Nucleoside analogs in ADAR guide strands targeting 5′-U![[A with combining low line]](https://www.rsc.org/images/entities/h2_char_0041_0332.gif) sites†
sites†
Received
11th July 2022
, Accepted 30th October 2022
First published on 31st October 2022
Abstract
Adenosine deaminases that act on RNA (ADARs) can be directed to predetermined sites in transcriptomes by forming duplex structures with exogenously delivered guide RNAs (gRNAs). They can then catalyze the hydrolytic deamination of adenosine to inosine in double stranded RNA, which is read as guanosine during translation. High resolution structures of ADAR2-RNA complexes revealed a unique conformation for the nucleotide in the guide strand base paired to the editing site's 5′ nearest neighbor (−1 position). Here we describe the effect of 16 different nucleoside analogs at this position in a gRNA that targets a 5′-U![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) -3′ site. We found that several analogs increase editing efficiency for both catalytically active human ADARs. In particular, 2′-deoxynebularine (dN) increased the ADAR1 and ADAR2 in vitro deamination rates when at the −1 position of gRNAs targeting the human MECP2 W104X site, the mouse IDUA W392X site, and a site in the 3′-UTR of human ACTB. Furthermore, a locked nucleic acid (LNA) modification at the −1 position was found to eliminate editing. When placed −1 to a bystander editing site in the MECP2 W104X sequence, bystander editing was eliminated while maintaining on-target editing. In vitro trends for four −1 nucleoside analogs were validated by directed editing of the MECP2 W104X site expressed on a reporter transcript in human cells. This work demonstrates the importance of the −1 position of the gRNA to ADAR editing and discloses nucleoside analogs for this site that modulate ADAR editing efficiency.
-3′ site. We found that several analogs increase editing efficiency for both catalytically active human ADARs. In particular, 2′-deoxynebularine (dN) increased the ADAR1 and ADAR2 in vitro deamination rates when at the −1 position of gRNAs targeting the human MECP2 W104X site, the mouse IDUA W392X site, and a site in the 3′-UTR of human ACTB. Furthermore, a locked nucleic acid (LNA) modification at the −1 position was found to eliminate editing. When placed −1 to a bystander editing site in the MECP2 W104X sequence, bystander editing was eliminated while maintaining on-target editing. In vitro trends for four −1 nucleoside analogs were validated by directed editing of the MECP2 W104X site expressed on a reporter transcript in human cells. This work demonstrates the importance of the −1 position of the gRNA to ADAR editing and discloses nucleoside analogs for this site that modulate ADAR editing efficiency.
Introduction
ADARs (Adenosine Deaminases that Act on RNA) catalyze adenosine deamination within duplex RNA to yield inosine. Since inosine is interpreted as guanosine due to its hydrogen bonding with cytidine, this modification introduced by ADARs can change codon meaning.1–3 Two ADAR genes encode catalytically active ADARs in humans (ADAR encoding ADAR1 proteins and ADARB1 encoding the ADAR2 protein).4 ADAR1 is expressed in two protein isoforms (p110 and p150) that differ in their N-terminal structures.5 Because ADARs require duplex structure for activity, their reaction can be directed to specific adenosines in different transcripts using complementary guide strands which form duplexes at the target sites.1 This approach is currently being pursued to develop therapeutic guide strands that recruit ADARs to correct disease-causing mutations in RNA.6–12 One promising variation on this strategy uses chemically modified oligonucleotides to recruit endogenously expressed human ADARs for directed RNA editing.8,10–12 Chemical modifications to the guide strand are used to facilitate uptake into target cells, to increase metabolic stability, and to increase efficiency and selectivity of the ADAR reaction.8,10,12–14 Our group has used structures of ADAR2 bound to RNA bearing an adenosine deamination transition state analog15 to inform the design of modifications that increase the efficiency11 and selectivity16 of the ADAR reaction. Here we focus on a nucleotide position in the guide strand near the editing site that adopts a highly unusual conformation in ADAR2-RNA structures.15 We show that several nucleoside analogs are effective at increasing the in vitro adenosine deamination rate for both ADAR2 and ADAR1 p110 when placed at this position in guide strands for three different target sequences. Importantly, in vitro trends observed for four nucleoside analogs were also validated by directed editing in human cells. Furthermore, we found that a locked nucleic acid (LNA) modification at this nucleotide position inhibits the ADAR reaction. This effect can also be used to block bystander editing while maintaining efficient on-target editing. These observations are rationalized based on the ability of the different nucleoside analogs to adopt the conformation observed in ADAR2-RNA structures.
Results
ADAR2-RNA structures indicate a nucleotide position in the guide strand adopts a highly unusual conformation
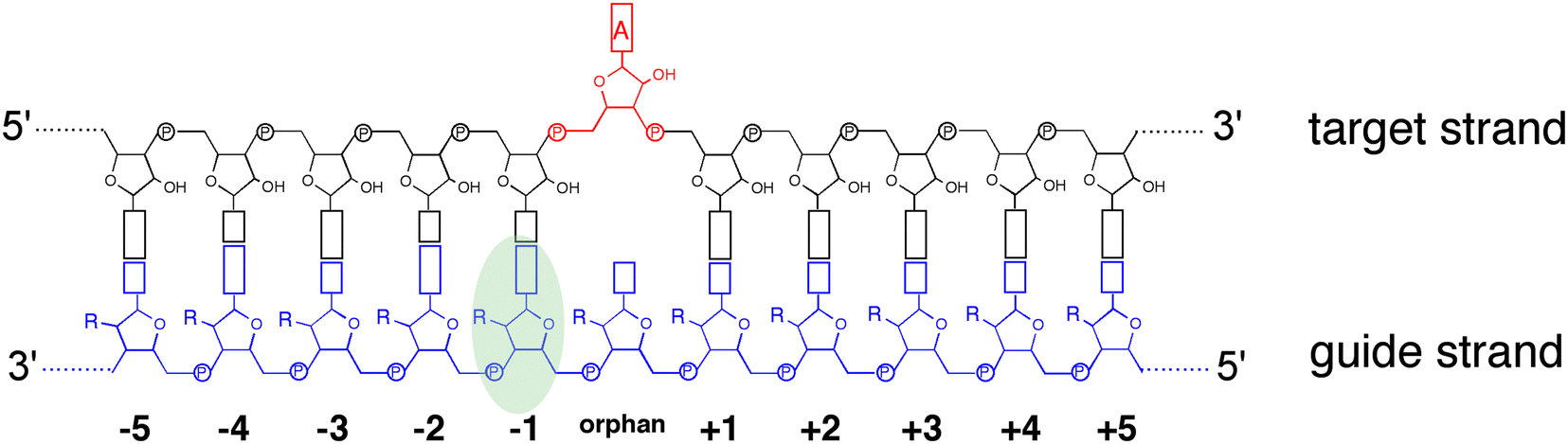
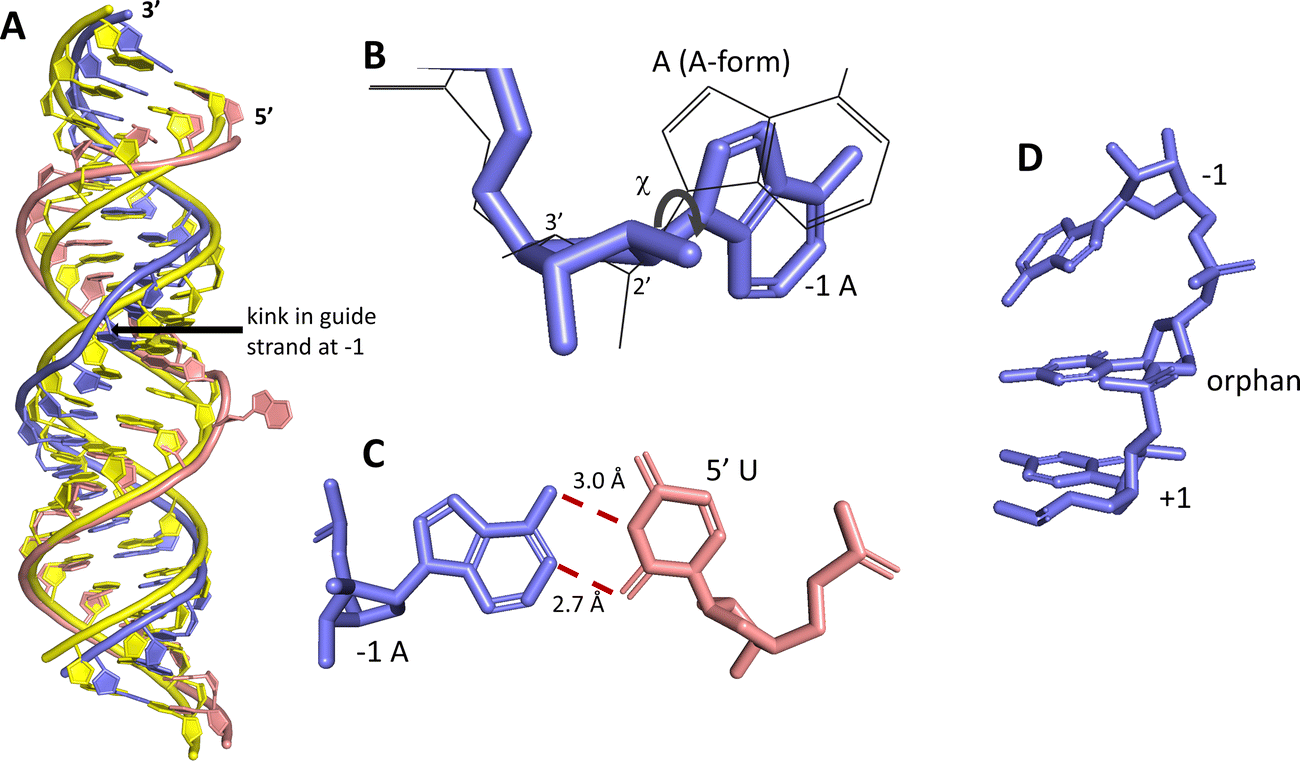
ADAR enzymes react exclusively with duplex substrates that adopt the A-form conformation (dsRNA or DNA–RNA hybrids).17 Thus, an adenosine within a transcript that would not normally react with ADARs can be induced to react by the formation of a duplex that is of sufficient length for ADAR recruitment. Chemically modified oligonucleotides can serve as ADAR guide strands (gRNAs) for this purpose.8,10–12 The different nucleotides of an ADAR guide are possible sites for optimization of properties such as cell uptake, metabolic stability, and ADAR reactivity. We have found it useful to designate these different nucleotides of the guide using positive or negative numbers emanating out from the position that opposes the editing site (Fig. 1). Crystal structures of ADAR2-RNA complexes have shown that the editing site nucleotide flips into the enzyme active site, allowing hydrolytic deamination to occur.15 The nucleotide opposite the flipped-out base is referred to as the orphan base since it lacks a pairing partner in the flipped-out conformation (Fig. 1). Another feature of the flipped-out conformation of the ADAR substrate RNA is a substantial kink in the backbone of the guide strand (Fig. 2A). This kink is a result of significant deviations from standard A-form conformation for the nucleotide paired with the editing site's 5′ nearest neighbor (guide −1 position). The sugar of the −1 position nucleotide adopts a C2′-endo pucker typical of a DNA nucleotide, not the C3′-endo sugar pucker found in A-form RNA (Fig. 2B).18 Additionally, the glycosidic bond angle (χ) is altered at the −1 position. Compared to the χ angle of −157° in the idealized A-form nucleotide conformation and the average χ angle of −160° for all other adenosines in a published structure of an ADAR2 deaminase domain-RNA complex, the χ angle for the −1 position is −90.6° (Table S1, ESI†).15 The base at the −1 position can also slide, adopting an A-U wobble base pair with its partner in one of the published ADAR2-RNA structures (Fig. 2C).15 Finally, the kink in the guide strand backbone pushes the major groove edge of the −1 base towards the orphan base, disrupting the base stacking at this location in the duplex (Fig. 2D). Thus, the −1 position of an ADAR guide undergoes a conformational change during the reaction that changes the sugar pucker and glycosidic bond angle, disrupts π-stacking and can alter base pairing. These observations suggested that nucleoside analogs at this site may substantially influence ADAR reactivity.
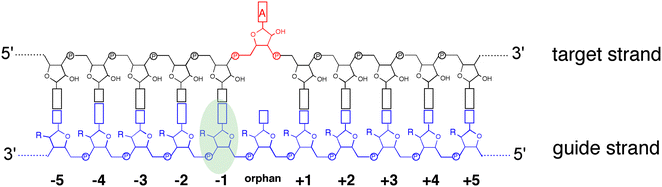 |
| | Fig. 1 Schematic of target:guide strand duplex formed in site directed RNA editing indicating nomenclature and numbering scheme used in this work. The −1 position (green oval) was optimized for target A (red) editing. | |
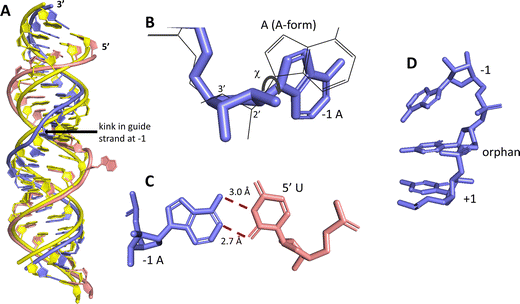 |
| | Fig. 2 Unusual conformation of guide strand near the −1 nucleotide indicated by X-ray crystallography.15 (A) Superposition of typical A-form duplex RNA (yellow strands) with RNA duplex conformation present in complex with ADAR2 (blue (guide) and pink (target) strands). (B) The −1 A has a C2′-endo sugar pucker with a high anti glycosidic bond angle (χ = −90.6°). Blue indicates conformation found in ADAR2-RNA structures, superimposed grey wire structure is A-form conformation. (C) The −1 A in a sheared pair with the 5′ U of a target strand. (D) The −1 A is unstacked from the orphan base in ADAR2-RNA structures. | |
Nucleoside analogs with modified sugars and modified bases enhance ADAR editing rate at the −1 position of the guide strand targeting MECP2 W104X
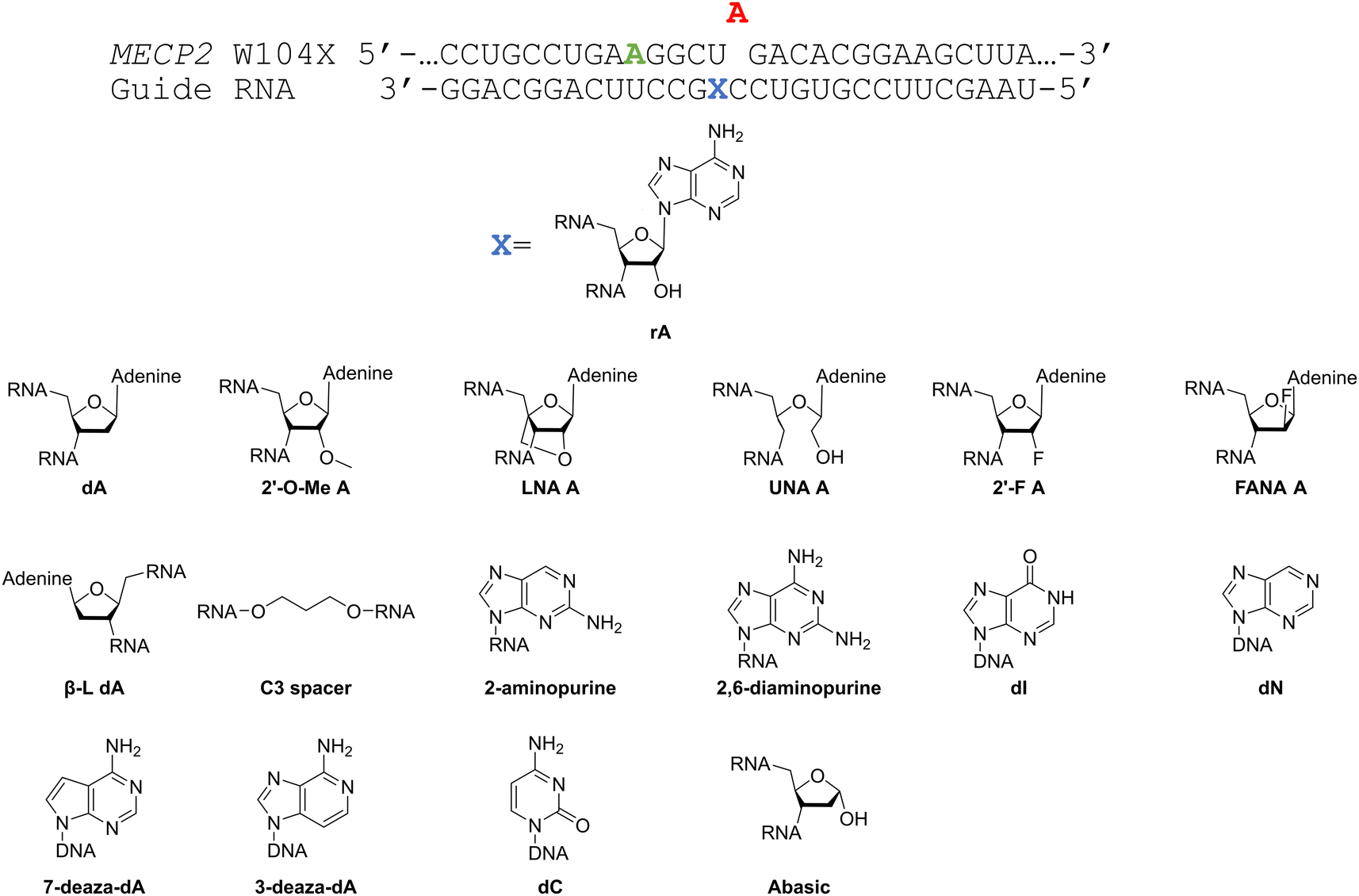
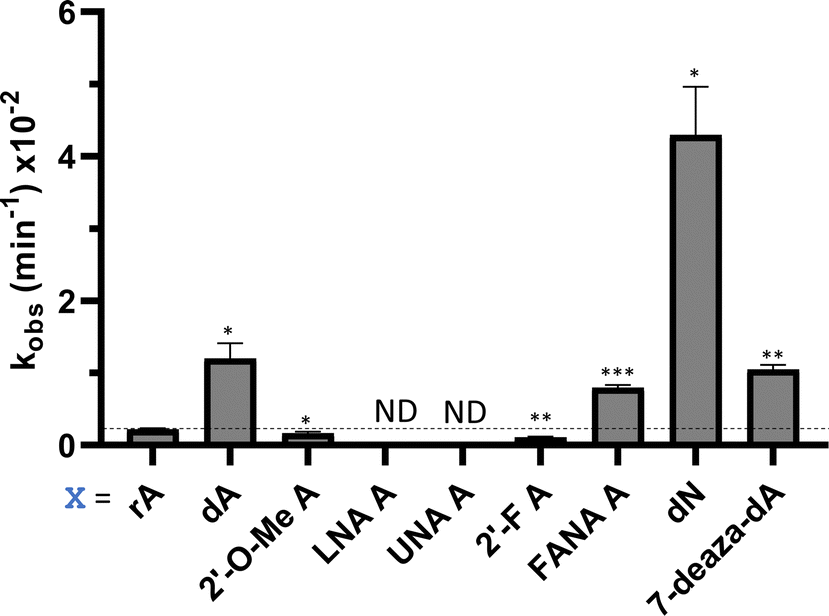
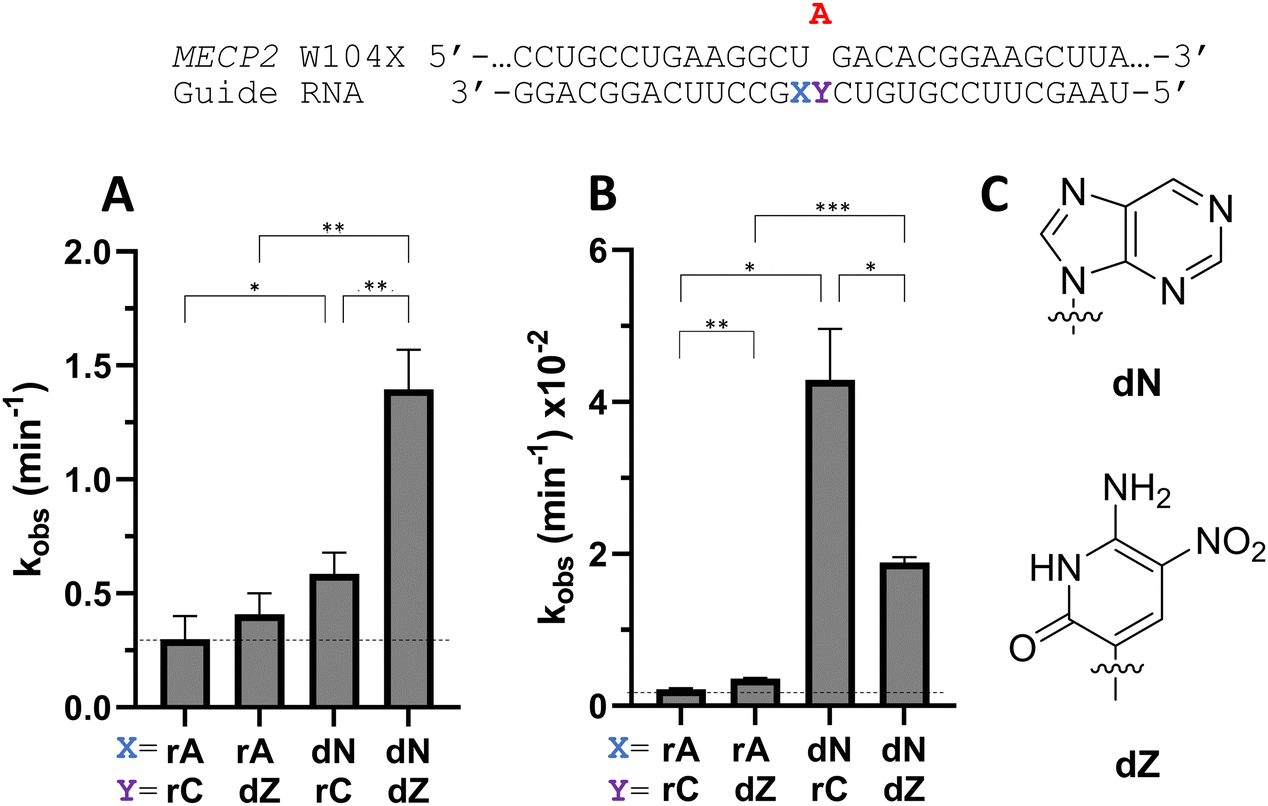
Considering the structural changes that the −1 position can undergo, sixteen different nucleoside analogs that vary in preferred sugar conformation and base pairing properties were tested for their effect on the ADAR2 deamination reaction when placed at the guide strand −1 position (Fig. 3). These were incorporated into a guide RNA (gRNA) targeting the human methyl CPG binding protein 2 (MECP2) W104X site, a G to A mutation that causes Rett syndrome.19 Each of the duplexes bearing different nucleoside analogs at guide position −1 were allowed to react in vitro with purified human ADAR2. The effect of each analog was determined by measuring the percent editing of the target site at the reaction end point and by comparing the rate of reaction under single turnover conditions (Table S2, ESI†). The rate constants (kobs) for editing were then plotted for each analog tested (Fig. 4). We also measured single turnover rates for target adenosine deamination by human ADAR1 p110 for these modified duplexes (Fig. 5 and Table S3, ESI†).
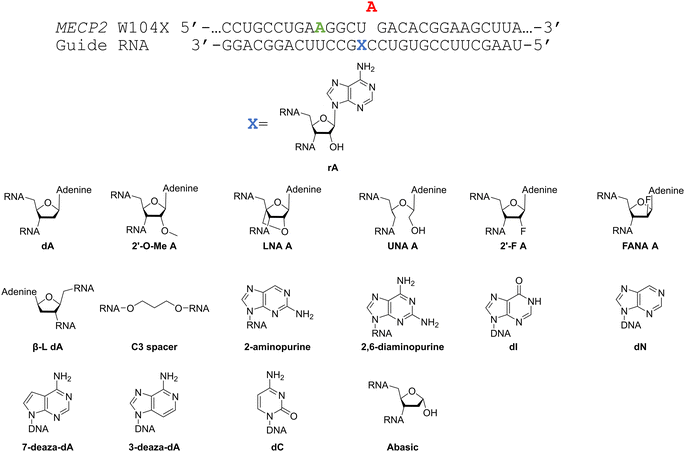 |
| | Fig. 3 (top) Target sequence derived from the mRNA for human MECP2 proximal to the W104X mutation associated with Rett syndrome and guide RNA used here to direct corrective editing at the premature stop codon. The target adenosine is indicated in red. X indicates the −1 position of the guide strand relative to the target adenosine. A bystander editing site in this strand is shown in green. (bottom) Nucleoside analogs tested at the −1 position for their effect on the ADAR2 deamination rate with the MECP2 W104X target duplex in vitro. | |
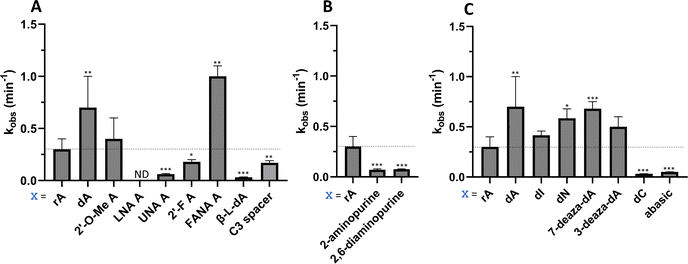 |
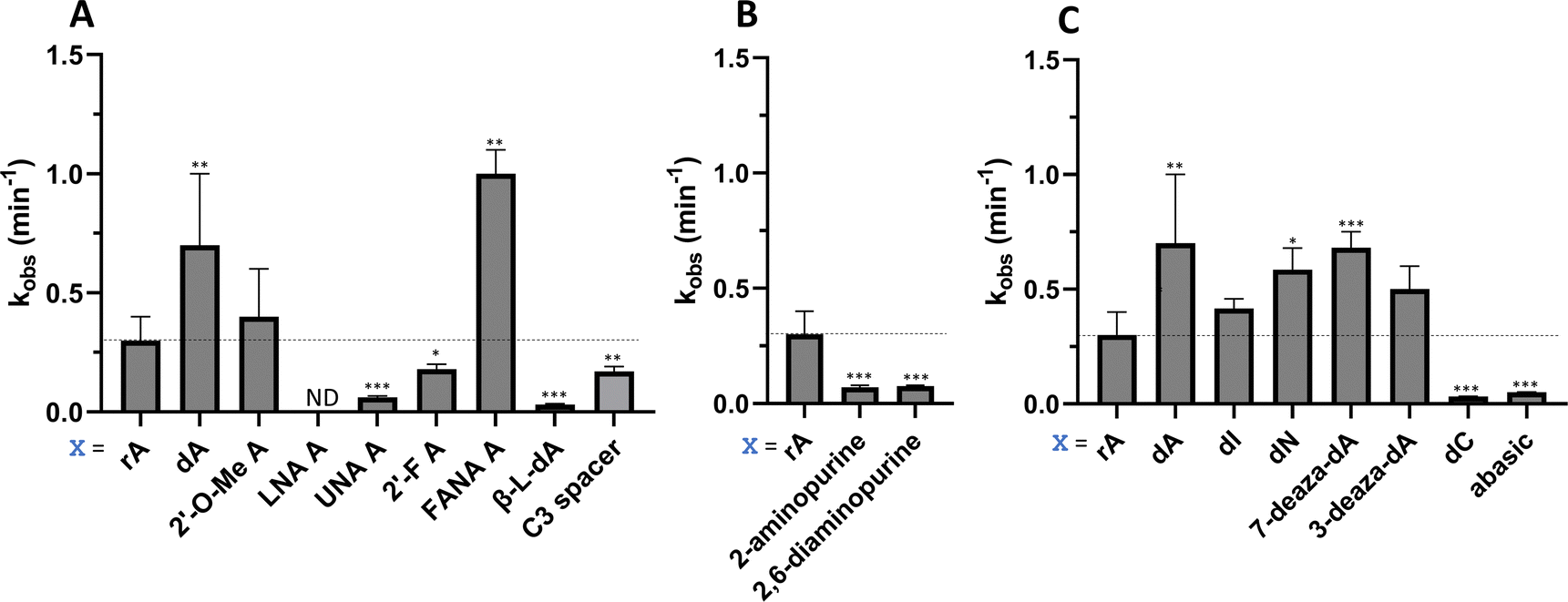
| | Fig. 4 Effect of guide strand −1 nucleotide modification on ADAR2 deamination rate for the MECP2 W104X site. (A) Comparison of different sugar modifications at −1 nucleotide on selective editing at target site plotted as observed rate constant. (B) Effect on selective editing by ADAR2 of ribopurine analogs at −1 nucleotide. (C) Effect on selective editing by ADAR2 of 2′-deoxyribonucleoside analogs. Reactions were carried out with 5 nM RNA and 15 nM ADAR2. Data were fitted to the equation [P]t = α[1 − exp(−kobs·t)]. ND = no detected editing. Error bars, s.d. (n ≥ 3 technical replicates). A two-tailed Welch's t test was conducted, where *p < 0.05, **p < 0.01, ***p < 0.001 from rA −1 gRNA. | |
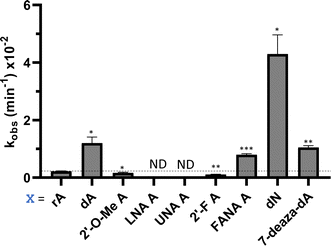 |
| | Fig. 5 Effect of guide strand −1 nucleotide modification on ADAR1 p110 deamination rate for the MECP2 W104X site. Rate constant for target site editing is shown for different nucleoside analogs at −1. Reactions were carried out with 15 nM RNA and 150 nM ADAR1 p110. Data were fitted to the equation [P]t = α[1 − exp(−kobs·t)], end point constrained to 65%. ND = no detected editing. Error bars, s.d. (n ≥ 3 technical replicates). A two-tailed Welch's t test was conducted, where *p < 0.05, **p < 0.01, ***p < 0.001 from rA −1 gRNA. | |
We found that an all RNA 29 nt guide strand, with adenosine (rA) at the −1 position complementary to the 5′ uridine in the target strand and cytidine (rC) at the orphan position, supported on-target editing by ADAR2 at the MECP2 W104X site (kobs = 0.3 ± 0.1 min−1) (Fig. 4 and Table S2, ESI†). Cytidine was chosen for the orphan position in this analysis since an A–C mismatch is the preferred pairing consisting of naturally occurring nucleosides for ADARs at an editing site.20 We then compared the editing outcomes for guides bearing adenosine analogs with sugar modifications (Fig. 4A) or base modifications (Fig. 4B) at the −1 position. We also tested several 2′-deoxyribose analogs bearing additional base modifications (Fig. 4C). For the sugar modifications alone, 2′-deoxyadenosine (dA) and 2'-deoxy-2'-fluoro-arabinonucleic acid (FANA) A increased the editing rate (i.e. increased kobs compared to rA) whereas locked nucleic acid (LNA) A, unlocked nucleic acid (UNA) A, 2′-deoxy-2′-fluoroadenosine (2′-F A), β-L-2′-deoxyadenosine (β-L-dA), and a C3 spacer at −1 showed reduced editing compared to rA. 2′-O-Methyladenosine (2′-O-Me A) showed no significant difference from rA (Fig. 4A). Interestingly, LNA A at the −1 position of the guide eliminated editing entirely at the target site for this sequence (Fig. 4A). Each of the base modified ribonucleosides tested inhibited editing at the target site—2-aminopurine ribonucleoside (2-aminopurine) and 2,6-diaminopurine ribonucleoside (2,6-diaminopurine) (Fig. 4B). In the group of 2′-deoxy analogs tested, 2′-deoxynebularine (dN) and 7-deaza-2′-deoxyadenosine (7-deaza-dA) both showed improvement over rA but no significant difference in deamination rate compared to dA. 2′-Deoxyinosine (dI) and 3-deaza-2′-deoxyadenosine (3-deaza-dA) showed no significant difference in deamination rate compared to rA. 2′-Deoxycytidine (dC) and 2′-deoxyribose (abasic) both inhibited editing at the target site compared to rA (Fig. 4C).
Eight of the −1 position-modified guide strands described above were also evaluated in reactions with ADAR1 p110 on the MECP2 W104X target (Fig. 5 and Table S3, ESI†). We found that dA, FANA A, dN, and 7-deaza-dA each substantially increased the rate of editing compared to rA, whereas 2′-O-Me A, LNA A, UNA A and 2′-F A decreased the rate of editing. Both LNA A and UNA A showed no editing with ADAR1 p110 for this target sequence.
A single LNA modification blocks bystander editing by ADAR2
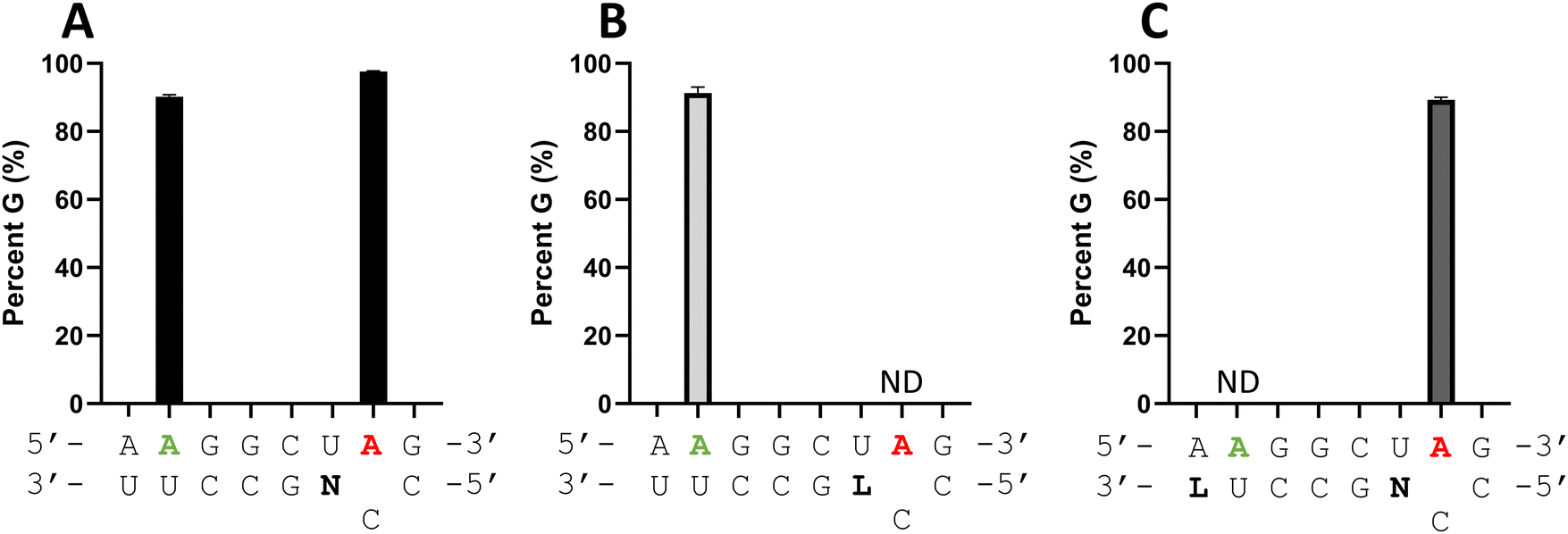
ADARs have preferences for editing adenosines that have specific 5′ and 3′ nearest neighbor nucleotides.20 Indeed, 5′-UG and 5′-AG are the two most efficiently edited triplet combinations by human ADAR2.21 On the MECP2 W104X sequence, the target editing site is found in a 5′-UG sequence with the nearby bystander editing site in a 5′-AG. While ADAR2 can deaminate the target site in an A–C mismatch at a higher rate than the bystander site, substantial bystander editing is observed at longer reaction times in vitro (Fig. 6A). Since it appeared that an LNA modification could block ADAR editing when paired with an editing site's 5′ nearest neighbor (Fig. 4A and 5), we generated a new modified guide strand to determine if bystander editing could be reduced with an LNA modification while maintaining enhanced on-target editing with a −1 nucleoside analog (Fig. 6). Thus, LNA U was introduced at the −6 position of the guide (−1 relative to the bystander site) while dN was placed at the −1 position relative to the target site. Editing with ADAR2 was measured at both the target and bystander sites. Bystander site editing was reduced from 90% to no detected editing, while the target site editing was maintained around 95% for both the dN −1 and the LNA U −6, dN −1 guide (Fig. 6C). This further demonstrates the position dependence of the LNA substitution in an ADAR guide, as well as its potential to be used to reduce off target editing with other gRNA modifications that enhance on target editing.
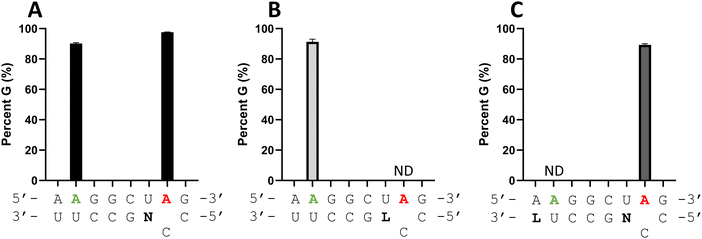 |
| | Fig. 6 LNA modification can block bystander editing. Editing yield for different guide strands with LNA (L) and dN (N) modifications at specific sites. (A) Guide strand with dN at the −1 position shows high levels of editing for both target and bystander. (B) LNA modification near target site reduces on target editing. (C) LNA modification near bystander site reduces bystander editing. ND = no detected editing. | |
Nucleoside analogs enhance ADAR editing rate at IDUA W392X and a position in the 3′-UTR of β-actin
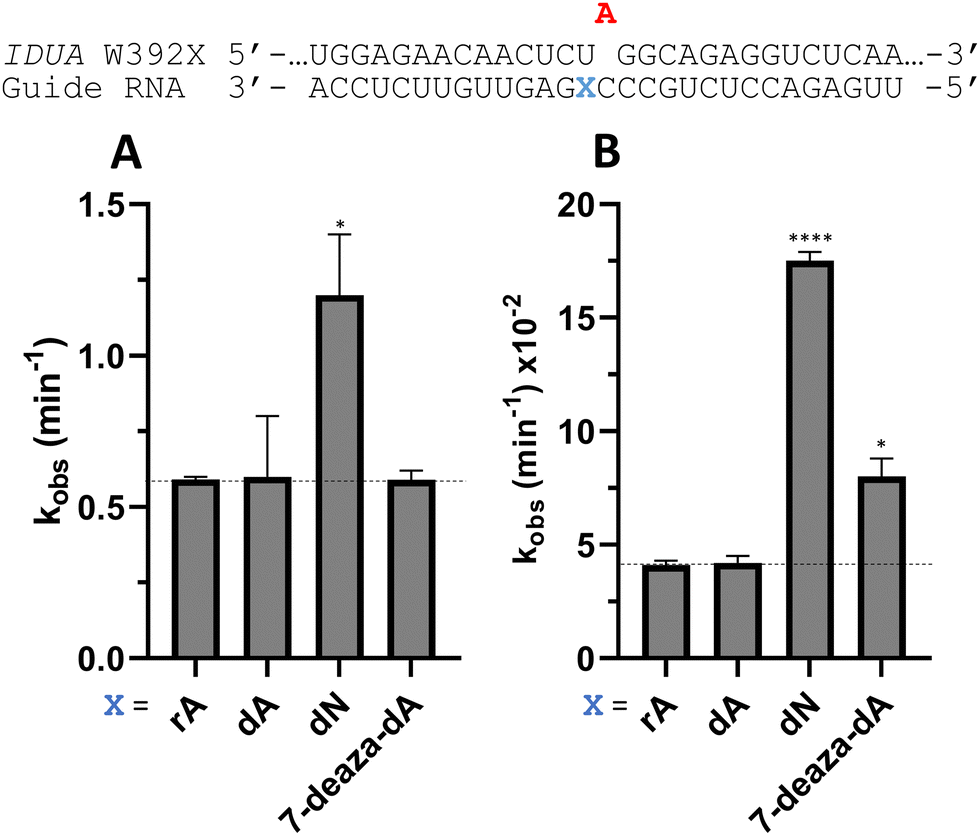
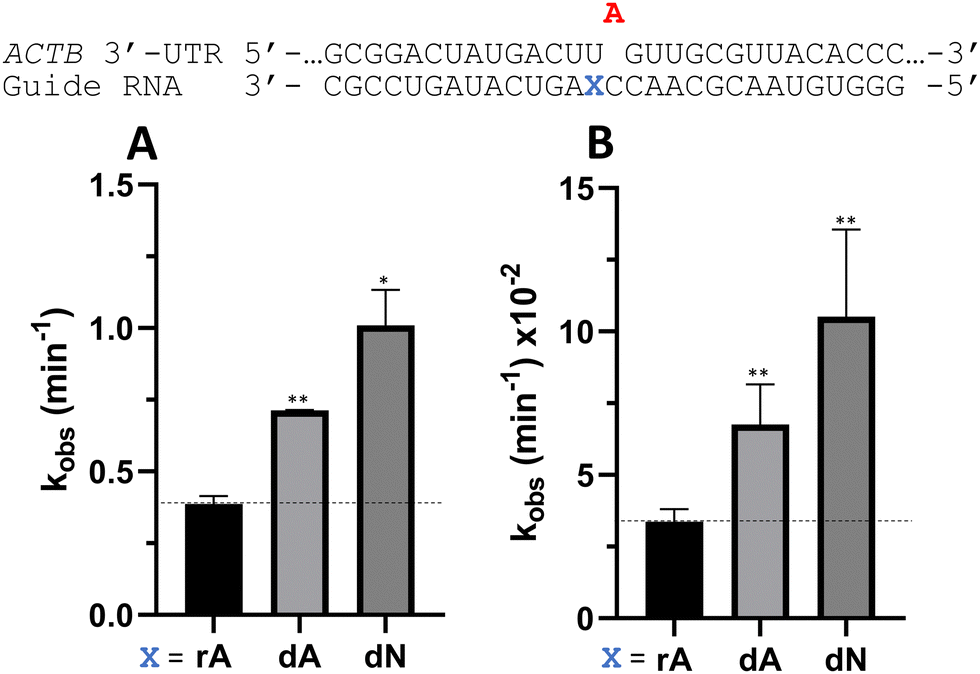
To determine whether the effects observed for editing the MECP2 W104X target with guides bearing nucleoside analogs at −1 would also be observed in the context of different sequences, we synthesized a set of four 29 nt guide strands targeting the mouse IDUA W392X mutation (Fig. 7) and a set of three 29 nt guides strands targeting a position in the 3′-untranslated region (UTR) of human β-actin (ACTB) (Fig. 8). Mutations within the human IDUA gene, which encodes α-L-iduronidase, can cause Hurler syndrome.22,23 The mouse IDUA W392X mutation mimics the Hurler-causing human W402X mutation that is reversed by ADAR-mediated editing.11,22 Adenosine analogs described above (dA, dN, and 7-deaza-dA for the IDUA target; dA and dN for the ACTB target) were introduced into the guide −1 position and compared to rA at these positions for both ADAR1 p110 and ADAR2. Single turnover rate constants were measured and compared (Fig. 7 and 8) as well as fitted end points (Tables S4–S7, ESI†). For both the MECP2 target and the ACTB target, dA stimulated editing compared to rA for ADAR1 and ADAR2. However, this was not the case for the IDUA target sequence. Nevertheless, dN showed a significant increase in deamination rate by ADAR2 on both IDUA and ACTB targets, while 7-deaza dA behaves like rA with ADAR2 on the IDUA target. The rate of editing by ADAR1 was increased by both dN (for the IDUA and ACTB targets) and 7-deaza dA (for the IDUA target).
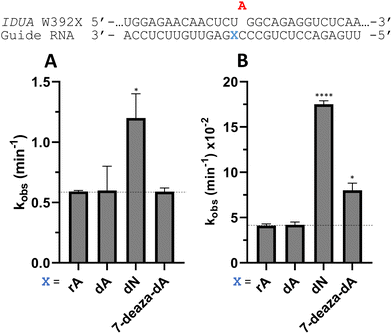 |
| | Fig. 7 Effect on deamination rate of nucleoside analogs at guide strand position −1 for the mouse IDUA W392X target sequence with (A) 5 nM RNA and 15 nM ADAR2 or (B) 15 nM RNA and 150 nM ADAR1 p110. Data were fitted to the equation [P]t = α[1 − exp(−kobs·t)]. Error bars, s.d. (n ≥ 3 technical replicates). A two-tailed Welch's t test was conducted, where *p < 0.05, ****p < 0.0001 from rA −1 gRNA. | |
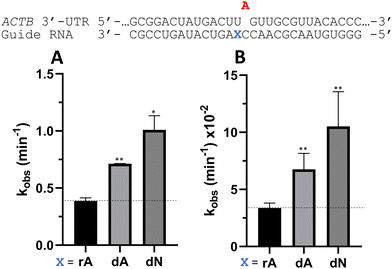 |
| | Fig. 8 Effect on deamination rate of nucleoside analogs at guide strand position −1 for the human ACTB 3′-UTR target sequence with (A) 5 nM RNA and 15 nM ADAR2 or (B) 15 nM RNA and 150 nM ADAR1 p110. Data were fitted to the equation [P]t = α[1 − exp(−kobs·t)]. Error bars, s.d. (n ≥ 3 technical replicates). A Grubbs test for outliers was conducted at the p = 0.05 significance level. A two-tailed Welch's t test was conducted, where *p < 0.05, **p < 0.01 from rA −1 gRNA. | |
Beneficial modifications at guide −1 and orphan positions can be combined for additional deamination rate enhancement
To determine whether beneficial −1 modifications could be combined with other rate enhancing guide strand modifications, a guide with dN −1 and dZ at the orphan position in MECP2 was tested with ADAR2 and ADAR1 (Fig. 9). dZ at the orphan position has been shown to enhance editing with ADARs relative to dC at the orphan position.11 While the addition of an orphan dZ alone did not significantly increase editing relative to the guide with rA at the −1 position and rC at the orphan position with ADAR2, the combination of an orphan dZ with dN at the −1 position increased editing relative to both the fully ribose guide and the guide with dN at −1 and rC at the orphan position (Fig. 9A and Table S2, ESI†). An increase in editing was seen for ADAR1 with the addition of dZ at the orphan position relative to fully ribose guide (Fig. 9B). With ADAR1, the combination of dZ at the orphan position with dN at the −1 position increased the observed rate constant relative to fully ribose and dZ orphan guides, but not compared to the rC orphan, dN −1 guide.
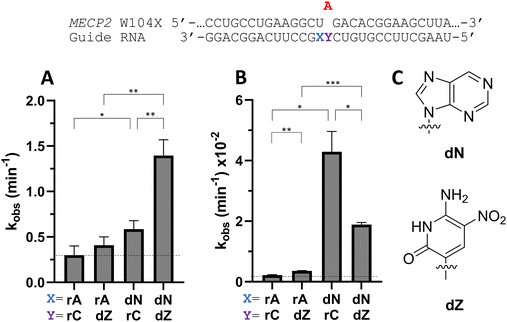 |
| | Fig. 9 Effect of combining two beneficial modifications on MECP2 W104X deamination rate with (A) 5 nM RNA and 15 nM ADAR2 or (B) 15 nM RNA and 150 nM ADAR1 p110. Data were fitted to the equation [P]t = α[1 − exp(−kobs·t)], end point constrained to 65% for (B). Error bars, s.d. (n ≥ 3 technical replicates). A two-tailed Welch's t test was conducted, where *p < 0.05, **p < 0.01, ***p < 0.001. (C) Structures of dN and dZ. | |
Nucleoside analogs affect cellular editing of MECP2 W104X
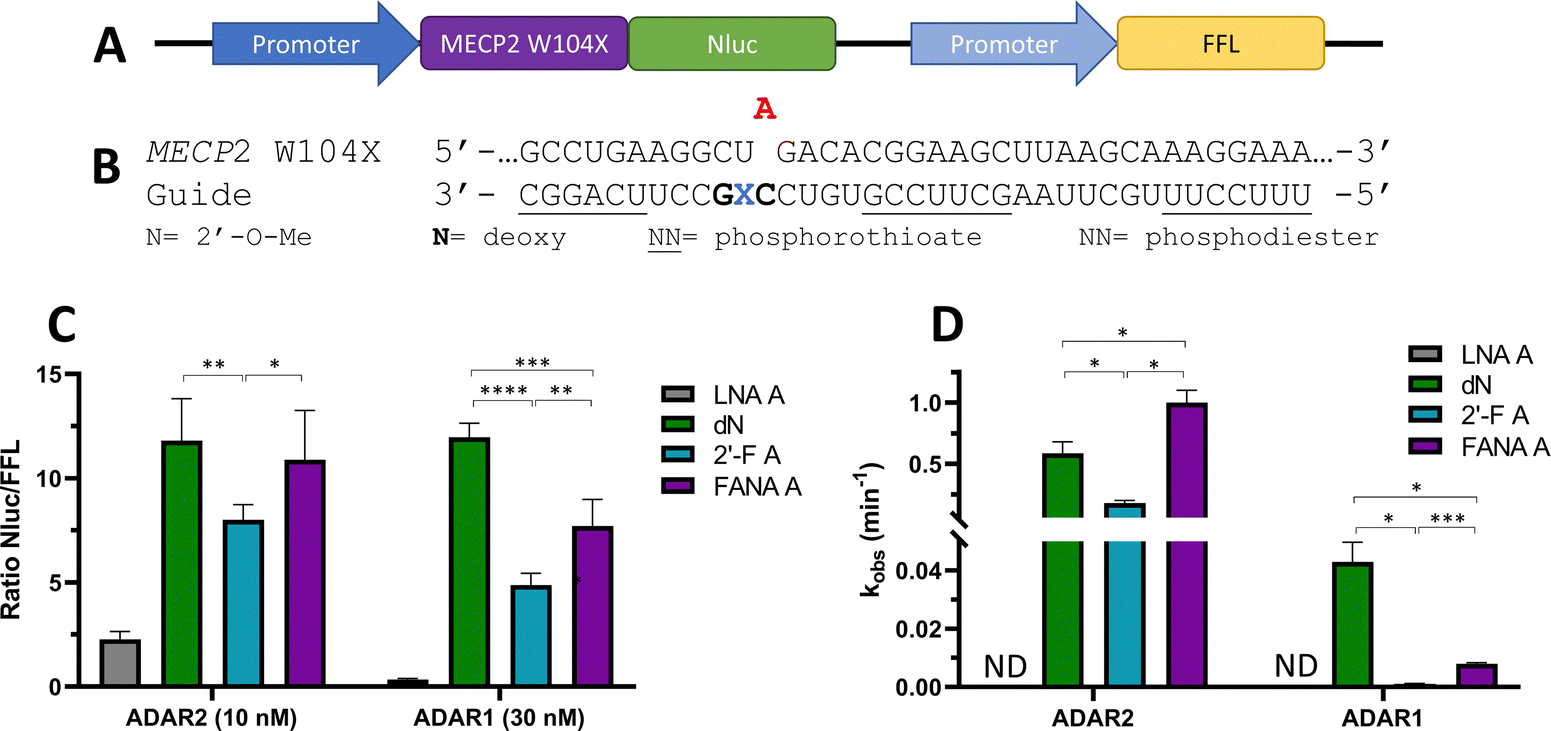
To ascertain whether trends observed in our deamination kinetics experiments were also seen in directed editing experiments in human cells, we used a dual luciferase assay in human embryonic kidney 293T (HEK293T) cells.24 For this purpose, we generated a reporter plasmid containing 90-nucleotides from the human MECP2 sequence bearing the W104X mutation downstream of a promoter and upstream of nanoluciferase (Nluc) coding sequence (Fig. 10A). Thus, translation of Nluc can occur only after RNA editing by ADAR at the W104X premature termination codon. Firefly luciferase (FFL) is expressed regardless of editing as a transfection control. ADAR editing is correlated to the ratio of luciferase signals observed.
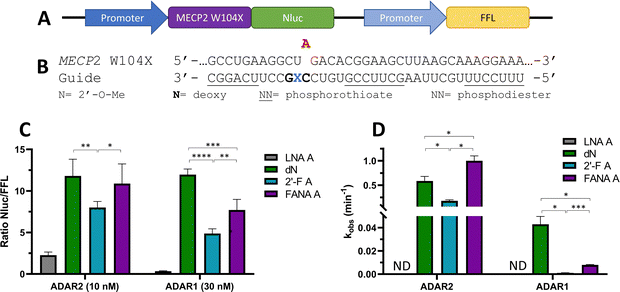 |
| | Fig. 10 Effect of −1 nucleoside analogs on cellular editing as measured by dual luciferase assay compared to effect on in vitro editing. (A) Schematic representation of dual luciferase plasmid with target site MECP2 W104X upstream of reporter luciferase Nluc. FFL expressed independently of editing. (B) Modification pattern for guide oligonucleotides used in cellular editing. (C) Relative Nluc/FFL luminescence signals observed for guide oligonucleotides with varying −1 nucleoside analogs and either 10 nM of guide and overexpressed ADAR2 or 30 nM of guide and overexpressed ADAR1 in HEK293T cells. (D) Comparison of in vitro deaminations of four −1 nucleoside analogs used for cell studies. gRNAs follow modification pattern found in Fig. 3. Information on in vitro deamination conditions and analysis can be found in Fig. 4 (ADAR2) or Fig. 5 (ADAR1). Error bars, s.d. (n ≥ 5 biological replicates). A Grubbs test for outliers was conducted at p = 0.05 significance level. A two-tailed Welch's t test was conducted, where *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | |
The guide oligonucleotides used for cellular editing contained 2′-OMe modifications at every position except the orphan, −1, and −2 positions, which contained 2′-deoxynucleotides (orphan, −2) or LNA A, dN, 2′-F A, or FANA A (−1) (Fig. 10B). Three blocks of phosphorothioate modifications were also included at each end and in the center for improved resistance to cellular nucleases as previously reported.11
After screening different conditions of guides (Fig. S1, ESI†), guide concentrations providing the best resolution between each −1 analog for each enzyme were chosen for comparisons (Fig. 10C). The LNA A −1 guide had the lowest Nluc/FFL ratio with both ADAR2 and ADAR1, indicating the lowest editing levels. dN −1 had the highest level of editing (ADAR1) or did not have a statistically significant difference in editing from the other highest −1 analog, FANA A −1 (ADAR2). The 2′-F A −1 guide had a lower ratio than the dN −1 and FANA A −1 guides with both ADAR2 and ADAR1. These trends are remarkably similar to those observed when comparing deamination rate constants measured in vitro using gRNAs bearing these analogs at the −1 position (Fig. 10D).
In order to confirm the correlation of the Nluc/FFL ratio to cellular editing, RNA isolation followed by reverse-transcription polymerase chain reaction (RT-PCR), nested polymerase chain reaction (PCR), and Sanger sequencing of ADAR1 editing of 30 nM LNA A −1 and dN −1 was conducted (Fig. S2, ESI†). The LNA A −1 guide sample showed no detectable levels of editing, while the dN −1 guide induced 62% editing at the target site.
Discussion
Oligonucleotide therapeutics require chemical modifications to modulate properties such as nuclease resistance, target engagement, enzymatic activity and cell uptake.25 Most of the published efforts describing optimization of ADAR guide strands have focused on the incorporation of modifications that improve metabolic stability or thermodynamic stability of RNA duplexes (e.g. 2′-O-methyl,8,10–14 2′-F,12,14 phosphorothioates,8,10–14 phosphoramidates,12 and LNA8) or GalNAc modification for uptake by hepatocytes.12 A few reports have emerged that use 2′-deoxy- or 2′-deoxy-2′-fluororibonucleotides around the editing site to facilitate editing, but no ADAR deamination kinetic parameters have been reported for these modifications.12,14 Indeed, nearly all published papers that have evaluated the impact of guide strand chemical modifications on directed RNA editing have used only cell-based assays. While these experiments are informative, it is difficult to discern the origin of the effects from these studies alone since chemical modifications can improve directed editing yields in cells in a variety of ways. To establish the effect on the ADAR reaction itself, we measured deamination rates for duplex RNA substrates bearing different modifications on the guide strand. In the work described here, we focused the modifications to a nucleotide position in the guide strand (i.e. position −1) that is highly distorted from A-form geometry in high-resolution structures of ADAR2 bound to RNAs harboring an adenosine deamination transition state analog.15,26 We found that sugar and base analogs that facilitated the unusual conformation at this position in ADAR2-RNA complexes, including those which favor the C2′-endo pucker and allow for A–U wobble pairing, typically increased the rate of ADAR-catalyzed adenosine deamination. This is likely why the dA −1 gRNA showed an increase in rate of editing compared to the rA −1 gRNA in the MECP2 and ACTB sequences, as 2′-deoxynucleotides preferentially adopt a C2′-endo sugar pucker.18 This can also be seen in the comparison of FANA A −1 and 2′-F A −1. The rate of FANA A −1 gRNA was 5.6-fold higher with ADAR2 and the rate with ADAR1 was 7.2-fold higher than 2′-F A −1 gRNA. The beneficial effect of FANA A at the −1 position compared to 2′-F A was maintained in human cells with both overexpressed ADAR1 and ADAR2. FANA A preferentially adopts a C2′-endo pucker,27 similar to the −1 sugar in the ADAR-bound conformation, unlike 2′-F A, which preferentially adopts a C3′-endo pucker.28 In contrast, the LNA A −1 gRNA, which is locked in the C3′-endo pucker29 showed no editing with ADAR2 and little editing with ADAR1 in vitro, and demonstrated little editing in cells. It seems unlikely that this is only a steric effect since 2′-O-Me A −1 gRNA showed editing more comparable to the rA −1 gRNA with both ADAR2 and ADAR1 but, unlike LNA, has conformational flexibility to adopt either sugar pucker.30 Recently, Monian et al. described the effect of 2′-O-methyl, 2′-F, and 2′-deoxy modifications at positions −1, orphan, and +1 in 30 nt ADAR guide strands using cell based assays.12 Interestingly, they also found that 2′-deoxy modification at −1 was significantly better than 2′-F modification at this site for supporting ADAR reactions. While some analogs, namely dN, showed consistent positive effects on ADAR reaction rate, the effects of −1 analogs are not the same across each sequence. Of note, the dA −1 analog is beneficial in the MECP2 and ACTB sequences, but not the IDUA sequence. There are likely other factors at work beyond sugar pucker for some sequences, but the results described here suggest these effects arise from the impact these modifications have on the ADAR reaction itself.
The dN modification at the −1 position had the most consistent positive impact on editing efficiency with ADARs compared to rA in this study, showing an increase in observed rate with all three in vitro sequences tested with both ADAR1 and ADAR2. Additionally, dN at the −1 position had the highest level of editing in cells of the four analogs tested (ADAR1) or was tied for the highest level of editing with FANA A −1 (ADAR2). dN lacks an exocyclic amine, possibly reducing steric clash when the base stacking at the −1 position is distorted. It is also possible that because there is one fewer hydrogen bond between dN and the uracil of the target strand, the disruption of the hydrogen-bonding pattern necessary to sheer the base pairing is easier, though the testing of other analogs at the −1 position with different Watson–Crick hydrogen bonding faces, such as dI or abasic, demonstrate that the benefit of dN at the −1 position is not hydrogen bond sheering alone. The beneficial effect of the dN gRNA analog at the −1 position was shown to be increasingly beneficial when combined with dZ at the orphan position with ADAR2. Considering that the addition of a dZ at the orphan position did not result in a higher ADAR2 deamination rate than the fully ribose gRNA, the beneficial effect of this combination is more than additive. However, this additive effect was not seen with ADAR1, as the dZ orphan, dN −1 gRNA was more reactive than the fully ribose gRNA, but not the rC orphan, dN −1 gRNA. However, it is important to note when comparing orphan rC vs. dZ that the beneficial effect of the base in dZ may be outweighed by the detrimental effect of its 2′-deoxyribose at the orphan position when measured by in vitro deamination kinetics. The relative impact of these effects may also be target sequence and ADAR dependent.
In summary, we have identified nucleoside analogs that enhance (e.g. dA, dN, FANA A) or inhibit (e.g. LNA) the rate of ADAR deamination when located at the −1 position of guide strands targeting 5′-U sites. We anticipate the use of these modifications will effectively modulate efficiency and selectivity in future site directed RNA editing applications.
Materials and methods
Target RNA synthesis
Double stranded DNA fragments corresponding to the target RNAs and including a T7 promoter sequence were purchased from IDT (see Table S8, ESI† for sequences). The DNA was amplified with NEB Q5 Hot Start high fidelity DNA polymerase and purified by phenol-chloroform extraction and ethanol precipitation according to manufacturer's protocol. Target RNAs were synthesized from corresponding DNA with NEB HiScribe T7 RNA polymerase according to the protocol. The target RNAs were then purified as described below.
gRNA synthesis
gRNAs were purchased from Horizon Dharmacon unless desired chemical modifications were unavailable from this vendor. gRNAs with nucleoside analogs not available for purchase from Horizon Dharmacon were synthesized on an ABI 394 DNA/RNA synthesizer. The unlocked nucleic acid A phosphoramidite was synthesized as previously reported31 and the dZ phosphoramidite was purchased from Firebird Biomolecular Sciences. All other reagents and phosphoramidites were purchased from Glen Research. gRNAs were synthesized on the appropriate cycle at 0.2 μmol scale, then dried under vacuum overnight (see Table S8, ESI† for sequences). If the gRNA contained dZ, the oligos were first incubated with 1 M DBU in anhydrous acetonitrile at room temperature overnight without exposure to light. All gRNAs were then cleaved from the controlled pore glass (CPG) column with 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ammonium hydroxide:ethanol at 55 °C overnight. After removing the remaining CPGs, the solution containing the gRNAs was dried down on a speed vacuum. They were then resuspended in 250 μL 1 M tetra-n-butylammonium fluoride in THF and allowed to incubate overnight at room temperature. The deprotected oligonucleotides were then precipitated with 1 mL 1-butanol and 25 μL 3 M sodium acetate before desalting through Amicon Ultra Ultracel-3 K centrifugal filters and purification as follows.
:1 ammonium hydroxide:ethanol at 55 °C overnight. After removing the remaining CPGs, the solution containing the gRNAs was dried down on a speed vacuum. They were then resuspended in 250 μL 1 M tetra-n-butylammonium fluoride in THF and allowed to incubate overnight at room temperature. The deprotected oligonucleotides were then precipitated with 1 mL 1-butanol and 25 μL 3 M sodium acetate before desalting through Amicon Ultra Ultracel-3 K centrifugal filters and purification as follows.
RNA purification
Target and guide RNAs were then purified by denaturing urea-polyacrylamide gel and visualized by UV shadowing. Bands were excised from gel and gel slices were crushed and soaked overnight at room temperature in 500 mM NH4OAc and 1 mM EDTA. Polyacrylamide fragments were removed using a 0.2 μm filter. Oligonucleotides were ethanol precipitated and lyophilized to dryness. Oligonucleotides were then resuspended in nuclease free water and desalted through Sartorius Vivaspin 500 5000 molecular weight cutoff spin filters. gRNA masses for in vitro deamination were confirmed by matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) at the University of California, Davis mass spectrometry facility on a Bruker UltraFlextreme MALDI TOF/TOF mass spectrometer. Oligonucleotide masses were determined by Mongo Oligo Calculator v2.06. Calculated mass was then compared to observed mass, where observed mass includes a correction for the mass of a standard DNA oligo of similar mass to gRNAs (standard DNA oligo was purchased from IDT). Guide oligo masses for cellular editing were confirmed by Electrospray Ionization (ESI) by Novatia LLC (Newtown, PA, USA). See Table S9 (ESI†) for calculated and observed masses.
Hybridization
Target and guide RNAs were combined in a 1:10 target:guide ratio to a final concentration of 180 nM duplex in 1× TE and 100 mM NaCl. The solution was heated to 95 °C for 5 min and then allowed to cool to room temperature over 1 h.
ADAR1 overexpression and purification
Human ADAR1 p110 with a C-terminal His10-tag was overexpressed in Saccharomyces cerevisiae BCY123 as previously described.32 Cells were lysed using a microfluidizer in the following buffer conditions: 20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 1000 mM KCl, 30 mM imidazole, 1 mM tris(2-carboxyethyl)phosphine-HCl (TCEP-HCl), 0.05% (v/v) Triton X-100, and 50 μM ZnCl2. The cell lysate was clarified by centrifugation at 18000 rpm, 4 °C for 1 h. The clarified lysate was then passed over a Ni-NTA column at a flow rate of 2 mL min−1 using an ÄKTA pure 25 FPLC system. The column was washed once with ten column volumes (10 CVs) of the lysis buffer, then twice with 10 CVs of wash buffer (20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 500 mM KCl, 30 mM imidazole, 1 mM TCEP-HCl, and 50 μM ZnCl2). Bound proteins were eluted in 2 mL fractions at a flow rate of 2 mL min−1 by gradient elution with 30 to 400 mM imidazole for 8 CVs, followed by linear elution with 400 mM imidazole for 3 CVs. Fractions containing the target protein were pooled and concentrated to ∼1 mg mL−1, then dialyzed against a storage buffer containing 50 mM Tris-HCl pH 8.0, 10% (v/v) glycerol, 400 mM KCl, 50 mM imidazole, 1 mM TCEP-HCl, and 0.01% (v/v) Nonidet P-40 (NP-40). Protein concentration was determined by running the sample alongside BSA standards in an SDS-PAGE gel, followed by SYPRO Orange staining.
ADAR2 overexpression and purification
Human ADAR2 was expressed as previously described.32 Purification was carried out by lysing cells in buffer containing 20 mM Tris-HCl, pH 8.0, 5% glycerol, 1 mM β-mercaptoethanol (BME), 750 mM NaCl, 35 mM imidazole, and 0.01% NP-40 using a French press. Cell lysate was clarified by centrifugation (19000 rpm for 1 h). Lysate was passed over a 3 mL Ni-NTA column, which was then washed in three steps with 20 mL lysis buffer, wash I buffer (20 mM Tris-HCl, pH 8.0, 5% glycerol, 1 mM BME, 750 mM NaCl, 35 mM imidazole, 0.01% NP-40), wash II buffer (20 mM Tris-HCl, pH 8.0, 5% glycerol, 1 mM BME, 35 mM imidazole, 500 mM NaCl), and eluted with 20 mM Tris-HCl, pH 8.0, 5% glycerol, 1 mM BME, 400 mM imidazole, 100 mM NaCl. Fractions containing the target protein were pooled and concentrated to 30–80 μM for use in biochemical assays. Protein concentrations were determined using BSA standards visualized by SYPRO orange staining of SDS-polyacrylamide gels. Purified protein was stored in 20 mM Tris-HCl pH 8.0, 100 mM NaCl, 20% glycerol and 1 mM BME at −70 °C.
In vitro deamination
For ADAR2 deaminations, hybridized target and guide RNA were diluted to 5 nM in 1× ADAR2 reaction buffer (15 mM Tris-HCl pH 7.5, 3% glycerol, 0.06 M KCl, 1.5 mM EDTA, 0.003% NP-40, and 3 mM MgCl2), 0.5 mM dithiothreitol (DTT), 0.16 units per mL Rnase inhibitor, 1 μg mL−1 yeast tRNA, and 15 nM ADAR2 WT. The reaction was conducted at 30 °C for 120 min, with time points taken at 1, 3, 5, 10, 15, 30, 60, and 120 min by quenching 8 μL reaction mixture in 190 μL 95 °C water for 5 min. For ADAR1 deaminations, hybridized target and guide RNA were diluted to 15 nM in 1× ADAR1 reaction buffer (15 mM Tris-HCl pH 7.5, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% NP-40, and 4% glycerol), 0.5 mM DTT, 0.16 units per mL RNAse inhibitor, 1 μg mL−1 yeast tRNA, and 150 nM ADAR1 p110. Due to the lower enzymatic activity of overexpressed and purified ADAR1, a higher concentration and molar ratio of ADAR1 than ADAR2 was used. The reaction was conducted at 30 °C for 120 min, with time points taken at 5, 30, 60, 90, and 120 min by quenching 8 μL reaction mixture in 190 μL 95 °C water for 5 min. A sample of each time point solution (5 μL) was then reverse transcribed with Access RT-PCR system (Promega) and purified by Zymo clean and concentrator PCR purification kit according to manufacturer protocol. DNA was sequenced with Azenta life sciences (Newtown, PA, USA). All statistical analyses and nonlinear fits were conducted in Microsoft Excel and GraphPad Prism.
Dual luciferase assay plasmid construction
The 90 nucleotide MECP2 target W104X mutation (see Table S8, ESI† for sequence) was ligated to sequences encoding nanoluciferase (Promega) and firefly luciferase (Promega), then subcloned into pcDNA3.1 vector (Invitrogen) using T4 DNA ligase (NEB). Plasmids were transformed into XL-10 Gold ultracompetent cells (Agilent) and purified by Promega PureYield Miniprep Kit according to manufacturer's protocol.
HEK293T cell culture and transfection
Full length hADAR2 WT or hADAR1 p110 were overexpressed using plasmids derived from pcDNA3.1 vector (Invitrogen) as previously described.26 Human embryonic kidney 293T (HEK293T) cells were cultured at 37 °C, 5% CO2 in Dulbecco's modified Eagle's medium (DMEM), 10% fetal bovine serum, and 1% anti–anti. HEK293T cells were used at less than 10 passages. Once cultivated cells reached 70–90% confluency, 1.6 × 103 cells were seeded into 96 well plates. Cells were transfected 24 h later using Lipofectamine 3000 (ThermoFisher Scientific). Transfection of plasmids and guides were as follows. For each well, 50 ng dual luciferase plasmid, 150 ng ADAR plasmid, 0.3 μL of P3000 and varying concentrations of chemically synthesized guide oligonucleotides were incubated with Lipofectamine 3000 transfection reagent (0.5 μL per well) in Opti-MEM Reduced Serum Media (ThermoFisher Scientific). The resulting solution was added to designated wells and incubated at 37 °C, 5% CO2 for 48 h.
Analysis of directed editing by dual luciferase assay
HEK293T cells were cultured and transfected according to the above procedures. After 48 h, lysis and luminescence reagents from Nano-Glo Dual-Luciferase Reporter Assay (Promega) were added to the wells according to manufacturer's protocol and luminescence readout was measured on a plate reader (BMG Labtech). All statistical analysis was conducted in Microsoft Excel.
Analysis of directed editing by Sanger sequencing
HEK293T cells were cultured and transfected according to the above procedures. After 48 h, total RNA was collected using RNAqueous Total RNA Isolation Kit (ThermoFisher Scientific). Isolated RNA was then reverse transcribed with Access RT-PCR system (Promega) followed by amplification by nested PCR with Q5 hot start high fidelity DNA polymerase (NEB). PCR product was purified by 1% agarose gel and Qiagen gel extraction kit. DNA was sequenced by Azenta Life Sciences (Newtown, PA, USA). All statistical analysis was performed in Microsoft Excel.
Conflicts of interest
P. A. B. is a consultant and holds stocks and stock options in ProQR Therapeutics and Beam Therapeutics, companies developing therapeutic editing technologies.
Acknowledgements
P. A. B. acknowledges financial support from the Rett Syndrome Research Trust and National Institutes of Health in the form of grant R35GM141907. E. E. D. was supported by F31CA265135-01 from the National Cancer Institute.
References
- H. M. Khosravi and M. F. Jantsch, RNA Biol., 2021, 18, 41–50 CrossRef CAS PubMed
 .
.
-
Y. Wang, Y. Zheng and P. A. Beal, Enzymes, Academic Press, 2017, vol. 41, pp. 215–268 Search PubMed .
- B. L. Bass, Annu. Rev. Biochem., 2002, 71, 817–846 CrossRef CAS PubMed .
- C. R. Walkley and J. B. Li, Genome Biol., 2017, 18, 205 CrossRef PubMed .
- E. A. Erdmann, A. Mahapatra, P. Mukherjee, B. Yang and H. A. Hundley, Crit. Rev. Biochem. Mol. Biol., 2021, 56, 54–87 CrossRef CAS PubMed .
- K. Nose, K. Hidaka, T. Yano, Y. Tomita and M. Fukuda, Nucleic Acid Ther., 2021, 31, 58–67 CrossRef CAS PubMed .
- J. Wettengel, P. Reautschnig, S. Geisler, P. J. Kahle and T. Stafforst, Nucleic Acids Res., 2017, 45, 2797–2808 CAS .
- T. Merkle, S. Merz, P. Reautschnig, A. Blaha, Q. Li, P. Vogel, J. Wettengel, J. B. Li and T. Stafforst, Nat. Biotechnol., 2019, 37, 133–138 CrossRef CAS PubMed .
- M. Fukuda, H. Umeno, K. Nose, A. Nishitarumizu, R. Noguchi and H. Nakagawa, Sci. Rep., 2017, 7, 41478 CrossRef CAS .
- L. Qu, Z. Yi, S. Zhu, C. Wang, Z. Cao, Z. Zhou, P. Yuan, Y. Yu, F. Tian, Z. Liu, Y. Bao, Y. Zhao and W. Wei, Nat. Biotechnol., 2019, 37, 1059–1069 CrossRef CAS .
- E. E. Doherty, X. E. Wilcox, L. Van Sint Fiet, C. Kemmel, J. J. Turunen, B. Klein, D. J. Tantillo, A. J. Fisher and P. A. Beal, J. Am. Chem. Soc., 2021, 143, 6865–6876 CrossRef CAS PubMed .
- P. Monian, C. Shivalila, G. Lu, M. Shimizu, D. Boulay, K. Bussow, M. Byrne, A. Bezigian, A. Chatterjee, D. Chew, J. Desai, F. Favaloro, J. Godfrey, A. Hoss, N. Iwamoto, T. Kawamoto, J. Kumarasamy, A. Lamattina, A. Lindsey, F. Liu, R. Looby, S. Marappan, J. Metterville, R. Murphy, J. Rossi, T. Pu, B. Bhattarai, S. Standley, S. Tripathi, H. Yang, Y. Yin, H. Yu, C. Zhou, L. H. Apponi, P. Kandasamy and C. Vargeese, Nat. Biotechnol., 2022, 40, 1093–1102 CrossRef CAS PubMed .
- P. Vogel, M. F. Schneider, J. Wettengel and T. Stafforst, Angew. Chem., Int. Ed., 2014, 53, 6267–6271 CrossRef CAS .
- P. Vogel, M. Moschref, Q. Li, T. Merkle, K. D. Selvasaravanan, J. B. Li and T. Stafforst, Nat. Methods, 2018, 15, 535–538 CrossRef CAS .
- M. M. Matthews, J. M. Thomas, Y. Zheng, K. Tran, K. J. Phelps, A. I. Scott, J. Havel, A. J. Fisher and P. A. Beal, Nat. Struct. Mol. Biol., 2016, 23, 426–433 CrossRef CAS PubMed .
- L. R. Monteleone, M. M. Matthews, C. M. Palumbo, J. M. Thomas, Y. Zheng, Y. Chiang, A. J. Fisher and P. A. Beal, Cell Chem. Biol., 2019, 26, 269–277 CrossRef CAS PubMed .
- Y. Zheng, C. Lorenzo and P. A. Beal, Nucleic Acids Res., 2017, 45, 3369–3377 CrossRef CAS PubMed .
- C. Altona and M. Sundarlingam, J. Am. Chem. Soc., 1972, 94, 8205–8212 CrossRef CAS PubMed .
- J. R. Sinnamon, S. Y. Kim, G. M. Corson, Z. Song, H. Nakai, J. P. Adelman and G. Mandel, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9395–E9402 CrossRef CAS PubMed .
- S. K. Wong, S. Sato and D. W. Lazinski, RNA, 2001, 7, 846–858 CrossRef CAS PubMed .
- A. Kuttan and B. L. Bass, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E3295–E3304 CrossRef CAS PubMed .
- D. Wang, C. Shukla, X. Liu, T. R. Schoeb, L. A. Clarke, D. M. Bedwell and K. M. Keeling, Mol. Genet. Metab., 2010, 99, 62–71 CrossRef CAS PubMed .
- H. S. Scott, T. Litjens, P. V. Nelson, P. R. Thompson, D. A. Brooks, J. J. Hopwood and C. P. Morris, Am. J. Hum. Genet., 1993, 53, 973–986 CAS .
- K. Fritzell, L. Di Xu, M. Otrocka, C. Andréasson and M. Öhman, Nucleic Acids Res., 2019, 47, e22 CrossRef PubMed .
- A. Khvorova and J. K. Watts, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed .
- A. S. Thuy-Boun, J. M. Thomas, H. L. Grajo, C. M. Palumbo, S. Park, L. T. Nguyen, A. J. Fisher and P. A. Beal, Nucleic Acids Res., 2020, 48, 7958–7972 CrossRef CAS .
- C. J. Wilds and M. J. Damha, Nucleic Acids Res., 2000, 28, 3625–3635 CrossRef CAS PubMed .
- W. Guschlbauer and K. Jankowskit, Nucleic Acids Res., 1980, 8, 1421–1433 CrossRef CAS .
- S. K. Singh, P. Nielsen, A. A. Koshkin and J. Wengel, Chem. Commun., 1998, 455–456 RSC .
- P. Lubini, W. Zürcher and M. Egli, Chem. Biol., 1994, 1, 39–45 CrossRef CAS PubMed .
- N. Langkjær, A. Pasternak and J. Wengel, Bioorganic Med. Chem., 2009, 17, 5420–5425 CrossRef PubMed .
- M. R. Macbeth and B. L. Bass, Methods Enzymol., 2007, 424, 319–331 CAS .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Victorio
Jauregui Matos
,
Victorio
Jauregui Matos