
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular presentation of bioinstructive peptides on soft multilayered nanobiomaterials stimulates neurite outgrowth†
Maria
Lopes
 a,
Marília
Torrado
bc,
Daryl
Barth
ad,
Sofia D.
Santos
b,
Melike
Sever-Bahcekapili
ef,
Ayse B.
Tekinay
e,
Mustafa O.
Guler
g,
Franck
Cleymand
d,
Ana P.
Pêgo
bc,
João
Borges
*a and
João F.
Mano
*a
a,
Marília
Torrado
bc,
Daryl
Barth
ad,
Sofia D.
Santos
b,
Melike
Sever-Bahcekapili
ef,
Ayse B.
Tekinay
e,
Mustafa O.
Guler
g,
Franck
Cleymand
d,
Ana P.
Pêgo
bc,
João
Borges
*a and
João F.
Mano
*a
aCICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal. E-mail: joaoborges@ua.pt; jmano@ua.pt
bINEB – Instituto de Engenharia Biomédica & i3S - Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Rua Alfredo Allen, 208, 4200-135 Porto, Portugal
cICBAS – Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal
dInstitut Jean Lamour, UMR 7198 CNRS – Université de Lorraine, Parc de Saurupt CS 50840, 54011 Nancy Cedex, France
eInstitute of Materials Science and Nanotechnology, National Nanotechnology Research Center (UNAM), Bilkent University, 06800 Ankara, Turkey
fInstitute of Neurological Sciences and Psychiatry, Hacettepe University, 06230 Ankara, Turkey
gThe Pritzker School of Molecular Engineering, The University of Chicago, Chicago, IL 60637, USA
First published on 13th June 2023
Abstract
Peptide amphiphiles (PAs) have emerged as effective molecular building blocks for creating self-assembling nanobiomaterials for multiple biomedical applications. Herein, we report a straightforward approach to assemble soft bioinstructive platforms to recreate the native neural extracellular matrix (ECM) aiming for neuronal regeneration based on the electrostatic-driven supramolecular presentation of laminin-derived IKVAV-containing self-assembling PA (IKVAV-PA) on biocompatible multilayered nanoassemblies. Spectroscopic and microscopic techniques show that the co-assembly of positively charged low-molecular-weight IKVAV-PA with oppositely charged high-molecular-weight hyaluronic acid (HA) triggers the formation of ordered β-sheet structures denoting a one-dimensional nanofibrous network. The successful functionalization of poly(L-lysine)/HA layer-by-layer nanofilms with an outer positively charged layer of self-assembling IKVAV-PA is demonstrated by the quartz crystal microbalance with dissipation monitoring and the nanofibrous morphological properties revealed by atomic force microscopy. The bioactive ECM-mimetic supramolecular nanofilms promote the enhancement of primary neuronal cells’ adhesion, viability, and morphology when compared to the PA without the IKVAV sequence and PA-free biopolymeric multilayered nanofilms, and stimulate neurite outgrowth. The nanofilms hold great promise as bioinstructive platforms for enabling the assembly of customized and robust multicomponent supramolecular biomaterials for neural tissue regeneration.
Introduction
Injuries inflicted on the central nervous system (CNS) often result in permanent neural tissue loss and damage to the circuits’ connectivity, impairing vital human body processes, such as motor function and sensory perception.1,2 Despite the growing knowledge in understanding the mechanisms and pathophysiology of lesions affecting the CNS, there is still not an effective clinically approved therapeutic strategy to improve the neurological outcomes and promote functional recovery. To support biological activities and achieve an efficient therapeutic solution for enabling neuronal regeneration, the molecular composition, structure, bioactive cues, fibrous architecture and dynamic nature of the native neural extracellular matrix (ECM) supramolecular landscape can be emulated by the self-assembled ECM-mimetic bioactive nanoarchitectures.3–6Recently, the rational design of supramolecular nanostructures together with tissue engineering efforts have paved the way for the development of neural ECM-mimetic supramolecular polymeric biomaterials, enlisted with biophysical, biochemical, mechanical and structural features to modulate the local lesion microenvironment, ultimately resulting in the development of biofunctional neural tissues.7–10 To date, a variety of methods have been employed to bioengineer relevant nanofibrous ECM-mimetic multicomponent scaffolds, including molecular self-assembly,11–14 phase separation,15,16 and electrospinning.4,17 Among the toolbox of building blocks, natural-origin polymers, natural fibrous proteins, and combinations thereof have attracted considerable attention in the build-up of neural ECM-mimetic biofunctional matrices owing to their ubiquitous presence in the native neural ECM. While the biocompatibility, biodegradability, non-cytotoxicity, and wide and ready availability of natural-origin polymers encourage their use,18,19 ECM fibrous proteins, namely fibronectin and laminin (LAM), are key components to enhance cell–ECM communication and repair tissue structure and function.20,21 However, ECM proteins entail high costs, batch-to-batch variability, risk of pathogenic contamination and are difficult to purify when obtained from mammalian-derived tissues, poor processability, and limited stability and availability, which extensively limit their use.22,23
Peptide amphiphiles (PAs) have shown to be very appealing alternatives to fibrous ECM proteins in designing tunable bioactive ECM-like nanostructures due to their key structural and biofunctional features. Those include ease of synthesis and functionality, inherent biocompatibility and biodegradability, high chemical diversity and versatility, increased stability, high surface density of bioactive epitopes, and self-assembling capability into one-dimensional (1D) fibrous nanostructures in aqueous medium. As such, the PAs emulate the intrinsic fibrillar topography, architecture and signalling cues of the neural ECM.24 Furthermore, their peptide sequence can be rationally designed and customized to impart any biofunctionality to enable desired cellular responses.6,25 In the literature one can find several studies that highlight the bottom-up nanofabrication of tunable PA-derived supramolecular matrices to control cell-biomaterial interactions for multiple tissue engineering strategies,26,27 including nerve tissue regeneration.28,29
The layer-by-layer (LbL) assembly technology has emerged as a simple, cost-effective, robust, and highly versatile nanofabrication technique to functionalize virtually any type of surface and build-up tunable ECM-mimetic multicomponent nanostructures by resorting to a repertoire of building blocks exhibiting complementary interactions.12,30,31 The fact that the LbL assembly process occurs under mild conditions in aqueous media enables the incorporation of biomolecules in the multilayered films, including polysaccharides, proteins, nucleic acids, or combinations thereof, without losing their biological activity, thus being very appealing to assemble biofunctional ECM-mimetic nanostructures.12,13,30,31
Herein, we report the electrostatic-driven supramolecular immobilization of LAM-derived isoleucine–lysine–valine–alanine–valine (IKVAV)-containing self-assembling PA on multilayered nanofilms encompassing oppositely charged biocompatible polymers to recreate the native neural ECM. High-molecular-weight (HMW) hyaluronic acid (HA) has been selected as an ingredient of the multilayered nanofilm owing to its ubiquitous presence in the native ECMs of many tissues and organs, including in the neural ECM, and key role in the regulation of neuronal processes, such as cell adhesion, migration and morphological maturation. In addition, it has proven biocompatibility and biodegradability, non-cytotoxicity, and non-immunogenic properties.32 As such, anionic HA has been LbL assembled with the cationic biopolymer poly(L-lysine) (PLL), widely used in in vitro studies as a standard substrate coating, into soft biomimetic multilayered thin films to modulate cell functions.19,33,34 However, their use for enabling bioinstructive ECM-like matrices to recapitulate the nanofiber architecture and biofunctionality of the native neural ECM has not yet been reported.
In this work, a straightforward methodology is proposed for the supramolecular presentation of self-assembling IKVAV-PA on soft PLL/HA multilayered nanofilms aiming to impart the nanocoatings with a nanofibrillar topography and biofunctional cues, reminiscent of the native neural ECM, to stimulate neurite outgrowth.
Experimental section
Materials
HMW HA sodium salt from Streptococcus equi (Mw = 1.5 MDa), PLL hydrobromide (Mw = 30–70 kDa), Trizma hydrochloride (TRIS-HCl), diisopropylethylamine (DIEA), dimethylformamide (DMF), acetonitrile, piperidine, acetic anhydride, triisopropylsilane (TIS), and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich (St Louis, MO, USA) and used as received. For the synthesis of PA molecules, protected amino acids, lauric acid, 4-(2′,4′-dimethoxyphenyl-Fmoc-aminomethyl)-phenoxyacetamido-norleucyl-4-methylbenzhydrylamine (MBHA) resin (rink amide MBHA resin) and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluorophosphate (HBTU) were purchased from Novabiochem®. Hank's balanced salt solution (HBSS), Neurobasal medium (NBM), B27 Supplement, L-glutamine, glutamate and gentamycin were all acquired from Gibco® Life Technologies (Grand Island, NY, USA). β3-Tubulin mouse monoclonal IgG antibody and Flash Phalloidin™ Red 594 were purchased from BioLegend (San Diego, CA, USA), and Alexa Fluor® 488 donkey anti-mouse IgG (H + L) secondary antibody Hoechst 33342 and Calcein, AM from Molecular Probes, Life Technologies (Grand Island, NY, USA). Fetal Bovine Serum (FBS), Propidium iodide (PI) solution and Fluoromount™ aqueous mounting medium were purchased from Sigma-Aldrich (St Louis, MO, USA).Synthesis and purification of PA molecules
K2PA (Lauryl–Val–Val–Ala–Gly–Lys–Lys–Am, Mw = 782.09 Da) and K2PA–IKVAV (Lauryl–Val–Val–Ala–Gly–Lys–Lys–Ile–Lys–Val–Ala–Val–Am, Mw = 1292.77 Da) were synthesized via Fmoc-protected solid-phase peptide synthesis method on rink amide MBHA resins, as previously described.35 The HBTU and N,N-diisopropylethylamine were used for peptide coupling. After each coupling step, 20% (v/v) piperidine/DMF solution was used to remove the Fmoc-protecting group and a 10% (v/v) acetic anhydride/DMF mixture was used to acetylate the unreacted amine groups. The peptides and side chain protecting groups were cleaved from the resin by using a TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TIS:H2O (95:2.5:2.5) solution. The peptides were precipitated overnight by using ice-cold diethyl ether. The peptide pellet was dissolved in ultrapure water, frozen at −80 °C and subsequently lyophilized.
:TIS:H2O (95:2.5:2.5) solution. The peptides were precipitated overnight by using ice-cold diethyl ether. The peptide pellet was dissolved in ultrapure water, frozen at −80 °C and subsequently lyophilized.
Liquid chromatography-mass spectrometry (LC-MS)
The mass spectra (MS) and liquid chromatogram (LC) of the non-bioactive and bioactive PAs were obtained with an Agilent 6530 Accurate-Mass Quadrupole time-of-flight LC-MS apparatus equipped with an electrospray ionization source connected to a reverse-phase high performance liquid chromatography (HPLC) system, as previously described.35 The PA molecules were dissolved in water at a concentration of 1 mg mL−1 and an Agilent Zorbax Extend-C18 column (2.1 mm × 50 mm) was used. A gradient of water (0.1% formic acid) and acetonitrile (0.1% formic acid) was used for LC-MS. The PAs were treated with 0.1 M HCl solution to remove residual TFA and further lyophilized. The purification of the synthesized PAs was carried out with an Agilent 1200 preparative reverse-phase HPLC system equipped with a Zorbax Extend-C18 column (21.2 mm × 150 mm). A gradient of (a) water (0.1% TFA) and (b) acetonitrile (0.1% TFA) was flowed at a rate of 0.65 mL min−1. The PAs were then freeze-dried prior to use.Circular dichroism (CD) spectroscopy
For the secondary structure analysis, 0.2 mg mL−1 K2PA, K2PA–IKVAV, HA, and HA/K2PA and HA/K2PA–IKVAV (1:1, v/v) mixtures were freshly prepared in ultrapure water. The CD spectra were recorded at room temperature in a JASCO J-815 spectrometer from 190 to 300 nm with a scanning speed of 100 nm min−1, a digital integration time of 4 s, a bandwidth of 1 nm, a data pitch of 0.1 nm and standard sensitivity. Measurements were repeated three times and averaged for each sample, and the final spectra were obtained after subtracting the background of the water sample.
Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy
The spectra of the dried films of the individual components (HA, K2PA, and K2PA–IKVAV), as well as of their mixtures (HA/K2PA and HA/K2PA–IKVAV) at a 1:1 (v/v) ratio were collected on a Bruker TENSOR 27 FTIR spectrometer fitted with a “Golden Gate” ATR module equipped with a diamond crystal. All spectra were obtained in the absorbance mode in the range of 4000–400 cm−1 by averaging 256 scans at a resolution of 4 cm−1. The collected data were baseline corrected and normalized in the OPUS spectroscopy software.
Powder wide-angle X-ray scattering (WAXS)
Prior to WAXS measurements, HA, K2PA, K2PA–IKVAV, HA/K2PA and HA/K2PA–IKVAV aqueous solutions were lyophilized for two days. Then, the diffraction patterns of the individual components (HA, K2PA, and K2PA–IKVAV) and co-assembled mixtures (HA/K2PA and HA/K2PA–IKVAV) in powder form were collected at room temperature on an Empyrean PANalytical diffractometer equipped with a PIXcel1D detector (active length = 3.35°) and a spinner flat sample holder in a Bragg–Brentano para-focusing optics configuration. Diffraction intensity data were collected in the 2θ range between 5–60° using CuKα radiation (λ ¼ 1.54 Å) filtered in Ni under beam conditions of 45 kV and 40 mA. Data were collected by the continuous counting method assuming a scanning step size of 0.0260° and a scanning rate of 0.02° min−1. Processing of the resultant diffractograms was performed with the HighScore Plus software and ICDD PDF4+ (2019 release) database. The Bragg's law was used to calculate the interplanar spacing distance (d).Zeta (ζ)-potential measurements
Prior to the preparation of the co-assembled systems and multilayer assemblies, the net electrical charge of the individual PAs, HA and PLL molecules was determined at 25 °C by ζ-potential measurements, using freshly prepared aqueous solutions at 0.5 mg mL−1 in 10 mM TRIS-HCl pH 7.4. The ζ-potential measurements were performed in triplicate and averaged for each sample using a Zetasizer Nano-ZS (Malvern Instruments Ltd, UK).Quartz crystal microbalance with dissipation monitoring (QCM-D)
The build-up of the supramolecular multilayered thin films encompassing oppositely charged biopolymers, PLL and HA, and further functionalized with an outermost PA layer was monitored in situ by the QCM-D apparatus operating in an automatic manner (Q-Sense Pro, Biolin Scientific, Sweden). Prior to the experiment, the gold (Au)-coated 5 MHz AT-cut quartz crystal substrates (AWS SNS 000043 A, Advanced Wave Sensors, Spain) were submitted to UV/ozone (UV/Ozone ProCleaner 220, BioForce Nanosciences, Inc.) treatment for 10 min, followed by immersion in a 5:1:1 ultrapure water:ammonia (NH4OH, 25%):hydrogen peroxide (H2O2, 30%) (v/v) cleaning solution in an ultrasound bath at 70 °C for 10 min. Subsequently, the quartz crystal substrates were rinsed with ultrapure water, dried under a soft stream of N2, and resubmitted to UV/ozone treatment for 10 min. Cleaned Au-coated quartz crystal substrates were then equilibrated in a 10 mM TRIS-HCl aqueous solution at pH 7.4 until a baseline was obtained. PLL and HA biopolymeric aqueous solutions at 0.5 mg mL−1 were alternately pumped at a constant flow rate of 50 μL min−1 into the QCM-D for 10 min each. In-between the adsorption of each biopolymeric material, a 10 min washing step was attempted to remove loosely adsorbed molecules and avoid the cross-contamination of the biopolymeric aqueous solutions. The assembly process was repeated five times until reaching a five PLL/HA multilayered film. At the end of the build-up of the (PLL/HA)5 multilayered film, the functionalization of the HA- and PLL-ending films with either 30 min adsorption of positively charged bioactive K2PA–IKVAV or non-bioactive K2PA was attempted. In the end of the assembly process, the assembled multilayered films were dried under a soft stream of N2. To study the stability of the films, similar multilayered films have been produced and exposed to cell culture medium (pH 7.4) for 96 h, at the end of the assembly process, to simulate the in vitro assays’ culture time. The Au-coated quartz crystal substrates were excited at multiple overtones (1st, 3rd, 5th, 7th, 9th, 11th, and 13th, corresponding to 5, 15, 25, 35, 45, 55, and 65 MHz, respectively) and the adsorption of the biopolymers and PAs solutions was followed in real time, at 25 °C, by monitoring the changes in the frequency (Δfn/n) and dissipation (ΔDn) shifts. The results presented herein correspond to the frequency and energy dissipation shifts obtained at the 7th overtone (n = 7; 35 MHz) since they presented the lowest level of noise. However, the results are representative of the other overtones.
The supramolecular multilayered thin films were similarly produced on freshly cleaned Au-coated glass substrates (IK4-Tekniker, Au layer: 50 nm; 1 × 1 cm2) as for the microscopy analysis and in vitro cell culture assays. Briefly, Au-coated glass substrates were firstly cleaned following the same procedure as reported for the QCM-D sensors. Then, a five bilayer PLL/HA film was assembled onto the Au-coated glass substrates by the alternate adsorption of PLL and HA aqueous solutions at 0.5 mg mL−1 in 10 mM TRIS-HCl at pH 7.4 for 10 min each. HA and PLL-ending multilayered films were further functionalized by the deposition of either K2PA or K2PA–IKVAV at 0.5 mg mL−1 in 10 mM TRIS-HCl at pH 7.4 (30 min). A rinsing step of 10 min in 10 mM TRIS-HCl at pH 7.4 was applied in-between the biopolymers deposition, as well as after the functionalization of the multilayered nanofilm with either the non-bioactive or bioactive PAs to remove unbounded molecules and avoid the cross-contamination of the biopolymeric solutions.
Transmission electron microscopy (TEM)
TEM analysis of each individual PA and HA/PA co-assemblies was performed on a field emission gun scanning transmission electron microscope (Hitachi STEM HD-2700, Hitachi High-Technologies, Japan), operating at an acceleration voltage of 200 kV. Prior to imaging, an aliquot of 10 μL of diluted aqueous solutions of K2PA, K2PA–IKVAV, HA/K2PA and HA/K2PA–IKVAV (1:1, v/v) mixtures at 0.2 mg mL−1 were separately prepared in ultrapure water, drop casted onto distinct carbon film-coated copper TEM grids (CF400-Cu-carbon film 400 square mesh copper grid), and incubated for 15 min. After removal of the excess of sample solution from the TEM grids by micropipette, the samples were negatively stained by placing 10 μL of uranyl acetate aqueous solution (0.5 wt%) on top, and the grids air dried in the fume hood overnight.
Atomic force microscopy (AFM)
Prior to analysis, the bare Au substrate, Au/(PLL/HA)5 and Au/(PLL/HA)5/K2PA–IKVAV multilayered thin films were rinsed with ultrapure water and dried in air at room temperature. Measurements were performed using a Dimension Icon atomic force microscope (Bruker, France) equipped with Nanoscope imaging software and operated in the ScanAsyst mode, using probes with a resonance frequency of 70 kHz and a spring constant of 0.4 N m−1. To evaluate the surface topographic details of the nanostructured thin film formulations, dried samples were scanned over 5 × 5 and 2 × 2 μm2 areas at a scan rate of 1 Hz and 512 × 512 pixel2 resolution. At least three samples and five distinct areas for each formulation were scanned.Primary neuronal cortical cells’ isolation and culture
Procedures involving animals were carried out with the permission of the i3S's animal ethical committee and in accordance with the European Union (EU) Directive (2010/63/EU) and Portuguese law (DL 113/2013). All the procedures were approved by the Portuguese official authority on animal welfare and experimentation (Direção-Geral de Alimentação e Veterinária; DGAV). All animals were maintained under a 12 h light/12 h dark cycle with free access to food and water.Primary mouse neuronal cortical cell cultures were prepared from the prefrontal cortex of E16.5 C57BL/6 mice embryos, as previously described.36 Briefly, embryos were obtained by caesarean section of pregnant C57BL/6 mice euthanized by cervical dislocation. The isolated prefrontal cortex was digested with trypsin (1.5 mg mL−1), washed with HBSS containing 10% (v/v) heat inactivated FBS (56 °C, 30 min) and then washed only with HBSS to remove FBS. The tissue was then dissociated in NBM using a pipette and cells were plated onto previously UV sterilized (30 min) Au-functionalized substrates at a density of 9 × 104 viable cells per cm2 (trypan blue assay). Bare Au substrates were used as blank controls. Cells were plated in a total volume of 50 μL in NBM supplemented with 2% (v/v) serum-free B27, L-glutamine (0.5 mM), glutamate (0.025 mM) and gentamycin (50 μg mL−1) and allowed to adhere for 2 h at 37 °C with 5% CO2. After that, 0.5 mL of complete medium was added per sample and cultured for 4 days.
Primary neuronal cortical cells’ viability
Cell viability was assessed by Calcein and PI staining, using image-specific black 24 well plates with flat and clear bottom (Ibidi, Germany). Cells cultured in the Au substrates were incubated with 4 μM Calcein for 10 min at 37 °C with 5% CO2. Counterstaining with 3 μM PI was performed for another 10 min in the same conditions. Thereafter, staining media was replaced by phosphate buffered saline (PBS), and Au substrates were turned upside down into each well. Fluorescence (Calcein: λex = 494, λem = 517 and PI: λex = 536, λem = 617) was analyzed in a microplate reader (SynergyMx, Biotek, VT, USA). Bare Au substrates were used as fluorescence controls. Results are expressed as the mean relative fluorescence units (RFU) ± standard deviation (SD) of three independent experiments (n = 3) performed with triplicates, except for the Au/(PLL/HA)5/PLL condition in which a n = 1 with triplicates was performed.Morphology of primary neuronal cortical cells
Cells were fixed with 4% (v/v) paraformaldehyde and permeabilized for 10 min in PBS with 0.1% (v/v) Triton X-100 (PBST), followed by a blocking step with 5% (v/v) bovine serum albumin (BSA) in PBST for 30 min at room temperature. Then, cells were O/N incubated with the primary antibody β3-tubulin (1:500) diluted in 1% (v/v) BSA at 4 °C. After incubation time, cells were rinsed in PBS and incubated with the respective fluorescent secondary antibody (488 anti-mouse IgG) diluted in 1% (v/v) BSA for 1 h at room temperature. After another washing step, cells were incubated with 594 flash-phalloidin (1:40) according to manufacturer's instructions and counterstained with Hoechst 33342. Substrates were mounted on slides with Fluoromount™ mounting medium and imaged in an inverted fluorescence microscope (20× objective; Axiovert 200M, Zeiss). Regarding the adhesion assay, images were analyzed using Ilastik (v1.3.2) and CellProfiler (v3.1.8) softwares to determine the number of nuclei per field of view (FOV) (3.8 × 105 μm2). At least 6 fields of each substrate were analysed, and results are shown as mean ± SD of three independent experiments (n = 3) performed with triplicates.
Neurite outgrowth assay
Firstly, the number of β3-tubulin positive cells presenting at least one neurite was determined per FOV (3.8 × 105 μm2). Subsequently, the average length of the longest neurite was calculated. The longest neurite of each cell was measured using the Simple Neurite Tracer plug-in of the image analysis software Fiji (ImageJ, NIH).37 For that, the colour channels were split and the green channel, corresponding to the β3-tubulin, was selected for analysis. Images from at least 6 randomly selected fields were analysed for each replicate. The mean ± SD of the above referred parameters was calculated based on three independent experiments (n = 3) with triplicates.Statistical analysis
Unless otherwise stated, all experiments were performed in triplicate (n = 3) and the data are presented as the mean ± SD. For statistical analysis, one-way ANOVA followed by Tukey's post-hoc multiple comparison test was performed using the GraphPad Prism 9.5.0 (GraphPad Inc.) software. Statistically significant differences were considered for *p < 0.05.Results and discussion
Characterization of biopolymers, self-assembling peptide amphiphiles, and supramolecular co-assemblies
In this study, biopolymeric multilayered thin films encompassing five PLL/HA bilayers were built-up on Au-coated substrates and further electrostatically functionalized with an outer layer of either non-bioactive or bioactive PA molecules for being used as an in vitro neuronal cell culture platform. Fig. 1A and B depicts the chemical structure of the biopolymers (HA and PLL), and synthesized bioactive (K2PA–IKVAV, chemical formula: Lauryl-VVAGKKIKVAV-Am) and non-bioactive (K2PA, chemical formula: Lauryl-VVAGKK-Am) self-assembling PA molecules to be used as building blocks in the development of supramolecular co-assemblies (Fig. 1C) and nanostructured multilayered thin films. HMW HA was chosen due to its higher viscosity, stronger hydrogen bonding-driven intermolecular association between HA chains, as well as faster exponential growth of PLL/HA multilayered films, thus leading to thicker, more viscoelastic, and stable films than their counterparts built with medium- and, mainly, low-molecular-weight HA.38 Moreover, it better emulates the native neural ECM and plays a vital role in regulating neuronal cell functions.32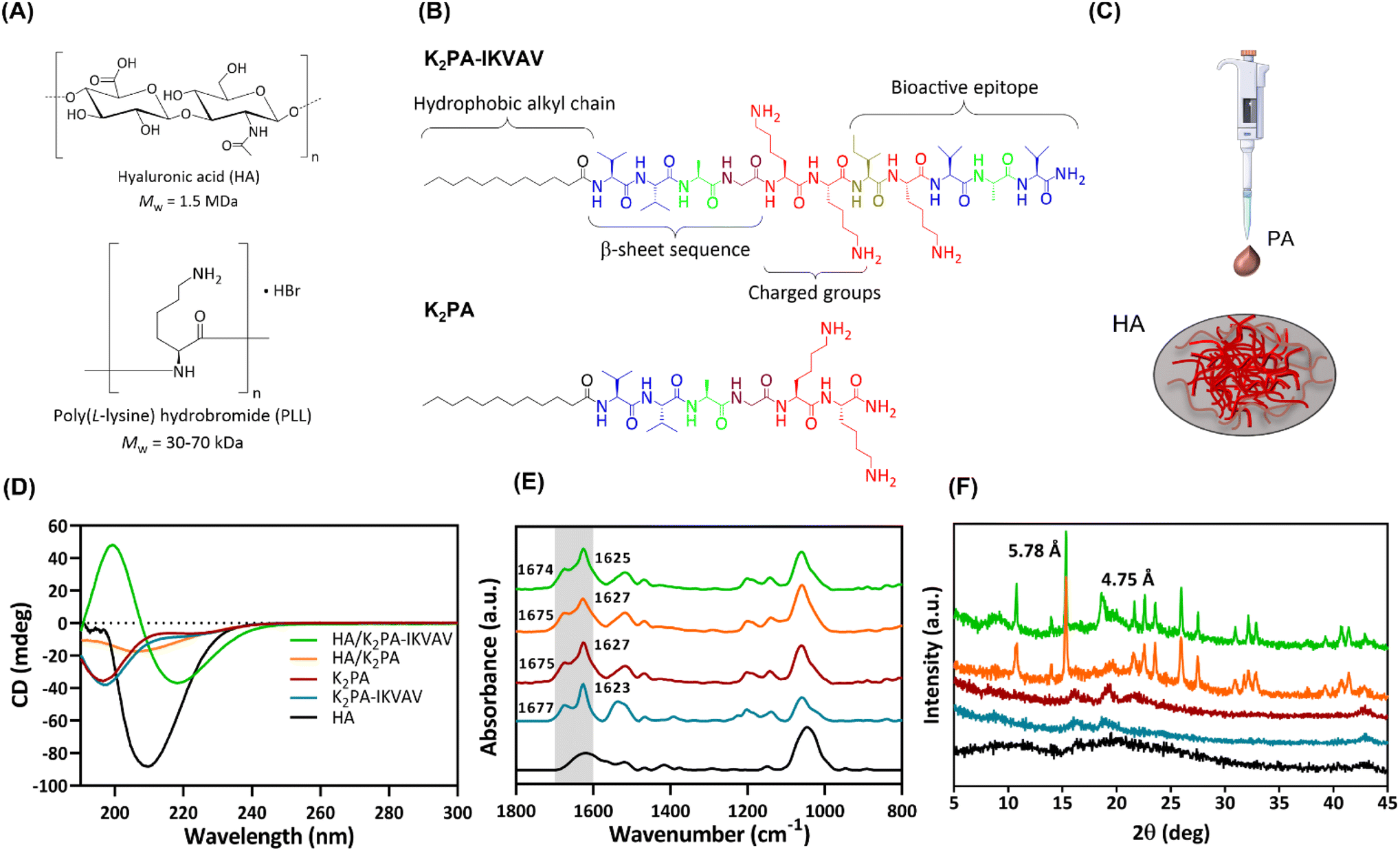 | ||
| Fig. 1 (A) Chemical structures of HA and PLL and (B) bioactive (K2PA–IKVAV) and non-bioactive (K2PA) PAs. (C) Schematic illustration of the preparation of either HA/K2PA or HA/K2PA–IKVAV supramolecular co-assemblies. Secondary structure analysis of the individual components and co-assembled systems using (D) CD, (E) ATR-FTIR, and (F) WAXS. | ||
Both PAs comprise a hydrophobic lauryl alkyl tail, which drives self-assembly and formation of peptide conformations, a short β-sheet sequence (VVAG) that promotes intermolecular hydrogen bonding,26,39 and a hydrophilic peptide headgroup, composed by lysine amino acid residues (KK), to enhance the solubility of the PAs in aqueous media. The bioactive peptide also denotes a LAM-derived IKVAV sequence attached to the headgroup to enhance neuronal cell growth.6,35,39 The amphiphilic peptides were synthesized by standard Fmoc solid-phase peptide synthesis method, characterized by LC-MS and purified by HPLC (Fig. S1 and S2, ESI†).
The analysis of the conformational arrangements of the PA molecules upon co-assembly with oppositely charged HA was assessed by spectroscopic techniques. The CD spectrum of HA revealed a negative band at 210 nm (Fig. 1D), assigned to the n–π* transitions of the N-acetylglucosamine chromophore.40 The individual PAs denoted a broad negative band in the far UV region (∼197 nm), characteristic of a major contribution of the disordered random coil conformation to the final secondary structure.41
The co-assembly of the cationic K2PA–IKVAV with the oppositely charged HA revealed the formation of an ordered β-sheet structure denoting a negative minimum at ∼218 nm and a positive maximum at ∼200 nm. On the other hand, the HA/K2PA system revealed a weak negative band at ∼204 nm, suggesting the presence of some contribution of β-sheet secondary structure. Noteworthy, the HA/K2PA–IKVAV co-assembled system showed a slight redshift from the typical canonical β-sheet band positions, which can be due to the presence of twists along the long axis of the fibers.42 The slight blueshift in the negative band of the HA/K2PA supramolecular system can suggest changes in intermolecular interactions that destabilize the β-sheet conformation.43 It has been reported that peptide sequences containing lysine amino acid tend to disrupt the effective packing of β-sheets due to electrostatic repulsion, while those exhibiting valine or isoleucine denote higher propensity to form β-sheet structures.44,45 The secondary structure of the individual PAs and HA/PA co-assemblies was predicted and quantified as previously described (Fig. S3, ESI†).46 It was found that, upon the co-assembly with HA, the PA's random coil content was lowered (44.2% for HA/K2PA and 43.1% for HA/K2PA–IKVAV), while the β-sheet and α-helix conformations (32.9 and 7.2% for HA/K2PA and 42.2 and 6.6% for HA/K2PA–IKVAV, respectively) prevailed.
ATR-FTIR measurements complemented the CD analysis on the molecular scale structure of the co-assemblies based on the analysis of the amide I region (Fig. 1E, highlighted in grey), primarily associated with the stretching vibrations of the peptide carbonyl groups.47 The individual spectra of both individual K2PA and K2PA–IKVAV molecules showcased a major strong and sharp peak at ∼1627 and 1623 cm−1, respectively, indicative of the presence of a β-sheet secondary structure (Fig. 1E),48,49 as well as a minor band at ∼1675 and ∼1677 cm−1, assigned to lysine side chains.50 The absence of a band at ∼1695 cm−1, characteristic of antiparallel β-sheet structures,51 suggests that the peptides adopt a β-sheet secondary structure with a parallel orientation. The band at ∼1540 cm−1, which was also observed on the spectra of HA and HA/PA co-assemblies, is assigned to the amide II band, mainly to N–H bending but also to C–N stretching.52
The HA/K2PA and HA/K2PA–IKVAV mixtures revealed broader absorption bands at ∼1625 cm−1, which suggest an increase of the hydrogen bonding β-sheet network. The ATR-FTIR spectrum of HA showed peaks in the range between 950–1200 cm−1, attributed to the typical skeletal vibrations of the saccharide units (asymmetric C–O–C stretching vibration),52 and at ∼1610 cm−1 and 1415 cm−1, assigned to the asymmetric and symmetric vibrations of the carboxylate anion (C–O stretching vibration).
To obtain further insights on the molecular arrangement of the individual HA and PAs, and co-assembled systems, we resorted to powder wide-angle X-ray scattering (WAXS) (Fig. 1F). As expected, the weak, broad and small number of diffraction peaks observed for HA and PA molecules reveal their amorphous and disordered structure. On the other hand, the HA/K2PA and HA/K2PA–IKVAV co-assembled systems displayed an intense and sharp Bragg peak at 15.3° (d-spacing of 5.78 Å). This peak is indicative of the formation of closed packed lauryl alkyl tails, which are responsible for the hydrophobic core formation within the β-sheet nanofiber networks.53 Noteworthy, the HA/K2PA–IKVAV co-assembled system denoted a Bragg peak at 18.6° (d-spacing of 4.75 Å), which was not observed in the case of the HA/K2PA system. This signal indicates crystalline ordering of the molecules in a β-sheet arrangement assigned to the β-sheet inter-strand spacing,6,54 suggesting a higher degree of β-sheet packing when compared with the HA/K2PA co-assembly.
The diffraction patterns corroborate the CD and ATR-FTIR findings, revealing the formation of ordered β-sheet secondary structures for both HA/K2PA and HA/K2PA–IKVAV co-assembled supramolecular systems, the latter system denoting a higher extent of β-sheet structure.
The morphology of the individual PAs and co-assembled supramolecular systems was investigated by TEM (Fig. 2 and Fig. S4, ESI†). The individual PAs self-assembled into supramolecular nanofibers in solution with 6.2 ± 1.2 nm and 17.2 ± 3.5 nm in width, for K2PA–IKVAV (Fig. S4A, ESI†) and K2PA (Fig. S4B, ESI†), respectively. However, while K2PA–IKVAV revealed the formation of long, thin, high-aspect-ratio cylindrical nanofibers with a few twisted nanofibers (red arrows in Fig. S4A, ESI†), the shorter K2PA denoted nanotape-like structures (Fig. S4B, ESI†). The formation of entangled networks of supramolecular nanofibers was denoted in the HA/K2PA–IKVAV supramolecular system, with 13.7 ± 2.5 nm in width (Fig. S4C, ESI†). This result can be explained by the interaction between the fibers exhibiting hydrophobic amino acids of the IKVAV epitope sequence, promoting aggregation.41 On the other hand, the HA/K2PA supramolecular system self-assembled into narrower nanobelt-like structures with 7.2 ± 1.6 nm in width (Fig. S4D, ESI†). These results corroborate the CD, ATR-FTIR and WAXS data once supramolecular nanofiber networks are the result of β-sheet structures.
 | ||
| Fig. 2 Representative TEM micrographs of the nanofibrous structure formed by (A) K2PA–IKVAV, (B) K2PA, (C) HA/K2PA–IKVAV, and (D) HA/K2PA systems (1:1 v/v ratio) at 0.2 mg mL−1. The magnification factor is ×100k. Scale bars: 300 nm. | ||
Real-time monitoring of the build-up of (PLL/HA)5/PAs supramolecular multilayered thin films
Prior to the build-up of the PA-functionalized biopolymer-based multilayered nanoassemblies, the net electrical charge of the freshly prepared 0.5 mg mL−1 PLL, HA, K2PA and K2PA–IKVAV aqueous solutions at pH 7.4 (10 mM TRIS-HCl) was assessed by zeta (ζ)-potential. The ζ-potential measurements revealed the cationic nature of PLL (+20.9 ± 0.5 mV), K2PA (+35.0 ± 1.8 mV) and K2PA–IKVAV (+46.2 ± 0.6 mV), and the anionic nature of HA (−32.7 ± 0.6 mV) aqueous solutions. Accordingly, we studied the possible build-up of electrostatic-driven soft PLL/HA multilayered thin films and their further functionalization with an outer layer of oppositely charged PAs (Fig. 3A) onto Au-coated quartz crystal sensors by QCM-D (Fig. 3B). This technique enable us to detect minute changes in the hydrodynamic mass (ng cm−2) due to changes in the resonance frequency of the quartz crystal sensor, as well as measure the viscoelastic properties of the adsorbed layers via the energy dissipated in the mechanical oscillation of the quartz sensors.55,56 Similar biopolymeric multilayered thin films, encompassing an extra PLL layer, i.e., (PLL/HA)5/PLL films, were also assembled to understand the influence of the outer PLL layer in the adsorption of the PAs.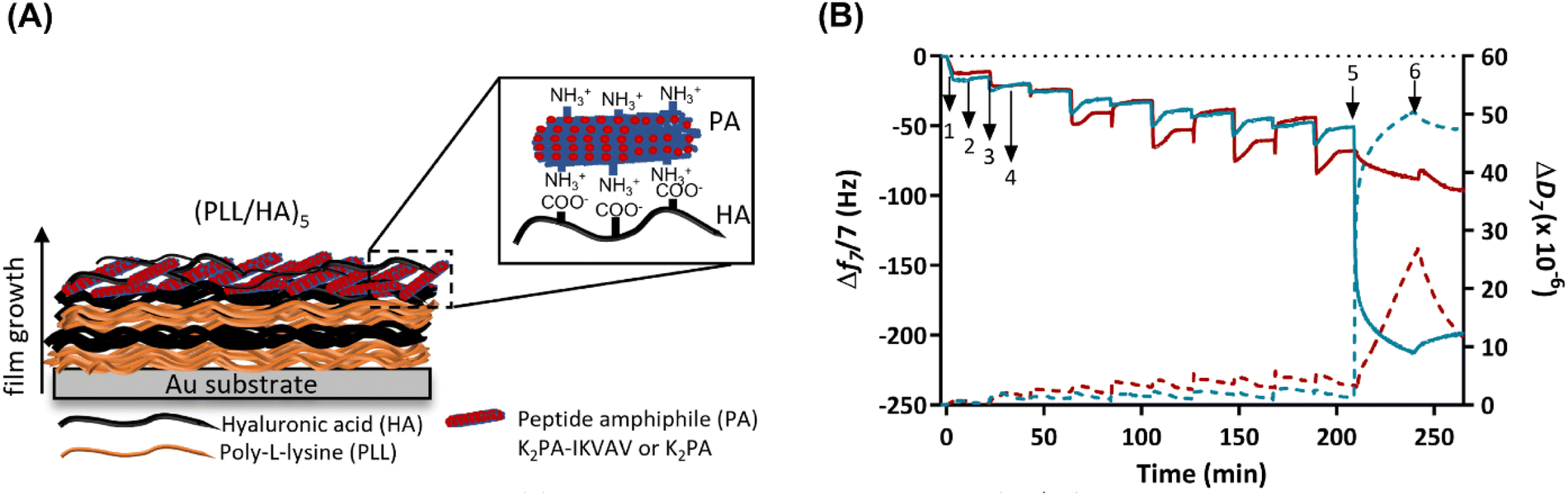 | ||
| Fig. 3 Build-up of the supramolecular multilayered thin films. (A) Schematic representation of the build-up of electrostatically driven (PLL/HA)5 multilayered thin films encompassing an outer PA layer. (B) Real time QCM-D monitoring of the normalized frequency (Δfn/n; solid lines) and dissipation (ΔDn; dashed lines) changes, obtained at the 7th overtone (n = 7; 35 MHz), as a function of time for the build-up of (PLL/HA)5 multilayered thin films functionalized with an outer K2PA (red) or K2PA–IKVAV layer (blue) onto Au-coated quartz crystal sensors. Numbers refer to the adsorption of PLL (1), HA (3), K2PA or K2PA–IKVAV (5), and intermediate rinsing steps (2, 4 and 6). | ||
As shown in the Fig. 3B, the sequential decrease in the normalized frequency shift at the 7th overtone (Δf7/7) as a function of time after the adsorption of each layered material indicates that both PLL and HA adsorbed successfully onto the Au-plated quartz crystal surface, as well as the successful interaction and stable LbL growth of the five PLL/HA bilayered film. However, the decrease in the Δf7/7 right after the adsorption of each HA layer experienced an overshoot, i.e., an increase towards more positive values, which means a decrease in the adsorbed mass. Such behaviour is assigned to the diffusion of free PLL chains “out” of the film (i.e., deswelling) and their complexation by the HA chains.57–59 On the other hand, the increase and decrease of ΔD7/7 after the adsorption of the PLL and HA layers, respectively, confirm the swelling and the deswelling behaviour of the soft and viscoelastic polymeric film. Moreover, the rinsing steps in-between the deposition of PLL and HA produced negligible changes in both Δf7/7 and ΔD7/7, thus revealing the strong association between the assembled biopolymers and the irreversible nature of the adsorption process. The build-up and characterization of PLL/HA multilayered thin films is well described in the literature.58,60 However, to the best of our knowledge, the adsorption of PA upon the precursor PLL/HA multilayered thin film has not been explored to date.
Protein adsorption can be sensed by QCM-D not only by the decrease of the frequency shift but also by the increase of the energy dissipation, which is also a typical behaviour of soft and hydrated polymeric films.61 In this work, the flushing of a positively charged K2PA–IKVAV aqueous solution onto the hydrophilic HA-ended multilayered film led to a significant decrease in the Δf7/7 by 2-fold (∼−150 Hz) when compared to the adsorption of the non-bioactive PA. Such behaviour reveals that the positively charged bioactive PA adsorbed strongly onto the underlying negatively charged HA layer when compared to the non-bioactive counterpart. On the other hand, an exponential increase in ΔD7/7 was observed upon the adsorption of the bioactive PA, which reveals the soft and viscoelastic nature of the bioactive PA layer. A similar behaviour has been reported for the adsorption of other biomolecules, such as bovine serum albumin, fibrinogen, and γ-globulin on polymeric layers adsorbed onto Au-coated quartz sensors, as monitored by the QCM-D apparatus.61,62 Moreover, we hypothesize that the higher ΔD7/7 values obtained after the deposition of the K2PA–IKVAV layer showcase its higher ability to bind and present water molecules on the outer film surface, i.e., form a surface hydration shell after the self-assembly of the PA nanofibers driven by charge neutralization, indicating that the film is not rigid showing damping properties. The adsorption of the positively charged K2PA onto similar HA-ended film also induced a decrease in the Δf7/7, i.e., increase in the adsorbed mass, and increase in the ΔD7/7, i.e., increase in the viscoelasticity but in much lower extent. This behaviour correlates well with the CD and WAXS data in which a higher degree of crystalline ordering in a β-sheet arrangement was found for the HA/K2PA–IKVAV co-assembled system when compared to the HA/K2PA counterpart, thus meaning that a higher extent of entangled nanofiber networks was formed by the former. Moreover, the rinsing step after the adsorption of either K2PA or K2PA–IKVAV led to negligible changes in the Δf7/7, thus revealing the irreversible nature of the adsorption process.
To understand the role of attractive electrostatic interactions on the successful deposition of the PAs, the adsorption of K2PA–IKVAV was attempted onto similarly positively charged PLL-ended multilayered films (Fig. S5, ESI†). Although both PLL- and HA-ended films present similar ΔD7/7 values after the adsorption of K2PA–IKVAV, and thus similar viscoelastic properties, the decrease in the PLL-ending films was lower (∼−75 Hz) than the one observed for its adsorption onto HA-ended films (∼−150 Hz). Such behaviour reveals that attractive electrostatic forces are not the sole interactions contributing to the build-up and stabilization of the supramolecular multilayered films. In fact, the hydrophobic collapse of the PA aliphatic chains, the hydrogen bonding interactions among the β-sheet forming peptide sequence, and the electrostatic repulsion between the charged lysine amino acid residues also play important roles on the assembly and stabilization of PAs.49,63 Moreover, the stability of the (PLL/HA)5/K2PA–IKVAV multilayered film was assessed by exposure to cell culture medium for 4 days (in vitro assays’ culture time). The injection of cell culture medium led to a continuous decrease of Δf7/7 and increase of ΔD7/7, thus suggesting the establishment of supramolecular interactions between the proteins present in the growth medium and the previously adsorbed K2PA–IKVAV molecules and the increase in the viscoelastic properties of the assembled film, respectively (Fig. S6, ESI†). Therefore, the native nanofilm showed to be stable in culture medium conditions for at least 4 days.
Morphologic characterization of the nanostructured supramolecular multilayered thin films
The surface morphological features, i.e., the surface roughness and morphology of the nanostructured (PLL/HA)5 multilayered thin films prior to and following the adsorption of the bioactive PA were evaluated by AFM. Fig. 4 displays the representative 2D topographical and deflection images of the bare Au, and (PLL/HA)5 and (PLL/HA)5/K2PA–IKVAV multilayered thin films assembled onto the Au substrate in which one could denote clear morphological and structural differences (Fig. 4A). The smooth surface topography exhibited by the bare Au surface revealed the presence of pinholes, i.e., surface defects which were covered after the assembly of five PLL/HA bilayers, leading to the formation of rougher biopolymeric islet-like structures over the surface, similarly to other studied biopolymeric systems.64 The subsequent adsorption of K2PA–IKVAV on top of the (PLL/HA)5 multilayered thin film led to the formation of randomly oriented 1D nanofibrous-like networks (Fig. 4A and Fig. S7, ESI†), uniformly distributed over the polymeric film surface, similar to the fibrillar surface morphology of collagen/alginate films.65 Such behaviour is corroborated by the sequential increase in the root-mean-square surface roughness (Rq, Fig. 4B) and average height (Ra, Fig. 4C). The AFM data correlates well with the previous results once negatively charged HA triggered the self-assembly of positively charged PA molecules via charge neutralization and led to the formation of nanofiber networks. Although both PAs denote an intrinsic capacity to fold into an organized β-sheet secondary structure upon interaction with HA, it is noteworthy that the bioactive PA displays the LAM-derived IKVAV pentapeptide sequence, which is relevant to instruct cell adhesion, migration, proliferation, differentiation, and neurite outgrowth.21 As such, we hypothesized that the nanostructured supramolecular multilayered thin films herein proposed could simultaneously provide an enhanced ECM-mimetic nanofibrillar-like environment and high density of bioactive cues for controlling neuronal growth. | ||
| Fig. 4 Morphological characterization of the supramolecular multilayered nanoassemblies. (A) Representative AFM topographic (top) and deflection (bottom) images taken in air for the bare Au substrate, and (PLL/HA)5 and (PLL/HA)5/K2PA–IKVAV functionalized Au substrate. The images scan sizes are 5 × 5 μm2 and 2 × 2 μm2 for main and inset images, respectively. (B) Root-mean-square surface roughness (Rq) and (C) average height (Ra) obtained for the bare Au, (PLL/HA)5 and (PLL/HA)5/K2PA–IKVAV functionalized Au substrate. Data is presented as mean ± SD. Significant differences were found for *p < 0.05. | ||
In vitro assays of supramolecular multilayered thin films
The in vitro biological performance of biomaterial scaffolds using primary cells provides crucial context for the applicability of the in vitro data assays by more accurately mimicking the behavior of neuronal cells in vivo.66 As such, they represent an important and functional tool to explore mechanisms behind the regeneration potential of biomaterials on neuronal tissues. Herein, primary cortical neuronal cells were used to infer about the neuro-compatibility of the biopolymeric nanostructured multilayer thin films functionalized with either bioactive or non-bioactive PAs by means of cell adhesion and viability, morphology, and neurite outgrowth studies.Primary cortical neuronal cells’ adhesion and viability
The process of cell adhesion is a source of specific cues involved in signal transduction, which influences cytoskeletal dynamics, gene expression and growth. As such, it is an important parameter to meet when designing biomaterials for tissue engineering and regenerative medicine applications.67 To quantitatively evaluate the cellular adhesion, primary cortical neurons were cultured for 4 days in vitro on top of the different substrate formulations, and further stained with a nuclear marker. As shown in Fig. 5A, all tested LbL formulations improved the adhesion of neuronal cells when compared to the bare Au substrate. Moreover, despite no significant differences were observed between distinct formulations, the K2PA–IKVAV-ended surfaces showed an increased ability to promote neuronal cell adhesion.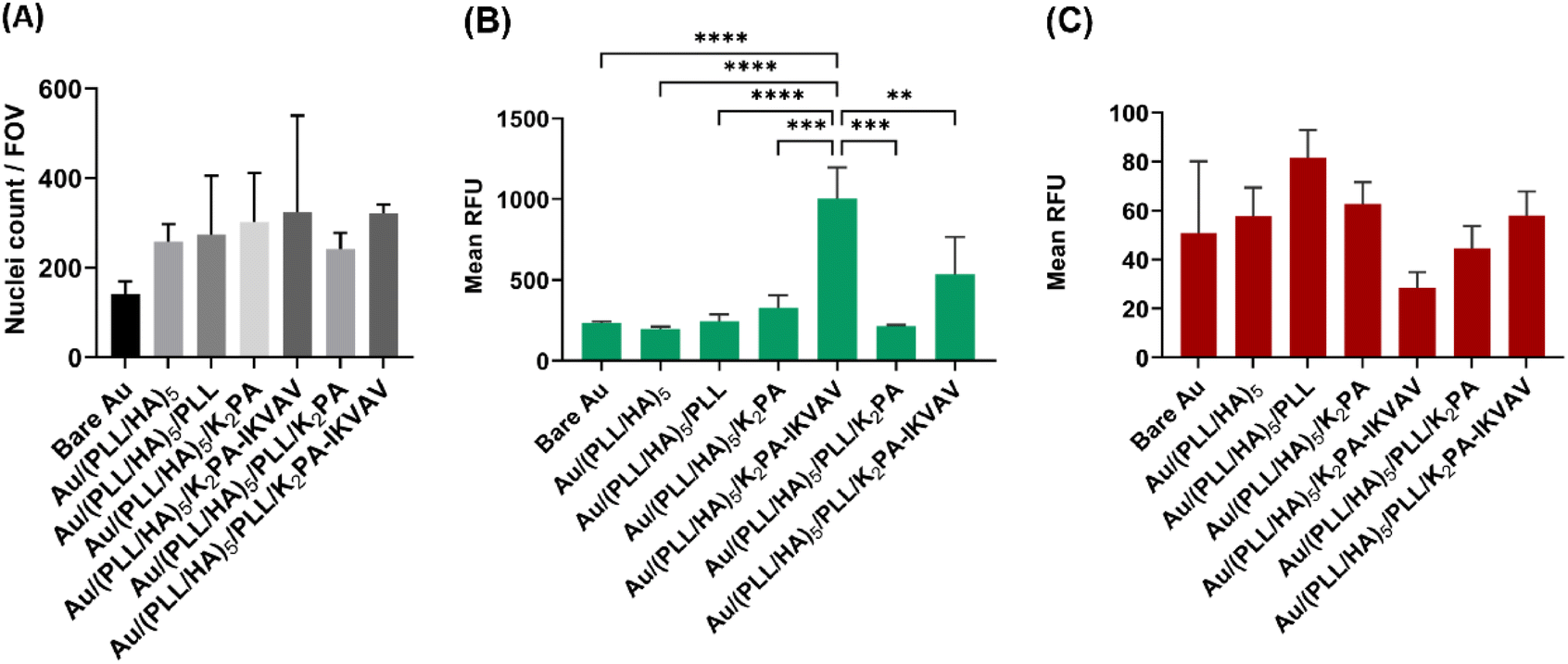 | ||
| Fig. 5 Adhesion and viability of primary neuronal cortical cells at 4 days of in vitro culture on uncoated and LbL coated Au surfaces. (A) Quantification of nuclei per field of view (FOV), with Hoechst as nuclear marker. Fluorescence quantification of (B) Calcein (live cells) and (C) PI (dead cells) represented as relative fluorescence units (RFU). Results are expressed as the mean ± SD of three independent experiments (n = 3) performed with triplicates. Significant differences were found for *p < 0.05 (****p < 0.0001, ***p = 0.0001, **p = 0.0065). There was no difference in the (A) number of nuclei per FOV and (C) dead cells between the different surface formulations. | ||
The cellular viability was also assessed after 4 days of culture in vitro via a live/dead assay. Consistent with the results of the cell adhesion studies, K2PA–IKVAV-ended multilayered films showed the best cell response in terms of primary neurons viability. However, only the Au/(PLL/HA)5/K2PA–IKVAV film showed a significant increase in the fluorescence of live cells when compared to the other conditions (Fig. 5B). Concomitantly, this condition also showed the lower values for cell death (Fig. 5C). The enhanced neuronal response registered for the cells cultured on the (PLL/HA)5/K2PA–IKVAV nanofilms when compared to the (PLL/HA)5/PLL/K2PA–IKVAV counterparts can be explained by the increased mass of PA adsorbed in the surface of the HA-ended films, as observed in QCM-D results, and thus higher density of IKVAV epitopes on the film surface. This result agrees with the findings reported in the literature where positively charged VEGF achieved higher surface concentration when adsorbed on a HA-ending film (1.5 ng cm−2) when compared to its adsorption on PLL-ending films (0.72 ng cm−2).68 Moreover, the higher density of IKVAV epitopes presented to the cells by PA nanofibers when compared to either LAM or IKVAV soluble peptide has been reported as a key factor for the preferential differentiation of neural progenitor cells into neurons instead of glial cells.69 These results highlight the importance of the availability of the LAM-derived IKVAV epitope in the multilayered film to enhance both the neuronal cell attachment via selective integrin binding and the cell viability.
Primary cortical neuronal morphology
Following up the differences observed on neuronal viability for cells cultured on the various multilayered nanofilms, distinct morphology was also noticed among the different film formulations. Immunostaining for β3-tubulin and F-actin allowed the evaluation of the cell morphology according to distinct neuronal developmental stages, as described by Banker.70 The bioactive nanostructured supramolecular multilayered nanofilms, i.e., Au/(PLL/HA)5/K2PA–IKVAV enhanced the overall neuronal morphology, in which cells displayed a polarized morphology and elongated processes, corresponding to the advanced stage of neuronal development. Such behavior allowed neurons to contact neighboring cells and establish early neuronal networks (Fig. 6).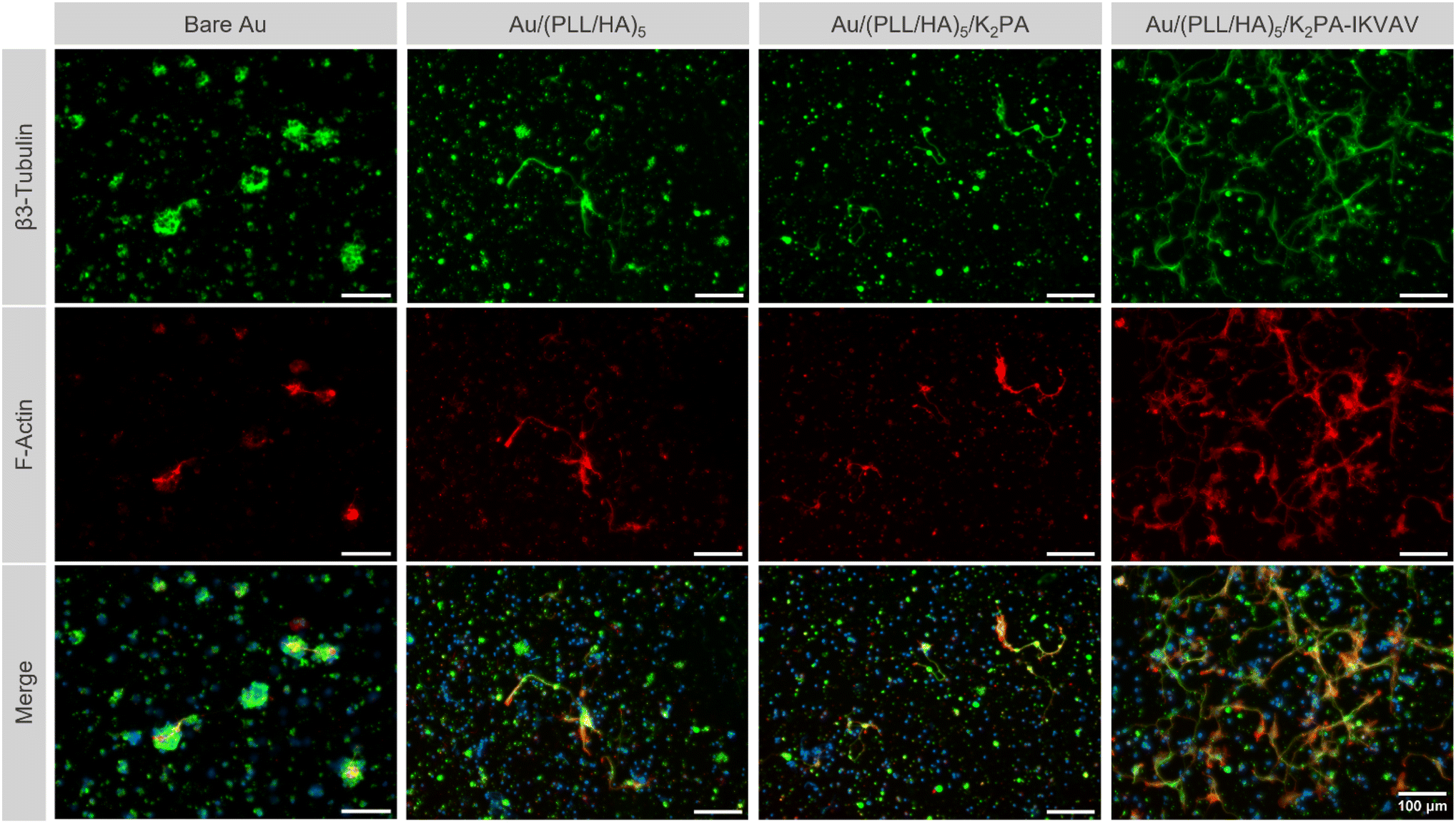 | ||
| Fig. 6 Morphology of primary neuronal cortical cells at 4 days of in vitro culture on uncoated and LbL coated Au surfaces. Representative fluorescence microscopy images of primary neuronal cortical cells at 4 days of culture on the bare Au substrate, and (PLL/HA)5, (PLL/HA)5/K2PA and (PLL/HA)5/K2PA–IKVAV functionalized Au substrate, after immunostaining with β3-tubulin (green), F-actin filaments (red), and nuclei counterstaining with Hoechst (blue). Scale bars: 100 μm. | ||
Contrarily, on the PA-free Au/(PLL/HA)5 and non-bioactive Au/(PLL/HA)5/K2PA multilayered films just a few sprouting neurites were visible and all were identical to one another, corresponding to the early-stage of neuronal development.70 Both multilayered thin films seem to be insufficient to promote cell polarization and cellular network connection to the same extent to the one observed on the multilayered nanofilms having the bioactive PA. Moreover, for all conditions, the cationic PLL-ended film proved to be disadvantageous for the development of neurites, even in the presence of the adsorbed bioactive peptide K2PA–IKVAV, denoting few neurites and the absence of cell polarization (Fig. S8, ESI†). In particular, the Au/(PLL/HA)5/PLL/K2PA supramolecular multilayered thin film promoted the formation of neuronal clusters, suggesting greater cell–cell interaction over cell–matrix adhesion. This behavior implies that the cells do not properly anchor to the matrix, generating cellular clustering that compromises their viability and maturation.
Neurite development of primary cortical neurons
Neurite outgrowth on the distinct formulations was evaluated at 4 days of culture through the quantification of the number of cells showing at least one neurite per FOV (Fig. 7A) and the average length of the longest neurite per cell (Fig. 7B). The results revealed that primary neuronal cortical cells cultured on Au/(PLL/HA)5/K2PA–IKVAV substrates developed significantly more neurites than the neurons cultured on top of all the other biopolymeric multilayered nanofilms and the bare Au substrate (Fig. 7A). Furthermore, the biopolymeric multilayered thin films having PLL as the outer layer, i.e., Au/(PLL/HA)5/PLL stands out for showcasing the lowest number of neurites, which correlates with the observed lower viability and higher mortality.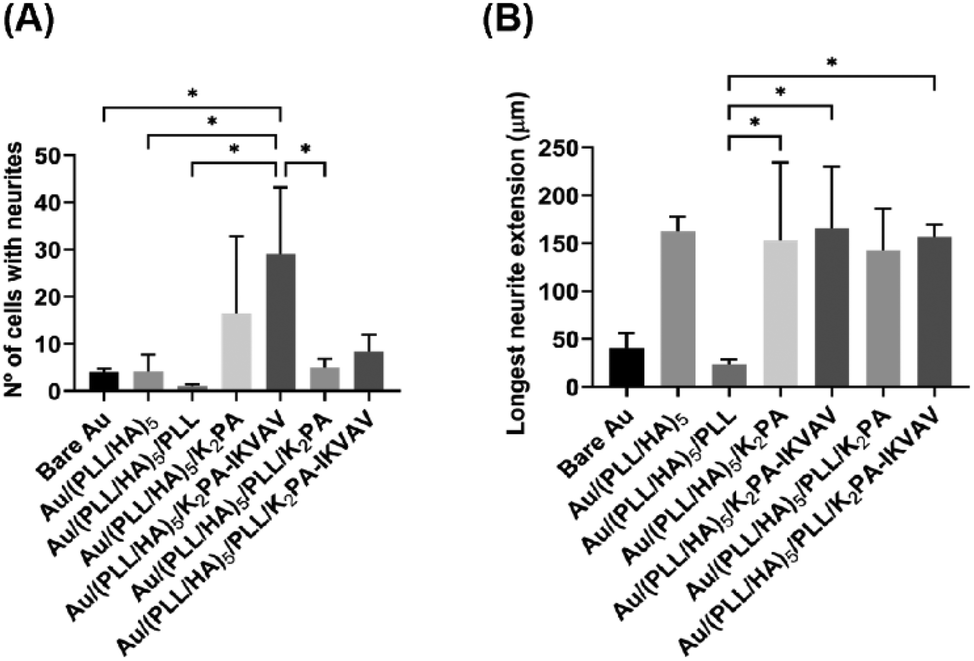 | ||
| Fig. 7 Effect of the distinct LbL film formulations on the outgrowth and length of neurites-bearing primary neuronal cortical cells. (A) Number of cells with at least one neurite per FOV. (B) Length of the longest neurite per cell. Results are shown as mean ± SD of three independent experiments (n = 3), performed with triplicates. Significant differences were found for *p < 0.05. | ||
The extension of the developed neurites was also measured. Apart from the Au/(PLL/HA)5/PLL nanofilm which demonstrated a significantly lower ability to promote neurite extension, the cells cultured on top of either bioactive or non-bioactive PA-functionalized multilayered nanofilms achieved neurite lengths in the range of ∼107–158 μm (Fig. 7B). The HA-ended multilayered nanofilms functionalized with either the non-bioactive K2PA or bioactive K2PA–IKVAV outer layers achieved neurite lengths of 154 ± 82 μm and 158 ± 77 μm, respectively. We hypothesize that the significant neurite outgrowth observed for the Au/(PLL/HA)5/K2PA–IKVAV nanofilm is assigned to the denser presentation of the bioactive LAM-derived IKVAV motif on top of the (PLL/HA)5 film, promoted by the interaction with HA. Overall, the results showed that the (PLL/HA)5/K2PA–IKVAV supramolecular nanofilms enhanced the neuronal cell network by significantly improving the neurite outgrowth, which corroborated the increased cell viability and development of neuronal morphology.
Conclusion
We have successfully demonstrated the electrostatic-driven supramolecular immobilization of LAM-derived IKVAV-containing PA on biocompatible PLL/HA nanostructured multilayered films for instructing neuronal cell functions. This straightforward methodology enabled the formation of neural ECM-mimetic supramolecular nanofiber matrices driven by charge neutralization and exhibiting a high-epitope density, which enhanced the cell viability and stimulated neurite outgrowth.We anticipate that this highly versatile bottom-up approach could be used to coat virtually any kind of surfaces, including implantable biomaterials, and be extended to other material combinations to improve the biological interactions with any cell type and modulate cell functions. In addition, it could enable bioengineering more robust neural ECM-mimetic scaffolds to stimulate specific cell-signaling pathways. Those include the development of self-standing 2D multilayered membranes and 3D bioarchitectures which can integrate a wide repertoire of molecular building blocks, including multiple bioactive molecules, and exhibit (multi)functional properties to trigger a desired cell response. Such biomaterial-based devices, which can be used as drug/therapeutic carriers, bioactive networks, or cell seeding scaffolds, could represent advanced therapeutic strategies to boost the regeneration in vivo, potentially opening new avenues in neural tissue engineering and regenerative medicine.
Author contributions
Maria Lopes: investigation, visualization, formal analysis, and writing – original draft. Marília Torrado: investigation, visualization, formal analysis, and writing – original draft. Daryl Barth: investigation, writing – original draft. Sofia D. Santos: supervision, validation, and writing – review & editing. Melike Sever-Bahcekapili: investigation, visualization, and writing – review & editing. Ayse B. Tekinay: resources, supervision, validation, and writing – review & editing. Mustafa O. Guler: resources, supervision, validation, and writing – review & editing. Franck Cleymand: validation and writing – review & editing. Ana P. Pêgo: funding acquisition, project administration, resources, supervision, validation, and writing – review & editing. João Borges: conceptualization, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, and writing – review & editing. João F. Mano: conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Union's Horizon Europe research and innovation programme under the grant agreement no. 101079482 (“SUPRALIFE”). This work was also supported by the Programa Operacional Regional do Centro – Centro 2020, in the component FEDER, and by national funds (OE) through Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Ensino Superior (FCT/MCTES), in the scope of the project “SUPRASORT” (PTDC/QUI-OUT/30658/2017, CENTRO-01-0145-FEDER-030658). M. T., S. D. S. and A. P. P. acknowledge FCT for funding through the project PTDC/BTM-MAT/4156/2021. M. L., M. T., S. D. S. and J. B. gratefully acknowledge FCT for the individual PhD grants (2020.05210.BD – M. L., SFRH/BD/146754/2019 – M. T.), individual contract under the Norma Transitória – DL57/2016/CP/CP1360/CT0013 (S. D. S.), and individual Assistant Researcher contract (2020.00758.CEECIND – J. B.) under the Scientific Employment Stimulus – Individual Call, respectively. This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020, UIDP/50011/2020 & LA/P/0006/2020, financed by national funds through the FCT/MCTES (PIDDAC). Optical and atomic force microscopy were conducted at the Bioimaging and Biointerfaces and the Nanotechnology i3S Scientific Platforms, respectively, both members of the PPBI (PPBI–POCI-01-0145-FEDER-022122).References
- S. G. Varadarajan, J. L. Hunyara, N. R. Hamilton, A. L. Kolodkin and A. D. Huberman, Cell, 2022, 185, 77–94 CrossRef CAS PubMed.
- C. S. Ahuja, J. R. Wilson, S. Nori, M. R. N. Kotter, C. Druschel, A. Curt and M. G. Fehlings, Nat. Rev. Dis. Primers, 2017, 3, 1–21 Search PubMed.
- B. S. Eftekhari, M. Eskandari, P. A. Janmey, A. Samadikuchaksaraei and M. Gholipourmalekabadi, Adv. Funct. Mater., 2020, 30, 1907792 CrossRef CAS.
- O. Y. Antonova, O. Y. Kochetkova, I. L. Kanev, E. A. Shlyapnikova and Y. M. Shlyapnikov, ACS Chem. Neurosci., 2021, 12, 2838–2850 CrossRef CAS PubMed.
- H. S. Azevedo and A. Mata, Biomater. Biosyst., 2022, 6, 100039 CrossRef CAS PubMed.
- Z. Álvarez, J. A. Ortega, K. Sato, I. R. Sasselli, A. N. Kolberg-Edelbrock, R. Qiu, K. A. Marshall, T. P. Nguyen, C. S. Smith, K. A. Quinlan, V. Papakis, Z. Syrgiannis, N. A. Sather, C. Musumeci, E. Engel, S. I. Stupp and E. Kiskinis, Cell Stem Cell, 2022, 30, 219–238 CrossRef PubMed.
- C. M. Dumont, M. A. Carlson, M. K. Munsell, A. J. Ciciriello, K. Strnadova, J. Park, B. J. Cummings, A. J. Anderson and L. D. Shea, Acta Biomater., 2019, 86, 312–322 CrossRef CAS PubMed.
- C. Fan, W. Yang, L. Zhang, H. Cai, Y. Zhuang, Y. Chen, Y. Zhao and J. Dai, Biomaterials, 2022, 288, 121689 CrossRef CAS PubMed.
- M. Isik, C. C. Eylem, T. Haciefendioglu, E. Yildirim, B. Sari, E. Nemutlu, E. Emregul, B. O. Okesola and B. Derkus, Biomater. Sci., 2021, 9, 8270–8284 RSC.
- L. R. Pires and A. P. Pêgo, Regener. Biomater., 2015, 2, 203–214 CrossRef PubMed.
- R. Jain and S. Roy, RSC Adv., 2019, 9, 38745–38759 RSC.
- J. Borges and J. F. Mano, Chem. Rev., 2014, 114, 8883–8942 CrossRef CAS PubMed.
- M. J. Landry, F. G. Rollet, T. E. Kennedy and C. J. Barrett, Langmuir, 2018, 34, 8709–8730 CrossRef CAS PubMed.
- K. E. Inostroza-Brito, E. Collin, O. Siton-Mendelson, K. H. Smith, A. Monge-Marcet, D. S. Ferreira, R. P. Rodríguez, M. Alonso, J. C. Rodríguez-Cabello, R. L. Reis, F. Sagués, L. Botto, R. Bitton, H. S. Azevedo and A. Mata, Nat. Chem., 2015, 7, 897–904 CrossRef CAS PubMed.
- H. Zhang, X. Liu, M. Yang and L. Zhu, Mater. Sci. Eng., C, 2015, 55, 8–13 CrossRef CAS PubMed.
- M. Criado-Gonzalez, J. R. Fores, A. Carvalho, C. Blanck, M. Schmutz, L. Kocgozlu, P. Schaaf, L. Jierry and F. Boulmedais, Langmuir, 2019, 35, 10838–10845 CrossRef CAS PubMed.
- H. Cheong, J. Kim, B. J. Kim, E. Kim, H. Y. Park, B. H. Choi, K. il Joo, M. la Cho, J. W. Rhie, J. I. Lee and H. J. Cha, Acta Biomater., 2019, 90, 87–99 CrossRef CAS PubMed.
- S. Wang, S. Guan, Z. Zhu, W. Li, T. Liu and X. Ma, Mater. Sci. Eng., C, 2017, 71, 308–316 CrossRef CAS PubMed.
- M. S. Niepel, B. K. Ekambaram, C. E. H. Schmelzer and T. Groth, Nanoscale, 2019, 11, 2878–2891 RSC.
- A. Sharma and J. E. Schwarzbauer, Mol. Neurobiol., 2022, 59, 1112–1123 CrossRef CAS PubMed.
- D. Barros, I. F. Amaral and A. P. Pêgo, Biomacromolecules, 2020, 21, 276–293 CrossRef CAS PubMed.
- A. E. Rodda, L. Meagher, D. R. Nisbet and J. S. Forsythe, Prog. Polym. Sci., 2014, 39, 1312–1347 CrossRef CAS.
- X. Li, X. Liu, B. Josey, C. J. Chou, Y. Tan, N. Zhang and X. Wen, Stem Cells Transl. Med., 2014, 3, 662–670 CrossRef CAS PubMed.
- Z. Álvarez, A. N. Kolberg-Edelbrock, I. R. Sasselli, J. A. Ortega, R. Qiu, Z. Syrgiannis, P. A. Mirau, F. Chen, S. M. Chin, S. Weigand, E. Kiskinis and S. I. Stupp, Science, 2021, 374, 848–856 CrossRef PubMed.
- S. C. Yuan, J. A. Lewis, H. Sai, S. J. Weigand, L. C. Palmer and S. I. Stupp, J. Am. Chem. Soc., 2022, 144, 16512–16523 CrossRef CAS PubMed.
- A. Mata, Y. Geng, K. J. Henrikson, C. Aparicio, S. R. Stock, R. L. Satcher and S. I. Stupp, Biomaterials, 2010, 31, 6004–6012 CrossRef CAS PubMed.
- K. Ban, H. J. Park, S. Kim, A. Andukuri, K. W. Cho, J. W. Hwang, H. J. Cha, S. Y. Kim, W. S. Kim, H. W. Jun and Y. S. Yoon, ACS Nano, 2014, 8, 10815–10825 CrossRef CAS PubMed.
- Z. Okur, O. I. Senturk, C. Yilmaz, G. Gulseren, B. Mammadov, M. O. Guler and A. B. Tekinay, Biomater. Sci., 2018, 6, 1777–1790 RSC.
- M. Sever, G. Gunay, M. O. Guler and A. B. Tekinay, Biomater. Sci., 2018, 6, 1859–1868 RSC.
- J. J. Richardson, M. Björnmalm and F. Caruso, Science, 2015, 348, aaa2491 CrossRef PubMed.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- K. Wolf and S. Kumar, ACS Biomater. Sci. Eng., 2019, 5, 3753–3765 CrossRef CAS PubMed.
- L. Richert, F. Boulmedais, P. Lavalle, J. Mutterer, E. Ferreux, G. Decher, P. Schaaf, J. C. Voegel and C. Picart, Biomacromolecules, 2004, 5, 284–294 CrossRef CAS PubMed.
- A. Schneider, G. Francius, R. Obeid, P. Schwinté, J. Hemmerlé, B. Frisch, P. Schaaf, J. C. Voegel, B. Senger and C. Picart, Langmuir, 2006, 22, 1193–1200 CrossRef CAS PubMed.
- G. Gunay, M. Sever, A. B. Tekinay and M. O. Guler, Biotechnol. J., 2017, 12, 1700080 CrossRef PubMed.
- A. P. Spencer, V. Leiro and A. P. Pêgo, Biomater. Sci., 2023, 11, 1499–1516 RSC.
- M. H. Longair, D. A. Baker and J. D. Armstrong, Bioinformatics, 2011, 27, 2453–2454 CrossRef CAS PubMed.
- S. Amorim, I. Pashkuleva, C. A. Reis, R. L. Reis and R. A. Pires, J. Mater. Chem. B, 2020, 8, 3880–3885 RSC.
- S. Sur, M. O. Guler, M. J. Webber, E. T. Pashuck, M. Ito, S. I. Stupp and T. Launey, Biomater. Sci., 2014, 2, 903–914 RSC.
- K. Matsuo, H. Namatame, M. Taniguchi and K. Gekko, Biosci. Biotechnol. Biochem., 2009, 73, 557–561 CrossRef CAS PubMed.
- J. E. Goldberger, E. J. Berns, R. Bitton, C. J. Newcomb and S. I. Stupp, Angew. Chem., Int. Ed., 2011, 50, 6292–6295 CrossRef CAS PubMed.
- C. Chothla, J. Mol. Biol., 1973, 75, 295–302 CrossRef PubMed.
- A. Laezza, A. Pepe and B. Bochicchio, Chem. – Eur. J., 2022, 28, e202201959 CrossRef CAS PubMed.
- M. Levittl, Preferences of amino acids, Biochemistry, 1978, 17, 4277–4285 CrossRef PubMed.
- H. Jiang, M. O. Guler and S. I. Stupp, Soft Matter, 2007, 3, 454–462 RSC.
- A. Micsonai, É. Moussong, F. Wien, E. Boros, H. Vadászi, N. Murvai, Y. H. Lee, T. Molnár, M. Réfrégiers, Y. Goto, Á. Tantos and J. Kardos, Nucleic Acids Res., 2022, 50, W90–W98 CrossRef CAS PubMed.
- D. M. Byler and H. Susi, Biopolymers, 1986, 25, 469–487 CrossRef CAS PubMed.
- P. I. Haris and D. Chapman, Biopolymers, 1995, 37, 251–263 CrossRef CAS PubMed.
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS PubMed.
- J. Borges, M. P. Sousa, G. Cinar, S. G. Caridade, M. O. Guler and J. F. Mano, Adv. Funct. Mater., 2017, 27, 1605122 CrossRef.
- J. T. Pelton and L. R. McLean, Anal. Biochem., 2000, 277, 167–176 CrossRef CAS PubMed.
- R. Yegappan, V. Selvaprithiviraj, A. Mohandas and R. Jayakumar, Colloids Surf., B, 2019, 177, 41–49 CrossRef CAS PubMed.
- A. Dehsorkhi, I. W. Hamley, J. Seitsonen and J. Ruokolainen, Langmuir, 2013, 29, 6665–6672 CrossRef CAS PubMed.
- I. W. Hamley, A. Dehsorkhi, V. Castelletto, J. Seitsonen, J. Ruokolainen and H. Iatrou, Soft Matter, 2013, 9, 4794–4801 RSC.
- F. Höök, B. Kasemo, T. Nylander, C. Fant, K. Sott and H. Elwing, Anal. Chem., 2001, 73, 5796–5804 CrossRef PubMed.
- K. A. Marx, Biomacromolecules, 2003, 4, 1099–1120 CrossRef CAS PubMed.
- D. Bütergerds, C. Cramer and M. Schönhoff, Adv. Mater. Interfaces, 2017, 4, 201600592 Search PubMed.
- C. Picart, P. Lavalle, P. Hubert, F. J. G. Cuisinier, G. Decher, P. Schaaf and J. C. Voegel, Langmuir, 2001, 17, 7414–7424 CrossRef CAS.
- C. Picart, J. Mutterer, L. Richert, Y. Luo, G. D. Prestwich, P. Schaaf, J.-C. Voegel and P. Lavalle, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12531–12535 CrossRef CAS PubMed.
- C. Porcel, P. Lavalle, G. Decher, B. Senger, J. C. Voegel and P. Schaaf, Langmuir, 2007, 23, 1898–1904 CrossRef CAS PubMed.
- G. V. Martins, E. G. Merino, J. F. Mano and N. M. Alves, Macromol. Biosci., 2010, 10, 1444–1455 CrossRef CAS PubMed.
- U. Ajdnik, T. Luxbacher and L. F. Zemljič, Colloids Surf., B, 2022, 221, 113011 CrossRef PubMed.
- K. L. Niece, J. D. Hartgerink, J. J. J. M. Donners and S. I. Stupp, J. Am. Chem. Soc., 2003, 125, 7146–7147 CrossRef CAS PubMed.
- M. J. Cardoso, S. G. Caridade, R. R. Costa and J. F. Mano, Biomacromolecules, 2016, 17, 1347–1357 CrossRef CAS PubMed.
- C. Chaubaroux, F. Perrin-Schmitt, B. Senger, L. Vidal, J. C. Voegel, P. Schaaf, Y. Haikel, F. Boulmedais, P. Lavalle and J. Hemmerlé, Tissue Eng., Part C, 2015, 21, 881–888 CrossRef CAS PubMed.
- R. Lozano, L. Stevens, B. C. Thompson, K. J. Gilmore, R. Gorkin, E. M. Stewart, M. in het Panhuis, M. Romero-Ortega and G. G. Wallace, Biomaterials, 2015, 67, 264–273 CrossRef CAS PubMed.
- L. M. Y. Yu, N. D. Leipzig and M. S. Shoichet, Mater. Today, 2008, 11, 36–43 CrossRef CAS.
- N. E. Vrana, O. Erdemli, G. Francius, A. Fahs, M. Rabineau, C. Debry, A. Tezcaner, D. Keskin and P. Lavalle, J. Mater. Chem. B, 2014, 2, 999–1008 RSC.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed.
- C. G. Dotti, C. A. Sullivan and G. A. Banker, J. Neurosci., 1988, 8, 1454–1468 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00438d |
| This journal is © The Royal Society of Chemistry 2023 |