2H, CH2), 2.13 (t, J = 7.6, 2H, CH2CO), 1.99 (q, J = 6.4, 4H, 2CH2), 1.68–1.41 (m, 8H, NH2, 3CH2), 1.27 (d, J = 9.4, 20H, 10 CH2), 0.87 (t, J = 6.4, 3H, CH3). The presence of the amide, the primary amine, and the aliphatic chain was confirmed. Spectra was processed with MestreNova (Mestrelab Research Inc., Spain). A molecular weight of 353.3 Da was obtained, in concordance with the expected one, confirming the successful reaction. Similar strategies have been previously used for the development of spermine and spermidine-conjugates.30,60–63 Moreover, it was postulated that the incorporation of a lipidic tail could increase the binding of the pDNA to the polyamine and improve its protection.64PSN were successfully prepared by an easy and mild methodology (Fig. 1), and the influence of different variables in composition, namely the amount of sphingomyelin (SM) and lipid-putrescine derivative (C18-Pt) (Table 1). In all cases, we obtained homogeneous formulations with mean sizes below 100 nm, and positive zeta potential values (>+50 mV). The hydrodynamic size is in concordance with other formulations containing polyamine–lipid conjugates.65,66 With respect to the zeta potential, cationic values lead to a better interaction with the negatively charged cell membranes, and thus, a higher internalization.67–69 Control formulations (nanosystems with only SM or C18-Pt or Pt (non-modified putrescine)) were also prepared and characterized (Table S1†). As expected, the formulation prepared in absence of C18-Pt (#G) showed relevant differences in the zeta potential value (negative). Nanosystems composed by only VitE and C18-Pt (#H, #I and #J) had a homogeneous size with a good PdI and a positive zeta potential. This confirms that C18-Pt is disposed at the interface stabilizing the oil droplets. An additional confirmation relates to the fact that, when putrescine is added instead of the lipidic derivative (C18-Pt), a nanometric formulation could not be obtained (#K). Overall, the successful addition of putrescine into SNs has been confirmed and its ability to associate nucleic acids and deliver them into cancer cells will be evaluated in the next experiments.
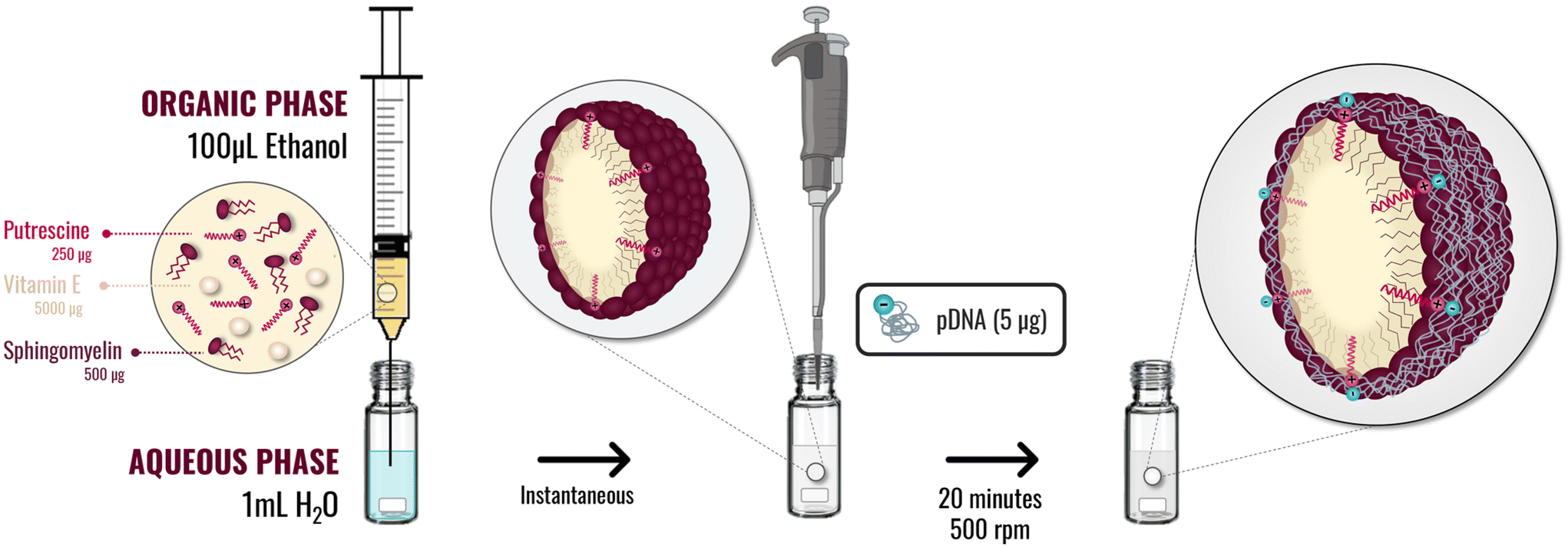 | ||
| Fig. 1 Schematic methodology implemented to putrescine nanosystems preparation by ethanol injection method, and association of the pDNA by incubation with the preformed nanosystems. | ||
Association of plasmid DNA
The use of pDNA has been widely explored for the expression of therapeutic proteins that stimulate apoptotic pathways in cancer cells.70,71 The facility to construct a plasmid with the sequence of the desired protein, and its remarkable stability under physiological conditions, make them promising candidates for the development of anticancer therapies.72,73The association of pDNA was attempted onto preformed PSN (Table 2). After the incorporation of the pDNA by gentle mixing, an increase in the mean size was observed, suggesting a successful association. Furthermore, there was a decrease in the zeta potential because of the neutralization of the positive charges of the amine groups in the putrescine by the phosphate groups of the pDNA. In the case of PSN prepared with the lowest amount of putrescine (formulations #A and #D), zeta potential values were close to neutral, and an increase in size was observed, probably due to partial aggregation, in agreement with other works in the field.74,75
The efficiency of the association was confirmed by agarose gel electrophoresis (Fig. 2). As observed, pDNA is efficiently associated when nanosystems are prepared with an amount of putrescine equal to or greater than 250 μg (formulations B, C, E and F). Oppositely, pDNA freely migrates when it is associated to formulations prepared with 50 μg of C18-Pt. This result shows a clear correlation with the physicochemical characterization, since these formulations have also shown an inversion of the zeta potential, suggesting an excess of pDNA.
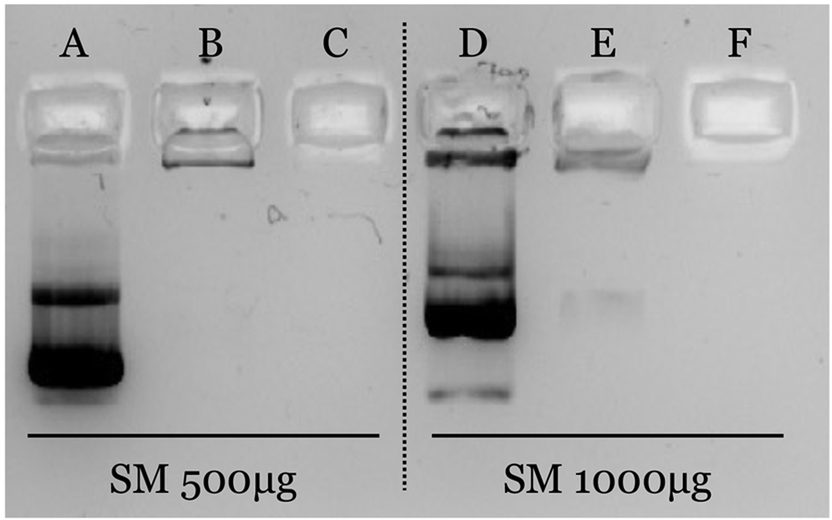 | ||
| Fig. 2 pDNA plasmid association to preformed nanosystems. An 1% agarose gel image was used to estimate the plasmid loading to PSN. Image of different ratios of VitE:SM:C18-Pt. A and D: 50 μg, B and E: 250 μg, C and F: 500 μg of C18-Pt. | ||
In view of the obtained results, formulation #B (ratio m/m VitE:SM:Pt 1:0.1:0.05) was selected for the next experiments and characterized by complementary methodologies. First, Nanoparticle Tracking Analysis (NTA) was used for a more precise analysis of the size distribution.76,77 As observed in Fig. 3a, PSN showed a clear population with an average size of 95 nm, whereas PSN-pDNA have a mean size of 150 nm. These results correlate well with results obtained by DLS (Tables 1 and 2). STEM examination (Fig. 3b and S3†) allows appreciation of a more circular and rigid structure after the association of the pDNA to PSN, a fact confirming its disposition onto the nanoparticle surface. Besides, the spherical shape has been postulated to have the fastest and greatest uptake.78 Moreover, the darker surface of PSN-pDNA reveals a higher electron dense surface in a similar fashion than other cationic lipid nanosystems.74,75
 | ||
| Fig. 3 Physicochemical characterization of PSN and PSN-pDNA (a) by NTA, and (b) by Scanning Transmission Electron Microscopy (STEM). Scale bar represents 200 nm (broad field) and 100 nm (magnification). | ||
The stability of the association of the pDNA was evaluated after storage at 4 °C, and upon incubation with cellular media. PSN-pDNA were colloidally stable, without significant changes in size, PdI and zeta potential (Fig. S4†). Moreover, as observed in Fig. 4a, results show a good association efficiency after 3 months of storage (there was no migration of released pDNA, as observed in the agarose gel). The stability in cell culture medium was similarly confirmed (Fig. 4b). Importantly, pDNA was not delivered after 4 h of incubation with the medium at 37 °C, conditions at which transfection experiments in vitro will be carried out. Similar results were obtained were incubated with supplemented medium (Fig. S5†). Although a continuous increase in size is observed, the PdI, indicative of aggregates, is maintained along the time, suggesting that the increase in size could most probably be related to the adsorption of serum proteins onto the nanoparticle surface. An inversion on the zeta potential was also reported, reinforcing this hypothesis.
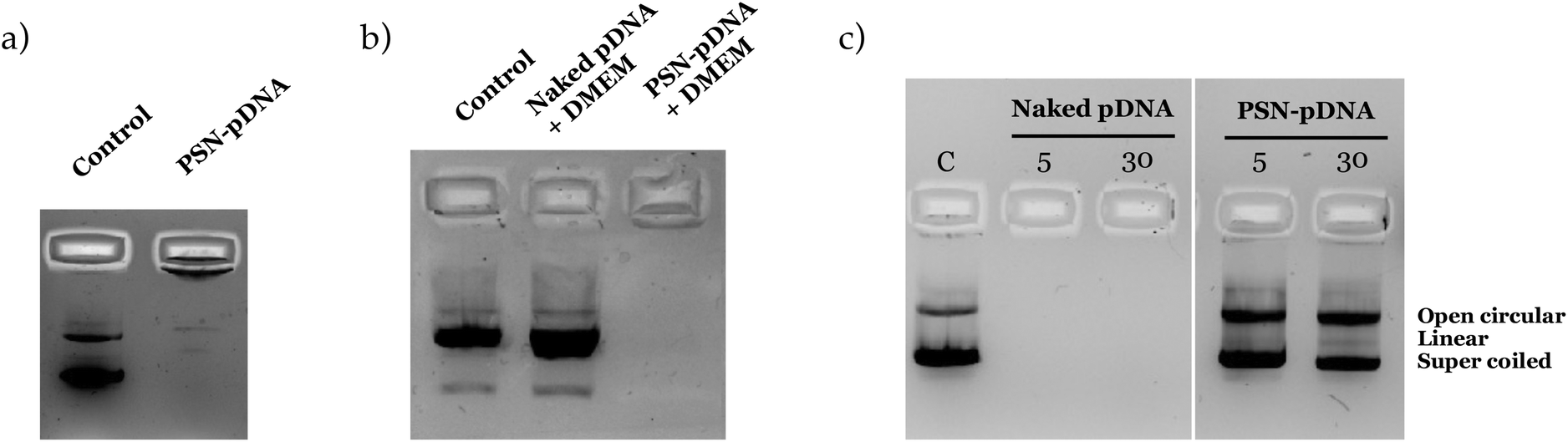 | ||
| Fig. 4 Stability of PSN-pDNA after (a) 12 weeks storage (4 °C), and incubation with (b) cellular media (DMEM, 4 h at 37 °C) and, (c) DNases (1 U/1 μg of pDNA, incubated for 5 and 30 min at 37 °C). | ||
Protection from DNases degradation was similarly evaluated. Relevantly, no degradation was observed after incubation of pDNA-PSN with PerfeCTa® DNase I (Fig. 4c), as only a minor conversion from the supercoiled to the open-circular form was observed.79 In this regard, the protective role of polyamine aggregates against nuclease degradation has already been described.25,80 Adsorption on nanostructures has also demonstrated to improve their stability against nucleases.74,81 In conclusion, we provide here the development and full characterization of a very simple, easy, and biodegradable formulation with high stability in different conditions.
Internalization and cellular uptake
The next step in the development of an efficient vehicle is to achieve a great cellular uptake and facilitate endosomal escape. Understanding the interaction of gene therapy nanosystems with the targeted cells, efficiency of internalization, endocytic pathway and intracellular trafficking, provides relevant information prior determining transfection efficiency.82–86 In agreement with other studies in the field, we observed that PSN-pDNA were efficiently internalized by triple negative MDA-MB-231 cells (Fig. S6†).14,58,87,88 To further characterize the intracellular uptake of TopFluor labelled PSN-pDNA, their colocalization with caveolin and clathrin was evaluated by confocal analyses at different time points (30 min, 1 h, 2 h, and 4 h) (Fig. 5c). Pinocytosis, a type of endocytic pathway which occurs in almost all cell types, is typically divided in macropinocytosis, caveolae-mediated endocytosis, clathrin-mediated endocytosis, and mechanisms independent of clathrin or caveolin.89–91 TopFluor-labelled PSN-pDNA mostly displayed with clathrin independently of the time point (around 90%). Furthermore, the colocalization rate with caveolin reached its maximum at 2 h (about 60%) (Fig. 5a). Therefore, these findings suggest that PSN-pDNA uptake is mainly due to clathrin-mediated endocytosis. These results are in line with previous studies, which describe the clathrin-mediated endocytosis for nanoparticles between 70–150 nm, in contrast with caveolin endocytosis for smaller nanoparticles.89 Additionally, positively charged nanoparticles have a higher preference by the clathrin pathway.69 However, it is noteworthy the high percentage of colocalization rate with caveolin, which could be related to a fusion between caveolin and clathrin vesicles.92 Next, the intracellular vesicle trafficking was assessed by means of confocal colocalization studies with different intracellular markers of the endosome (EEA1), lysosome (LAMP1), Golgi apparatus (GM130) and endoplasmic reticulum (calreticulin) to identify the intracellular destiny of PSN-pDNA. The colocalization between TopFluor-labelled PSN, Cy5-labelled pDNA (loaded pDNA) and the above-mentioned intracellular markers was measured after 4 h of PSN-pDNA treatment following 2 or 4 h of culture (Fig. 5b and d). Besides, we also tested the colocalization of Cy5-labelled pDNA; which does not colocalize with TopFluor-labelled PSN (free pDNA) to follow the pDNA delivery. Analysis of vesicle trafficking revealed that although about 30% of PSN-pDNA might be degraded by the lysosomal pathway (LAMP1), the rest of PSN-pDNA could enter the maturation pathways (GM130 and calreticulin) or might be released in the cytosol (EEA1, about 30% in loaded pDNA) (Fig. 5b). Our findings support that a large part of PSN-pDNA could be released from the endosomes, a key factor for the successful development of gene therapies.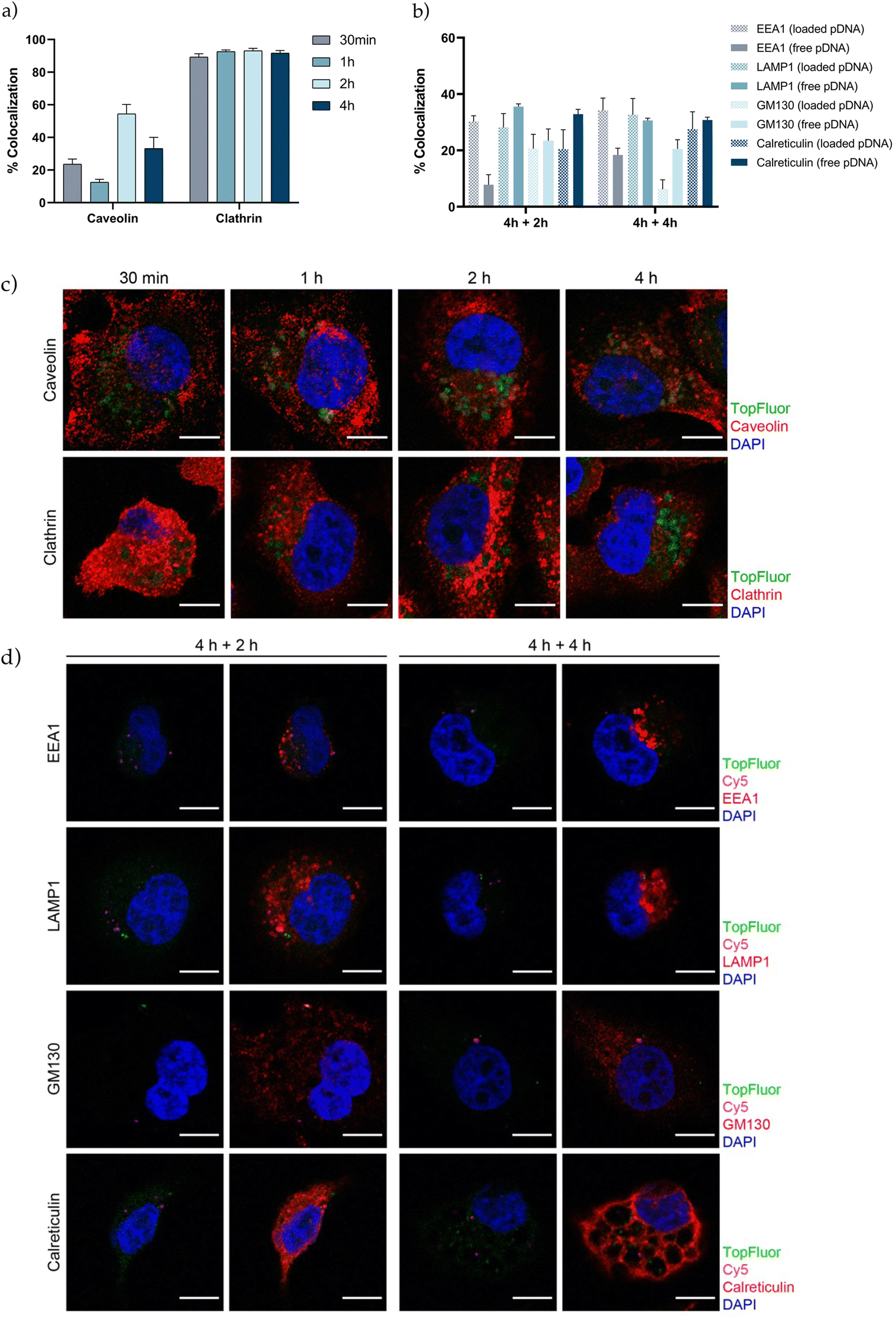 | ||
| Fig. 5 Intracellular delivery of PSN-pDNA in MDA-MB-231 cells treated for 30 min, 1 h, 2 h and 4 h with 0.44 mg mL−1 of TopFluor-labelled PSN-pDNA. (a) Evaluation of the endocytic routes involved in the uptake of PSN-pDNA measured by the quantification of colocalization percentage of intracellular TopFluor-labelled PSN with caveolin or clathrin. Colocalization of TopFluor-labelled PSN with each marker was performed in independent slides. For each endocytic marker, the colocalization rate was calculated as follows: number of TopFluor-labelled PSN dots co-localizing with the marker per field, divided by total number of TopFluor-labeller PSN dots per field (a minimum of three fields were taken per each condition); (b) intracellular trafficking of PSN-pDNA was evaluated through their colocalization with different organelle markers (EEA1, early endosome; LAMP1, lysosome; GM130, Golgi apparatus; calreticulin, endoplasmic reticulum). For each endocytic marker, the percentage of colocalization was calculated as follows: number of Cy5-labelled pDNA dots co-localizing with TopFluor-labelled PSN and each trafficking marker per field (loaded pDNA) or only colocalizing with each intracellular marker (free pDNA). (c) Representative confocal images of the colocalization between TopFluor-labeled PSN-pDNA (green) and the endocytic markers caveolin and clathrin (red); (d) representative confocal images of the subcellular localization of TopFluor-labeled PSN (green)/Cy5-labeled pDNA (deep red) and their colocalization with the different intracellular markers (red), (performed in independent slides). A minimum of three fields were taken per each condition. Bars represent average colocalization per microscope field ± SEM. Scale bar, 10 μm. | ||
Transfection efficiency and therapeutic effect
Preliminary results obtained with a plasmid DNA encoding for the red protein mCherry validated the capacity of PSN-pDNA to transfect MDA-MB-231 cells, with a transfection efficiency comparable to the commercial reagent for in vitro testing, Lipofectamine 2000 (Fig. S7†). Next experiments were carried out with a therapeutic plasmid that expresses the Fas Ligand (FasL) protein, member of the tumour necrosis factor superfamily, whose main function is the induction of apoptosis.42 In this regard, pDNA-control and pDNA-FasL constructs were associated to PSN and added to MDA-MB-231 cells. Importantly, cells transfected with PSN-pDNA-FasL showed a significant reduction in cell viability (Fig. 6a) concomitant with an increase in cell deaths rates (Fig. 6b), compared with the control cells transfected with the empty plasmid (PSN-pDNA-control). This effect was concentration dependent. For validation, similar results were obtained with HCC38 cells (Fig. S8†). These data indicate that the intracellular uptake of PSN-pDNA allows the correct release of the functional plasmid leading to the expression of the FasL protein and, consequently, to the expected therapeutic effect.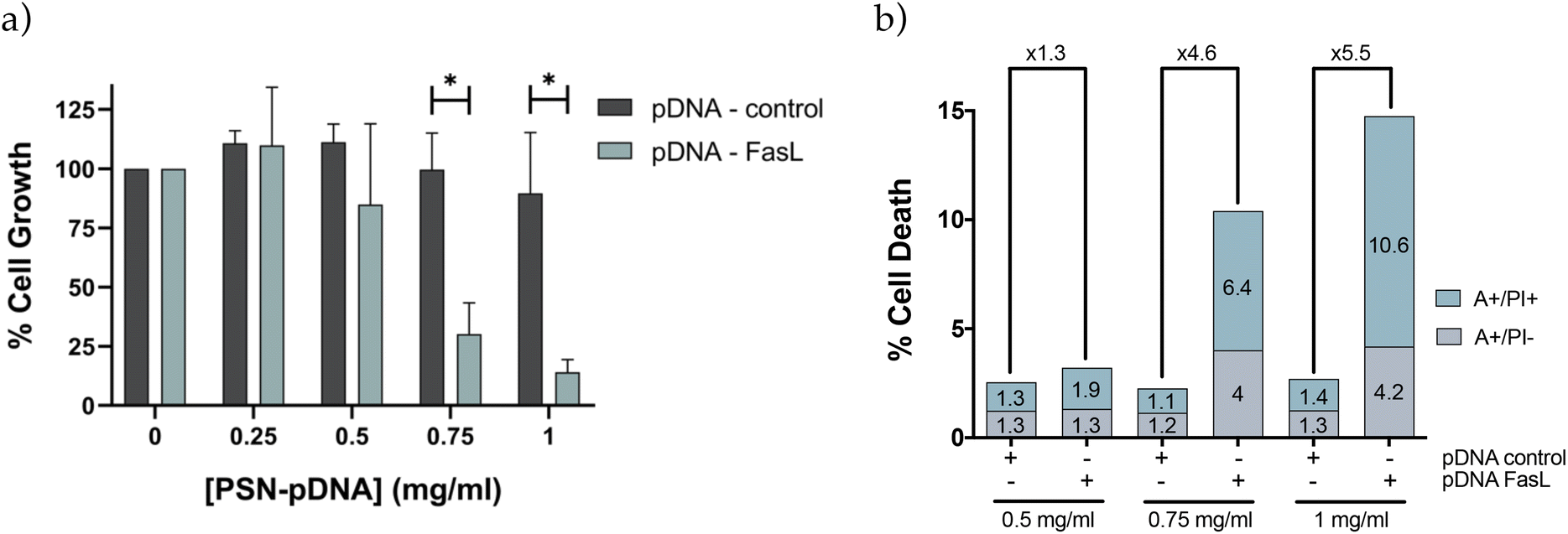 | ||
| Fig. 6 Cytotoxic effect of PSN-pDNA-FasL treatment in MDA-MB-231 cells was evaluated with different concentrations (from 0.25 to 1 mg mL−1) by means of (a) cell viability assays and (b) Annexin V-FITC plus PI. MDA-MB-231 cells were treated with PSN-pDNA-control or PSN-pDNA-FasL for 4 h when treatment medium was replaced by fresh medium, and cells were cultured for 72 h. Statistical significance was determined by two-tailed unpaired t-test (*P < 0.05; **P < 0.01). Data are shown as the mean ± SEM. Annexin V-FITC positive cells (A+/PI−) and Annexin V-FITC and PI doubled stained (A+/PI+) were defined as apoptotic cells. The number over the bars indicate the fold increase in cell death between the indicated conditions. Three independent experiments with similar results were performed. | ||
Validation was further performed in a xenograft zebrafish model, as an intermediate step between in vitro and in vivo validation, which allows obtaining a first validation of the potential of new therapeutic approaches with a minor cost and fewer restrictions in relation to mice models.93 Overall, zebrafish embryos are nowadays recognized as a valuable model particularly suitable for the evaluation of nanotherapies.4,94–97 Some of the intrinsic characteristics of the model enable not only to perform toxicological assessment but also to perform functional assays and real-time monitoring of nanocarriers along the fish circulation.4,98 For example, their transparency allows to visualize the biodistribution of fluorescent nanosystems or the real-time tracking of tumour growth.4,99 Besides, their remarkable grade of homology with the human genome (around 70%) and with orthologous human disease-related genes, higher than 80%,98,100 make of them an attractive model for gene therapy approaches.
GFP-transduced MDA-MB-231 cells were injected into the yolk sac, the most recommended site to study cellular survival.101 After 24 h, PSN-pDNA-control or PSN-pDNA-FasL were injected at a concentration of 26 mg mL−1. Each 24 h, zebrafish were analysed in terms of GFP positive pixels. A significant decrease in the fluorescent attributed to cancer cells was observed for fish treated with PSN-pDNA-FasL treated cells in relation to those treated with PSN-control (Fig. 7a). Representative confocal pictures of xenograft zebrafish embryos after 72 h of treatment are shown in Fig. 7b. These results are highly encouraging, providing the first evidence of the potential of the newly developed gene nanotherapy in an in vivo model. Indeed, results are comparable to previous studies employing this procedure, which have shown the ability of metal nanoparticles to induce apoptosis and inhibit the tumour growth of cancer cells injected into the yolk sac of zebrafish embryos.102
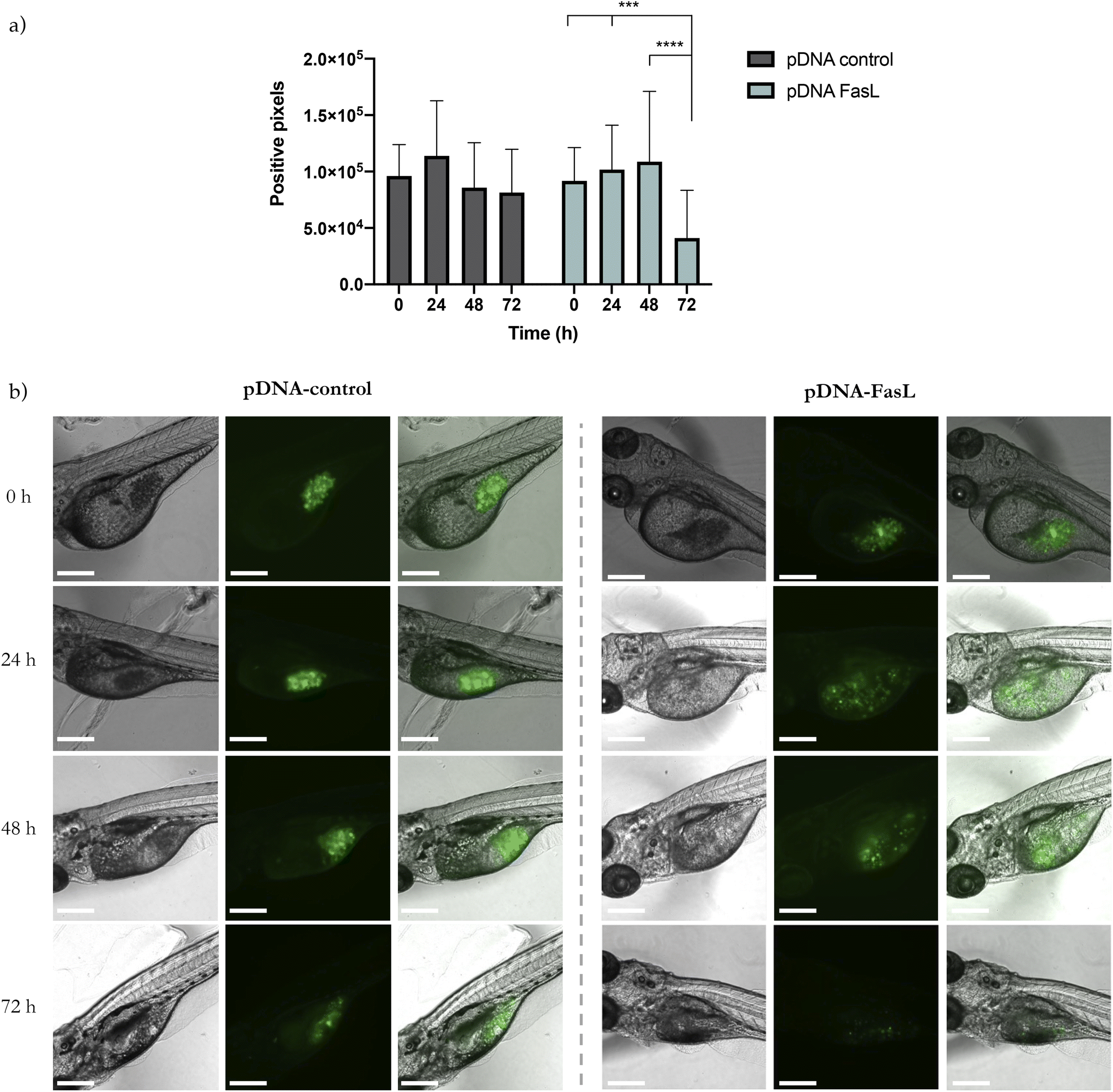 | ||
| Fig. 7 Cytotoxic effect of PSN-pDNA-FasL treatment in zebrafish embryos. Fluorescence pixels from GFP labelled MDA-MB-231 previously injected in the yolk sac show (a) the decrease in the cellular viability, which can be observed in (b) the representative pictures from confocal microscopy. 33 zebrafish were treated per condition. Bars represent mean ± SD. ***p < 0.001; ****p < 0.0001 compared to time 0 h of the same group. Scale bars represent 300 μm. | ||
Final experiments were conducted to validate the potential of the newly developed PSN-loaded with a therapeutic plasmid (pDNA-FasL) in mice bearing orthotopically injected MDA-MB-231 breast cancer cells. Two different routes of administration were explored, intratumorally, injecting the formulations directly in the tumour, as reported for different types of nanosystems that are validated in vivo for the first time,103,104 and intraperitoneally, adding additional biological barriers, an accepted route of administration for cancer therapeutics suitable for repeated doses of treatment,105,106 which we have used previously for testing novel nanotherapies loaded with biomolecules for the treatment of cancer.52,107 As observed (Fig. 8a and S9a, S10†), and irrespective of the route of administration, a significant reduction of tumour growth was reported after treatment with PSN-pDNA-FasL in relation to control treatment (PSN-pDNA-control). Furthermore, ex vivo tissue biodistribution of PSN-pDNA confirmed that TopFluor fluorescent signal (attributed to the fluorescently labelled PSN), was mostly detected in the breast xenografts (Fig. 8b and S9b, S10†). As expected, this accumulation was more exacerbated in the case of the intratumoral injection. Differences for PSN-FasL and PSN-control were not reported. Therefore, these findings prove that pDNA-FasL is able to correctly target the tumour, and lead to the expected therapeutic effect, validating the previously reported results in cell culturing and in zebrafish models, and opening up new venues for the development of novel gene therapies for triple-negative breast cancer, a currently unmet clinical need that has to be urgently addressed.
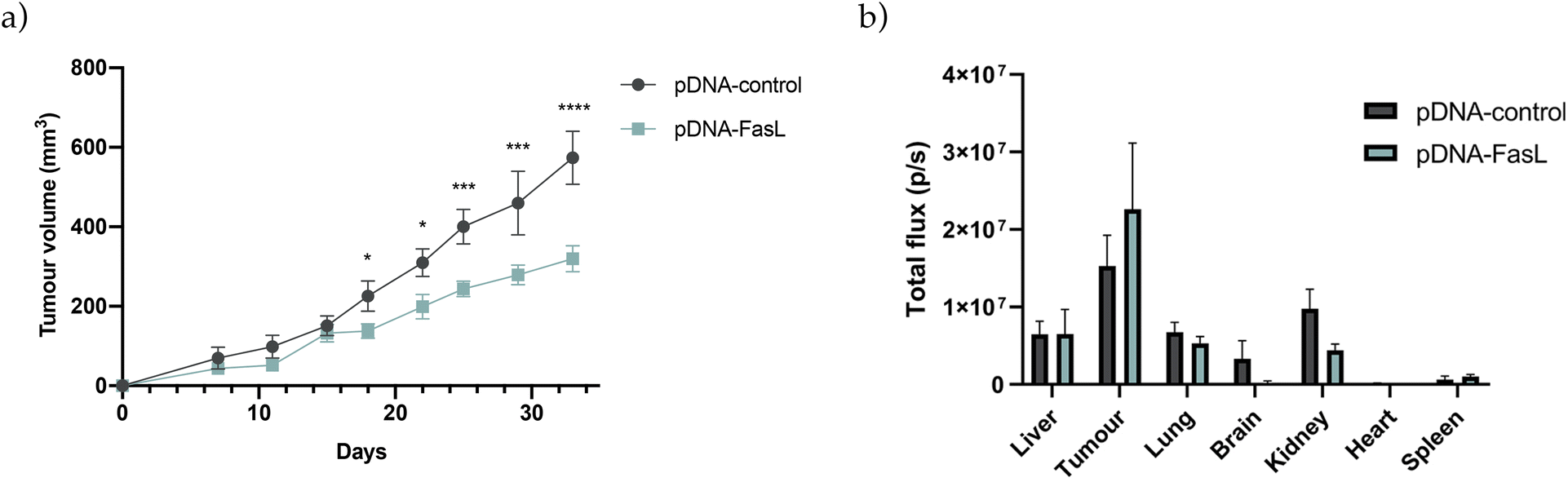 | ||
| Fig. 8 Cytotoxic effect of PSN-pDNA-FasL treatment in mice. (a) Quantification of tumour volume evolution of MDA-MB-231 mouse xenografts, treated intraperitoneal (pDNA-control n = 6; pDNA-FasL n = 9). Statistical significance was determined by multiple unpaired t-test – comparing both treatments at every time point. (b) Biodistribution of TopFluor-labelled pDNA-control or pDNA-FasL treated via intraperitoneal. Quantification of the TopFluor fluorescence of the indicated organs ex vivo was performed by normalizing it to the organs’ background from untreated mice. Bars represent mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. | ||
Conclusions
We described for the first time the development of a new type of gene therapy nanocarriers (PSN) based on putrescine, a polyamine for which cancer cells have a high avidity, in combination with biodegradable and biocompatible lipids (sphingomyelin and vitamin E). We proved that PSN can efficiently associate pDNA and are stable in several conditions. Importantly, internalization and cell trafficking experiments revealed a successful delivery of the associated pDNA to cancer cells via clathrin-mediated endocytosis, endosomal escape, and the capacity to mediate the expected therapeutic effect. In vivo studies in different preclinical models confirm the potential of the developed gene nanotherapy. Overall, our results indicate that PSN-pDNA-FasL represent a promising therapeutic approach opening new ways for the targeted therapy of TNBCs which constitute one of the main challenges in the current oncology due to the lack of this kind of treatment.Author contributions
S. L. has performed experimental design, conducted the experiments, prepared the figures, and wrote the manuscript. M. G. C., M. C., C. R. N., and P. H. contributed to experimental design and experiments, prepared figures, and corrected the manuscript. S. A. performed cell experiments. M. G. C. performed cell trafficking and in vivo experiments in mice models. All authors discussed the results presented on the manuscript. R. L. L., G. M. B., R. P., and M. d. l. F. supervised the project. M. d. l. F. designed and administer the project. G. M. B. designed cell trafficking and in vivo experiments in mice models. All authors read and approved the final manuscript.Conflicts of interest
M. d. l. F. is the co-founder and CEO of DIVERSA Technologies SL.Acknowledgements
This research was funded by Instituto de Salud Carlos III (ISCIII) and the European Regional Development Fund (FEDER) (grant numbers: CB16/12/00328, PI18/00176; AC18/00045; CIBERONC; CB16/12/00295), by ERA-NET EURONANOMED III project METASTARG (grant number JTC2018-045), by Roche-Chus Joint Unit, Axencia Galega de Innovación (GAIN), Consellería de Economía, Emprego e Industria (IN853B 2018/03), by the Spanish Ministry of Economy and Innovation (PID2019-104644RB-I00), and by the AECC Scientific Foundation (FC_AECC PROYE19036MOR). S. L. and P. H. also acknowledge the funding given by GAIN (IN606A-2019/003 and IN606A-2018/019). G. M. B. thanks the financial support given. We gratefully thank Raquel Antón Segurado for her technical support in scanning transmission electron microscopy, Montserrat García Lavandeira and Marta Picado for their technical support with confocal imaging, Lidia Martínez and Saleta Morales for their technical support cell culture maintenance and treatment, and all the co-workers in our laboratories for the help and discussion day by day. Funding for open access charge: Universidade de Santiago de Compostela/CISUG.References
- U.S. Food & Drug Administration, What is gene therapy?|FDA, https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/what-gene-therapy.
- Z. Zhou, X. Liu, D. Zhu, Y. Wang, Z. Zhang, X. Zhou, N. Qiu, X. Chen and Y. Shen, Nonviral cancer gene therapy: Delivery cascade and vector nanoproperty integration, Adv. Drug Delivery Rev., 2017, 115, 115–154, DOI:10.1016/j.addr.2017.07.021.
- E. B. Harrison, S. H. Azam and C. V. Pecot, Targeting Accessories to the Crime: Nanoparticle Nucleic Acid Delivery to the Tumor Microenvironment, Front. Pharmacol., 2018, 9, 1–15, DOI:10.3389/fphar.2018.00307.
- C. Gutiérrez-Lovera, A. J. Vázquez-Ríos, J. Guerra-Varela, L. Sánchez and M. de la Fuente, The potential of zebrafish as a model organism for improving the translation of genetic anticancer nanomedicines, Genes, 2017, 8, 1–20, DOI:10.3390/genes8120349.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Non-viral vectors for gene-based therapy, Nat. Rev. Genet., 2014, 15, 541–555, DOI:10.1038/nrg3763.
- M. H. Amer, Gene therapy for cancer: present status and future perspective, Mol. Cell. Ther., 2014, 2, 27, DOI:10.1186/2052-8426-2-27.
- N. Nayerossadat, P. Ali and T. Maedeh, Viral and nonviral delivery systems for gene delivery, Adv. Biomed. Res., 2012, 1, 27, DOI:10.4103/2277-9175.98152.
- M. Ramamoorth and A. Narvekar, Non viral vectors in gene therapy – An overview, J. Clin. Diagn. Res., 2015, 9, GE01–GE06, DOI:10.7860/JCDR/2015/10443.5394.
- M. Morille, C. Passirani, A. Vonarbourg, A. Clavreul and J. P. Benoit, Progress in developing cationic vectors for non-viral systemic gene therapy against cancer, Biomaterials, 2008, 29, 3477–3496, DOI:10.1016/j.biomaterials.2008.04.036.
- N. Somia and I. M. Verma, Gene therapy: Trials and tribulations, Nat. Rev. Genet., 2000, 1, 91–99, DOI:10.1038/35038533.
- D. Adams, A. Gonzalez-Duarte, W. D. O'Riordan, C.-C. Yang, M. Ueda, A. V. Kristen, I. Tournev, H. H. Schmidt, T. Coelho, J. L. Berk, K.-P. Lin, G. Vita, S. Attarian, V. Planté-Bordeneuve, M. M. Mezei, J. M. Campistol, J. Buades, T. H. Brannagan, B. J. Kim, J. Oh, Y. Parman, Y. Sekijima, P. N. Hawkins, S. D. Solomon, M. Polydefkis, P. J. Dyck, P. J. Gandhi, S. Goyal, J. Chen, A. L. Strahs, S. V. Nochur, M. T. Sweetser, P. P. Garg, A. K. Vaishnaw, J. A. Gollob and O. B. Suhr, Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis, N. Engl. J. Med., 2018, 379, 11–21, DOI:10.1056/nejmoa1716153.
- Nanomedicine and the COVID-19 vaccines, Nat. Nanotechnol., 2020, 15, 963, DOI:10.1038/s41565-020-00820-0.
- J. K. L. Wong, R. Mohseni, A. A. Hamidieh, R. E. MacLaren, N. Habib and A. M. Seifalian, Will Nanotechnology Bring New Hope for Gene Delivery?, Trends Biotechnol., 2017, 35, 434–451, DOI:10.1016/j.tibtech.2016.12.009.
- S. Nagachinta, B. L. Bouzo, A. J. Vazquez-Rios, R. Lopez and M. de la Fuente, Sphingomyelin-Based Nanosystems (SNs) for the Development of Anticancer miRNA Therapeutics, Pharmaceutics, 2020, 12, 189, DOI:10.3390/pharmaceutics12020189.
- C. Liu, L. Zhang, W. Zhu, R. Guo, H. Sun, X. Chen and N. Deng, Barriers and Strategies of Cationic Liposomes for Cancer Gene Therapy, Mol. Ther. – Methods Clin. Dev., 2020, 18, 751–764, DOI:10.1016/j.omtm.2020.07.015.
- C. K. Chen, P. K. Huang, W. C. Law, C. H. Chu, N. T. Chen and L. W. Lo, Biodegradable polymers for gene-delivery applications, Int. J. Nanomed., 2020, 15, 2131–2150, DOI:10.2147/IJN.S222419.
- B. R. Olden, Y. Cheng, J. L. Yu and S. H. Pun, Cationic polymers for non-viral gene delivery to human T cells, J. Controlled Release, 2018, 282, 140–147, DOI:10.1016/j.jconrel.2018.02.043.
- Q. Zheng, D. Lin, L. Lei, X. Li and S. Shi, Engineered non-viral gene vectors for combination cancer therapy: A review, J. Biomed. Nanotechnol., 2017, 13, 1565–1580, DOI:10.1166/jbn.2017.2489.
- M. Farriol, T. Segovia-silvestre, J. M. Castellanos, Y. Venereo and X. Orta, Role of Putrescine in Cell Proliferation in a Colon Carcinoma Cell Line, Nutrition, 2001, 934–938 CrossRef CAS PubMed.
- T. R. Murray-Stewart, P. M. Woster and R. A. Casero, Targeting polyamine metabolism for cancer therapy and prevention, Biochem. J., 2016, 473, 2937–2953, DOI:10.1042/BCJ20160383.
- S. L. Nowotarski, P. M. Woster and R. A. Casero Jr., Polyamines and cancer: Implications for chemoprevention and chemotherapy, Expert Rev. Mol. Med., 2014, 15, 1–28, DOI:10.1017/erm.2013.3.
- E. Bignon, C. H. Chan, C. Morell, A. Monari, J. L. Ravanat and E. Dumont, Molecular Dynamics Insights into Polyamine–DNA Binding Modes: Implications for Cross-Link Selectivity, Chem. – Eur. J., 2017, 23, 12845–12852, DOI:10.1002/chem.201702065.
- T. J. Thomas, H. A. Tajmir-Riahi and T. Thomas, Polyamine–DNA interactions and development of gene delivery vehicles, Amino Acids, 2016, 48, 2423–2431, DOI:10.1007/s00726-016-2246-8.
- G. Iacomino, G. Picariello and L. D'Agostino, DNA and nuclear aggregates of polyamines, Biochim. Biophys. Acta, Mol. Cell Res., 2012, 1823, 1745–1755, DOI:10.1016/j.bbamcr.2012.05.033.
- H. C. Ha, N. S. Sirisoma, P. Kuppusamy, J. L. Zweier, P. M. Woster and R. A. Casero, The natural polyamine spermine functions directly as a free radical scavenger, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11140–11145, DOI:10.1073/pnas.95.19.11140.
- M. Peters, A. Minton, O. Phanstiel IV and S. Gilmour, A Novel Polyamine-Targeted Therapy for BRAF Mutant Melanoma Tumors, Med. Sci., 2018, 6, 3, DOI:10.3390/medsci6010003.
- J. M. Barret, A. Kruczynski, S. Vispé, J. P. Annereau, V. Brel, Y. Guminski, J. G. Delcros, A. Lansiaux, N. Guilbaud, T. Imbert and C. Bailly, F14512, a potent antitumor agent targeting topoisomerase II vectored into cancer cells via the polyamine transport system, Cancer Res., 2008, 68, 9845–9853, DOI:10.1158/0008-5472.CAN-08-2748.
- S. Ganesh, A. K. Iyer, D. V. Morrissey and M. M. Amiji, Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors, Biomaterials, 2013, 34, 3489–3502, DOI:10.1016/j.biomaterials.2013.01.077.
- H. Hosseinkhani, T. Azzam, Y. Tabata and A. J. Domb, Dextran-spermine polycation: An efficient nonviral vector for in vitro and in vivo gene transfection, Gene Ther., 2004, 11, 194–203, DOI:10.1038/sj.gt.3302159.
- N. Niyomtham, N. Apiratikul, K. Suksen, P. Opanasopit and B. E. Yingyongnarongkul, Synthesis and in vitro transfection efficiency of spermine-based cationic lipids with different central core structures and lipophilic tails, Bioorg. Med. Chem. Lett., 2015, 25, 496–503, DOI:10.1016/j.bmcl.2014.12.043.
- G. Torrieri, F. Fontana, P. Figueiredo, Z. Liu, M. Ferreira, V. Talman, J. P. Martins, M. Fusciello, K. Moslova, T. Teesalu, V. Ceullo, J. Hirvonen, H. Ruskoaho, V. Balasubramanian and H. A. Santos, Dual-Peptide Functionalized Acetalated Dextran-Based Nanoparticles for Sequential Targeting of Macrophages during Myocardial Infarction, Nanoscale, 2020, 2350–2358, 10.1039/c9nr09934d.
- R. P. Shukla, S. Urandur, V. T. Banala, D. Marwaha, S. Gautam, N. Rai, N. Singh, P. Tiwari, P. Shukla and P. R. Mishra, Development of putrescine anchored nano-crystalsomes bearing doxorubicin and oleanolic acid: deciphering their role in inhibiting metastatic breast cancer, Biomater. Sci., 2021, 9, 1779–1794, 10.1039/D0BM01033B.
- B. L. Bouzo, M. Calvelo, M. Martin-Pastor, R. Garcia-Fandino and M. de la Fuente, In Vitro-in Silico Modelling Approach to Rationally Design Simple and Versatile Drug Delivery Systems, J. Phys. Chem. B, 2020, 124, 5788–5800, DOI:10.1021/acs.jpcb.0c02731.
- S. Nagachinta, G. Becker, S. Dammicco, M. E. Serrano, N. Leroi, M. A. Bahri, A. Plenevaux, C. Lemaire, R. Lopez, A. Luxen and M. de la Fuente, Radiolabelling of lipid-based nanocarriers with fluorine-18 for in vivo tracking by PET, Colloids Surf., B, 2020, 188, 110793, DOI:10.1016/j.colsurfb.2020.110793.
- S. Díez-Villares, M. A. Ramos-Docampo, A. da Silva-Candal, P. Hervella, A. J. Vázquez-Ríos, A. B. Dávila-Ibáñez, R. López-López, R. Iglesias-Rey, V. Salgueiriño and M. de la Fuente, Manganese Ferrite Nanoparticles Encapsulated into Vitamin E/Sphingomyelin Nanoemulsions as Contrast Agents for High-Sensitive Magnetic Resonance Imaging, Adv. Healthcare Mater., 2021, 10, e2101019, DOI:10.1002/adhm.202101019.
- B. L. Bouzo, S. Lores, R. Jatal, S. Alijas and M. J. Alonso, Sphingomyelin nanosystems loaded with uroguanylin and etoposide for treating metastatic colorectal cancer, Sci. Rep., 2021, 11, 1–12, DOI:10.1038/s41598-021-96578-z.
- R. Jatal, S. Mendes Saraiva, C. Vázquez-Vázquez, E. Lelievre, O. Coqueret, R. López-López and M. de la Fuente, Sphingomyelin nanosystems decorated with TSP-1 derived peptide targeting senescent cells, Int. J. Pharm., 2022, 617, 121618, DOI:10.1016/j.ijpharm.2022.121618.
- S. Díez-Villares, J. Pellico, N. Gómez-Lado, S. Grijalvo, S. Alijas, R. Eritja, F. Herranz, P. Aguiar and M. de la Fuente, Biodistribution of (68/67)Ga-Radiolabeled Sphingolipid Nanoemulsions by PET and SPECT Imaging, Int. J. Nanomed., 2021, 16, 5923–5935, DOI:10.2147/IJN.S316767.
- N. Bidan, S. Lores, A. Vanhecke, V. Nicolas, S. Domenichini, R. López, M. de la Fuente and S. Mura, Before in vivo studies: In vitro screening of sphingomyelin nanosystems using a relevant 3D multicellular pancreatic tumor spheroid model, Int. J. Pharm., 2022, 617, 121577, DOI:10.1016/j.ijpharm.2022.121577.
- R. C. Rowe, P. J. Sheskey and M. E. Quinn, Handbook of Pharmaceutical Excipients, Pharmaceutical Press, 6th edn, 2009, pp. 506–509 Search PubMed.
- J. P. Slotte and B. Ramstedt, The functional role of sphingomyelin in cell membranes, Eur. J. Lipid Sci. Technol., 2007, 109, 977–981, DOI:10.1002/ejlt.200700024.
- M. J. Pinkoski and D. R. Green, Fas ligand, death gene, Cell Death Differ., 1999, 6, 1174–1181, DOI:10.1038/sj.cdd.4400611.
- T. Kolben, U. Jeschke, T. Reimer, N. Karsten, E. Schmoeckel, A. Semmlinger, S. Mahner, N. Harbeck and T. M. Kolben, Induction of apoptosis in breast cancer cells in vitro by Fas ligand reverse signaling, J. Cancer Res. Clin. Oncol., 2018, 144, 249–256, DOI:10.1007/s00432-017-2551-y.
- P. Guo, J. Yang, J. Huang, D. T. Auguste and M. A. Moses, Therapeutic genome editing of triple-negative breast tumors using a noncationic and deformable nanolipogel, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18295–18303, DOI:10.1073/pnas.1904697116.
- A. Sorolla, E. Wang, T. D. Clemons, C. W. Evans, J. H. Plani-Lam, E. Golden, B. Dessauvagie, A. D. Redfern, K. Swaminathan-Iyer and P. Blancafort, Triple-hit therapeutic approach for triple negative breast cancers using docetaxel nanoparticles, EN1-iPeps and RGD peptides, Nanomedicine, 2019, 20, 102003, DOI:10.1016/j.nano.2019.04.006.
- J. Yu, J. Zayas, B. Qin and L. Wang, Targeting DNA methylation for treating triple-negative breast cancer, Pharmacogenomics, 2019, 20, 1151–1157, DOI:10.2217/pgs-2019-0078.
- S. L. Ginn, I. E. Alexander, M. L. Edelstein, M. R. Abedi and J. Wixon, Gene therapy clinical trials worldwide to 2012 – an uptate, J. Gene Med., 2013, 15, 65–77, DOI:10.1002/jgm.
- A. Afghahi, M. L. Telli and A. W. Kurian, Genetics of triple-negative breast cancer: Implications for patient care, Curr. Probl. Cancer, 2016, 40, 130–140, DOI:10.1016/j.currproblcancer.2016.09.007.
- C. M. McCrudden and H. O. McCarthy, Current status of gene therapy for breast cancer: Progress and challenges, Appl. Clin. Genet., 2014, 7, 209–220, DOI:10.2147/TACG.S54992.
- M. E. Tanenbaum, L. A. Gilbert, L. S. Qui, J. S. Weissman and R. D. Vale, A protein tagging system for signal amplification in gene expression and fluorescence imaging, Cell, 2015, 159, 635–646, DOI:10.1016/j.cell.2014.09.039.A.
- D. R. Stirling, O. Suleyman, E. Gil, P. M. Elks, V. Torraca, M. Noursadeghi and G. S. Tomlinson, Analysis tools to quantify dissemination of pathology in zebrafish larvae, Sci. Rep., 2020, 10, 3149, DOI:10.1038/s41598-020-59932-1.
- A. Molina-Crespo, A. Cadete, D. Sarrio, M. Gamez-Chiachio, L. Martinez, K. Chao, A. Olivera, A. Gonella, E. Díaz, J. Palacios, P. K. Dhal, M. Besev, M. Rodríguez-Serrano, M. L. G. Bermejo, J. C. Triviño, A. Cano, M. García-Fuentes, O. Herzberg, D. Torres, M. J. Alonso and G. Moreno-Bueno, Intracellular delivery of an antibody targeting gasdermin-b reduces her2 breast cancer aggressiveness, Clin. Cancer Res., 2019, 25, 4846–4858, DOI:10.1158/1078-0432.CCR-18-2381.
- M. Hergueta-Redondo, D. Sarrió, Á. Molina-Crespo, D. Megias, A. Mota, A. Rojo-Sebastian, P. García-Sanz, S. Morales, S. Abril, A. Cano, H. Peinado and G. Moreno-Bueno, Gasdermin-B promotes invasion and metastasis in breast cancer cells, PLoS One, 2014, 9, e90099, DOI:10.1371/journal.pone.0090099.
- M. Maqbool, F. Bekele and G. Fekadu, Treatment Strategies Against Triple-Negative Breast Cancer: An Updated Review, Breast Cancer: Targets Ther., 2022, 14, 15–24, DOI:10.2147/BCTT.S348060.
- R. Yang, Y. Li, H. Wang, T. Qin, X. Yin and X. Ma, Therapeutic progress and challenges for triple negative breast cancer: targeted therapy and immunotherapy, Mol. Biomed., 2022, 3, 8, DOI:10.1186/s43556-022-00071-6.
- M. Montaño-Samaniego, D. M. Bravo-Estupiñan, O. Méndez-Guerrero, E. Alarcón-Hernández and M. Ibáñez-Hernández, Strategies for Targeting Gene Therapy in Cancer Cells With Tumor-Specific Promoters, Front. Oncol., 2020, 10, 1–18, DOI:10.3389/fonc.2020.605380.
- T. M. Belete, The current status of gene therapy for the treatment of cancer, Biol.: Targets Ther., 2021, 15, 67–77, DOI:10.2147/BTT.S302095.
- F. Masoumi, S. M. Saraiva, B. L. Bouzo, R. López-López, M. Esteller, Á. Díaz-Lagares and M. de la Fuente, Modulation of colorectal tumor behavior via lncrna tp53tg1-lipidic nanosystem, Pharmaceutics, 2021, 13, 1–16, DOI:10.3390/pharmaceutics13091507.
- A. Arruabarrena-Aristorena, A. Zabala-Letona and A. Carracedo, Oil for the cancer engine: The cross-talk between oncogenic signaling and polyamine metabolism, Sci. Adv., 2018, 4, 1–12, DOI:10.1126/sciadv.aar2606.
- P. A. Puchkov and M. A. Maslov, Lipophilic Polyamines as Promising Components of Liposomal Gene Delivery Systems, Pharmaceutics, 2021, 13, 920, DOI:10.3390/pharmaceutics13060920.
- T. Fujiwara, S. Hasegawa, N. Hirashima, M. Nakanishi and T. Ohwada, Gene transfection activities of amphiphilic steroid-polyamine conjugates, Biochim. Biophys. Acta, Biomembr., 2000, 1468, 396–402, DOI:10.1016/S0005-2736(00)00278-9.
- A. J. Geall and I. S. Blagbrough, Homologation of polyamines in the rapid synthesis of lipospermine conjugates and related lipoplexes, Tetrahedron, 2000, 56, 2449–2460, DOI:10.1016/S0040-4020(99)01082-0.
- S. Grijalvo, G. Puras, J. Zárate, M. Sainz-Ramos, N. A. L. Qtaish, T. López, M. Mashal, N. Attia, D. Díaz, R. Pons, E. Fernández, J. L. Pedraz and R. Eritja, Cationic niosomes as non-viral vehicles for nucleic acids: Challenges and opportunities in gene delivery, Pharmaceutics, 2019, 11, 1–23, DOI:10.3390/pharmaceutics11020050.
- J. R. Viola, H. Leijonmarck, O. E. Simonson, I. I. Oprea, R. Frithiof, P. Purhonen, P. M. D. Moreno, K. E. Lundin, R. Strömberg and C. I. E. Smith, Fatty acid-spermine conjugates as DNA carriers for nonviral in vivo gene delivery, Gene Ther., 2009, 16, 1429–1440, DOI:10.1038/gt.2009.108.
- O. Paecharoenchai, N. Niyomtham, L. Leksantikul, T. Ngawhirunpat, T. Rojanarata, B. Yingyongnarongkul and P. Opanasopit, Nonionic Surfactant Vesicles Composed of Novel Spermine-Derivative Cationic Lipids as an Effective Gene Carrier In Vitro, AAPS PharmSciTech, 2014, 15, 722–730, DOI:10.1208/s12249-014-0095-x.
- T. Dewa, T. Asai, N. Oku and M. Nango, Polyamine – Lipid Conjugates as Effective Gene Carriers: Chemical Structure, Morphology, and Gene Transfer Activity, Non-Viral Gene Ther, 2011, pp. 243–266. DOI:10.5772/18929.
- R. Cortesi, M. Campioni, L. Ravani, M. Drechsler, M. Pinotti and E. Esposito, Cationic lipid nanosystems as carriers for nucleic acids, New Biotechnol., 2014, 31, 44–54, DOI:10.1016/j.nbt.2013.10.001.
- S. Prabha, G. Arya, R. Chandra, B. Ahmed and S. Nimesh, Effect of size on biological properties of nanoparticles employed in gene delivery, Artif. Cells, Nanomed., Biotechnol., 2014, 44, 83–91, DOI:10.3109/21691401.2014.913054.
- D. Manzanares and V. Ceña, Endocytosis: The nanoparticle and submicron nanocompounds gateway into the cell, Pharmaceutics, 2020, 12, 1–22, DOI:10.3390/pharmaceutics12040371.
- R. Kircheis, E. Ostermann, M. F. Wolschek, C. Lichtenberger, C. Magin-Lachmann, L. Wightman, M. Kursa and E. Wagner, Tumor-targeted gene delivery of tumor necrosis factor-α induces tumor necrosis and tumor regression without systemic toxicity, Cancer Gene Ther., 2002, 9, 673–680, DOI:10.1038/sj.cgt.7700487.
- N. Serna, L. Sánchez-García, U. Unzueta, R. Díaz, E. Vázquez, R. Mangues and A. Villaverde, Protein-Based Therapeutic Killing for Cancer Therapies, Trends Biotechnol., 2018, 36, 318–335, DOI:10.1016/j.tibtech.2017.11.007.
- M. A. Liu, A Comparison of Plasmid DNA and mRNA as Vaccine Technologies, Vaccines, 2019, 7, 37, DOI:10.4172/978-1-63278-004-1-005.
- S. Uchida, K. Itaka, H. Uchida, K. Hayakawa, T. Ogata, T. Ishii, S. Fukushima, K. Osada and K. Kataoka, In Vivo Messenger RNA Introduction into the Central Nervous System Using Polyplex Nanomicelle, PLoS One, 2013, 8, 4–12, DOI:10.1371/journal.pone.0056220.
- É. Martini, E. Fattal, M. C. de Oliveira and H. Teixeira, Effect of cationic lipid composition on properties of oligonucleotide/emulsion complexes: Physico-chemical and release studies, Int. J. Pharm., 2008, 352, 280–286, DOI:10.1016/j.ijpharm.2007.10.032.
- H. Yasar, A. Biehl, C. De Rossi, M. Koch, X. Murgia, B. Loretz and C. M. Lehr, Kinetics of mRNA delivery and protein translation in dendritic cells using lipid-coated PLGA nanoparticles, J. Nanobiotechnol., 2018, 16, 1–19, DOI:10.1186/s12951-018-0401-y.
- V. Filipe, A. Hawe and W. Jiskoot, Critical evaluation of nanoparticle tracking analysis (NTA) by NanoSight for the measurement of nanoparticles and protein aggregates, Pharm. Res., 2010, 27, 796–810, DOI:10.1007/s11095-010-0073-2.
- M. Y. Chan, Q. M. Dowling, S. J. Sivananthan and R. M. Kramer, Particle sizing of nanoparticle adjuvant formulations by dynamic light scattering (DLS) and nanoparticle tracking analysis (NTA), Methods Mol. Biol., 2017, 239–252, DOI:10.1007/978-1-4939-6445-1_17.
- Y. Li, M. Kröger and W. K. Liu, Shape effect in cellular uptake of PEGylated nanoparticles: Comparison between sphere, rod, cube and disk, Nanoscale, 2015, 7, 16631–16646, 10.1039/c5nr02970h.
- M. S. Levy, P. Lotfian, R. O’Kennedy, M. Y. Lo-Yim and P. A. Shamlou, Quantitation of supercoiled circular content in plasmid DNA solutions using a fluorescence-based method, Nucleic Acids Res., 2000, 28, 12, DOI:10.1093/nar/28.12.e57.
- G. Iacomino, G. Picariello, F. Sbrana, A. Di Luccia, R. Raiteri and L. D'Agostino, DNA is wrapped by the nuclear aggregates of polyamines: The imaging evidence, Biomacromolecules, 2011, 12, 1178–1186, DOI:10.1021/bm101478j.
- A. R. Chandrasekaran, Nuclease resistance of DNA nanostructures, Nat. Rev. Chem., 2021, 5, 225–239, DOI:10.1038/s41570-021-00251-y.
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics, Nat. Nanotechnol., 2021, 16, 266–276, DOI:10.1038/s41565-021-00858-8.
- M. Durymanov and J. Reineke, Non-viral delivery of nucleic acids: Insight into mechanisms of overcoming intracellular barriers, Front. Pharmacol., 2018, 9, 1–15, DOI:10.3389/fphar.2018.00971.
- D. J. Brock, H. M. Kondow-McConaghy, E. C. Hager and J.-P. Pellois, Endosomal Escape and Cytosolic Penetration of Macromolecules Mediated by Synthetic Delivery Agents, Bioconjugate Chem., 2018, 30, 293–304, DOI:10.1021/acs.bioconjchem.8b00799.
- A. Ahmad, J. M. Khan and S. Haque, Strategies in the design of endosomolytic agents for facilitating endosomal escape in nanoparticles, Biochimie, 2019, 160, 61–75, DOI:10.1016/j.biochi.2019.02.012.
- T. F. Martens, K. Remaut, J. Demeester, S. C. De Smedt and K. Braeckmans, Intracellular delivery of nanomaterials: How to catch endosomal escape in the act, Nano Today, 2014, 9, 344–364, DOI:10.1016/j.nantod.2014.04.011.
- S. Wang, M. Shao, Z. Zhong, A. Wang, J. Cao, Y. Lu, Y. Wang and J. Zhang, Co-delivery of gambogic acid and TRAIL plasmid by hyaluronic acid grafted PEI-PLGA nanoparticles for the treatment of triple negative breast cancer, Drug Delivery, 2017, 24, 1791–1800, DOI:10.1080/10717544.2017.1406558.
- X. X. Ma, J. L. Xu, Y. Y. Jia, Y. X. Zhang, W. Wang, C. Li, W. He, S. Y. Zhou and B. Le Zhang, Enhance transgene responses through improving cellular uptake and intracellular trafficking by bio-inspired non-viral vectors, J. Nanobiotechnol., 2020, 18, 26, DOI:10.1186/s12951-020-0582-z.
- R. M. Pearson, H. J. Hsu, J. Bugno and S. Hong, Understanding nano-bio interactions to improve nanocarriers for drug delivery, MRS Bull., 2014, 39, 227–237, DOI:10.1557/mrs.2014.9.
- I. U. Khan, R. U. Khan, H. Asif, Alamgeer, S. H. Khalid, S. Asghar, M. Saleem, K. U. Shah, S. U. Shah, S. A. A. Rizvi and Y. Shahzad, Co-delivery strategies to overcome multidrug resistance in ovarian cancer, Int. J. Pharm., 2017, 533, 111–124, DOI:10.1016/j.ijpharm.2017.09.060.
- I. A. Khalil, K. Kogure, H. Akita and H. Harashima, Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery, Pharmacol. Rev., 2006, 58, 32–45, DOI:10.1124/pr.58.1.8.
- K. He, X. Yan, N. Li, S. Dang, L. Xu, B. Zhao, Z. Li, Z. Lv, X. Fang, Y. Zhang and Y. G. Chen, Internalization of the TGF-β type I receptor into caveolin-1 and EEA1 double-positive early endosomes, Cell Res., 2015, 25, 738–752, DOI:10.1038/cr.2015.60.
- C. Delvecchio, J. Tiefenbach and H. M. Krause, The Zebrafish: A powerful platform for in vivo, HTS drug discovery, Assay Drug Dev. Technol., 2011, 9, 354–361, DOI:10.1089/adt.2010.0346.
- M. Cascallar, S. Alijas, A. Pensado-López, A. J. Vázquez-Ríos, L. Sánchez, R. Piñeiro and M. de la Fuente, What Zebrafish and Nanotechnology Can Offer for Cancer Treatments in the Age of Personalized Medicine, Cancers, 2022, 14, 1–28, DOI:10.3390/cancers14092238.
- S. Lin, Y. Zhao, A. E. Nel and S. Lin, Zebrafish: an in vivo model for nano EHS studies, Small, 2013, 9, 1608–1618, DOI:10.1002/smll.201202115.
- J. Crecente-Campo, J. Guerra-Varela, M. Peleteiro, C. Gutiérrez-Lovera, I. Fernández-Mariño, A. Diéguez-Docampo, Á. González-Fernández, L. Sánchez and M. J. Alonso, The size and composition of polymeric nanocapsules dictate their interaction with macrophages and biodistribution in zebrafish, J. Controlled Release, 2019, 308, 98–108, DOI:10.1016/j.jconrel.2019.07.011.
- S. George, T. Xia, R. Rallo, Y. Zhao, Z. Ji, S. Lin, X. Wang, H. Zhang, B. France, D. Schoenfeld, R. Damoiseaux, R. Liu, S. Lin, K. A. Bradley, Y. Cohen and A. E. Nel, Use of a High-Throughput Screening Approach Coupled with In Vivo Zebrafish Embryo Screening To Develop Hazard Ranking for Engineered Nanomaterials, ACS Nano, 2011, 5, 1805–1817, DOI:10.1021/nn102734s.
- S. Zhao, J. Huang and J. Ye, A fresh look at zebrafish from the perspective of cancer research, J. Exp. Clin. Cancer Res., 2015, 34, 1–9, DOI:10.1186/s13046-015-0196-8.
- M. Cully, Zebrafish earn their drug discovery stripes, Nat. Rev. Drug Discovery, 2019, 18, 811–813, DOI:10.1038/d41573-019-00165-x.
- C. Santoriello and L. I. Zon, Hooked! Modeling human disease in zebrafish, J. Clin. Invest., 2012, 122, 2337–2343, DOI:10.1172/JCI60434.
- P. Cabezas-Sáinz, A. Pensado-López, B. Sáinz and L. Sánchez, Modeling Cancer Using Zebrafish Xenografts: Drawbacks for Mimicking the Human Microenvironment, Cells, 2020, 9, 1978, DOI:10.3390/cells9091978.
- S. W. Wang, C. H. Lee, M. S. Lin, C. W. Chi, Y. J. Chen, G. S. Wang, K. W. Liao, L. P. Chiu, S. H. Wu, D. M. Huang, L. Chen and Y. S. Shen, ZnO nanoparticles induced caspase-dependent apoptosis in gingival squamous cell carcinoma through mitochondrial dysfunction and p70s6K signaling pathway, Int. J. Mol. Sci., 2020, 21, 1–16, DOI:10.3390/ijms21051612.
- Z. J. Deng, S. W. Morton, E. Ben-Akiva, E. C. Dreaden, K. E. Shopsowitz and P. T. Hammond, Layer-by-Layer Nanoparticles for Systemic Codelivery of an Anticancer Drug and siRNA for Potential Triple-Negative Breast Cancer Treatment, ACS Nano, 2013, 7, 1–26, DOI:10.1021/nn4047925.
- S. Goklany, P. Lu, S. Godeshala, A. Hall, E. Garrett-Mayer, C. Voelkel-Johnson and K. Rege, Delivery of TRAIL-expressing plasmid DNA to cancer cells: In vitro and in vivo using aminoglycoside-derived polymers, J. Mater. Chem. B, 2019, 7, 7014–7025, 10.1039/c9tb01286a.
- A. Al Shoyaib, S. R. Archie and V. T. Karamyan, Intraperitoneal Route of Drug Administration: Should it Be Used in Experimental Animal Studies?, Pharm. Res., 2020, 37, 12, DOI:10.1007/s11095-019-2745-x.
- S. Mannucci, F. Boschi, B. Cisterna, E. Esposito, R. Cortesi, C. Nastruzzi, E. Cappellozza, P. Bernardi, A. Sbarbati, M. Malatesta and L. Calderan, A correlative imaging study of in vivo and ex vivo biodistribution of solid lipid nanoparticles, Int. J. Nanomed., 2020, 15, 1745–1758, DOI:10.2147/IJN.S236968.
- A. J. Vázquez-Ríos, Á. Molina-Crespo, B. L. Bouzo, R. López-López, G. Moreno-Bueno and M. de la Fuente, Exosome-mimetic nanoplatforms for targeted cancer drug delivery, J. Nanobiotechnol., 2019, 17, 85, DOI:10.1186/s12951-019-0517-8.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01456d |
| This journal is © The Royal Society of Chemistry 2023 |