
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow molecular weight poly((D,L)-lactide-co-caprolactone) liquid inks for diluent-free DLP printing of cell culture platforms†
Sandra
Ramos-Díez
*a,
Garazi
Larrañaga-Jaurrieta
*ab,
Leire
Iturriaga
a,
Ander
Abarrategi
 bc and
Sandra
Camarero-Espinosa
ac
bc and
Sandra
Camarero-Espinosa
ac
aBioSmarTE Lab, POLYMAT, University of Basque Country UPV / EHU, Donostia / San Sebastián 20018, Gipuzkoa, Spain. E-mail: sandra.camarero@ehu.eus
bRegenerative Medicine Lab, CICbiomaGUNE, Donostia / San Sebastián 20014, Gipuzkoa, Spain
cIkerbasque, Basque Foundation for Science, Euskadi Pl., 5, 48009, Bilbao, Spain
First published on 3rd July 2023
Abstract
Digital light processing (DLP) printing offers the possibility of fabricating complex objects in a fast and reproducible manner. A main requirement for DLP printing is the use of inks with low viscosities that can flow under the printing platform in a short period of time. Its exploitation in tissue engineering applications has been centered on the use of hydrogel forming materials diluted in aqueous solutions or the use of polyesters in combination with diluents and heating platforms that aid in the reduction of their viscosity. The use of diluents, however, modifies the mechanical properties and reduces the shape fidelity of the printed objects and, the use of heating platforms results in vats with heterogeneous temperatures and ink viscosities. Here, we report on the synthesis of a library of methacrylated low molecular weight (<3000 g mol−1) homopolymers ((P(D,L)LA and PCL) and copolymers (P((D,L)LA-co-CL)) of 2- and 3-arms based on (D,L)-lactide and ε-caprolactone. The resulting inks possessed low viscosity that made them printable in the absence of diluents and heating elements. DLP printing of cubical and cylindrical patterns resulted in objects with a higher shape fidelity than their counterparts fabricated using diluents and with printed features on the order of 300 μm. The printed materials were biocompatible and supported the growth of human mesenchymal stem cells (hMSCs). Moreover, the variations in the composition resulted in polymers that enabled the attachment of hMSCs to different extents, leading to the formation of well-adhered cell monolayers or loosely adhered cell aggregates.
1. Introduction
Additive manufacturing, in particular three-dimensional (3D) printing, has attracted exceptional interest in the last decade due to its undeniable advantages, such as the easy and fast fabrication of objects with complex structures at reduced costs and has found application in numerous and varying fields including dental applications, prototyping, tissue engineering and tissue regeneration.1–3 Furthermore, the wide scope of different available 3D printing technologies enables the use of materials with different characteristics as inks derived from metals4 to hydrogels,2 passing through ceramics and polymers.5 Light based 3D printing technologies such as 2-photon polymerization (2PP), digital light processing (DLP) and stereolithography (SLA) offer the possibility of printing most complex structures commonly required in the field of tissue regeneration and great advances have been made in the last few years, including volumetric 3D bioprinting.6–8 The main difference between them is that the last two, DLP and volumetric 3D bioprinting, use a light projector to create 3D objects instead of a laser, reducing significantly the printing time. While volumetric 3D bioprinting permits the fabrication of objects in extremely short times, it has a very restricted window of materials that can be used, as they have to be solid or gel-like during the fabrication process.The DLP printing technique has aroused great interest since it has proven to be a fast (from 25 to 1000 mm min−1) and reproducible manufacturing method with high efficiency and theoretical resolutions of a few microns (∼10 μm).9–11 Nevertheless, these parameters vary in function of the experimental set-up, specifically in function of the viscoelastic properties of the selected resin, the intensity of the light source, the photosensitivity of the ink and the overall concentration and the nature of cross-linkable groups contained in the ink.3
In this regard, the choice of inks used for DLP printing is crucial; it will not only determine the spatial resolution, surface chemistry or mechanical properties, but will also define the biocompatibility and degradability of the final model. Most of the studies using DLP printing for tissue engineering constructs have been focused on the use of hydrogel forming materials such as those based on polyethylene glycol (PEG), gelatin, hyaluronic acid or silk.8–10,12 Polyester-based polymers such as poly(lactide) (PLA),13,14 poly(ε-caprolactone) (PCL)15–17 and poly(trimethylene carbonate) (PTMC)18 are commonly used in tissue engineered scaffolds including those fabricated by DLP, as they can be synthesised by ring-opening polymerization (ROP) allowing the control of the polymer structure and chain length.19 One of the main requirements for good printability is the low viscosity (<10 Pa s) of the resin and a Newtonian response to shear,3,19 as it will determine the ability of the material to flow under the crosslinked layer in between the sliced projections. Polymers with a high molecular weight or semicrystalline structures, like the aforementioned polyesters, generally result in viscous (or even solid) resins which are incompatible with DLP printing. Thus, they are combined with reactive or non-reactive diluents and heating elements to lower their viscosity into the printable regime. Reactive diluents such as 2-(2-ethoxyethoxy)ethyl acrylate (EOEOEA) or N-vinyl-2-pyrrolidone (NVP) are incorporated in the bulk of the printed objects compromising their mechanical properties and altering the preselected chemistry of the inks;14,18 non-reactive diluents such as ethylene lactate or dioxane are washed away after printing, compromising the structural stability of the object17,20 and, heating stages require the design of home-made set-ups with vague control of the applied temperatures and size limitations in the printed object derived from the heterogeneous temperatures in larger vats.21
Melchels et al.20 synthesised star-shaped methacrylated P(D,L)LA inks with 2–, 3- and 6-arms and DLP printed them using ethylene lactate as non-reactive diluent. All the reported mixtures had viscosities below 10 Pa s. They selected those with the highest diluent concentration (19 wt%) for printing, yielding inks with viscosities of approximately 1 Pa s. While the resulting materials after DLP printing proved biocompatible, the use of a non-reactive diluent led to a shrinkage of 10% (in all directions) when complex gyroid structures were created. Similarly, Paunović et al.16 synthesised 4-arm methacrylated P((D,L)lactide-co-caprolactone) (P((D,L)LA-co-CL)-MA low molecular weight (<9000 g mol−1) copolymers by ROP. The neat polymer showed a viscosity of over 40 Pa s when heated at 60 °C, which decreased to below 5 Pa s when mixed with 7.5 wt% NVP reactive diluent. The printed objects possessed structural features on the mm scale, probably due to the still high viscosity of the inks that yield low-resolution objects. The mechanical properties of the printed polymer with NVP were of 4 MPa, which appears to be very low for a P((D,L)LA-co-CL copolymer, probably due to the incorporation of the reactive diluent in the bulk.
Kuhnt et al.18 reported on the synthesis of DLP inks based on poly(caprolactone-co-trimethylenecarbonate) urethane acrylates and used 30 wt% EOEOEA as the reactive diluent and a heating platform to further decrease the viscosity of some of their ink compositions.21 They neatly showed the capability of tuning the mechanical properties of their inks from 0.2–5.4 MPa by varying the ratio of caprolactone to trimethylenecarbonate, which also affected the morphology of adhered cells. Nevertheless, the mechanical properties were lower than those expected for films fabricated from these types of polyester urethanes, which could be explained by the large amount of reactive diluent required for DLP printing. The same group recently reported on the scaled-up synthesis of P((D,L)LA-co-CL)-MA inks for DLP printing. In this work, they studied resins with molecular weights ranging from 1000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1.22 Despite reporting on the inks being liquid, a 30 wt% EOEOEA reactive diluent was added to the polymer and a heating stage was used for DLP processing.
000 g mol−1.22 Despite reporting on the inks being liquid, a 30 wt% EOEOEA reactive diluent was added to the polymer and a heating stage was used for DLP processing.
Due to the lack of biocompatible resins with a suitable viscosity that can be DLP printed without the use of diluents or heating stages, in this work, we strove to develop liquid biocompatible inks based on (D,L)-lactide and ε-caprolactone monomers. We hypothesised that low molecular polymers could result in liquid inks. We created a library of homopolymers and copolymers with 2- and 3-arms and investigated the effect of the different monomers and the steric hindrance of the chains on the viscosity of the resulting ink.
2. Experimental section
2.1. Materials
Triethylene glycol (TEG), tin(II) 2-ethylhexanoate (Sn(Oct)2), methacrylic anhydride (MMA), sodium bicarbonate (NaHCO3), diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO), sodium chloride (NaCl), and triethanolamine (TEA) were purchased from Sigma-Aldrich. (D,L)-lactide and hydroquinone were obtained from Acros. Dichloromethane (CH2Cl2) anhydrous and methanol were purchased from Scharlau. ε-caprolactone, dimethyl sulfoxide (DMSO), and 2-ethyl-2-(hydroxymethyl)-1,3-propanediol (TMP) were supplied by Alfa-Aesar. Deuterated chloroform (CDCl3) was obtained from VWR Chemicals.2.2. Synthesis of polymers and methacrylation procedure
All polymers were synthesised by ring-opening polymerization. To synthesise 2- and 3-arm-P(D,L)LA, 25.85 g (0.18 mol) of (D,L)-lactide was added to a round-bottom flask and heated to 130 °C under vigorous stirring until molten, under a N2 atmosphere. Then, 3 mL of TEG (0.022 mol) or 2 g of TMP (0.015 mol) was added for 2- and 3-arm polymers, respectively. Yields of 64.9% and 71.3% were obtained respectively. For the synthesis of 2- and 3-arm-PCL, 19.0 mL of ε-caprolactone (0.18 mol) was added to a round-bottom flask under vigorous stirring and a N2 atmosphere. Then, 3 mL of TEG (0.022 mol) or 2 g of TMP (0.015 mol) was added. Yields of 78.0% and 83.5% were obtained respectively. For the synthesis of 2-arm-P((D,L)LA-co-CL), 5.43 g of (D,L)-lactide (0.037 mol) was melted in a round-bottom flask at 130 °C under a N2 atmosphere. Then, 15.0 mL of ε-caprolactone (0.14 mol) and 3 mL of TEG (0.022 mol) were added to the flask. A yield of 98.2% was obtained. For the synthesis of 3-arm-P((D,L)LA-co-CL), 5.36 g of (D,L)-lactide (0.037 mol) were melted in a round-bottom flask at 130 °C together with 2 g of TMP (0.015 mol). Later, 14.8 mL of ε-caprolactone (0.14 mol) was incorporated. A yield of 98.7% was obtained.To all reactions, 0.28 mL of 1:10 (vol:vol) Sn(Oct)2:toluene catalyser solution (0.088 mmol) was subsequently added. The reactions were carried out for 24 hours at 130 °C. After the reaction, the product was washed three times by precipitation in methanol and dried under vacuum. For the methacrylation step, 5 g of purified polymer was dissolved in 5 mL of anhydrous CH2Cl2 by gentle ultrasonication for 10 minutes. The solution was then poured over 5 mg of hydroquinone (0.045 mmol) in a round-bottom flask in the dark and under a N2 atmosphere. Afterwards, 3:3 or 4:4 ratio (mol:mol) of TEA:MAA per mol of oligomer was added to 2- and 3-arm polymers, respectively. The reaction was left to proceed for 48 hours at room temperature. The product was recovered by phase separation with both saturated NaHCO3 and brine solutions, consecutively. The organic extract was firstly concentrated in a rotary evaporator at 60 °C, precipitated three times in methanol at 4 °C and dried under vacuum. Yields between 70% and 90% were obtained after the purification step in all the methacrylated polymers.
2.3. Characterization of polymers
200 g ml−1. The temperature of the columns was 35 °C.
2.4. Cell adhesion and biocompatibility studies
To study cell biocompatibility and adhesion, films of 7 × 7 × 0.4 mm were prepared using the DLP Titan 2 HR (KUDO 3D) printer for both P((D,L)LA-co-CL)-MA copolymers of 2- and 3-arms. Homopolymer films were prepared by adding 500 mg of each sample between two coverslips and curing them by UV exposure with a lamp operating at 405 nm wavelength and 48 W power (post-curing LED Lamp KUDO 3D). All samples were submerged in 1:2 (vol:vol) isopropanol:acetone solution for 10 minutes to remove the non-cured ink. Afterwards, the films were sterilised in ethanol at 70% for 15 minutes. Samples were then rinsed three times with PBS and placed in non-treated 24-well plates containing complete culture media 30 min before use.
000 cells per cm2.
:500 was added to each sample in a 1:1 ratio, mixed and incubated at RT for 1 hour to degrade the cellular RNA. In parallel, the GR-dye stock solution was diluted 200-fold into the lysis buffer. Finally, 100 μl of each sample was transferred to a well of a black bottom microplate in triplicate. After adding 100 μl of the GR-dye solution and incubating for 15 minutes at RT, fluorescence was measured at 480 nm excitation and 520 nm emission.
:100) in PBS for 1 h, rinsed twice with PBS and stained for 5 min with Hoechst 33342 (1:500) (Invitrogen, Thermo Scientific).
The cell viability was evaluated by LIVE/DEAD™ viability/cytotoxicity kit for mammalian cells (Invitrogen) following manufacturer's instructions. Briefly, 24 h after cell seeding in the films the culture media was removed and the films were washed 2 times with DPBS. Afterwards, 1 ml of 6 μM ethidium homodimer and 1 μl of 1 mM Calcein were combined and 300 μL of the solution were used to cover each polymer film. The cells were incubated for 45 min protected from light and the staining solution was replaced with DPBS. The samples were visualized immediately under a fluorescence microscope at 518 nm and 540 nm for live and dead cells, respectively.
2.5 DLP printing of structures with micro-topographical features
The models were designed using Solid Edge (Siemens) CAD software and exported as STL file for subsequent treatment with Kudo 3D slicing software. PNG images were created for each slice of 15 μm height composed of 32 bits per pixel. Models of cubes and cylinders had a base of 7 × 7 mm and 0.35 mm in height. The surface pattern was created as a matrix of 8 × 8 elements of 0.3 × 0.3 × 0.3 mm cubes or 0.3 ∅ × 0.3 mm pillars. In all cases, the first layer was irradiated for 50 seconds to ensure the attachment of the model to the platform. The rest of the layers of the base were irradiated for 5 seconds and the layers corresponding to the topography for 8 seconds.The printing error of the models was measured from the SEM images, taking into account the width of the cubes or the diameter of the pillars and the height of the topography, following the equation:
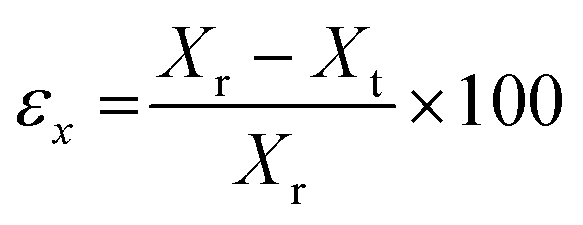
2.6. Statistical analysis
The statistical analysis was performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). One-way and two-way analyses of variance (ANOVA) were carried out for complex viscosity, storage modulus G′ values, Young's modulus, elongation at break, maximum stress and cell viability assays to calculate the significance and including a post-hoc Tukey's multiple comparison test. All the results are expressed as mean values ± standard deviation.3. Results and discussion
3.1. Synthesis and characterization of low molecular weight PCL- and P(D,L)LA-based polymers and copolymer inks
Polylactide (P(D,L)LA) and polycaprolactone (PCL) are among the most commonly used polyesters for the biofabrication and additive manufacturing of tissue engineering and regeneration scaffolds due to their biocompatibility, their ease to be processed and shaped into complicated structures, their structural stability and their relatively high mechanical properties. However, their application in DLP printing is restricted due to their high viscosity at room temperature, requiring the use of (reactive) diluents or heating platforms that make the fabrication process difficult and limit the resolution of the obtained structures.13,16,18,21 We hypothesised, that low molecular weight P(D,L)LA and PCL homopolymers and copolymers thereof, would result in materials of lower viscosity and that after methacrylation, could be exploited in the DLP biofabrication process without the use of heating platforms and/or diluents. Hence, low molecular weight (<3000 g mol−1) resins were synthesised from (D,L)-lactide (LA) and ε-caprolactone (CL) monomers. Homopolymers and copolymers were synthesized as 2- and 3-arm polymers to obtain, after methacrylation, a high density of photoreactive motifs that could speed up the printing process and yield materials with higher cross-linking densities and printing resolutions. 2-arm and 3-arm polymers and copolymers were synthesised by ring-opening polymerization using triethylene glycol (TEG) or 2-ethyl-2-(hydroxymethyl)-1,3-propanediol (TMP) at 8:1 and 12:1 monomer to initiator ratios, respectively (Scheme 1A). P((D,L)LA-co-CL) copolymers were synthesized using the same monomer to initiator ratio with a 1:3.76 LA:CL ratio and with the addition of stannous octanoate as catalyst (Table 1). H1-NMR spectroscopy was used to evaluate the successful polymerization of the polymers (Fig. S1†). For 2-arm polymers, chemical shifts at δ = 3.65 ppm, corresponding to CH2 adjacent to the terminal monomer-bound TEG, were observed as a triplet, while terminal –OH groups corresponding to free TEG were not detected (expected as a triplet at δ = 3.56 ppm). For 3-arm polymers, monomer-bound TMP showed a singlet at δ = 4.02 ppm as opposed to the singlet at δ = 3.39 ppm of TMP-free terminal –OH groups. Moreover, in the case of PCL homopolymers and copolymers, a peak at δ = 2.23 ppm corresponding to the CH2 nearest to the formed ester bond was detected (labelled in grey in Fig. S1†). The ratio of LA:CL in the copolymers was calculated using the integrals of CH protons appearing at δ = 3.51 ppm for P(D,L)LA and CH2 nearest to carboxylic proton, appearing at δ = 2.23 ppm for PCL (shown respectively as black and grey dots in Fig. 2). This ratio was slightly lower than the feed ratio and we hypothesised that LA has a higher reactivity than CL monomers.
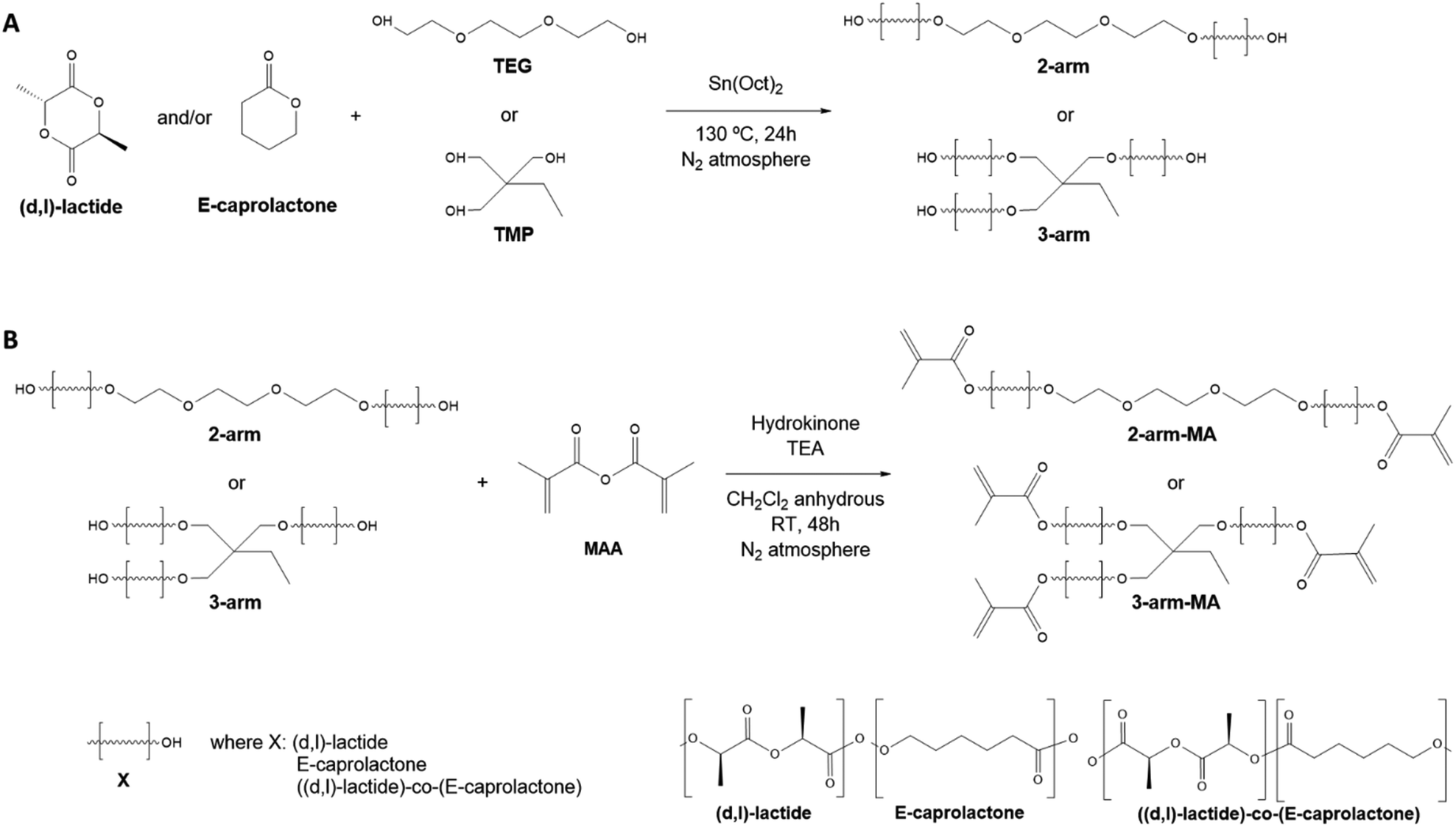 | ||
| Scheme 1 (A) Ring-opening polymerization reaction to synthesize 2- and 3-arm homopolymers and copolymers of lactide and caprolactone using TEG or TMP as co-initiator, respectively, and Sn(Oct)2 as catalyst. (B) Methacrylation reaction that introduces photocross-linkable terminal groups on 2- (top) and 3-arm (bottom) homopolymers and copolymers employing hydroquinone as catalyst. | ||
| Polymer | Theor·LA:CL ratio (mol:mol) |
Product LA:CL ratio (mol:mol) |
D of methacrylation (%) | Theor. Mw (g mol−1) | M w (g mol−1) | M n (g mol−1) | M w/Mn | Appearance |
|---|---|---|---|---|---|---|---|---|
| 2-arm-P(D,L)LA | 1:0 |
1:0 |
88 | 1303.18 | 1264 | 1572 | 1.24 | Very viscous liquid |
| 2-arm-PCL | 0:1 |
0:1 |
61 | 1063.33 | 1670 | 2310 | 1.38 | Waxy solid |
| 2-arm-P((D,L)LA-co-CL) | 1:3.76 |
1:2.93 |
98 | 1123.29 | 1559 | 2197 | 1.40 | Viscous liquid |
| 3-arm-P(D,L)LA | 1:0 |
1:0 |
100 | 1863.69 | 2093 | 2559 | 1.22 | Very viscous liquid |
| 3-arm-PCL | 0:1 |
0:1 |
96 | 1503.9 | 2187 | 2606 | 1.19 | Waxy solid |
| 3-arm-P((D,L)LA-co-CL) | 1:3.76 |
1:3.12 |
99 | 1593.85 | 2188 | 2918 | 1.33 | Viscous liquid |
Gel permeation chromatography revealed that all polymers had a low molecular weight ranging from ≈1200 to 2200 g mol-1 (Table 1 and Fig. S2†), which varied slightly with respect to the calculated theoretical value. This low molecular weight resulted in polymers with lower viscosities as compared to the traditional solid powders obtained for high molecular weight PLA, PCL and copolymers thereof. Indeed, 2- and 3-arm P(D,L)LA appeared as very viscous liquids, while 2- and 3-arm PCL resulted in waxy solids and, the 2- and 3-arm P((D,L)LA-co-CL) copolymers in viscous liquids.
Analysis of the thermal properties of the synthesised inks via dynamic scanning calorimetry (DSC, Fig. 1) showed, as expected, that 2- and 3-arm P(D,L)LA resulted in amorphous polymers with no melting transitions (Tm) and with glass transition temperatures (Tg) of −8 °C and 17 °C, respectively. 2- and 3-arm PCL homopolymers presented Tg at −65 °C and −58 °C, respectively. PCL homopolymers showed in both cases two distinct Tm of 32 °C and 42 °C for the 2-arm-PCL and 15 °C and 28 °C for the 3-arm-PCL. The double melting peaks observed in PCL polymers are relatively common in polyesters. We hypothesised that these two peaks correspond to the reorganization of the crystal structures during the heating process whereas the thinner lamellar structures formed during the cooling process melt, recrystallizing into thicker lamellae that melt at a higher temperature. Another possibility would be the formation of extended-chain and folded-chain crystal structures, due to the low molecular weight polymers, that results again in lamellae of different thicknesses. This behaviour is not observed during the cooling process, where a single crystallization peak is detected at 16 °C and −8 °C for the 2- and 3-arm homopolymers respectively, as this one is generally dominated by the density of active nucleating sites. 2- and 3-Arm P((D,L)LA-co-CL) copolymers showed a thermal response characteristic of amorphous polymers with Tg at −55 °C and −48 °C, respectively and no associated melting transitions, further confirming the formation of copolymers.
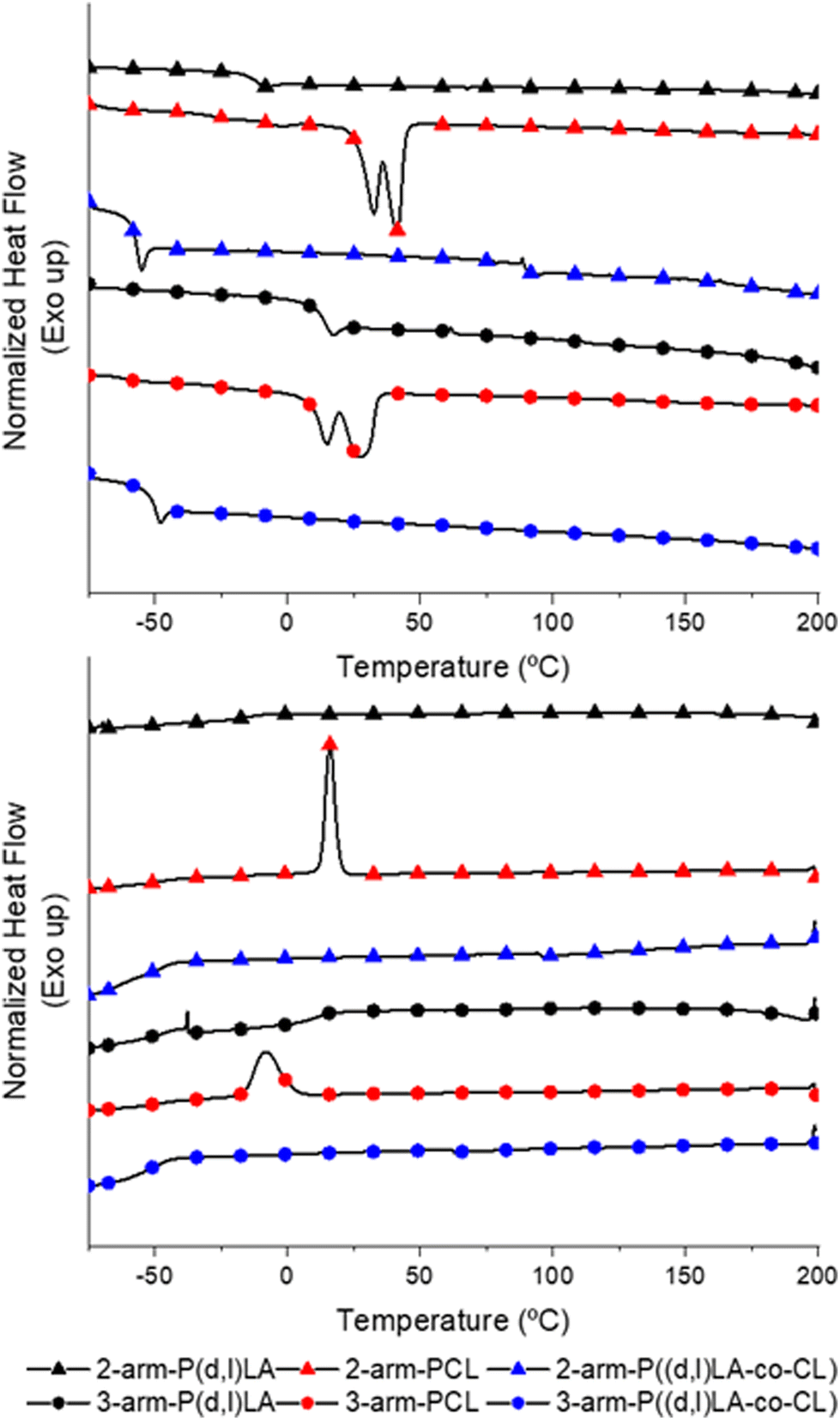 | ||
| Fig. 1 DSC thermograms of second heating scans (top) and cooling scans (bottom) of synthetized 2-arm (▲) and 3-arm (●) P(D,L)LA, 2-arm (▲) and 3-arm (●) PCL and 2-arm (▲) and 3-arm (●) P((D,L)LA-co-CL) polymers. | ||
Having characterized the homopolymers and copolymers synthesized, we proceeded with the methacrylation of the terminal –OH groups (Scheme 1B). For that, the dry polymers were reacted with methacrylic anhydride in the presence of triethanolamine and hydroquinone. The methacrylated polymers were then analysed by H1-NMR (Fig. 2), where the characteristic doublets associated to the CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the methacrylic group were detected in all compounds. These protons associated to the methacrylic groups in methacrylated P(D,L)LA and PCL (P((D,L)LA-MA and PCL-MA) presented slightly different shifts at δ = 6.23 ppm and δ = 5.69 ppm, and δ = 6.15 ppm and δ = 5.60 ppm, respectively. These peaks were also detected in the methacrylated copolymers (P((D,L)LA-co-CL)-MA), indicating that the arms had different compositions and terminal repeating units. We hypothesized that these mild differences are due to the charge deshielding that carboxylic groups from PLA repeating units induce into methacrylic protons, which results in an increase in their chemical shifts. The degree of methacrylation of 2- and 3-arm methacrylated polymers was calculated from the ratio of the mentioned peaks and both CH2 signals from TEG at δ = 3.65 ppm and δ = 4.33 ppm, or CH3 signal from TMP at δ = 0.91 ppm, ranging from 61 to 100% (Table 1).
C bond of the methacrylic group were detected in all compounds. These protons associated to the methacrylic groups in methacrylated P(D,L)LA and PCL (P((D,L)LA-MA and PCL-MA) presented slightly different shifts at δ = 6.23 ppm and δ = 5.69 ppm, and δ = 6.15 ppm and δ = 5.60 ppm, respectively. These peaks were also detected in the methacrylated copolymers (P((D,L)LA-co-CL)-MA), indicating that the arms had different compositions and terminal repeating units. We hypothesized that these mild differences are due to the charge deshielding that carboxylic groups from PLA repeating units induce into methacrylic protons, which results in an increase in their chemical shifts. The degree of methacrylation of 2- and 3-arm methacrylated polymers was calculated from the ratio of the mentioned peaks and both CH2 signals from TEG at δ = 3.65 ppm and δ = 4.33 ppm, or CH3 signal from TMP at δ = 0.91 ppm, ranging from 61 to 100% (Table 1).
 | ||
| Fig. 2 1H-NMR spectrum of 2- (A) and 3-arm (B) methacrylated polymers in CDCl3. (top) P(D,L)LA-MA, (middle) PCL-MA and (bottom) P((D,L)LA-co-CL)-MA. The identified proton signals are associated to the molecular structure with coloured circles. | ||
3.2 Printability and viscoelastic properties of DLP resins
To evaluate the printability of the synthesized resins via DLP or lithographic techniques, the viscoelastic properties were measured by rheological analysis (Fig. 3). Too viscous resins require the use of diluents that reduce the viscosity and enable the fast penetration of the ink underneath the printing platform and object when the platform is raised between the printed layers. Moreover, inks presenting a non-Newtonian response to shear–stresses could change their viscosity upon displacement of the printing platform, reducing their capability to flow and cover the printing vat and the speed at which the platform can be displaced. All resins presented a Newtonian response, as evidence by the constant complex viscosity recorded for the tested oscillation rates and the linear increase in oscillation stress with the oscillation rate (Fig. 3A). Complex viscosity values ranged from 1–70 Pa s, depending on the polymer composition and the number of arms. The complex viscosity appeared to decrease for all polymers and copolymers with an increasing number of arms (although not statistically significant for all), probably as a result of the higher electrostatic hindrance between adjacent arms in a same polymer chain (Fig. 3A). PCL-based polymers presented the highest viscosity with values of 65 ± 16 Pa s for the 2-arm-PCL-MA and decreasing to 1.5 ± 0.1 Pa s when presenting 3-arms. PLA-based inks presented a viscosity of 16 ± 2 and 5 ± 3 Pa s for the 2-arm and 3-arm-PLA-MA, respectively. Copolymers of P((D,L)LA-co-CL)-MA showed the lowest viscosities with values of 1.4 ± 0.2 and 0.72 ± 0.05 Pa s for the 2- and 3-arm versions. The optimal viscosity of inks for DLP and SLA printing has been set in the range of 0.1–10 Pa s.3 Thus, 2-arm-PCL-MA and 2-arm-P(D,L)LA-MA polymers appeared to be too viscous to be printed without the incorporation of diluents or the use of heating sources (Fig. 3C).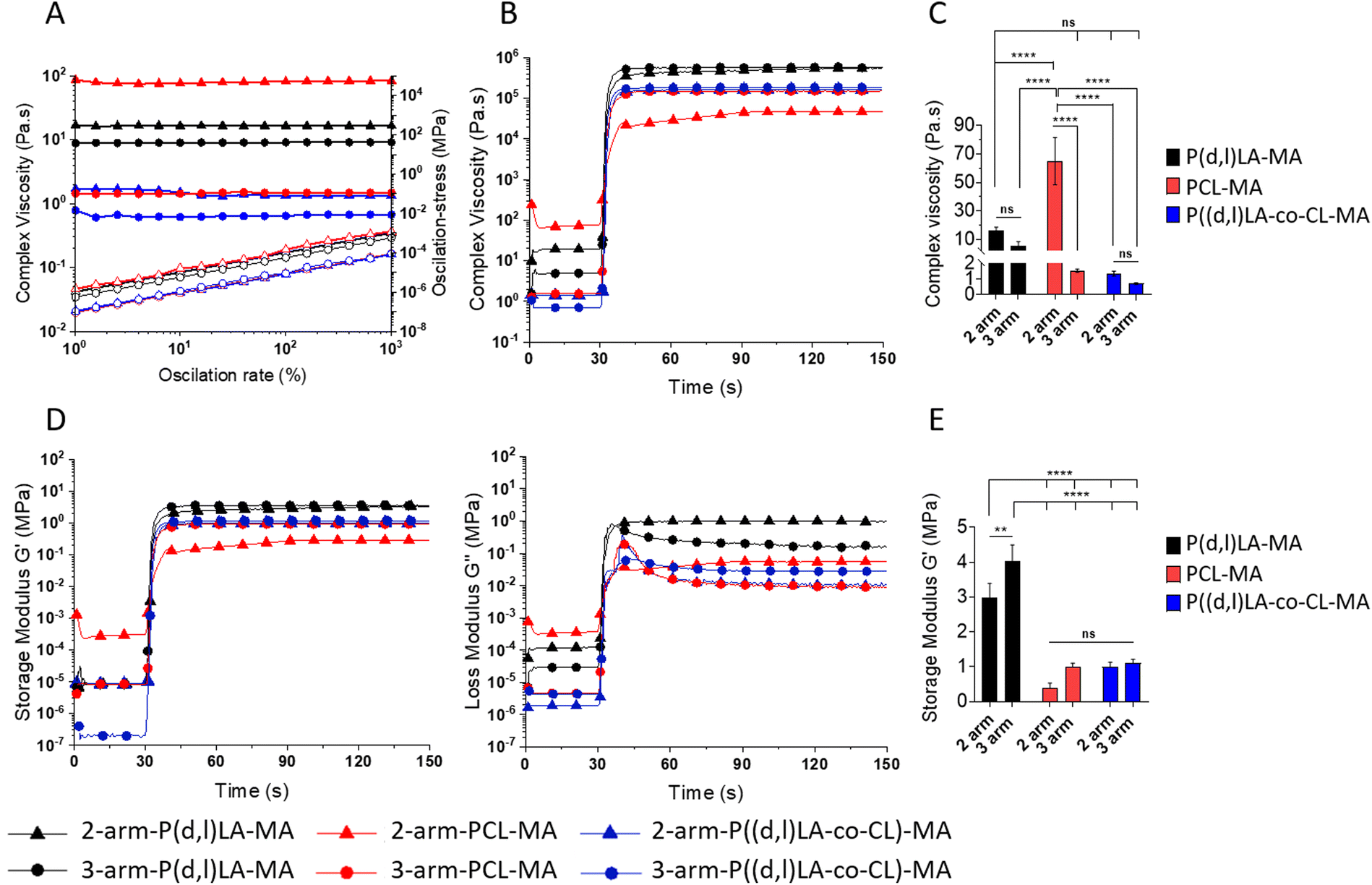 | ||
| Fig. 3 Viscosity measurements of the resins as a function of the oscillation rate (strain sweep) (A), from 1 to 1000% of deformation, and (B) variation of the complex viscosity as a function of time and upon irradiation with UV light. (C) Statistical analysis of the average complex viscosity of the inks before photocross-linking. Statistical significance was calculated from two-way ANOVA with Tukey's multiple comparison test. For complex viscosity: interaction F(2,12) = 35.28, p < 0.0001; arm number F(1,12) = 58.04, p < 0.0001; material type F(2,12) = 33.90, p < 0.0001. Adjusted p-values: p < 0.1 (ns), p < 0.0322(*), p < 0.0021 (**), p < 0.0002 (***) and <0.0001 (****). (D) Elastic (left, Storage Modulus G′) and viscoelastic (right, Loss Modulus G′′) response of the inks upon irradiation with 200 mW cm−2 UV light module (365 nm) for 60 seconds after a settling time of 30 seconds. (E) Storage Modulus G′ of crosslinked materials at plateau. Statistical significance was calculated from two-way ANOVA with Tukey's multiple comparison test. For complex viscosity: interaction F(2,12) = 4.098, p = 0.0440; arm number F(1,12) = 20.04, p = 0.0008; material type F(2,12) = 183.2, p < 0.0001. Adjusted p-values: p < 0.1 (ns), p < 0.0322 (*), p < 0.0021 (**), p < 0.0002 (***) and <0.0001 (****). | ||
Oscillatory measurements were carried out during UV exposure to evaluate the kinetics of the curing process. Upon irradiation with UV light, the viscosity of all the materials increased rapidly and exponentially, reaching a plateau as soon as 1–2 s after the irradiation was started, demonstrating the fast crosslinking speed of the resins (Fig. 3B and Fig. S3†). The complex viscosity of the photocross-linked materials was the highest for 2- and 3-arm-P(D,L)LA-MA polymers, followed by 2- and 3-arm-PCL and P((D,L)LA-co-CL) copolymers (Fig. 3C). In all cases, 3-arm polymers displayed a higher complex viscosity after cross-linking than their 2-arm counterparts, as a result of the higher cross-linking density. P(D,L)LA-MA, PCL-MA and P((D,L)LA-co-CL)-MA films prepared from 2-arm and 3-arm polymers were attributed for a complex viscosity of 502 ± 36 and 572 ± 9, 42 ± 6and 150 ± 0.9 and, 162 ± 1 and 185 ± 1 kPa s, respectively.
To evaluate the viscoelastic properties of the liquid resins and the cross-linked materials, the storage (G′) and loss moduli (G′′) of the resins were measured before and after UV irradiation during oscillatory photorheological experiments (Fig. 3D and E). G′ and G′′ represent the elastic and viscous components of a viscoelastic material, respectively. Readily upon UV irradiation both, G′ and G′′, increased over five orders of magnitude in all inks demonstrating the cross-linking of the chains. 2-arm-PCL-MA, which was initially a waxy solid, suffered an increase of G′ and G′′ of only 3 orders of magnitude, yet, indicating the cross-linking of the resin. 2- and 3-arm P((D,L)LA-MA, 2-arm PCL-MA and P((L,D)LA-co-CL)-MA inks displayed a G′ that was lower than G′′ at the beginning of the measurement and before UV irradiation, which increased upon UV irradiation crossing at the gelling point, to give rise to solid resins with an elastomeric response (G′ above G′′) (Fig. S4†). 3-arm-PCL-MA and 2-arm-P((D,L)LA-co-CL)-MA inks however, displayed a G′ above G′′ along the entire experiment, before and after UV irradiation, showing a higher elastic component in the resin already before irradiation. This was ascribed to certain chain ordering in the liquid resins or to a partial cross-linking of the ink.
The G′ after cross-linking of the formed films was 3 ± 0.4 and 4 ± 0.5 MPa, of 0.4 ± 0.2 and 1 ± 0.1 MPa and of 0.9 ± 0.1 and 1.1 ± 0.1 MPa for the 2- and 3-arm- P(D,L)LA-MA, PCL-MA and P((D,L)LA-co-CL)-MA materials (Fig. 3E), showing again a higher G′ for materials presenting 3-arms than the 2-arm counterparts. The flexibility of this system allowed us to create materials with a wide range of mechanical properties which were well on the range of several soft tissues such as the arterial wall, hyaline cartilage or the skin.23
3.3 Tensile properties of 3D printed and cross-linked materials
The mechanical properties of the 3D printed 2- and 3-arm copolymers and UV cross-linked 2- and 3-arm homopolymers were tested under tension (Fig. 4). All materials showed a rather stiff and brittle response, without plastic deformation, as observed by the absence of a clear strain hardening or necking before sample rupture (Fig. 4A).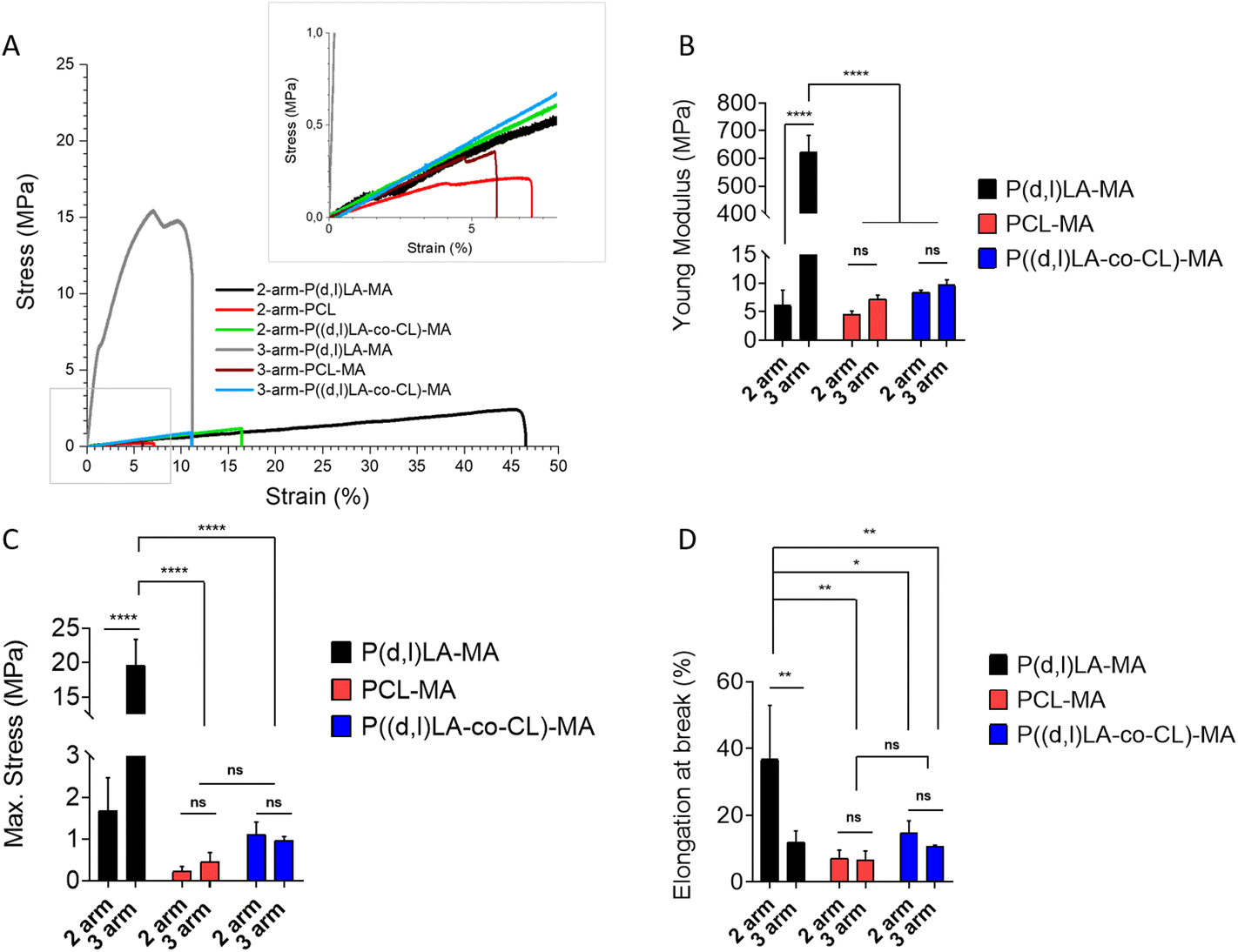 | ||
| Fig. 4 Representative tensile test traces (A) of 2- and 3-arm copolymers and homopolymer films fabricated via 3D printing and UV cross-linking, respectively. Inset shows a zoom-in of the region labelled with a grey box. n > 3 for all samples. (B) Young's modulus, (C) maximum stress and (D) elongation at break calculated from tensile tests. Statistical significance was calculated from two-way ANOVA with Tukey's multiple comparison. For Young's modulus: test interaction F(2,12) = 329.9, p < 0.0001; arm number F(1,12) = 336.3, p < 0.0001; material type F(2,12) = 329.3, p < 0.0001. Adjusted p-values: p < 0.1 (ns), p < 0.0322 (*), p < 0.0021 (**), p < 0.0002 (***) and <0.0001 (****). For maximum stress: interaction F(2,12) = 65.48, p < 0.0001; arm number F(1,12) = 66.37, p < 0.0001; material type F(2,12) = 81.43, p < 0.0001. Adjusted p-values: p < 0.1 (ns), p < 0.0322 (*), p < 0.0021 (**), p < 0.0002 (***) and <0.0001 (****). For elongation at break: interaction F(2,12) = 5.230, p = 0.0233; arm number F(1,12) = 8.738, p = 0,0120; material type F(2,12) = 9.613, p = 0,0032. Adjusted p-values: p < 0.1 (ns), p < 0.0322 (*), p < 0.0021 (**), p < 0.0002 (***) and <0.0001 (****). | ||
The 2-arm based polymer films displayed in all cases a lower Young's modulus than their 3-arm counterparts, although this was only significantly different for the P((D,L)LA-MA based inks, with values of 6.2 ± 2.6 MPa and 624.6 ± 58.7 MPa for 2- and 3-arm P((D,L)LA-MA, 4.5 ± 0.6 MPa and 7.2 ± 0.7 MPa for 2- and 3-arm PCL-MA and 8.4 ± 0.3 MPa and 9.7 ± 0.9 MPa for 2- and 3-arm P((D,L)LA-co-CL)-MA films (Fig. 4B). As expected, P((D,L)LA-MA films showed the highest stiffness and PCL-MA the lowest, with P((D,L)LA-co-CL)-MA films showing an intermediate response. Maximum stresses followed the same trend with values of 1.7 ± 0.8 MPa and 19.6 ± 3.7 MPa for 2- and 3-arm P((D,L)LA-MA films; 0.2 ± 0.1 MPa and 0.4 ± 0.2 MPa for 2- and 3-arm PCL-MA films and 1.1 ± 0.3 MPa and 0.9 ± 0.1 MPa for 2- and 3-arm P((D,L)LA-co-CL)-MA films, respectively (Fig. 4C).
The maximum strain measured followed the opposite trend, with elongation at breaks that were the highest for the 2-arm based inks as compared to their 3-arm counterparts, although this was only significantly different for P(D,L)LA-MA films. Again P(D,L)LA-MA films showed the highest elongation at break and PCL-MA the lowest, with P((D,L)LA-co-CL)-MA copolymers showing an intermediate behaviour. 2- and 3-arm P(D,L)LA-MA films showed an elongation at break of 38.9 ± 18.5% and 8.5 ± 1.3%, respectively; 2- and 3-arm PCL-MA films of 7.1 ± 2.4% and 6.6 ± 2.7%, respectively and 2- and 3-arm P((D,L)LA-co-CL)-MA films of 14.8 ± 3.4% and 10.7 ± 0.3%, respectively.
Melchels et al. synthesised P(D,L)-LA based resins using ethyl lactate as non-reactive diluent.20 They investigated the impact of the number of arms (2-, 3- or 6-arm) of the synthesised resin on the mechanical properties of the prepared films by 3-point bending mechanical analysis. In contrast to our results, no relationship between the number of arms and the mechanical properties was observed. However, they observed a clear decrease in the flexural modulus with increasing molecular weight of the inks, that ranged from 2.5 to 3.6 GPa, approximately. Moreover, the resulting films were brittle, as expected for PLA-based inks, with the maximum elongation at break of 6.2% approximately. Similarly, Elomaa et al. prepared resins from PCL oligomers with molecular weights ranging from 800 to 6000 g mol−1 and showed a clear decrease of the tensile Young's modulus from 15 to 6.7 MPa with increasing molecular weight, which appear to be well on the range of the PCL based homopolymers synthesised here.15 As expected, PCL based films accounted for an elongation at break higher than that reported for P(D,L)-LA based resins, with values ranging from 19 to 78% that were lowest for lower molecular weight inks.
3.4 Biocompatibility and cell adhesion support of the cross-linked materials
In order to evaluate the applicability of the synthesised inks as tissue engineering scaffolds and cell culture platforms, their capability to support cell adhesion and survival was tested in vitro with human bone marrow mesenchymal stem cells (hMSCs). hMSCs are known to readily respond to differences in their chemical and mechanical environment, making them ideal candidates to evaluate biocompatibility, adhesion and growth on novel materials.24,25 To do so, films of the different resins were fabricated by DLP printing, in the case of 2- and 3-arm-P((D,L)LA-co-CL) copolymers, and via casting and UV cross-linking in the case of 2- and 3-arm-PCL-MA and P(D,L)LA-MA polymers. Cell–material interactions are a key parameter determining the integration of biomaterial scaffolds. The first step towards guiding cell processes through material interactions is the adhesion to the materials’ surface. Analysis of the material's capability to support cell adhesion (Fig. 5) showed that after 24 h of culture PCL-MA films supported the highest cell adhesion with a total of 38709 ± 9100 and 36164 ± 2203 cells per film for 2- and 3-arm polymers, respectively. P(D,L)LA-MA showed the poorest cell adhesiveness with a total of 5643 ± 1000 and 5603 ± 388 cells per film for 2- and 3-arm polymers. PLA is known to display poor cell adhesion due to the hydrophobicity of its surface that limits the absorption of growth factors and proteins (and hence, cells) and multiple strategies have been developed to introduce negative charges through functionalization or formation of surface radicals.26–28 Moreover, previously reported comparative studies of PLA and PCL polymers demonstrate the highest cell attachment supported by PCL-based materials.29 2- and 3-arm copolymers showed a behavior intermediate of that of the two homopolymers, with attached cell numbers of 9115 ± 2846 and 23928 ± 1304 for 2- and 3-arm-P((D,L)LA-co-CL)-MA copolymers, respectively. Interestingly, this intermediate behavior made evident a higher cell attachment when the resins presented 3- instead of 2-arms, probably due to the higher storage modulus of the materials.
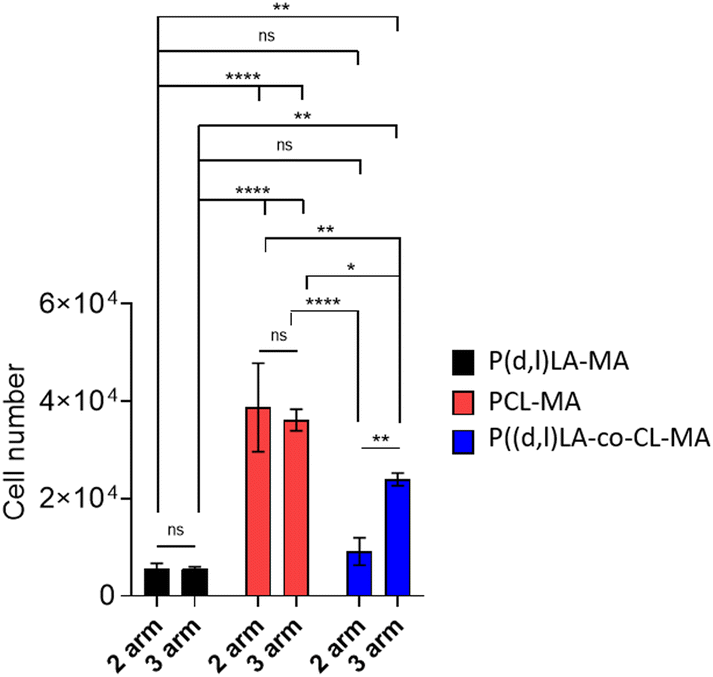 | ||
| Fig. 5 hMSC adhesion after 24 h of culture on 2- and 3-arm-P(D,L)LA-MA, PCL-MA and P((D,L)LA-co-CL)-MA polymer films. Error bars represent standard deviation; n = 3. Statistical significance was calculated from two-way ANOVA with Tukey's multiple comparison test. For complex viscosity: interaction F(2,12) = 8.049, p = 0.0061; arm number F(1,12) = 4.558, p = 0.054; material type F(2,12) = 95.62, p < 0.0001. Adjusted p-values: p < 0.1 (ns), p < 0.0322 (*), p < 0.0021 (**), p < 0.0002 (***) and <0.0001 (****). | ||
After initial adhesion (generally within 4 h), cells continue sensing their microenvironment and can adopt various cell morphologies and orientations, a process in which the substrate mechanical properties and topographical features play an important role.30,31 Cell observation via fluorescence microscopy revealed that, despite having a higher number of adhere cells, hMSCs cultured in PCL-MA films for 24 h presented a rounded morphology together with the formation of large cell aggregates, suggesting that cell–cell interactions were stronger than cell–material interactions (Fig. 6). Cells cultured in P(D,L)LA-MA films, however, presented an elongated and well-spread morphology in both, 2-arm and 3-arm-based polymer films, which could be ascribed to the higher mechanical properties of the substrates (Fig. 3E and 4). Cells cultured on P((D,L)LA-co-CL)-MA copolymers presented a cell morphology that was intermediate to the observed for the two homopolymers. In fact, hMSCs cultured on 2-arm copolymers displayed rounded cells forming aggregates, while hMSCs cultured on 3-arm-copolymers presented a morphology more similar to the observed in PCL-MA substrates, with higher cell spread areas. After 24 h, cells cultured in all ink types were mostly viable with few cells dead, as demonstrated by the live/dead stain with red cells denoting compromised membranes and green stain showing live cells (Fig. 6B and Fig. S5†).
 | ||
| Fig. 6 Fluorescence microscopy images of hMSCs after 24 h of culture in homopolymer and copolymer films. (A) Morphology of the cells stained for F-actin (green, cytoskeleton) and nuclear DNA (blue, nucleus). Scale bar is 50 μm. (B) Live/Dead images of hMSCs stained for ethidium homodimer (red, dead), and calcein (green, alive). Scale bar is 50 μm. | ||
3.5 Shape fidelity in DLP printed topographies
After proving the biocompatibility of (P(D,L)LA-co-CL)-MA inks, the shape fidelity post printing was evaluated. Shape fidelity is dependent on the printing resolution, cross-linking efficiency and structural stability of the fabricated objects.3 For DLP printing and SLA techniques, the printing resolution is determined by the equipment (theoretical) but, is affected by the absorbance and scattering of light by the ink and the diffusion of radicals within the resin. To this end, light blockers that absorb light at the irradiation wavelength and limit scattering are very often included in the ink mixture. However, the inclusion of such molecules compromises the biocompatibility of the ink and supposes an extra component that can be released to the culture media when the fabricated structures are biodegradable. The cross-linking efficiency determines the light exposure time required to cross-link the object and the presence of unreacted species (monomers or prepolymers) in the final object. The latter affects the shape stability after washing and leaching the final object. This effect is further enhanced when non-reactive diluents are included in the reaction mixture, which will escape or leach during washing steps (or during culture in media).The biocompatible P((D,L)LA-co-CL)-MA inks developed here were printed in the absence of light blockers and diluents, which facilitated shape stability after printing. A comparison between structures printed with reactive, non-reactive and without diluent revealed that the objects printed without diluent achieved a higher shape fidelity than those printed with diluent (Fig. S6 and Table S1†). Objects printed with reactive diluent presented also a good shape fidelity but, as explained earlier, included the reactive diluent in the final formulation which would alter the predesigned mechanical and chemical properties of the substrates.
To further investigate the achievable shape fidelity and resolution, repeating patterns of cubes, cylinders and cones were fabricated via DLP printing (Fig. 7). Cubes and cylinders were designed to have dimensions of 300 × 300 μm (with × depth) and 300 μm diameter, respectively, with a constant height of 300 μm. The objects appeared to reproduce the designed pattern successfully across the entire surface of the chips, with printing errors lower than 21% in all dimensions (Table 2). Of note was that 3-arm-copolymers appeared to have an overall lower printing error than 2-arm-copolymers, probably due to the higher density of photoreactive groups. Fabrication of conical structures was designed with decreasing size, informing about the printing resolution of these challenging shapes. These structures usually require to set a gradient of exposure times that decreases as the height of the cone increases and the printed circle decreases in size. Hence, the highest error measured was for the height of the cones, reaching values as high as 126 ± 3% for the smallest printed cones. Indeed, the height printing error increased with decreasing size of the printed cone and was again smallest for the topographies printed with the 3-arm copolymer. Nevertheless, further optimization of the printing parameters would yield lower errors. The printing error at the base of the cones was much smaller than that of the height, with errors that ranged from 2–19% and increased again with the smaller cone sizes.
 | ||
| Fig. 7 (A) Computational design and dimensions of micro-topography models with cubes (left) and cylinders (right). (B and C) SEM images of top-view (top) and cross-view (bottom) of micro-topography using 3-arm- (left) and 2-arm-P((D,L)LA-co-CL) (right) as printable resins: (B) cube- and (C) pillar-surface structures were shown. Scale bar is 2 mm in all images. (D and E) SEM images of cones on gradient structures printed with 3-arm- (D) and 2-arm-P((D,L)LA-co-CL)-MA (E). Scale bars of 2 mm and 500 μm are shown in the left and right images, respectively. | ||
| Cubes | Cylinders | Cones | ||||
|---|---|---|---|---|---|---|
| 2-arm | 3-arm | 2-arm | 3-arm | 2-arm | 3-arm | |
| ε w (%) ± SD | 15 ± 14 | 12 ± 11 | — | — | — | — |
| ε ∅ (%) ± SD | — | — | 21 ± 15 | 9 ± 7 | 2 ± 1 Cone 1 | 5 ± 2 Cone 1 |
| 5 ± 1 Cone 2 | 8 ± 1 Cone 2 | |||||
| 19 ± 7 Cone 3 | 5 ± 7 Cone 3 | |||||
| ε h (%) ± SD | 21 ± 4 | 5 ± 4 | 11 ± 4 | 9 ± 5 | 35 ± 0.4 Cone 1 | 35 ± 1 Cone 1 |
| 57 ± 1 Cone 2 | 42 ± 1 Cone 2 | |||||
| 126 ± 3 Cone 3 | 75 ± 23 Cone 3 | |||||
4 Conclusions
Here, we report the synthesis of a library of P(D,L)LA and PCL-based low molecular weight homopolymer and copolymer inks with 2- and 3-arms. Methacrylation of the polymers yielded 2- and 3-arm-PCL-MA, P(D,L)LA-MA and P((D,L)LA-co-CL)-MA with high degrees of methacrylation that enabled a fast photocross-linking of the resins in 1–2 s. Analysis of the viscoelastic properties demonstrated that 3-arm PCL-MA, P(D,L)LA-MA, P((D,L)LA-co-CL)-MA and 2-arm-P((D,L)LA-co-CL)-MA accounted for a low viscosity and met the main requirements to be used in DLP printing, that is to behave as Newtonian fluids with a viscosity <10 Pa s. 2- and 3-arm-P((D,L)LA-co-CL)-MA copolymers were selected for DLP printing as they presented the lowest viscosities (1.4 ± 0.2 and 0.72 ± 0.05 Pa s). We showed that these inks were printable at room temperature and without the use of diluents, resulting in structures with higher shape fidelity and resolution as compared to prints of the same inks using non-reactive diluents. The inks were exploited to print micro-topographies in the shape of cubes, cylinders and cones, showing printing errors that decreased with increasing number of arms in the copolymer. Analysis of the biocompatibility and cell adhesion of the resulting materials showed that the materials supported hMSC adhesion and viability. hMSCs cultured in P((D,L)LA-co-CL)-MA copolymer films presented an intermediate behaviour to that of cells cultured in PCL-MA and P(D,L)LA-MA films. PCL-MA films showed the highest cell adhesion followed by P((D,L)LA-co-CL)-MA and P(D,L)LA-MA films. The apparent cell spread area appeared to be highest in P(D,L)LA-MA films, followed by P((D,L)LA-co-CL)-MA and PCL-MA films. Being hMSCs highly sensitive to their chemical, structural and mechanical microenvironment and having shown good biocompatibility to the developed materials, extrapolation of these resuls to other cell and tissue types is expected. Altogether, these data demonstrate the synthesis of novel DLP biocompatible inks that can be processed at room temperature and in a solvent free environment, yielding high resolution objects that could be further exploited as cell culture platforms or tissue engineering scaffolds. Extrapolation of these findings to 3D architectures for cell immunomodulation would be an interesting possibility as it would allow for precise control of sizes and topographies up to a scale that could enable cell polarization on command.Author contributions
Sandra Ramos-Díez and Garazi Larrañaga-Jaurrieta contributed equally to this work: methodology, investigation, writing, visualization and formal analysis. Leire Iturriaga: Methodology and investigation. Ander Abarrategi: Supervision, writing and funding acquisition. Sandra Camarero-Espinosa: Conceptualization, methodology, investigation, supervision, writing, visualization, formal analysis and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding bodies and support through the EMAKIKER grant. S. C.-E. acknowledges the Spanish Ministry of Science and Innovation (MICINN) – State Investigation Agency (AEI) (PID2020-114901RA-I00). S. C.-E. and S. R.-D. acknowledge the Basque Government (PIBA_2022_1_0006). G. L.-J. acknowledges the Basque Government Predoctoral grant PRE_2021_1_0403. S. C.-E. and L. I. acknowledge the Provincial Council of Guipuzcoa. The project that gave rise to these results received the support of a fellowship from the “laCaixa” Foundation (ID100010434). The fellowship code is 117145. S. C.-E. acknowledges funding from the University of the Basque Country UPV/EHU within the framework of Grupos de Investigación (GIU21/033). A. A. acknowledges funding from PID2021-127191OB-I00 and RTI2018-101708-A-I00 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”. Grant RYC2018-025502-I funded by MCIN/AEI/10.13039/501100011033 and by “ESF Investing in your future”.References
- L. Moroni, T. Boland, J. A. Burdick, C. De Maria, B. Derby, G. Forgacs, J. Groll, Q. Li, J. Malda, V. A. Mironov, C. Mota, M. Nakamura, W. Shu, S. Takeuchi, T. B. F. Woodfield, T. Xu, J. J. Yoo and G. Vozzi, Trends Biotechnol., 2018, 36, 384–402 CrossRef CAS PubMed
.
- C. Mota, S. Camarero-Espinosa, M. B. Baker, P. Wieringa and L. Moroni, Chem. Rev., 2020, 120, 10547–10607 CrossRef CAS PubMed
- A. Schwab, R. Levato, M. D'Este, S. Piluso, D. Eglin and J. Malda, Chem. Rev., 2020, 120, 11028–11055 CrossRef CAS PubMed
- T. Duda and L. V. Raghavan, IFAC-PapersOnLine, 2016, 49, 103–110 CrossRef
- Z. Chen, Z. Li, J. Li, C. Liu, C. Lao, Y. Fu, C. Liu, Y. Li, P. Wang and Y. He, J. Eur. Ceram. Soc., 2019, 39, 661–687 CrossRef CAS
- P. N. Bernal, P. Delrot, D. Loterie, Y. Li, J. Malda, C. Moser and R. Levato, Adv. Mater., 2019, 31, 1904209 CrossRef CAS PubMed
- P. N. Bernal, M. Bouwmeester, J. Madrid-Wolff, M. Falandt, S. Florczak, N. G. Rodriguez, Y. Li, G. Größbacher, R.-A. Samsom, M. van Wolferen, L. J. W. van der Laan, P. Delrot, D. Loterie, J. Malda, C. Moser, B. Spee and R. Levato, Adv. Mater., 2022, 34, 2110054 CrossRef CAS PubMed
- K. S. Lim, J. H. Galarraga, X. Cui, G. C. J. Lindberg, J. A. Burdick and T. B. F. Woodfield, Chem. Rev., 2020, 120, 10662–10694 CrossRef CAS PubMed
- D. Xue, J. Zhang, Y. Wang and D. Mei, ACS Biomater. Sci. Eng., 2019, 5, 4825–4833 CrossRef CAS PubMed
- S. H. Kim, Y. K. Yeon, J. M. Lee, J. R. Chao, Y. J. Lee, Y. B. Seo, M. T. Sultan, O. J. Lee, J. S. Lee, S.-i. Yoon, I.-S. Hong, G. Khang, S. J. Lee, J. J. Yoo and C. H. Park, Nat. Commun., 2018, 9, 1620 CrossRef PubMed
- H. Goodarzi Hosseinabadi, D. Nieto, A. Yousefinejad, H. Fattel, L. Ionov and A. K. Miri, Appl. Mater. Today, 2023, 30, 101721 CrossRef
- A. Thomas, I. Orellano, T. Lam, B. Noichl, M.-A. Geiger, A.-K. Amler, A.-E. Kreuder, C. Palmer, G. Duda, R. Lauster and L. Kloke, Acta Biomater., 2020, 117, 121–132 CrossRef CAS PubMed
- S. Pal and S. K. Asha, Macromol. Chem. Phys., 2022, 223, 2200139 CrossRef CAS
- J. Jansen, F. P. W. Melchels, D. W. Grijpma and J. Feijen, Biomacromolecules, 2009, 10, 214–220 CrossRef CAS PubMed
- L. Elomaa, S. Teixeira, R. Hakala, H. Korhonen, D. W. Grijpma and J. V. Seppälä, Acta Biomater., 2011, 7, 3850–3856 CrossRef CAS PubMed
- N. Paunović, J. Marbach, Y. Bao, V. Berger, K. Klein, S. Schleich, F. B. Coulter, N. Kleger, A. R. Studart, D. Franzen, Z. Luo and J.-C. Leroux, Adv. Sci., 2022, 9, 2200907 CrossRef PubMed
- B. J. Green, K. S. Worthington, J. R. Thompson, S. J. Bunn, M. Rethwisch, E. E. Kaalberg, C. Jiao, L. A. Wiley, R. F. Mullins, E. M. Stone, E. H. Sohn, B. A. Tucker and C. A. Guymon, Biomacromolecules, 2018, 19, 3682–3692 CrossRef CAS PubMed
- T. Kuhnt, R. Marroquín García, S. Camarero-Espinosa, A. Dias, A. T. ten Cate, C. A. van Blitterswijk, L. Moroni and M. B. Baker, Biomater. Sci., 2019, 7, 4984–4989 RSC
- Y. Bao, N. Paunović and J.-C. Leroux, Adv. Funct. Mater., 2022, 32, 2109864 CrossRef CAS
- F. P. W. Melchels, J. Feijen and D. W. Grijpma, Biomaterials, 2009, 30, 3801–3809 CrossRef CAS PubMed
- T. Kuhnt, F. L. C. Morgan, M. B. Baker and L. Moroni, Addit. Manuf., 2021, 46, 102102 CAS
- R. Wang, F. Damanik, T. Kuhnt, A. Jaminon, S. Hafeez, H. Liu, H. Ippel, P. J. Dijkstra, N. Bouvy, L. Schurgers, A. T. ten Cate, A. Dias, L. Moroni and M. B. Baker, Adv. Healthcare Mater., 2023, 2202648 CrossRef CAS PubMed
-
F. H. Silver and D. L. Christiansen, in Biomaterials Science and Biocompatibility, ed. F. H. Silver and D. L. Christiansen, Springer New York, New York, NY, 1999, pp. 187–212, DOI:10.1007/978-1-4612-0557-9_7
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed
- S. Camarero-Espinosa and J. J. Cooper-White, Biomaterials, 2019, 210, 105–115 CrossRef CAS PubMed
- M. Schroepfer, F. Junghans, D. Voigt, M. Meyer, A. Breier, G. Schulze-Tanzil and I. Prade, ACS Omega, 2020, 5, 5498–5507 CrossRef CAS PubMed
- B. N. Teixeira, P. Aprile, R. H. Mendonça, D. J. Kelly and R. M. d. S. M. Thiré, J. Biomed. Mater. Res., Part B, 2019, 107, 37–49 CrossRef CAS PubMed
- R. A. Quirk, W. C. Chan, M. C. Davies, S. J. B. Tendler and K. M. Shakesheff, Biomaterials, 2001, 22, 865–872 CrossRef CAS PubMed
- S. Camarero-Espinosa and L. Moroni, Nat. Commun., 2021, 12, 1031 CrossRef CAS PubMed
- A. D. Doyle and K. M. Yamada, Exp. Cell Res., 2016, 343, 60–66 CrossRef CAS PubMed
- J. Zonderland and L. Moroni, Biomaterials, 2021, 268, 120572 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00581j |
| This journal is © The Royal Society of Chemistry 2023 |