
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasound-responsive glycopolymer micelles for targeted dual drug delivery in cancer therapy†
Shanmeng
Lin‡
ab,
Liwei
Zhu‡
 cd,
Zhiying
Li
a,
Siyuan
Yue
a,
Zhaohan
Wang
a,
Youwei
Xu
a,
Yichuan
Zhang
a,
Quan
Gao
a,
Jie
Chen
a,
Ting
Yin
a,
Lili
Niu
a and
Jin
Geng
*a
cd,
Zhiying
Li
a,
Siyuan
Yue
a,
Zhaohan
Wang
a,
Youwei
Xu
a,
Yichuan
Zhang
a,
Quan
Gao
a,
Jie
Chen
a,
Ting
Yin
a,
Lili
Niu
a and
Jin
Geng
*a
aShenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China. E-mail: jin.geng@siat.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cDepartment of General Surgery, Nanfang Hospital, The First School of Clinical Medicine, Southern Medical University, Guangzhou, Guangdong 510515, China
dGuangdong Provincial Key Laboratory of Precision Medicine for Gastrointestinal Tumor, Nanfang Hospital, The First School of Clinical Medicine, Southern Medical University, Guangzhou, Guangdong 510515, China
First published on 24th July 2023
Abstract
Controlled drug release of nanoparticles was achieved by irreversibly disrupting polymer micelles through high-intensity focused ultrasound (HIFU) induction. An ultrasound-responsive block copolymer was synthesized, comprising an end-functional Eosin Y fluorophore, 2-tetrahydropyranyl acrylate (THPA), and acrylate mannose (MAN). The block copolymer was then self-assembled to produce micelles. The chemotherapy drug dasatinib (DAS) and the sonodynamic therapy agent methylene blue (MB) were encapsulated by the self-assembly of the block copolymer. This targeted nanoparticle enables sonodynamic therapy through high-intensity focused ultrasound while triggering nanoparticle disassembly for controlled drug release. The ultrasound-mediated, non-invasive strategy provides external spatiotemporal control for targeted tumour treatment.
Introduction
Ultrasound (US) plays an increasingly important role in delivering therapeutic drugs, such as the genetic material, proteins, and chemotherapy drugs. It uses the energy of non-interacting concentrated pressure waves to generate forces that penetrate cell membranes and disrupt vesicles that carry drugs.1–5 Vesicles are usually microbubbles or gas-containing liposomes.6–8 Ultrasound has high spatial resolution and can deliver stimuli precisely to deep locations in the body. The principle of ultrasound-triggered drug release in the presence of oscillating bubbles is cavitation, while diffusion-enhanced drug transport occurs through the transient formation of pores in the cell membrane. However, the stability of these delivery vehicles is always a challenge due to unexpected stimuli and the internal environment because they mainly consist of soluble gases.9,10 Therefore, it is important to develop a release enhancer with improved thermodynamic stability and high sensitivity to ultrasonic waves while minimizing ultrasonic power and the exposure time.It is worth noting that the use of high-intensity or power ultrasound in chemistry has dramatically increased over the decades. The scope of synthetic procedures and process methods discovered has benefited greatly from sonication. Sonochemistry degradation of polymers has proven to be an attractive process due to the fact that it only simply divides the most susceptible chemical bonds. For example, low frequency ultrasound (typically 20 kHz) has been used for mechanoresponsive dendritic and polymeric organogels.11 Chen et al. demonstrated the formation of dual ultrasound- and pH-responsive polymer vesicles from a diblock copolymer containing tetrahydrofuranyloxy ethyl methacrylate as ultrasound sensitive moieties, enabling the controlled release of a loaded anticancer drug by both triggers.12,13 Although low frequency ultrasounds have greater penetration depth in tissues and are non-invasive, they cannot be focused into a small spot. As the frequency increases, the focus point of the ultrasound becomes smaller and the intensity at the focal spot becomes higher. Therefore, the high-intensity focused ultrasound (HIFU, frequency range 0.8–3.5 MHz) technology has attracted a great deal of attention because it allows external spatiotemporal control for targeted treatment, either through direct application or via controlled release of anticancer drugs from nanocarriers. Rapoport's group used a Pluronic P-105 copolymer to form DOX-loaded micelles in water. Focused 1 MHz ultrasound triggered a local release of anticancer drugs at tumour sites through the mechanism of droplet-to-bubble transition.14 Likewise, an S–S bond was embedded in the centre of the polymer chain, and the reverse DA reaction was triggered by sonomechanical force, enabling the effective release and activation of conjugated furosemide and DOX.15 Some mechanoresponsive polymers contain chemical bonds that break under ultrasonic conditions.16,17 Over the past few decades, researchers have made significant progress in the development of ultrasound-responsive polymeric nanoparticles for drug delivery applications. These nanoparticles are designed to release their payload in response to external ultrasound stimulation, which can provide spatiotemporal control over drug delivery.13,18 The aforementioned studies demonstrate that HIFU is a very attractive and promising technique for the spatiotemporally controlled release of drug payloads from nanocarriers, and the development of novel polymeric materials with excellent HIFU degradation properties is required to produce more reliable ultrasound-responsive polymeric nanoparticles for controlled drug delivery applications.
Herein, we report on a novel class of HIFU responsive block copolymer self-assembled nanocarriers with a reporter system, capable of successively releasing the anticancer therapy drug dasatinib (DAS) and the sonodynamic therapy agent methylene blue (MB) upon HIFU application. Sonodynamic therapy (SDT) is a method of enhancing the cytotoxic effect of drugs (sonosensitizers) on tumour cells by exposing them to ultrasound (US). Methylene blue is an inexpensive phenothiazine dye with low toxicity and has been approved for clinical use. It has been demonstrated that MB has a sonodynamic antitumor effect.19 An ultrasound-responsive block copolymer was synthesized, comprising an end-functional Eosin Y fluorophore, 2-tetrahydropyranyl acrylate (THPA), and acrylate mannose (MAN). The block copolymer was then self-assembled to produce micelles. HIFU-induced selective scission of the hydrophobic THPA group into hydrophilic methacrylic acid groups led to the disruption of the polymer micelles and the release of the cargo (Scheme 1). Drug activation was confirmed by tracking MB fluorescence as well as Eosin Y from the polymer, which occurs in the cytoplasm inside cells. To achieve targeted delivery, we employed mannose to form the hydrophilic block and as a targeting moiety in the system. As we reported previously, the introduction of mannose groups can enhance cellular uptake in human hepatoma (HepG2) cells.20 To outline the scope of this system, we investigated the activation and release of drug payloads for safe and effective treatment with a tumor model in mice.
 | ||
| Scheme 1 Schematic illustration of the preparation of DAS/MB@M for ultrasound mediated tumour treatment. | ||
Results and discussion
To achieve controlled HIFU degradability and multifunctionality of polymers, we first designed and synthesized a novel Eosin Y terminated RAFT agent (1), acrylate mannose (2) and a THPA monomer (3), which could be polymerized by RAFT polymerization (shown in Fig. 1a). RAFT polymerization is particularly appealing because of its versatility and experimental simplicity. A RAFT chain transfer agent, 1, was used to mediate the polymerization of 2 at 25 °C under 365 nm illumination using phenylbis(2,4,6-trimethylbenzoyl)-phosphine oxide (BAPO) as a photoinitiator, and the resulting polymer, a macro-RAFT agent, was used to polymerize 3 to produce the block copolymer P1, denoted as PMAN-b-PTHPA. 1H NMR spectroscopy (ESI Fig. S7–S10†) revealed that the conversion values of the PMAN-RAFT polymer and the PMAN-b-PTHPA polymer were 68.0% and 76.3% respectively, the polymer had a comonomer composition of 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :61 (MAN:THPA) and the presence of the pendant mannose and tetrahydropyranyl functional groups was confirmed.
:61 (MAN:THPA) and the presence of the pendant mannose and tetrahydropyranyl functional groups was confirmed.
 | ||
| Fig. 1 Synthesis and characterization of DAS/MB@M. (a) Preparation steps of PMAN24-b-PTHPA61. (b) The change of the 1H spectrum of DAS/MB@M before and after ultrasound irradiation in water. (c) TEM images of DAS/MB@M before and after ultrasound irradiation; the scale bar is 50 nm. (d) The critical micelle concentration (CMC) of polymeric micelles. (e) UV-vis absorbance spectra of 7.73 μmol L−1 free dasatinib (DAS), 5.85 μmol L−1 free methylene blue (MB), 2.83 μmol L−1 PMAN-b-PTHPA and DAS/MB@M. (f) DAS and MB release from micelles under ultrasound irradiation in PBS (1.25 MHz, 4.2 W cm−2, 50% duty cycle). | ||
To test the ultrasound responsiveness of THPA pendants along the polymer chain, we recorded the 1H NMR spectra of the copolymer P1 solution (in DMSO-d6) before and after the ultrasound treatment (Fig. 1b). Upon HIFU application (1.25 MHz, 4.2 Wcm−2), the integral of THPA 5.84 ppm showed reduced intensities over time due to the cleavage of the THP side groups.
At room temperature, the P(MAN)-b-P(THPA) copolymer was self-assembled into micelles in a DMF/H2O mixture (1/9, v/v) at a concentration of 0.25 mg mL−1, followed by dialysis against water. The TEM images in Fig. S11a† confirm the morphology as micelles had a diameter of approximately 36 nm. The zeta potential of the micelles remained almost unchanged before and after sonication (Fig. S11b†), indicating that the ultrasound treatment had a negligible effect on the surface charge of the micelles. A hydrodynamic diameter (Dh) of 65 nm was confirmed by dynamic light scattering (DLS, Fig. S11c†). To load DAS and MB, the block copolymer P1 was used to self-assemble into core–shell micelles (DAS/MB@M) in aqueous media under the same conditions, where the drug-containing P(THPA) blocks form a hydrophobic core, surrounded by a hydrophilic P(MAN) corona, enabling cell targeting ligands to be presented on the exterior of the micelles (Fig. 1c). Upon US irradiation, the micelles were observed to enlarge to 75 nm due to ultrasonic cavitation, which produced instantaneous energy and disrupted P(THPA) chains, leading to the rearrangement of the self-assembled micelles (Fig. 1c). The critical micelle concentration (CMC) of PMAN-b-PTHPA polymeric micelles was 4.01 × 10−4 mg mL−1 (Fig. 1d).
The loading contents of DAS and MB were determined by UV spectrophotometry, which were 19.6% and 11.0% respectively, and the encapsulating efficiencies of DAS and MB were 78.34% and 43.89% respectively (Fig. 1e), further illustrating the successful loading of DAS and MB. The release of DAS and MB from DAS/MB@M was evaluated in the absence and in the presence of ultrasound. DAS/MB@M did not release drugs in PBS buffer (pH 7.4) at 37 °C. However, upon ultrasound exposure, both DAS and MB were gradually released from the micelles at different rates under the same conditions (Fig. 1f). The concentrations reached 19.03 μM and 45.89 μM after 10 min sonication, respectively. These results confirmed that the micelles responded to the presence of ultrasound, allowing for controlled and accelerated release of DAS and MB.
Targeted micelles were evaluated in cell-based assays. The cytotoxicity of free micelles and drug loaded DAS/MB@M was assessed to determine their effects on cell viability in the concentration range used for further treatment in the absence of HIFU. This is essential for using these micelles as a drug delivery platform. After incubating HepG2 (human liver hepatocellular carcinoma) cells with DAS/MB@M for 4 h, no obvious cell death was induced by the micelles at the concentrations from 0 μM to 10 μM without sonication. However, the cell viability was obviously inhibited at the concentration of 10 μM under ultrasound irradiation (Fig. S13†). The cellular uptake of Eosin Y functionalised DAS/MB@M was evaluated, where cells were incubated for 2 h with the same concentration of DAS/MB@M (10 μM dasatinib) and imaged by confocal laser scanning microscopy (CLSM). The micelles showed comparable uptake in both LO2 (human normal liver cells) and HeLa cells (human cervical cancer cells), but a much higher signal intensity was observed in HepG2 cells, indicating enhanced delivery of micelles (Fig. 2a). The flow cytometry results matched the CLSM observations, as DAS/MB@M showed better cellular internalization in HepG2 cells. The signal intensity from FITC or Cy5.5 in the micelle treated groups gradually increased with the incubation time (Fig. 2b) in all three cell lines, with a preference for cellular uptake in HepG2 cells. Notably, HepG2 cells treated with micelles became fluorescent faster than LO2 and HeLa cells (Fig. 2b). Further incubation of the micelles with HepG2 cells confirmed that cellular uptake reached a plateau after 4 h, where the signal intensity increased faster and more strongly than that of free MB in HepG2 cells (Fig. 2c and d). These results suggest that the internalization of the micelles was mediated, at least in part, by the high expression of mannose receptor (MR) on HepG2 cells.21,22
 | ||
| Fig. 2 Evaluation of the targeting ability of DAS/MB@M. (a) Representative CLSM images for observing the DAS/MB@M uptake in LO2, HeLa and HepG2 cells after incubation with DAS/MB@M for 2 h; the scale bar is 50 nm. (b) Flow cytometry quantification of LO2, HeLa and HepG2 cells after incubation with DAS/MB@M for 0, 15, 30, 60, 120 and 240 min 0. (c) Representative CLSM images for observing the uptake of MB and DAS/MB@M in HepG2 cells after incubation with DAS/MB@M for 4 h; the scale bar is 100 nm. (d) Flow cytometry analysis in HepG2 cancer cells incubated with MB and DAS/MB@M for 0, 0.5, 2, 4, 6, and 12 h (n = 3). | ||
The accumulation and release of Eosin Y-labelled micelles in HepG2 cells were assessed by CLSM imaging after irradiation by HIFU (0.25 MHz, 1.7 W cm−2, 10 min) at 0.5 h post-treatment. Both MB fluorescence and Eosin Y fluorescence were detectable in HepG2 cells, and efficient MB release was observed upon HIFU application. As shown in Fig. 3a, different in cellulo distribution profiles were observed with and without HIFU. Without sonication, MB and micelles (Eosin Y labelled) were colocalised well, but not after 10 min of HIFU treatment. This result indicated the release of drugs from the micelles, as most of the Eosin Y fluorescence derived from MB accumulation in cells. Previous reports have demonstrated that polymer self-assembled micelles can be internalised via endocytosis. To confirm this, we stained the micelle-treated cells with LysoTracker™ Red. The yellow colour in Fig. 3b represents the overlap of micelles (green) and endo/lysosomes (red), indicating the colocalization of endo/lysosomes and polymer micelles.
 | ||
| Fig. 3 Intracellular drug release and localization of DAS/MB@M. (a) Visualisation of the intracellular drug release before and after ultrasound irradiation. (b) and (c) Intracellular localization of DAS/MB@M in HepG2 cells. | ||
In vitro studies were conducted to evaluate the enhanced anticancer effect of the drug-loaded micelles. The viability of HepG2 cells treated with DAS/MB@M with or without sonication was assessed using the CCK-8 assay (Fig. 4a). At a concentration of 10 μM dasatinib, the cell viability in the presence of DAS/MB@M + US was lower than that of free DAS/MB + US, US alone, and DAS. 74.38% cell kill was achieved with HIFU treatment in the presence of DAS/MB@M, compared with 48.84% with free MB and DAS + US, indicating that the micelles could increase the intracellular efficiency and enhance cancer treatment. To further evaluate the effect of MB and DAS loaded in micelles on phosphorylated Src levels, DNA damage, or apoptosis, the protein levels of related genes were analysed by immunoblotting. The results indicated that DAS and micelles loaded with DAS significantly inhibited the phosphorylation of Src (Fig. 4b), suggesting that DAS/MB@M could suppress the phosphorylation of Src and further induce apoptosis.23 Similarly, high levels of γ-H2AX were observed in free DAS/MB + US and DAS/MB@M + US (Fig. 4c), indicating that M, as a drug carrier, could induce DNA damage.24
 | ||
| Fig. 4 In vitro SDT therapy of DAS/MB@M against HepG2 cancer cells. (a) In vitro growth inhibition effects of DAS, MB and DAS/MB@M toward HepG2 cancer cells under US irradiation. (b and c) Western blot analysis of the expression of the apoptosis-related proteins (p-Src, Src, γ-H2AX). | ||
The efficacy of targeted micelles in cancer therapy was evaluated in NOD/SCID mice bearing HepG2 tumours. Intravenous injection of DAS/MB@M, free DAS/MB, with or without US (1.0 W cm−2, 5 min) was performed 4 h prior to US treatment, which was repeated every other day for 4 cycles. Tumour volumes were measured for over 20 days (Fig. 5a), with drugs administered at a dose of 5 mg kg−1 based on DAS for micelles when tumours reached 200 mm3. The results showed that DAS/MB@M effectively suppressed tumour growth when used in combination with US (Fig. 5c). Tumour progression was markedly reduced compared to free DAS/MB under US irradiation and US treatment alone, which also increased the survival of tumour-bearing mice (Fig. 6a). The enhanced tumor therapy achieved through this combination treatment strategy can be attributed to the synergistic effects of chemotherapy, sonodynamic therapy mediated by MB, and high-intensity focused ultrasound (HIFU). By encapsulating MB within the micelles, we harnessed its sonosensitizing properties, which enhance the cytotoxic effects on tumor cells when exposed to ultrasound. It is important to note that throughout the study, the micelle formulation showed no reduction in the body weights of the mice, indicating good tolerability of the treatment at the doses used (Fig. 5b). These results highlight the promising potential of MB as a sonodynamic therapy agent when combined with targeted micelles for tumor treatment. The combination of chemotherapy, sonodynamic therapy, and HIFU provides a multifaceted approach to achieving enhanced therapeutic outcomes.
 | ||
| Fig. 5 In vivo SDT treatment efficiency of DAS/MB@M against tumors. (a) Schematic illustration of the therapeutic procedure on a NOD/SCID mouse bearing HepG2 subcutaneous tumor. (b) Body weight changes and (c) Tumor volume after different treatments (control, US, DAS, MB + DAS + US, DAS/MB@M and DAS/MB@M + US, n = 4, mean ± SD) for 20 days. (d) In vivo fluorescence images of HepG2 tumor bearing NOD/SCID mice after intravenous injection of MB and DAS/MB@M. (e and f) Biodistribution of free MB and DAS/MB@M in tumor, heart, liver, spleen, lungs and kidneys 48 h after iv injection. (g). Microscopy images of tunel and H&E-stained tumor slices (the scale bars are 150 μm). | ||
 | ||
| Fig. 6 Survival and biosafety evaluation. (a) Survival curbs of HepG2 tumor bearing NOD/SCID mice with various treatments (n = 3). (b–d) Blood biochemistry analysis (alanine aminotransferase (ALT), aspartate aminotransferase (AST) and blood urea nitrogen (BUN)). (e) H&E staining of major organ sections collected from healthy C57 mice after 48 h. | ||
The distribution of DAS/MB@M in tumours and organs over time was evaluated in mice bearing HepG2 tumours using fluorescence imaging of MB and MB loaded micelles (8 mg kg−1 based on MB concentrations). Whole-body imaging of mice revealed a higher accumulation of DAS/MB@M in tumours compared to free MB after intravenous injection of micelles (Fig. 5d). Ex vivo imaging also confirmed that DAS/MB@M had approximately a 2-fold higher tumour accumulation than free MB (Fig. 5e and f). The pathological changes of tumors were investigated by TdT-mediated dUTP nick-end labeling (TUNEL) and hematoxylin and eosin (H&E) staining (Fig. 5g), and more severe apoptosis of tumor cells was observed after treatment with DAS/MB@M and US irradiation. It revealed that DAS/MB@M with US irradiation effectively inhibited tumour growth by promoting the apoptosis of tumor cells. Moreover, no significant accumulation of micelles was observed in the liver tissue, kidneys, heart, lungs and spleen.
The biosafety of DAS/MB@M was evaluated and compared to that of free DAS/MB and HIFU treatment alone. Body weight measurements showed negligible weight reduction in all groups (Fig. 5b). Healthy BALB/c mice were treated with PBS, DAS/MB@M, and free DAS/MB, with or without US treatment. Blood and major organs were collected and analysed. Hematology tests and key organ histological abnormalities showed no gross toxicities in mice treated with the micelles, with or without sonication (Fig. 6b–e and ESI Fig. S12a–c†). These results demonstrate that the treatment had negligible toxicity for the tumour bearing mice.
Conclusions
In summary, the development of an ultrasound-responsive dual drug delivery system with visualization and targeting capabilities has been described. This system utilizes PMAN-b-PTHPA micelles loaded with dasatinib and methylene blue to effectively suppress tumour growth by inducing DNA damage and inhibiting Src phosphorylation in cancer cells. The targeted micelles are rapidly internalized by HepG2 cells and enriched in tumour tissues while exhibiting negligible toxicity in vivo. These results indicate that this drug delivery system has great potential for the treatment of cancer with the combined therapy of chemotherapy, sonodynamic therapy, and HIFU. The use of RAFT polymerisation in the synthesis of the block copolymer and the detailed characterization of the micelles provide insight into the design of future drug delivery systems for enhanced cancer therapy.Experimental section
Sonodynamic performance of PMAN-b-PTHPA
PMAN-b-PTHPA (40.00 mg) was dissolved in DMSO (0.20 mL), and then the mixture was added to 8.00 mL of deionized water. The mixture solution was exposed to US irradiation (1.25 MHz, 4.2 W cm−2, 50% duty cycle) for 15 min. The dry sample was obtained by lyophilization. The 1H spectrum was recorded in DMSO-d6.Fabrication of DAS/MB@M micelles
PMAN-b-PTHPA (4.00 mg), DAS (dasatinib, 1.00 mg) and MB (methylene blue, 1.00 mg) were dissolved in 1.60 mL of DMF and then added dropwise to 14.40 mL of deionized water with stirring at room temperature for 30 min. According to the literature,25,26 the solution was dialysed against deionized water (MWCO: 2000 Da) and then concentrated with an ultrafiltration centrifugal tube (MWCO: 3000 Da) at 4000 rpm and 4 °C.Characterization
The zeta potential and hydrodynamic particle size of DAS/MB@M micelles were determined on a Malvern Zetasizer Nanoseries (Nano ZS90). The loading of dasatinib (DAS) and methylene blue (MB) was determined by UV spectrophotometry.27 The DAS/MB@M suspension was lyophilized and re-dissolved in DMSO. The content of dasatinib and methylene blue was determined by the absorbance at 324 nm and 670 nm. The encapsulation efficiency (EE) and drug loading content (LC) of dasatinib and methylene blue were calculated using the following formulas:
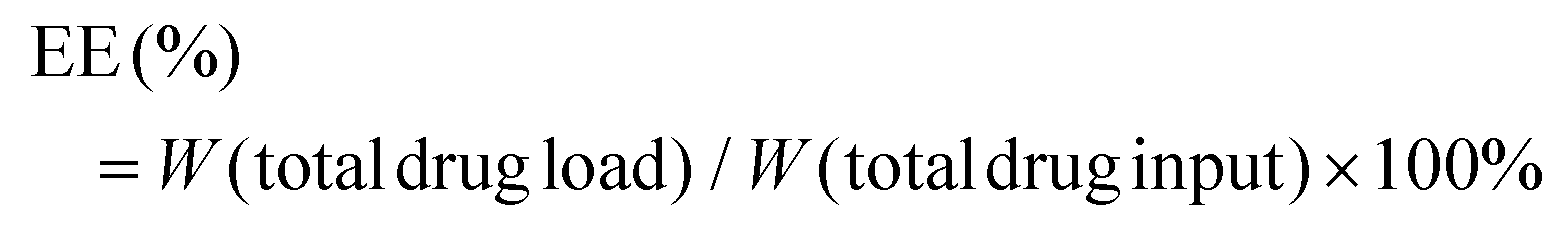
Determination of the critical micelle concentration (CMC)
The CMC of polymers was determined using a fluorescence spectrometer (FLS920, Edinburgh Instruments, England) using pyrene as the probe. Pyrene was dissolved in acetone, divided and added to glass vials, and acetone was allowed to evaporate. Micellar solutions of different concentrations (0.0001–100 μg mL−1) were incubated with pyrene and it was ensured that the final concentration of pyrene was 6 × 10−7 mol L−1.28 The solution was stirred overnight at room temperature. The fluorescence excitation was fixed at 335 nm, and the emission spectra were recorded from 350 nm to 450 nm. The excitation and the emission slits were set at 2 nm and 1 nm, and the intensity ratio (I382/I371) of pyrene fluorescence was plotted against the logarithm of micelle concentration.29Cell culture and the cell viability test
HepG2 cells were cultured in a DMEM (high glucose) containing 10% fetal bovine serum and 1% antibiotics at 37 °C and under 5% CO2. HepG2 cells were pre-seeded into a 96-well plate (0.8 × 104 cells per well) and incubated overnight. Then, the cells were incubated with PBS, DAS/MB@M (10.0 μM dasatinib, 8.50 μM), dasatinib (10.0 μM) and methylene blue (8.50 μM) for 4 h or the cells were incubated with different concentrations of DAS/MB@M for 4 h. The cells were washed with PBS three times, incubated with a medium, treated with or without ultrasound irradiation (1.25 MHz, 4.2 W cm−2, 50% duty cycle) for 15 min and then incubated for 24 h. Finally, the cells were incubated with 10 μL of CCK-8 solution in 100.0 μL of DMEM for 1 h and the absorbance at 450 nm was measured.Cellular uptake of DAS/MB@M micelles
HepG2, HeLa and LO2 cells were grown in a 24-well plate (5 × 104 cells per well), incubated overnight, and then incubated with DAS/MB@M (10.0 μM dasatinib). At 0, 15, 30, 60, 120 and 240 min, the cells were washed with PBS three times, designated by trypsin, and centrifuged at 2000 rpm for 5 min at 4 °C; the collected cells were resuspended in 0.30 mL of cooled PBS and analysed by flow cytometry. In addition, the cells were seeded on Ibidi μ-slide 8 well (Ibidi GmbH, Germany) plates (3.5 × 104 cells per well), incubated overnight, and then incubated with DAS/MB@M (10.00 μM dasatinib). At 120 min, the cells were washed with PBS three times and then analysed under a confocal fluorescent microscope. λex/em (Eosin Y) = 488/540 nm and λex/em (Hoechst 33342) = 405/460 nm.Subcellular distribution of DAS/MB@M micelles observed by CLSM
HepG2 cells were grown in an Ibidi μ-slide 8 well plate (3.5 × 104 cells per well), incubated overnight, and then incubated with DAS/MB@M (10.0 μM dasatinib) for 4 h. The cells were washed with PBS three times and then incubated with LysoTracker™ Red (0.20 μM) in DMEM for 20 min and then Hoechst 33342 (1.00 μM) was added to the medium. After 10 min, the cells were washed with PBS three times and then imaged using a confocal fluorescent microscope. λex/em (Eosin Y) = 488/540 nm, λex/em (Hoechst 33342) = 405/460 nm and λex/em (LysoTracker™ Red) = 560/600.Observation of intracellular drug release of DAS/MB@M micelles
HepG2 cells were pre-seeded on confocal dishes (BeyoGold™ 35 mm) at 5 × 104 cells per well and incubated overnight. The cells were treated with DAS/MB@M (10.0 μM dasatinib) for 4 h and washed with PBS three times. The cells were treated with or without ultrasound irradiation (0.25 MHz, 1.7 W cm−2, 20% duty cycle) for 10 min and then incubated for 30 min. Then, the cells were stained with Hoechst 33342 (1.00 μM) for 10 min and imaged using a confocal fluorescent microscope. λex/em (Eosin Y) = 488/540 nm, λex/em (Hoechst) = 405/460 nm and λex/em (methylene blue) = 630/694.Immunoblotting analysis
HepG2 cells were grown in 35 mm cell culture dishes at a density of 3.5 × 105 cells per well and incubated overnight. The cells were incubated with different concentrations of PBS, DAS/MB@M (10.0 μM dasatinib, 8.5 μM methylene blue), dasatinib (10.0 μM) and methylene blue (8.50 μM) for 4 h. The cells were washed with PBS three times, incubated with a medium, treated with or without ultrasound irradiation (0.25 MHz, 1.7 W cm−2, 20% duty cycle) for 15 min and then incubated for 12 h. Then whole cells were collected and the samples were prepared with RIPA lysis buffer which contained phosphatase inhibitors. The samples were separated on 8%–13.5% SDS-PAGE gel and transferred to a PVDF membrane. The marker and desired protein were detected by primary incubation with an antibody (diluted 1:1000), followed by horseradish peroxidase-conjugated secondary antibodies (diluted 1:5000). The membrane was developed using BeyoECL Moon (Beyotime) and imaged using the ChemiDoc XRS + System (Bio-Rad).
Tumor suppression experiments in vivo
All animal studies were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Shenzhen Institutes of Advanced Technology (SIAT-IACUC-220419-YYS-GJ-A2140). For tumor inoculation, 100.0 μL of HepG2 cells (4.5 × 106) was subcutaneously injected into the right rear side of the NOD/SCID mice. HepG2 tumor bearing mice were randomly divided into six groups (n = 4 per group) for treatment with PBS, PBS + US, DAS, DAS + MB + US, DAS/MB@M and DAS/MB@M + US (DAS: 5.00 mg kg−1, MB: 2.00 mg kg−1, DAS/MB@M: 5.00 mg kg−1 for DAS, intravenous injection). In the HIFU irradiation groups, the tumors were treated with HIFU irradiation (1.0 MHZ, 1.0 W cm−2, 5 min) 4 h after the intravenous injection. During the treatment, the tumor size and body weight were measured every two days for 20 days. Tumor volume = length × width2/2. For H&E staining and TUNEL immunostaining analysis, the tumor tissues of mice were collected.In vivo NIR fluorescence imaging
HepG2 tumor-bearing mice were intravenously injected with MB (200.0 μL, 8.0 mg kg−1) and DAS/MB@M (200.0 μL, 8.0 mg kg−1 for methylene blue) respectively. At 0, 2, 4, 8, 12, 24 and 48 h post-injection, the mice were imaged using an IVIS Spectrum imaging system. Then, the mice were sacrificed and the tumor tissues and main organs were collected. The fluorescence intensities of the tumor tissues and main organs were finally analyzed using the IVIS Spectrum imaging system. λex/em (methylene blue) = 640/700.Biosafety study
Healthy BALB/c mice were randomly divided into 6 groups (n = 3) for treatment with PBS, PBS + US, DAS, DAS + MB + US, DAS/MB@M and DAS/MB@M + US (DAS dose: 5.0 mg kg−1, MB dose: 2.0 mg kg−1, DAS/MB@M dose (DAS dose: 5.0 mg kg−1), intravenous injection).30 The BALB/c mice were treated with or without US irradiation (1.0 MHZ, 1.0 W cm−2, 5 min) 4 h after the intravenous injection. At 48 h, the mice were sacrificed, and the blood serum and main organs including the heart, liver, spleen, lungs and kidneys were collected for further analysis of blood chemistry and H&E.Author contributions
J. G. conceived, designed, and directed the project. S. L., Z. W., Y. X., Y. Z., Z. L., and J. C. conducted the compound synthesis, polymerization optimization, polymer extraction, and characterization. S. L. and Q. G. conducted the CCK-8 assays, western blotting and flow cytometry. S. L., L. Z., S. Y., T. Y., and L. N conducted the in vivo experiments. S. L. and J. G. co-wrote the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge support from the National Natural Science Foundation of China (22071263), the Natural Science Foundation of Guangdong Province, P. R. China (2020A1515010994) and the Shenzhen Fundamental Research Program (JCYJ20200109110215774).References
- S. M. Chowdhury, T. Lee and J. Willmann, Ultrasonography, 2017, 36, 171–184 CrossRef PubMed.
- T. Miyazaki, S. Chen, S. Florinas, K. Igarashi, Y. Matsumoto, T. Yamasoba, Z.-Q. Xu, H. Wu, C. Gao, K. Kataoka, R. J. Christie and H. Cabral, ACS Nano, 2022, 16, 12290–12304 CrossRef CAS PubMed.
- D. Ye, X. Zhang, Y. Yue, R. Raliya, P. Biswas, S. Taylor, Y. C. Tai, J. B. Rubin, Y. Liu and H. Chen, J. Controlled Release, 2018, 286, 145–153 CrossRef CAS PubMed.
- D. Y. Zhang, C. Dmello, L. Chen, V. A. Arrieta, E. Gonzalez-Buendia, J. R. Kane, L. P. Magnusson, A. Baran, C. D. James, C. Horbinski, A. Carpentier, C. Desseaux, M. Canney, M. Muzzio, R. Stupp and A. M. Sonabend, Clin. Cancer Res., 2020, 26, 477–486 CrossRef CAS PubMed.
- J. M. Wasielewska, J. C. S. Chaves, R. L. Johnston, L. A. Milton, D. Hernández, L. Chen, J. Song, W. Lee, G. Leinenga, R. M. Nisbet, A. Pébay, J. Götz, A. R. White and L. E. Oikari, Theranostics, 2022, 12, 6826–6847 CrossRef CAS PubMed.
- X. Li, S. Khorsandi, Y. Wang, J. Santelli, K. Huntoon, N. Nguyen, M. Yang, D. Lee, Y. Lu, R. Gao, B. Y. S. Kim, C. de Gracia Lux, R. F. Mattrey, W. Jiang and J. Lux, Nat. Nanotechnol., 2022, 17, 891–899 CrossRef CAS PubMed.
- Y. Guo, H. Lee, Z. Fang, A. Velalopoulou, J. Kim, M. B. Thomas, J. Liu, R. G. Abramowitz, Y. Kim, A. F. Coskun, D. P. Krummel, S. Sengupta, T. J. MacDonald and C. Arvanitis, Sci. Adv., 2021, 7, eabf7390 CrossRef CAS PubMed.
- J. Xu, N. Solban, Y. Wang, H. Ferguson, S. Perera, K. Lin, M. Cai, M. Paul, E. G. Schutt, C. T. Larsen, R. Li, R. Saklatvala, B. J. Long, S. Ranganath, A. T. Procopio, S. Mittal and A. C. Templeton, Adv. Ther., 2021, 4, 2170031 CrossRef.
- R. H. Abou-Saleh, A. Delaney, N. Ingram, D. V. B. Batchelor, B. R. G. Johnson, A. Charalambous, R. J. Bushby, S. A. Peyman, P. L. Coletta, A. F. Markham and S. D. Evans, ACS Appl. Bio Mater., 2020, 3, 7840–7848 CrossRef CAS PubMed.
- B. Park, S. Yoon, Y. Choi, J. Jang, S. Park and J. Choi, Pharmaceutics, 2020, 12, 1089 CrossRef CAS PubMed.
- Z.-X. Liu, Y. Feng, Z. Yan, Y. He, C.-Y. Liu and Q. Fan, Chem. Mater., 2012, 24, 3751–3757 CrossRef CAS.
- W. Chen and J. Du, Sci. Rep., 2013, 3, 2162 CrossRef PubMed.
- J. Wang, M. Pelletier, H. Zhang, H. Xia and Y. Zhao, Langmuir, 2009, 25, 13201–13205 CrossRef CAS PubMed.
- N. Y. Rapoport, A. M. Kennedy, J. E. Shea, C. L. Scaife and K. H. Nam, J. Controlled Release, 2009, 138, 268–276 CrossRef CAS PubMed.
- Z. Shi, J. Wu, Q. Song, R. Göstl and A. Herrmann, J. Am. Chem. Soc., 2020, 142, 14725–14732 CrossRef CAS PubMed.
- H. M. Klukovich, T. B. Kouznetsova, Z. S. Kean, J. M. Lenhardt and S. L. Craig, Nat. Chem., 2013, 5, 110–114 CrossRef CAS PubMed.
- J. Xuan, O. Boissière, Y. Zhao, B. Yan, L. Tremblay, S. Lacelle, H. Xia and Y. Zhao, Langmuir, 2012, 28, 16463–16468 CrossRef CAS PubMed.
- P. Wei, E. J. Cornel and J. Du, Drug Delivery Transl. Res., 2021, 11, 1323–1339 CrossRef CAS PubMed.
- C. Komori, K. Okada, K. Kawamura, S. Chida and T. Suzuki, Anticancer Res., 2009, 29, 2411–2415 CAS.
- Y. Xu, S. Lin, R. He, Y. Zhang, Q. Gao, D. K. P. Ng and J. Geng, Chem. – Eur. J., 2021, 27, 11268–11272 CrossRef CAS PubMed.
- M. Assali, N. Kittana, S. Dayyeh and N. Khiar, Nanotechnology, 2021, 32, 205101 CrossRef CAS PubMed.
- H. Wang, C. A. Thorling, X. Liang, K. R. Bridle, J. E. Grice, Y. Zhu, D. H. G. Crawford, Z. P. Xu, X. Liu and M. S. Roberts, J. Mater. Chem. B, 2015, 3, 939–958 RSC.
- I. Amata, M. Maffei and M. Pons, Front. Genet., 2014, 5, 181 Search PubMed.
- M. Podhorecka, A. Skladanowski and P. Bozko, J. Nucleic Acids, 2010, 2010, 9 Search PubMed.
- M. Xu, C. Y. Zhang, J. Wu, H. Zhou, R. Bai, Z. Shen, F. Deng, Y. Liu and J. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 5701–5713 CrossRef CAS PubMed.
- L. Zhu, S. Lin, W. Cui, Y. Xu, L. Wang, Z. Wang, S. Yuan, Y. Zhang, Y. Fan and J. Geng, Biomater. Sci., 2022, 10, 3624–3636 RSC.
- L. Li, J. Fu, X. Wang, Q. Chen, W. Zhang, Y. Cao and H. Ran, ACS Appl. Mater. Interfaces, 2021, 13, 3605–3621 CrossRef CAS PubMed.
- J. Tian, L. Xu, Y. Xue, X. Jiang and W. Zhang, Biomacromolecules, 2017, 18, 3992–4001 CrossRef CAS PubMed.
- R. Yang, S. Zhang, D. Kong, X. Gao, Y. Zhao and Z. Wang, Pharm. Res., 2012, 29, 3512–3525 CrossRef CAS PubMed.
- X. Zeng, Y. Zhang, X. Xu, Z. Chen, L. Ma, Y. Wang, X. Guo, J. Li and X. Wang, Drug Delivery, 2022, 29, 792–806 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01101a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |