
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmulating nonribosomal peptides with ribosomal biosynthetic strategies
Silja
Mordhorst†‡
 a,
Fleur
Ruijne‡
b,
Anna L.
Vagstad
a,
Oscar P.
Kuipers
*b and
Jörn
Piel
*a
a,
Fleur
Ruijne‡
b,
Anna L.
Vagstad
a,
Oscar P.
Kuipers
*b and
Jörn
Piel
*a
aInstitute of Microbiology, Eidgenössische Technische Hochschule (ETH), Zürich, Vladimir-Prelog-Weg 4, 8093 Zürich, Switzerland. E-mail: jpiel@ethz.ch
bDepartment of Molecular Genetics, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 7, 9747, AG Groningen, The Netherlands. E-mail: o.p.kuipers@rug.nl
First published on 6th December 2022
Abstract
Peptide natural products are important lead structures for human drugs and many nonribosomal peptides possess antibiotic activity. This makes them interesting targets for engineering approaches to generate peptide analogues with, for example, increased bioactivities. Nonribosomal peptides are produced by huge mega-enzyme complexes in an assembly-line like manner, and hence, these biosynthetic pathways are challenging to engineer. In the past decade, more and more structural features thought to be unique to nonribosomal peptides were found in ribosomally synthesised and posttranslationally modified peptides as well. These streamlined ribosomal pathways with modifying enzymes that are often promiscuous and with gene-encoded precursor proteins that can be modified easily, offer several advantages to produce designer peptides. This review aims to provide an overview of recent progress in this emerging research area by comparing structural features common to both nonribosomal and ribosomally synthesised and posttranslationally modified peptides in the first part and highlighting synthetic biology strategies for emulating nonribosomal peptides by ribosomal pathway engineering in the second part.
 Silja Mordhorst | Silja Mordhorst studied Pharmaceutical Sciences (BSc) at the University of Freiburg, Germany, before she joined the Fast-Track programme for her doctoral studies. Working in the lab of Professor Jennifer Andexer, she implemented various modular enzyme cascades for cofactor regeneration. In 2019, she started her postdoc as a Feodor-Lynen fellow of the Alexander-von-Humboldt foundation in the natural product lab of Professor Jörn Piel at ETH Zurich, Switzerland, where she investigated novel peptide-modifying enzymes from RiPP biosynthetic gene clusters. In 2022, she was appointed assistant professor for Pharmaceutical Biology at the University of Tübingen, Germany. |
 Fleur Ruijne | Fleur Ruijne obtained her Bsc and Msc in Biomolecular Sciences from the University of Groningen, The Netherlands. During her studies she developed great interest in using synthetic biology to tackle the major challenges of our world. She started her PhD in 2018 at the University of Groningen in the group of Professor Oscar Kuipers, where she is focusing on the in vivo production of new-to-nature nonribosomal peptide-emulating antibiotics by novel combinatorial RiPP biosynthetic pathways. |
 Anna L. Vagstad | Anna Vagstad is a senior scientist (and former postdoc) in the group of Jörn Piel at the ETH Zurich Institute of Microbiology, where she leads a small subgroup focused on the enzymology of posttranslational modifications in the maturation of ribosomally synthesized peptide natural products and use of peptide tailoring enzymes for biotechnology applications. She earned her PhD in 2012 deconvoluting the intricacies of fungal polyketide biosynthesis under supervision of Professor Craig Townsend at Johns Hopkins University as a member of the National Institute of Health Chemistry-Biology Interface training program before moving to Switzerland. |
 Oscar P. Kuipers | Oscar Kuipers is Professor in Molecular Genetics and heads the Department of Molecular Genetics at the University of Groningen since 1999. He was trained in molecular biology and biochemistry and moved to the direction of molecular genetics and synthetic biology after his PhD. Central topics in his research are the development of novel antimicrobials by synthetic biology approaches, studying population heterogeneity and bistability of clonal bacterial populations at the single cell level, and bacterial gene regulation and genomics. He is member of the Royal Netherlands Academy of Arts and Sciences and of the European Academy of Microbiology. |
 Jörn Piel | Jörn Piel received a PhD in Chemistry at the University of Bonn, Germany, and conducted postdoctoral studies with Bradley Moore and Heinz Floss at the University of Washington, Seattle. Subsequently, he led a research group at the Max Planck Institute for Chemical Ecology in Jena, Germany, and was Associate Professor of Bioorganic Chemistry at the University of Bonn. In 2013 he became Full Professor of Microbiology at ETH Zurich. His lab studies natural products from uncultivated and other poorly explored bacteria, their role in host-microbe interactions, the identification and utilization of new enzymes, and ecology- and genome-based natural product discovery. |
1 Introduction
Natural products are highly complex secondary metabolites produced by many different types of organisms such as bacteria, fungi, and plants. The scaffolds of natural products continue to be a major source for drug discovery and drug development studies. Many natural products consist of a peptide backbone decorated by specific functional features. These features are often important for the biological activity of the compounds: they might increase the affinity between the peptide natural product and the target molecule or cell, promote the formation of distinct secondary structures, improve the membrane permeability, or lead to an increased stability.1–3The two largest groups of peptide natural products are nonribosomal peptides (NRPs) and ribosomally synthesised and posttranslationally modified peptides (RiPPs). For NRPs, hundreds of different building blocks are available, whereas only the limited set of 20 proteinogenic amino acids (22 including selenocysteine and pyrrolysine) is available for ribosomal biosynthesis. Several biosynthetic pathways of highly modified peptide natural products remained elusive for years, since the products were assumed to be synthesised by nonribosomal peptide synthetases (NRPS), although no corresponding NRPS gene cluster could be found in the genome. Prominent example are thiopeptides and polytheonamides, the latter of which were for a long time considered as the largest NRPs known.4 However, in 2009, thiopeptides were established as ribosomal peptides,5–7 and in 2012, a ribosomal origin from an uncultivated sponge symbiont was demonstrated for polytheonamides.8 Patellamides9 are moderately cytotoxic peptides produced by various cyanobacterial strains and were assumed to be NRPS products as well. In 2005, the ribosomally encoded biosynthetic genes for patellamide A and C were first identified in Prochloron didemni.10 There are also examples for fungal natural products previously considered to be NRPS products. The omphalotins are cyclopeptides that resemble the NRPS product cyclosporine A, but are synthesised via a ribosomal pathway.11 Only recently, the decades-old mystery of the biochemical origin of victorin, a toxin from the oat pathogen Cochliobolus victoriae, was solved: the cyclic hexapeptides are also members of the RiPP superfamily.12
As their names suggest, the two biosynthetic pathways differ substantially (Fig. 1). NRPs are synthesised by large multienzyme complexes (type I NRPS) consisting of different biosynthetic modules that produce the peptide chain in an assembly-line-like manner by a conserved thiotemplate mechanism.13 Each module typically incorporates one amino acid building block into the final product. In the first step, the adenylation domain (A domain) selects the specific building block, which can be a proteinogenic or nonproteinogenic amino acid, the latter synthesised by additional enzymes. A domains normally exhibit a high substrate specificity and activate amino acids as acyl adenylates with adenosine 5′-monophosphate (AMP) from an adenosine 5′-triphosphate (ATP) donor. In the next step, the aminoacyl adenylate is transferred to the peptidyl carrier protein (PCP) by forming a covalent link with the free thiol group of the PCP-bound 4′-phosphopantetheinyl cofactor. At this stage, different modifications can be installed on the substrate by optional domains, such as epimerisation (E domain) to result in D-amino acid residues, peptide-bond N-methylation (NMT domain), cyclisation (Cy domain) to yield 5-membered heterocycles, or oxidation (Ox domain). The condensation domain (C domain) mediates peptide bond formation between the PCP-bound growing peptide chain and the newly activated amino acid leading to chain elongation. The last module contains domain(s) that catalyse release of the peptide. This is typically a C-terminal thioesterase domain (TE domain) that releases the peptide chain by hydrolysis or a (macro)cyclisation reaction, though other off-loading mechanisms are known, such as reduction by a reductase domain (Red domain). The released peptide is often further modified by so-called tailoring reactions, such as glycosylation, acylation, halogenation, or hydroxylation, to yield the mature natural product(s).
 | ||
| Fig. 1 General biosynthetic principles of NRP (type I, A) and RiPP (B) pathways. | ||
In RiPP biosynthetic pathways, one or more precursor peptides and posttranslational maturation enzymes are encoded in a gene cluster. The ribosomally synthesised precursor peptide usually consists of a core peptide region and an additional N-terminal leader region and/or a C-terminal follower region. The maturation enzymes install modifications in the core peptide region prior to proteolytic release. The posttranslational modifications (PTMs) installed by RiPP maturases greatly expand the structural diversity of the canonical proteinogenic amino acids.
The ribosomal pathway uses mRNA as a template for peptide biosynthesis, whereas the A domains of NRPS enzyme complexes act as a template for NRP biosynthesis. These templates are substantially larger with approximately 100 kDa of protein machinery per incorporated amino acid, instead of one base triplet of mRNA in the ribosomal biosynthesis. Therefore, NRPS products are limited in size and most often consist of <10 amino acids.14 Syringopeptin 25A, a virulence factor of Pseudomonas syringae, is the largest NRPS compound described so far, comprising 25 amino acid building blocks.15,16 In contrast, the largest RiPP product described until now contains 70 amino acid residues: the head-to-tail cyclised bacteriocin uberolysin A from Streptococcus uberis.17 Bacteriocins are gene-encoded antimicrobial peptides produced by bacteria that traditionally exhibit narrow spectrum antibiotic activity against closely related strains.18
In general, NRPSs involve complex enzyme systems encoded by large gene clusters and the assembly-line production route makes them difficult targets for engineering. In comparison, RiPP pathways are genetically simple, since they encode separate enzymes and the modularity of RiPP enzymes facilitates manipulation to generate designer peptides.19
Extensive reviews on both NRPS3,16,20–23 and RiPP24,25 biosynthetic machineries have been published in the past. This review focusses on noncanonical amino acids and single structural features common to both of these peptide natural product classes and the great potential of the enzymology found in ribosomal pathways to emulate NRPS natural products.
2 RiPP enzymology that emulates structural features of NRPS products
2.1 D-Amino acids
D-Amino acids are a common feature of both NRPS and RiPP natural products (Fig. 2). These nonproteinogenic residues lend stability against proteolytic degradation and offer stereochemical constraints that promote downstream biosynthetic steps and the resulting tertiary structure of the peptide natural product. Thus, the biosynthetic processing and bioactivity of the peptide is often dependent on the presence of the D-amino acid, which cannot simply be substituted by the L-configured counterpart.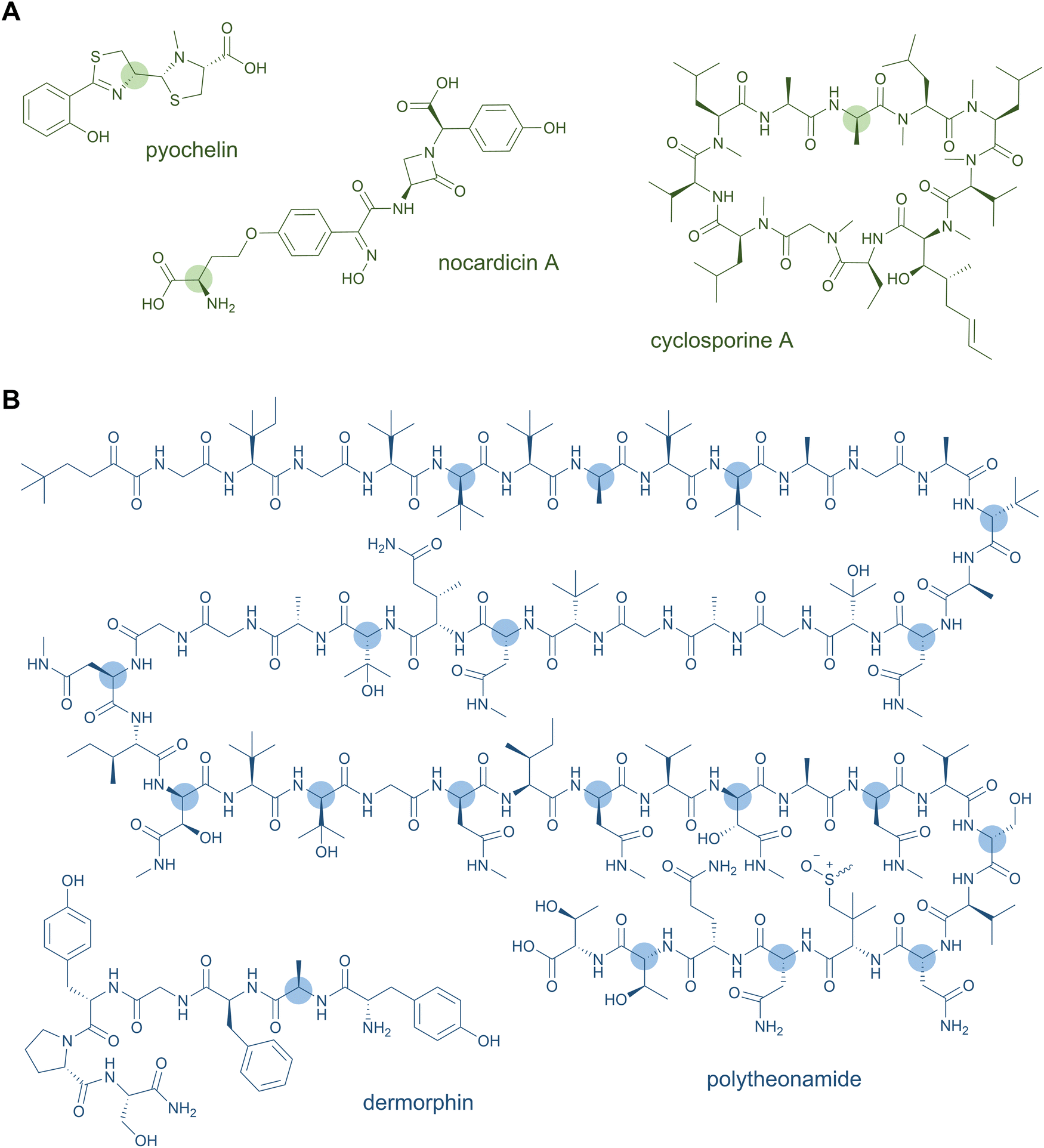 | ||
| Fig. 2 D-Amino acids are common structural features in both NRP (A) and RiPP (B) natural products. | ||
In NRPSs, most D-amino acids are converted from L-amino acids in situ by module-embedded cis-domains. Commonly, an E domain located between the PCP and C domains racemises the PCP-bound intermediate at the α-carbon of the most-recently added monomer to generate an equilibrium of D- and L-configured products.26 The downstream C domain exhibits strict stereochemical control for the D-aminoacyl or -peptidyl variant in peptide bond formation, thus syphoning the preferred D-stereoisomer into the growing peptide chain.26 Bifunctional C domains are also known that catalyse both the condensation and epimerisation of the amino acid and tend to form distinct phylogenetic clades to standard C domains.27,28 More exotic examples of in situ mechanisms for D-amino acid incorporation in NRPS products exist. The C-terminal TE domain of the nocardicin NRPS catalyses both epimerisation of the terminal phenylglycine residue and hydrolytic release of the monolactam-containing pentapeptide product.29 In pyochelin and yersiniabactin biosynthesis, a so-called “stuffed” E domain embedded within the A domain and resembling a defunct methyltransferase enables formation of a D-thiazoline moiety.30 Alternatively, D-amino acids, typically generated by free-standing pyridoxal 5'-phosphate (PLP)-dependent racemases, can be selectively activated by A domains for direct incorporation into the peptide. This is the case for selection of D-Ala by the first module of cyclosporine A synthase31 and by the stand-alone NRPS module involved in a post-synthase tailoring modification of the ansatrienin polyketides.32
D-Amino acids have also been recognised as important structural features of many ribosomal-origin peptides, as several different enzyme classes have convergently evolved to posttranslationally epimerise RiPPs. Peptidyl isomerases have long been known from bioactive eukaryotic peptide biosynthetic processes predominantly found in venoms or nervous tissues. Although they favour production of the D-amino acid, they generally yield a mixture of D- and L-stereoisomers, relying on deprotonation/reprotonation chemical steps. Examples include D-Ala in the potent μ-opioid agonist dermorphin from frog skin,33D-Ser in ω-agatoxin from funnel web spider,34 and various D-residues from mollusc conotoxins,35,36 among others.37 Although the responsible isomerases catalyse similar acid-base chemistry, the enzymes characterised to date are largely unrelated structurally to each other and contain no cofactors.
Some lanthipeptides contain D-Ala or D-amino butyric acid residues, which arise from a two-enzyme process. In the first step, Ser or Thr residues undergo a formal dehydration to dehydroalanine (DhA) or dehydrobutyrine (DhB) intermediates, respectively, by the first dehydratase component of lanthionine synthetases. Although these dehydroamino acids are typically subject to conjugate addition of Cys sulfhydryls to form the namesake lanthionine thioether bridges, here, dehydration is followed by subsequent diastereoselective hydrogenation to produce the D-stereoisomers. The hydrogenation enzyme types differ and include zinc-dependent dehydrogenases termed LanJA, members of the flavin-dependent oxidoreductase superfamily termed LanJB, and the most recently discovered F420H2-dependent reductases termed LanJC from, for example, lacticin 3147,38 carnolysin,39 and lexapeptide40 biosyntheses, respectively.
Two subfamilies of radical S-adenosylmethionine (rSAM) enzymes that utilise oxygen-sensitive [4Fe–4S]-cluster cofactors and SAM co-substrates are known to install diverse D-amino acids. The cytotoxic, 48-mer polytheonamides contain 18 D-amino acids (natively at Val, Ala, Asn, Ser, and Thr residues) installed by a single enzyme, i.e., the rSAM peptide epimerase PoyD.8 Characterisation of PoyD and several homologues, as well as various mutagenesis studies, demonstrated this enzyme class catalyses epimerisation on a wide range of peptide targets and residue types.41–46 A second class of rSAM epimerases with distinct domain architecture to the PoyD-type comes from epipeptide RiPPs first characterised in Bacillus subtilis.47 The epimerase YydG installs D-allo-Ile and D-Val in the YygF precursor protein. These posttranslational epimerisations are essential for the peptide's function to induce a major component of the bacterial cell envelope stress-response. Radical-mediated epimerisation is advantageous over racemases that use acid-base catalysis in that D-amino acids are installed irreversibly via a radical mechanism involving abstraction of the α-hydrogen of the target amino acid residue and hydrogen donation on the back-face likely from a conserved Cys side chain.45,47,48 However, D-amino acid patterns installed by a rSAM epimerase vary greatly depending on the core sequence and it is not yet possible to predict the product stereochemistry.42
Single representatives of other epimerase types have been characterised in additional RiPPs. In the biosynthesis of the antibiotic bottromycin, Asp is converted from the L- to D-configuration by α-β hydrolase family member BotH.49 The C-terminal D-Tyr of the lasso peptide MS-271 was recently shown to be installed by a metal- and cofactor-independent enzyme MslH belonging to the metallo-dependent phosphatase family.50 In other RiPPs, the source of D-amino acids remains elusive. Salinipeptins, for example, contain up to nine D-amino acids at diverse residues by an as-yet undefined mechanism, since the biosynthetic gene cluster lacks genes for any known class of epimerase.51 Recent work suggests that the gene sinL from the salinipeptin biosynthetic gene cluster encodes a novel peptide epimerase, which is also found in other type-A linaridins such as grisemycin and cypemycin.52 The diversity and prevalence of epimerases and the importance of D-amino acids to RiPP bioactivities suggest that additional new epimerase families will be discovered in the coming years.
2.2 Macrocycles
Head-to-tail macrocycles as well as internal cyclised moieties of various ring sizes form a large group of NRPs. Cyclisation constrains the conformation of the peptide, which is often beneficial for efficient binding to a biological target and its subsequent activity, and it increases the proteolytic stability as well. Some cyclic peptides even possess exceptional cell permeability and oral bioavailability.53,54 These features of macrocyclic NRPs have resulted in their use as peptide drugs. The ring closure can occur via a peptide bond (peptidolactam formation) or via an ester bond (peptidolactone formation).2Examples for macrocyclised NRPS products include the antibiotics daptomycin,55,56 gramicidin S,57 and tyrocidin A (Fig. 3A),58 the immunosuppressant cyclosporine A,59 the thrombin inhibitor cyclotheonamide,60,61 the antifungal lipopeptides echinocandin B62 and fengycin,63,64 and the biosurfactant surfactin A,65 among many others. An unusual macrocyclic structure is found in the weakly cytotoxic nostocyclopeptides, in which an imino linkage is responsible for the ring closure.66,67 Further examples of peptide-containing macrocycles are hybrid polyketide synthase (PKS)-NRP products, such as streptogramins (pristinamycin68 and virginiamycin69), rapamycin,70,71 and tricholide A and B.72
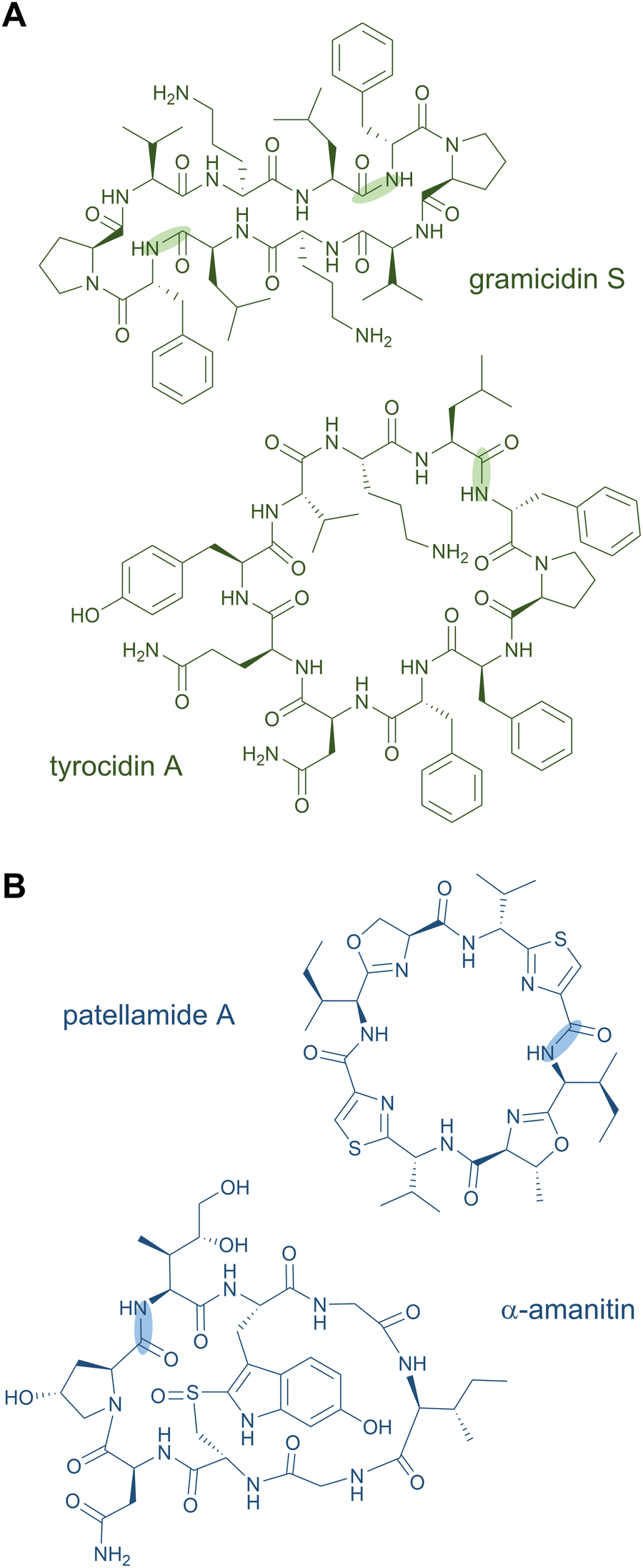 | ||
| Fig. 3 Macrocyclic peptide natural products biosynthesised by NRPS (A) and RiPP (B) pathways. | ||
In NRPSs, the macrocyclisation reaction is usually catalysed by the C-terminal TE domain. It is included in the final module of the enzyme complex and responsible for off-loading the peptide product. For the cyclisation reaction, a nucleophilic group of the nascent peptide attacks the thioester bond to the PCP domain and releases the cyclic peptide. In case of imine formation in nostocyclopeptides, an NAD(P)H-dependent Red domain is predicted to be involved in the ring closure.67
Several RiPP natural products also contain macrocycles. Circular bacteriocins are large antimicrobial peptides forming monocyclic skeletons; prominent representatives are enterocin AS-48 from Enterococcus faecalis, uberolysin A from Streptococcus uberis, and circularin A from Clostridium beijerincki.17,73–76 Other common examples for macrocycles are found in the class of cyanobactins including the patellamides (Fig. 3B) and trunkamides.24 The plant-derived RiPP classes of orbitides (e.g. cyclolinopeptide A24) and cyclotides (e.g. kalata B177,78) include macrocyclic structures as well. The fungal peptide omphalotin A also features a head-to-tail cyclised peptide produced by the basidiomycete Omphalotus olearius. Further fungal examples are amatoxins (α-amanitin) and phallotoxins (phalloidin).24 The telomerase inhibitor telomestatin, a potential therapeutic agent for cancer treatment, was only recently identified as a RiPP and contains eight heterocyclised amino acids in a macrocyclic ring.79
In contrast to NRPS macrocyclisation reactions, which are commonly catalysed by TE domains, RiPP macrocyclisation reactions are accomplished by diverse enzymes and some remain elusive to date. For example, the biosynthetic mechanism and the role of the leader peptide resulting in the circular bacteriocins are not well understood yet and the nature and type of enzyme remains enigmatic. It has been suggested that no single protein, but rather a group of four to five gene products are responsible for their circular maturation and secretion.76 On the contrary, the cyanobactin macrocyclases are well-studied enzymes. They are dual-action subtilisin-like serine proteases that cleave the C-terminal follower peptide upstream of the recognition sequence and concomitantly catalyse the N-to-C cyclisation.80 The crystal structure of PatG from the patellamide biosynthetic gene cluster has been solved,81 and a homologue, PagG from prenylagaramide biosynthesis has been used to generate a large peptide library highlighting the biotechnological potential of cyanobactin macrocyclases.82 A different family of macrocyclases are cysteine proteases of the asparaginyl endoprotease family found in plants; these enzymes cleave C-terminally to Asn or Asp residues, generating an acyl-enzyme intermediate that is intra- or intermolecularly captured to yield cyclisation or ligation products, respectively.25 They are typically involved in the biosynthesis of cyclotides and orbitides. The macrocyclisation of fungal amatoxins and omphalotins is proposed to be catalysed by a prolyl oligopeptidase performing two reaction steps: first, cleavage at the N-terminus and second, a transpeptidation reaction to cleave the C-terminus of the core peptide and form the cyclic peptide.83,84
Besides macrocycles, many other cyclic structures are formed by both NRPS and RiPP pathways. Various NRPS intramolecular crosslinks are found in, for example, glycopeptide antibiotics, such as vancomycin, teicoplanin, and kistamycin, which contain multiple crosslinks via side chains of aromatic amino acids. These crosslinks are introduced by cytochrome P450 enzymes.85,86 In RiPP products, many different cyclisation variants have been discovered.24,25 Common crosslinks are thioether bridges which are found in lanthipeptides,87 sactipeptides,88 and ranthipeptides.89 Disulphide bonds occur in cyclotides90 and in class I, II, and IV lassopeptides.91 In addition to disulphide bridges, lassopeptides contain a macrolactam cycle formed by an isopeptide bond between the N-terminal α-amino group and an Asp or Glu side chain.91 A Cys-Trp crosslink is found in amatoxins leading to a tryptathionine structure.25 C–C crosslinks have been discovered in streptides (Lys–Trp crosslink)25 and in ryptides (Arg–Tyr crosslink),92 and ether bonds (C–O) in rotapeptides (Thr–Gln crosslink).93 Both C–C and C–O crosslinks have recently been identified in indole- or phenyl-bridged cyclophanes.94 Most of these crosslinks are installed by rSAM enzymes, while the lanthionine and methyllanthionine bridges are introduced between dehydrated Ser or Thr residues and Cys by different classes of lanthionine synthetases.25
2.3 Lipopeptides
NRPS-generated lipopeptides form a diverse group of cyclic and linear species with antimicrobial, antitumour, immunosuppressant, and surfactant activities, among others.95 In most cases, the hydrophobic lipid group is either essential or strongly enhances the bioactivity. Canonically, fatty acids (typically of medium chain lengths) act as starter units that are extended by the mega-synthetase assembly line, either NRPS or PKS-NRPS hybrids, to produce peptides with lipid tails capping their N-termini.96 Depending on the biosynthetic machinery, acyl groups can be directly donated from an acyl-coenzyme A or the acylated phosphopantetheine prosthetic group of a holo acyl-carrier protein (ACP). Dedicated domains and standalone proteins belonging to the ANL superfamily97 activate the carboxylic acid of fatty acids and ligate them as thioesters to coenzyme A or holo-ACP. The fatty acyl groups can undergo further modifications prior to condensation with amino acid extender units, resulting in a large diversity of lipid structures. NRP lipopeptides are particularly common in Bacillus and Pseudomonas bacteria but have been isolated from a range of other bacterial and fungal species, highlighting their ecological importance. Prominent examples have been exploited in human medicine, including the membrane-active antibiotics daptomycin and polymyxin B (Fig. 4A) used as last-line treatments against multidrug-resistant Gram-positive and Gram-negative bacteria, respectively.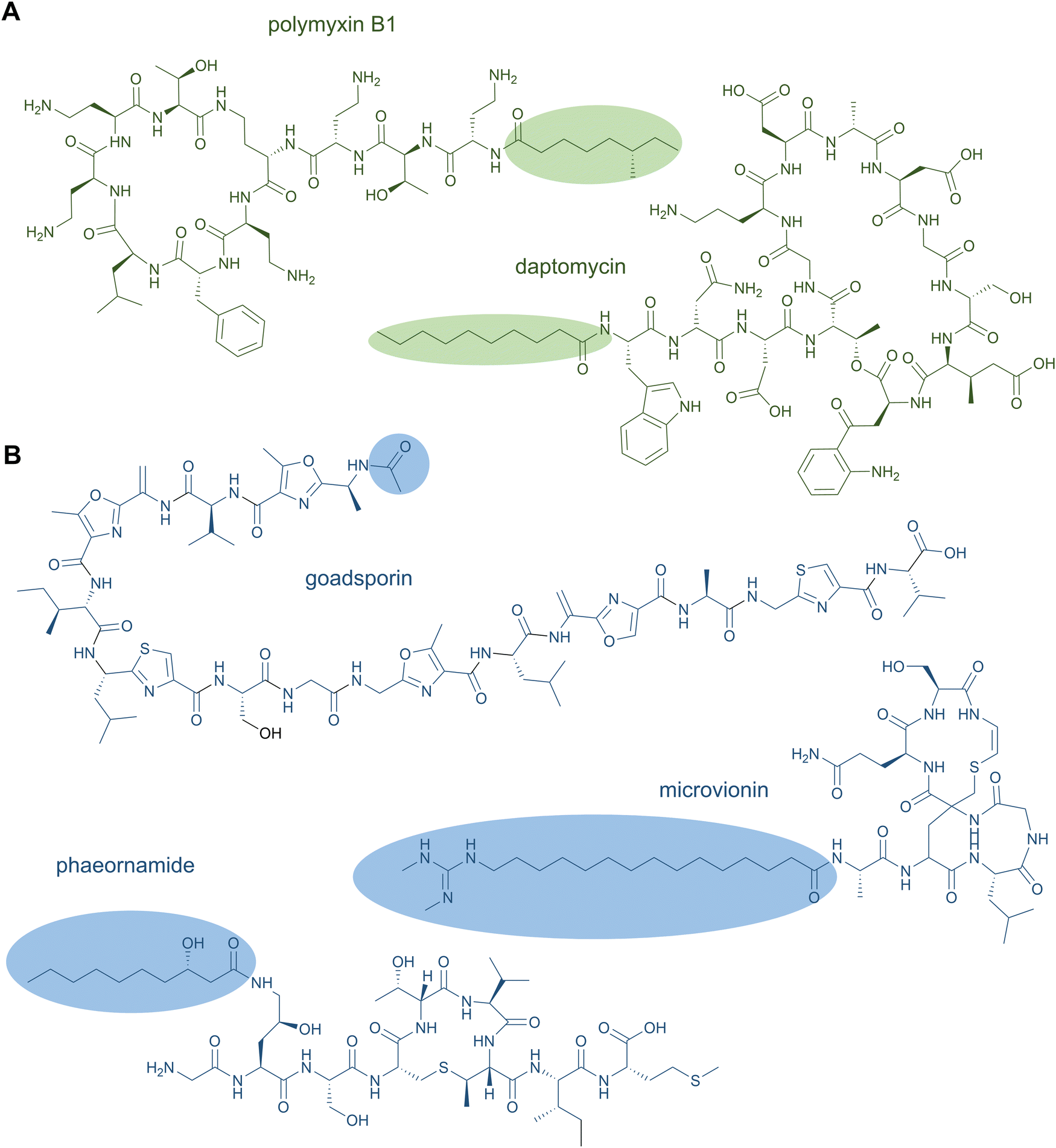 | ||
| Fig. 4 Nonribosomal (A) and ribosomal (B) peptides carrying a lipid tail. | ||
Lipid modifications can also be added by post-synthase tailoring reactions, as in the lipoglycopeptides. The representatives teicoplanin and A40926 isolated from Actinomadura species are clinically used against Gram-positive bacterial infections where they bind the D-Ala-D-Ala moiety of peptidoglycan with high affinity.98 Their crosslinked, heptapeptide backbones are synthesised by NRPSs in concert with trans-acting oxidases and are sequentially modified by glycosyl- and acyltransferases to attach lipid-modified amino sugars to the 4-hydroxyphenylglycine at residue 4. Other examples for post-synthase lipidation are prenylated NRPs, e.g. the antitumour fungal metabolite terrequinone A from Aspergillus sp. Terrequinone A is built by head-to-tail dimerisation of indole pyruvate followed by bisprenylation.99
N-Acetylations were identified in a number of RiPPs—including microviridin toxins from various cyanobacteria,100 the linear azol(in)e-containing peptide goadsporin (Fig. 4B),101 and the lasso peptide albusnodin102—but until recently, longer chain-length acylations were unknown. In 2018, the founding members of the anti-staphylococcal lipolanthines, i.e., microvionin and nocavionin, containing N-terminal N,N′-bismethylated guanidine fatty acid modifications were reported (Fig. 4B).103 Genome mining and biosynthetic studies indicated an extended family of lipolanthine-type peptides occur in Actinobacteria. The presence of genes encoding fatty acid synthase (FAS), PKS, and NRPS biosynthetic machineries clustered adjacent to the RiPP precursor and maturase encoding genes are suggestive of diverse acyl group appendages.104 In 2020, the structures and biosynthetic route to another type of lipidated lanthipeptide, the goadvionins, were reported.105 Goadvionin and goadpeptin analogues slightly vary in their peptide sequence (encoded by slightly different precursor cores) and the oxidation state of the lipid moieties. They contain trimethylammonio 32-carbon, polyhydroxylated polyketide acyl groups generated by a dedicated PKS. A Gcn5-related N-acetyltransferase superfamily (GNAT)-member, GdvG, catalyses the polyketide-RiPP coupling step in an ACP-dependent manner from the PKS to the N-terminus of the modified core peptide following proteolytic cleavage of the precursor leader. Lipolanthine clusters also contain GNAT homologues thought to be responsible for diverse N-acylations.
Whereas to date most lipid modifications in lipopeptides of NRP or RiPP origins are located at the peptide N-termini, the newest family of lipopeptide RiPPs, termed selidamides, are N-acylated at Lys or ornithine side chains.106 Ornithines arise from modification of Arg by peptide arginases, as described in the ornithine section (see Section 2.8 Ornithine residues).107,108 Three founding members were recently characterised by heterologous expression of pathways in E. coli: kamptornamide and nostolysamides from cyanobacteria, and phaeornamide from the marine alphaproteobacterium Phaeobacter arcticus. Phaeornamide was furthermore directly detected in the host, indicating it is an authentic natural product. Acyltransferases belonging to the GNAT superfamily, but with distinct phylogenetic lineage to the lipolanthine and goadvionin type GNATs, are responsible for catalysing the side-chain fatty acylations. The acyl groups appear to be recruited from the primary metabolic lipid pools, and the GNATs exert tight chain-length specificity, transferring C12, C10–OH and C16(–OH) fatty acids to kamptornamide, phaeornamide, and nostolysamides, respectively. This specificity stands in contrast to many NRP products, which are generated in difficult-to-separate mixtures of differentially fatty-acylated congeners. Bioinformatic analyses of the novel subclass of GNATs and their respective biosynthetic gene clusters indicate a wide distribution of selidamides in bacteria with diverse precursor sequences. Their compact gene clusters and propensity to make a single fatty acylated product lends the selidamides well to synthetic biology strategies for peptide engineering.
In addition to N-acylations, isoprenoid alkylations of diverse carbon and heteroatom positions in linear and cyclic cyanobactin RiPPs are known.109 The responsible prenyltransferases tend to attach 5-carbon prenyl units, but 10-carbon geranyl transferases have also been characterised.110 Tyr, Ser, and Thr O-prenylations were the earliest examples and recent genome mining efforts have expanded the scope to C- and N-prenylations of Trp, bis-N-prenylation of Arg,111C-geranylation of His,110 and even prenylations at the amide nitrogen or carboxy oxygen of linear peptide termini.112,113 Crystal structures of several peptide prenyltransferases have been solved, providing a mechanistic basis for their broad substrate specificity toward alternative peptide and even small-molecule substrates containing suitable acceptor moieties in a largely leader-independent manner.110,114 Furthermore, the preference for prenyl versus geranyl units can be toggled by a single point mutation in the prenyltransferase, as shown for interconversion between C5 and C10 isoprenoid preferences for PagF from prenylagaramide and PirF from piricyclamides RiPPs.115–117 In a distinct class of prenylated RiPPs, ComX pheromones from Bacillus subtilis are geranylated at C3 of Trp4 within a 6-residue core peptide.118
2.4 Heterocyclisations
A rather dramatic chemical modification is the formation of a heterocycle in a peptide chain. Like Pro residues, heterocycles lead to severe changes in the peptide by altering the backbone connectivity and hence the three-dimensional structure of the peptide product. Heterocyclisations occur on Cys, Ser, and Thr residues yielding five-membered rings of thiazolines and oxazolines.3 These structural features can further be oxidised to thiazoles and oxazoles or reduced to thiazolidines and oxazolidines, respectively.119 The heterocycles can function as metal-chelating and intercalating groups and are thought to be important for the biological function of the peptide products,119e.g., in the case of the antitumour drug bleomycin, a tandem bis-heterocycle forms the DNA-intercalating moiety (Fig. 5A).120 | ||
| Fig. 5 Heterocyclisations are common in both nonribosomal (A) and ribosomal (B) peptides. | ||
In NRPS products (and NRPS-PKS hybrids), heterocycles of the oxazole series are, for example, found in the iron-chelating siderophores vibriobactin (Fig. 5A) from Vibrio cholerae121 and mycobactin from Mycobacterium tuberculosis122 (both contain phenyloxazoline moieties), and in the antiviral thiangazole123,124 (β-methyloxazole). Two adjacent, cyclodehydrated and dehydrogenated Ser residues occur in the cytotoxic compound diazonamide A (monochloro-bioxazole) and in the antiviral hennoxazole A (4,2-linked bisoxazole). Heterocycles of the thiazole series are found in the siderophores yersiniabactin125 (two thiazolines, one thiazolidine) and pyochelin126,127 (one thiazoline, one thiazolidine), as well as in the antibiotic bacitracin128 (thiazoline). Analogously to bisoxazoles, bithiazoles can be formed by two adjacent Cys residues, as discovered in myxothiazole129 and bleomycin.130
Cyclodehydration during NRPS biosynthesis is catalysed by Cy domains, which are variants of C domains (sometimes referred to as C’). The reaction occurs during elongation of the covalently bound substrate. It is presumed that Cy domains first catalyse the peptide bond condensation reaction and subsequently cyclise the thiol or hydroxyl side chain of Cys or Ser/Thr against the peptide backbone onto the previously formed peptide bond yielding (thio)hemiaminal intermediates that are dehydrated to form the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in the thiazoline and oxazoline rings. These may subsequently be oxidised to thiazoles and oxazoles by oxidative (Ox) domains or reduced to thiazolidines and oxazolidines by reductive (Red) domains.
N bond in the thiazoline and oxazoline rings. These may subsequently be oxidised to thiazoles and oxazoles by oxidative (Ox) domains or reduced to thiazolidines and oxazolidines by reductive (Red) domains.
Products of ribosomal pathways can also contain heterocycles derived from Cys, Ser, or Thr residues. The family of linear azol(in)e-containing peptides (LAPs) is characterised by the presence of such heterocycles.25 A prominent representative is the antibiotic microcin B17 with four oxazoles and four thiazoles, where two of each form mixed tandem pairs of biazoles. Other RiPP families with azol(in)e heterocycles include thiopeptides (e.g. thiostrepton, nosiheptide, sulfomycin), cyanobactins (e.g. patellamide, trunkamide), and bottromycins (e.g. bottromycin A1).25 A RiPP product consisting solely of heterocycles is the telomerase inhibitor telomestatin (Fig. 5B), which has only recently been identified as a ribosomal product.79
While heterocyclisation in NRPS products occurs “co-translationally”, it happens posttranslationally in RiPPs. Oxazole and thiazole installations proceed in two steps: in the first step, the oxazoline or thiazoline is installed in an ATP-dependent reaction and in the second step, the formed heterocycle is oxidised to an oxazole or thiazole moiety in a flavin mononucleotide (FMN)-dependent reaction. The whole process is catalysed by a trimeric synthetase, whereby the first step requires an association of a YcaO enzyme and an E1-ubiquitine activating protein; these two are often fused in one protein (cyclodehydratase/heterocyclase). The second step is optional and catalysed by an FMN-dependent dehydrogenase.25,131 Distinctly, a parallel pathway towards thio(seleno)oxazole has been identified recently that relies on rSAM chemistry.132
2.5 Glycopeptides
Glycopeptides are peptide natural products conjugated with sugar moieties, which are located at the amino acid side chains or at the peptide's C- or N-termini. Glycosylation changes the physiological properties of the peptide and glycosyl residues may improve the bioavailability of therapeutic peptides.133 This effect is either achieved by an improved uptake (facilitating the penetration of membranes)134 or by an increased metabolic stability (lowering the clearance rate or preventing side-chain oxidation).135,136 Moreover, glycosylation can be used to target specific organs and to enhance receptor binding.133,137,138 If the glycosylation is crucial for the bioactivity, the peptides are regarded as being “glycoactive”.139Glycosylation is ubiquitous in secondary metabolite biosynthetic pathways. Approximately 20% of all bacterial natural products are glycosides.140 For a detailed overview of glycosylated peptides, the reader is referred to chapter 15 of the review article entitled “A comprehensive review of glycosylated bacterial natural products” by Elshahawi et al.140 In this section, we list a few examples of the most common NRPS glycopeptides and the (still) rather small number of RiPP glycopeptides.
Glycopeptide antibiotics are clinically used as cell wall biosynthesis inhibitors to fight infections by Gram-positive bacteria. Famous representatives are vancomycin from Amycolatopsis orientalis and teicoplanin from Actinoplanes teichomyceticus, which both contain O-glycosylations (Fig. 6A).141,142 Vancomycin carries two sugar moieties, i.e.D-glucose and L-vancosamine,141 whereas teicoplanin contains the carbohydrates N-acyl-β-D-glucosamine, N-acetyl-β-D-glucosamine, and D-mannose. Teicoplanin is a mixture of several compounds that differ in the fatty acyl side chain attached to β-D-glucosamine.142 Another O-glycoside natural product is the cytotoxin bleomycin, which is conjugated to a disaccharide of D-mannose and L-gulose.130 The sugar moieties can also be attached to nitrogen atoms; examples for this are the cyclic glycopeptide antibiotics mannopeptimycins. These structures contain the unusual amino acid α-amino-β-[4′-(2′-iminoimidazolidinyl)]-β-hydroxypropionic acid that is fused to D-mannose.143
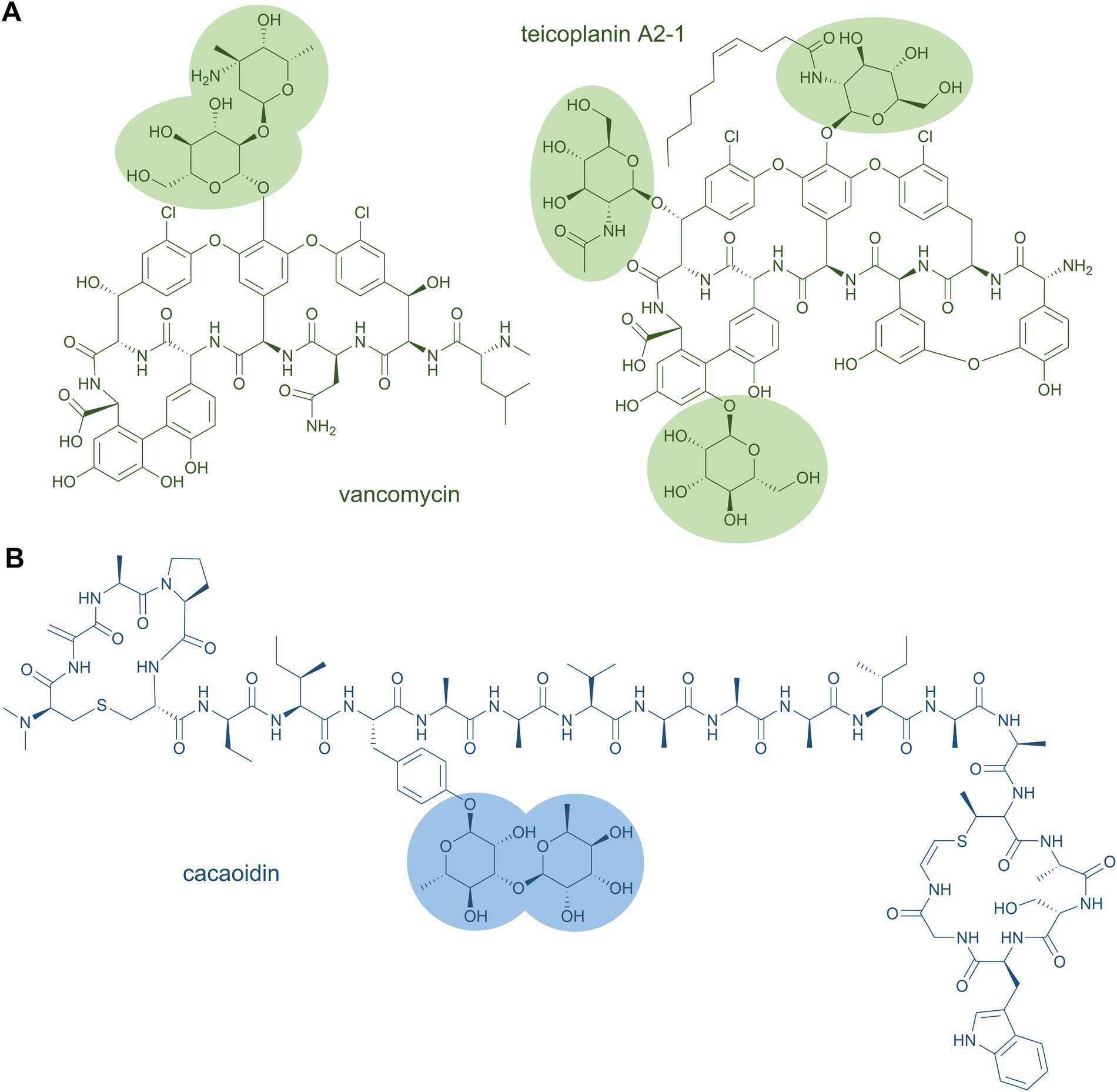 | ||
| Fig. 6 Selected examples for glycopeptide natural products from NRPS (A) and RiPP (B) pathways. | ||
In NRP biosynthesis, glycosylation is a tailoring reaction, which occurs after peptide release from the NRPS assembly line. In general, this regio- and stereospecific bond formation is catalysed by glycosyltransferases. Many biosynthetic pathways comprise several glycosyltransferases, each enzyme being specific for the glycosylated position and the transferred sugar moiety. Some glycosyltransferases are promiscuous and have been employed for in vitro glycorandomisation to generate libraries of peptides with diverse glycosylation patterns.144–146
The RiPP class of glycocins (glycosylated bacteriocins) is defined by the PTM glycosylation. Sugar moieties are found on Cys, Ser, or Thr residues yielding S- or O-glycosides.25 Representatives are, amongst others, glycocin F from Lactobacillus plantarum, sublancin 168 from Bacillus subtilis 168, and pallidocin from Aeribacillus pallidus.139,147 Sublancin 168 and pallidocin contain a single glycosylated Cys residue, whereas glycocin F contains both glycosylated Ser and Cys residues. There are also diglycosylated examples of glycocins; listeriocytocin and enterocin 96 both contain a diglucosylated Ser.148
Glycosylation has also been reported for a few other RiPP classes, such as thiopeptides (e.g., glycothiohexide α, philipimycin, nocathiacin I), lanthipeptides (NAI-112), lanthidins (cacaoidin), lassopeptides (pseudomycoidin), and sulfatyrotides (plant peptide PSY1).25,149–152 These examples are distinguished from the glycocins by the residue carrying the sugar moiety. Cacaoidin features an O-glycosylated Tyr (Fig. 6B),150 and PSY1 a hydroxyl-Pro residue.152 In the case of NAI-112, a Trp residue is glycosylated and it is the indole nitrogen that forms the glycosidic bond. This N-glycoside is to date unique to RiPPs.149 A special case is also found in pseudomycoidin; a C terminal phosphorylated Ser is glycosylated at the phosphate group.151
The sugar moieties attached to RiPPs range from glucose (sublancin, pallidocin, listeriocytocin) and arabinose (PSY1) to rhamnose and gulose (both cacaoidin). Analogously to NRPS biosynthesis, they are generally installed by glycosyltransferases. Some of these enzymes are able to transfer the sugar moiety onto different acceptor molecules, for example the glycosyltransferases from the biosynthetic gene clusters of thurandacin and glycocin F glycosylate both Cys and Ser residues.139,153 For some glycosylation steps, the enzymes are not characterised to date, such is the case for nocathiacin I.154 The glycosylation of the phosphate group in pseudomycoidin biosynthesis is hypothesised to be catalysed by a nucleotidyltransferase.151 The class-defining enzyme of glycocins is a SunS-like glycosyltransferease (protein family PF00535) in combination with a peptidase-containing ATP-binding cassette transporter.25
2.6 β-Amino acids
β-Amino acids are noncanonical residues with a one-carbon extension of the standard α-amino acid backbone. They are found in diverse NRPs and their polyketide hybrid natural products.155 β-Lactam rings are small, cyclic variants of β-amino acids and are also found in some classes of NRPs, like the penicillins/cephalosporins antibiotics that inhibit cell-wall biosynthesis and the monolactams such as nocardicin A and tabtoxin;156–158 however, such structures have so-far not been mimicked by RiPP biosynthetic machinery and are hence beyond the scope of this review. Here, we will focus on β-amino acids that are incorporated into the backbone of cyclic or linear peptides; these structures have parallels in RiPP biosynthesis. For a holistic, but slightly dated, overview of β-amino acids in different structural classes of natural products, we recommend the 2014 review by Kudo et al.155 As with most of the peptide modifications covered in this review, β-amino acids can render peptides more resistant to proteolysis and yield alternative structures with distinct properties and activities to their standard α-amino acid analogues.In NRP or NRP-PKS hybrid products, β-amino acids are commonly synthesised prior to incorporation and are selected by β-amino acid specific A domains. In a typical scenario, the β-amino acid is activated by the A domain and transferred to the PCP domain. The β-amino group then acts as nucleophile for condensation with the growing peptide chain thioester. Examples include the incorporation of β-Ala in bleomycin,159 (2R,3S)-MeAsp in microcystin (Fig. 7A),160 and (R)-β-Tyr in chondramides161 by modular cis-acting A domains, whereas representative discrete A domains select 3-aminononanoic acid in cremimycin,162 (S)-β-Phe in andrimid,163 and (S)-β-Tyr in the enediyne antibiotic C-1027.164,165 Many of these monomers are catalytic rearrangements of standard α-amino acids. A variety of enzyme types catalyse rearrangements from α- to β-amino acids, but PLP-dependent transaminases and aminomutases are the major classes.
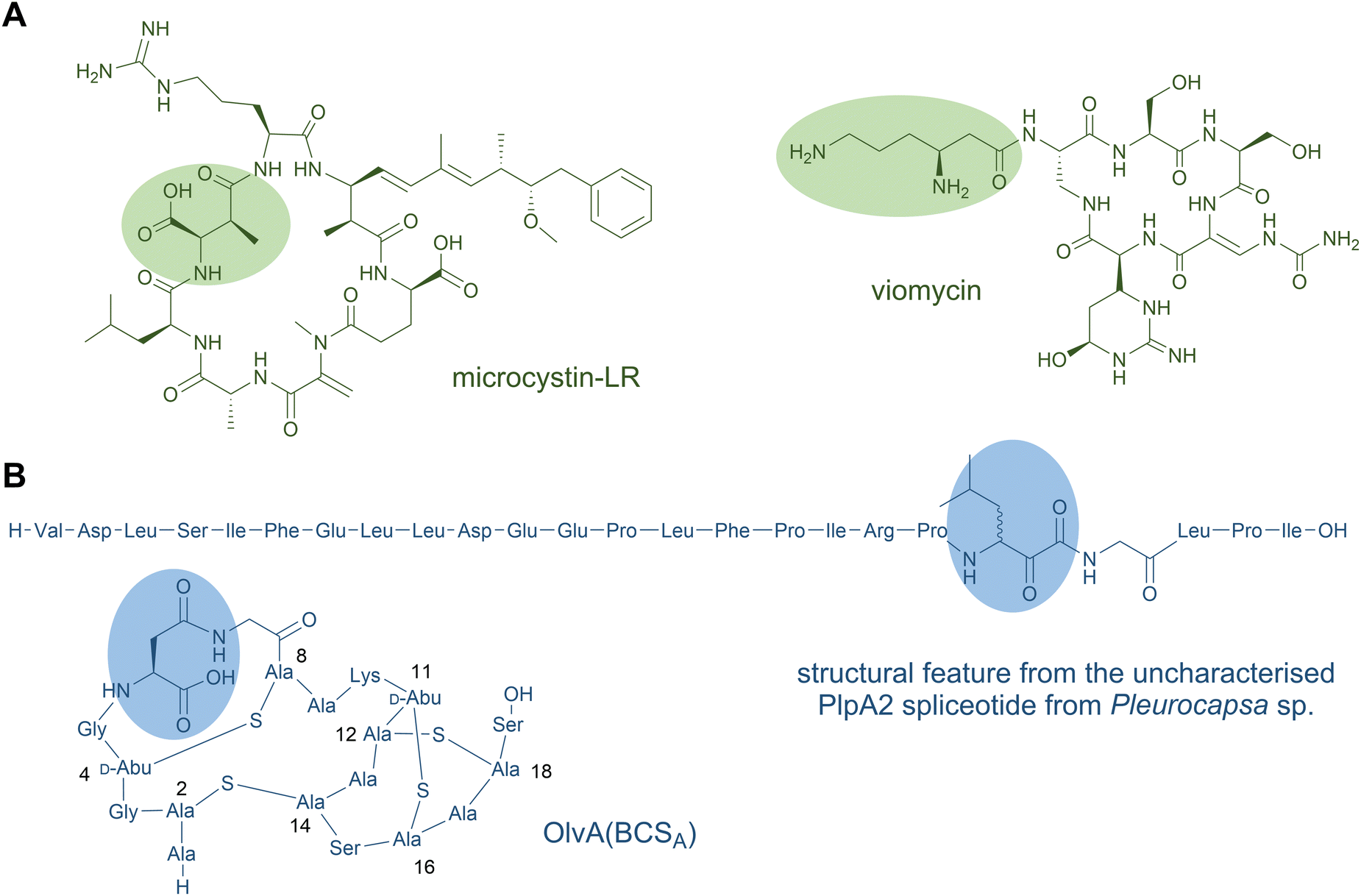 | ||
| Fig. 7 β-Amino acid residues occur in NRP (A) and in ribosomal peptides (B). The amino acid residues of the first structure shown in panel B is expected based on the PlpA2 core peptide. The common three-letter amino acid code is employed. The formation of the highlighted β-amino acid has been confirmed experimentally.166 | ||
β-Amino acids may also be formed in situ as NRPS-bound intermediates. In the antitumour drug bleomycin, on-line condensation of the N1 of Asp with the C3 of a Ser-derived α,β-unsaturated dehydroalanine in an aza-Michael addition reaction is proposed to yield the β-amino acid residue 2,3-diaminopropionate.
Examples of β-amino acids being added as late-stage tailoring reactions in NRPs are also known, as in the biosynthesis of the anti-tuberculosis antibiotics viomycin (Fig. 7A) and capreomycins. Here, a β-lysyl-carrier protein intermediate is generated from L-Lys by a minimalistic NRPS module containing only A and PCP domains in concert with a lysine-2,3-aminomutase. A discrete C domain is proposed to append the β-Lys to the main cyclic NRP structure.167,168
β-Amino fatty acids are also common starter units or in situ generated intermediates of lipopeptides. The β-amine is typically generated in situ on the mega-synthetase-bound thioester intermediate. The canonical mechanism for this is exemplified in the microcystin-type fatty-acylated cyclic peptides.169 In these, a PKS-extended β-keto-ACP fatty acyl unit is converted by an embedded PLP-dependent transaminase domain to form the analogous β-aminoacyl-ACP. This β-aminoacyl-intermediate is then elongated by NRPS modules. The β-amino group may be involved in macrocyclisation to release the mature macrolactam peptide product from the synthetase, as occurs in microcystin170 and the related PKS-NRP mycosubtilin, among others.171
In RiPPs, only a few examples of β-amino acids are known, and the biosynthetic logic for their production is unrelated. The spliceotides are a wide-spread family of RiPPs with posttranslationally installed α-keto-β-amides at diverse residues within distinct classes of precursor proteins.166,172 Such ketoamides are known pharmacophores that inhibit serine and cysteine proteases naturally found in some NRPs—such as cyclotheonamides, cyclotheonellazoles,173 orbiculamide,174 jahnellamides,175 and calyxamides176—suggesting that spliceotides may serve a similar function in nature. In further support of this function, three synthetically generated spliceotide representatives exhibited selective and potent inhibitory activity against pharmaceutically relevant proteases.172 Furthermore, the α-keto-moiety is uniquely electrophilic in a protein environment, facilitating chemical derivatisation at these modified sites, exemplified by the fluorescent tagging of a modified precursor protein in the original study.166 To date, the final spliceotide natural products from native hosts remain elusive, but heterologous expression in E. coli or alternative hosts have established the activity of the rSAM superfamily member enzymes—termed “spliceases”—responsible for catalysing α-ketoamide formation in their cognate precursor substrates. Labelling studies revealed that spliceases excise all but C1 of a Tyr residue (equivalent to removal of tyramine) within an “XYG” motif. In an unusual rearrangement of the peptide backbone, a new C–C bond is formed between C1 of the former Tyr and C1 of the upstream adjacent residue. In the originally characterised type I spliceotide systems from Pleurocapsa spp. Cyanobacteria, the resulting β-amino acid (“X” from the target motif) was natively Met or Leu, but mutagenesis studies and natural precursor variants from other spliceotide pathways showed a variety of α-keto-β-amino acids can be formed.166,172 Jahnellamides also contain α-keto-β-methionine as a direct structural parallel in an NRP product.175
In a second example of posttranslational installation of a β-amino acid, a cryptic methylation of the carboxylate side chain of a conserved L-Asp by an O-methyltransferase is key for its rearrangement to L-isoaspartate in the lanthipeptide OlvA(BCSA).177 The olv pathway from Streptomyces olivaceus NRRL B-3009 was studied by heterologous expression in E. coli. The former Asp side chain becomes integrated by a rearrangement into the backbone, introducing an extra methylene to form a β-amino acid with a carboxylate side chain. Mechanistic studies support that a succinamide intermediate is formed when nitrogen of the neighbouring Gly cyclises upon the carboxyl of the methyl ester within the precursor core. Hydrolytic opening of the succinamide yields isoaspartate. It is unclear if the electrophilic succinamide group is the true enzymatic product as multiple rounds of methylation occur on the same substrate residues. Homologues of the responsible O-methyltransferase are abundant in lanthipeptide pathways in Actinobacteria, which contain precursor proteins with a conserved Asp core residue, indicating this modification is widespread.
In cinnamycin RiPP biosynthesis, the C-terminal Lys of the core peptide attacks a dehydroalanine intermediate in a β-amino acid-generating aza-Michael-addition macrocyclisation reaction.178 This general type of reaction parallels the way β-amino acids are formed in some NRPs. β-Amino acids are commonly generated by Michael addition of ammonia or an amino-acid amino group to α,β-unsaturated carboxylates that can occur on either free monomers or mega-synthetase-bound intermediates.
2.7 N-Methylamides
The methylation of backbone amide nitrogen atoms is a highly interesting structural feature for pharmaceutical applications. Backbone N-methylation enhances the proteolytic stability of peptides and proteins, it increases their membrane permeability, their target selectivity and affinity, as well as oral bioavailability.53,179,180 This modification is a characteristic property of many NRPs; it has been identified in both cyclic peptides, such as cyclosporine A (Fig. 8A),59 dictyonamide,181 enniatin A,182 bouvardin,183 PF1022A,184 metamarin,185 and linear peptides, such as pyochelin,127 hemiasterlin186 and dolastatin 10.187 The backbone of NRPs can also be methylated at different positions than the amide nitrogen, as exemplified by the C-methylation of yersiniabactin.188 | ||
| Fig. 8 N-Methylamides. Peptide natural products with backbone N-methylation produced by NRPS (A) and RiPP (B) pathways. | ||
Backbone N-methylation in NRPs is commonly introduced by NMT domains, which are integrated in A domains and share sequence similarity with class I SAM-dependent methyltransferases.189 Hence, N-methylation occurs on the activated aminoacyl adenylate substrate prior to the condensation reaction.190–193
Until recently, peptide backbone N-methylation has been an exclusive characteristic of NRPs, However, in 2017, the first posttranslationally modified α-N-methylated peptides from the mushroom Omphalotus olearius were characterised: the omphalotins. In contrast to NRPS biosynthesis, where methylation occurs before amide bond formation, in RiPP biosynthesis the N-methyl group is introduced after amide bond formation. Omphalotins contain nine (out of the twelve in total) methylated backbone amide nitrogen atoms (Fig. 8B) that are installed by an unprecedented precursor-fused methyltransferase domain that acts iteratively on the core region in an autocatalytic fashion.194 Thereby, for NRPS as well as RiPP biosynthetic pathways, SAM serves as the methyl donor for N-methylation. Interestingly, the omphalotin precursors form homodimers, where each α-N-methyltransferase acts on the C-terminal core peptide of the other precursor.194,195 Upon completion of methylation, the core peptide is cleaved off and macrocyclised by OphP to yield the final omphalotin (Fig. 8B).11,194 The nematotoxic omphalotins are the founding members of the RiPP family of borosins that have recently been expanded and divided into different classes with other fungal representatives such as the gymnopeptides,196 lentinulins and dendrothelins.83 Even more recently, the N-methylated family of peptides was expanded by a model type IV borosin RiPP system: the more distantly related “split borosins”.197 As the name indicates, split borosins are characterised by a separately encoded α-N-methyltransferase and precursor peptide, with their gene clusters predominantly identified in bacteria. In vitro biochemical characterisation of the methyltransferase SonM and precursor peptide SonA from the bacterium Shewanella oneidensis showed that SonA is composed of an N-terminal five-helix-bundle (named the borosin binding domain) that serves as the leader peptide, which is connected to the core peptide by a linker. Besides borosins, backbone N-methylation has also been found in the RiPP family of proteusins to which the polytheonamides also belong: The FkbM-like methyltransferase EreM from the pythonamide pathway installs up to six backbone N-methylations, mainly on Val residues. EreM is the first member of the FkbM family member that displays N-methyltransferase activity instead of O-methyltransferase activity.198
In in vivo co-expression experiments, EreM showed activity with a variety of core analogues.198 Likewise, for both the fused and split borosins, the N-methyltransferase showed moderate sequence flexibility for the core peptide, which is a promising rationale for engineering cleavable, N-methylated linear peptides. Initial results indicate that NRP-related cores corresponding to cyclosporine A-like and dictyonamide-like peptide sequences can be modified by the omphalotin N-methyltransferase with up to five and nine methylations, respectively.194
2.8 Ornithine residues
The nonproteinogenic amino acid ornithine and its derivatives are common structural features of bioactive NRPs. Unmodified ornithine residues are found in the glycopeptide antibiotic ramoplanin (Fig. 9A), in the lipopeptide antibiotic daptomycin,56 as well as in gramicidin S57 and tyrocidine,58 while ornithine derivatives are found in the siderophores erythrochelin, marinobactin, and pyoverdin.199 | ||
| Fig. 9 Ornithine-containing peptides can be produced nonribosomally (A) or by a PTM of ribosomally synthesised peptides (B). | ||
In NRPS biosynthesis, ornithine residues are installed by an ornithine-specific A domain. The formation of ornithine from Arg takes place before loading onto the NRPS module. Some modifications, such as N-acylation, occur when ornithine is bound to the PCP domain.20 Other modifications of ornithines can take place before loading onto the A domain. These are for example catalysed by δ-N-ornithine monooxygenase (hydroxylation) and δ-N-hydroxy-L-ornithine acetyltransferase (acetylation) during erythrochelin biosynthesis.200
Until recently, the incorporation of ornithine residues was thought to be limited to NRPSs. A ribosomal pathway could potentially be used to install tRNA-bound ornithine residues, but the tRNA aminoacyl ester is highly unstable since ornithine has the ideal chain length to form a δ-lactam ring. Likewise, this lactamisation reaction is the reason for the instability of ornithine-containing peptides and proteins: the formation of the lactam-ring can lead to spontaneous cleavage of the backbone. This is probably the reason why many ornithine residues are further modified, e.g., by hydroxylations and/or acylations, where the induced steric hinderance would prevent intramolecular lactamisation.
It has been shown that ornithine residues can be mimicked by Lys in RiPP biosynthesis; the designed NRPS analogues of brevicidine—containing three Lys residues instead of ornithines—showed similar anti-Gram-negative bacterial activity.201 Nevertheless, ornithine residues were recently discovered in the RiPP natural product landornamide A (Fig. 9B).107 In its biosynthesis, an Arg residue is posttranslationally converted to ornithine by an enzyme termed peptide arginase. Several representatives of this new enzyme family (PF12640) that modify a variety of precursor types have been identified in bioinformatics analyses and a few representatives were characterised by coexpression experiments conducted in E. coli. In general, peptide arginases seem to be promiscuous toward diverse precursor sequences, which makes them an interesting family for synthetic biology approaches.108 A distinct lineage of peptide arginases was recently identified from the biosynthetic pathway for enteropeptins, unusual sactipeptides containing an N-methylated ornithine residue and possessing self-bacteriostatic activity isolated from the gut microbe Enterococcus cecorum.202
In several RiPP products, ornithine residues have also been identified to be further modified. Similarly to NRPS products, peptide arginase-generated ornithines can also be hydroxylated and acylated in RiPP biosynthetic pathways, such as was observed in phaeornamide that contains an ornithine residue with a C10–OH fatty acid attached to the side-chain amino group (see Section 2.3 Lipopeptides).106 Likewise, the ornithines from enteropeptins are further modified by N-methylation of the side chain and thioether crosslinking to a neighbouring Cys at the backbone α-carbon.202
3 Synthetic biology approaches and considerations for emulating NRPs with RiPP biosynthetic machinery
3.1 Synthetic biology successes and challenges
As the previous sections have shown, much of the chemical space that is offered by NRPs exists in RiPPs as well. Rather, it is the combination of different modifications, such as the presence of D-amino acids, macrocycles, and lipid tails in a single product that varies between NRPs and RiPPs. Both biosynthetic pathways can be manipulated to yield products differing from the naturally produced peptide. NRPS modules have been re-engineered to produce novel peptide analogues and general strategies for engineering NRPSs (or NRPS/PKS hybrids) have been reviewed elsewhere.203–206 In brief, the specificity-conferring code of A domains can be altered to change substrate selection by high throughput methods such as yeast surface display,207,208 or complete modules, multiple domains or exchange units (XUs) can be swapped between different NRPSs to generate new peptides.209,210 Despite recent successes and advances in NRPS redesign, it remains challenging to engineer and clone a functional hybrid NRPS, and thereby produce novel NRP analogues, due to the mere size of these megasynthetases and inter-domain and inter-module protein interactions that have to remain intact for a functional NRPS. The rational design of specific NRP analogues will therefore require custom NRPS redesign.211,212 In contrast, compared to the multi-modular assembly-line production route of NRPSs, ribosomal pathways seem to be an easier target for engineering as their gene-encoded nature allows for the rapid generation of novel variants by simple mutagenesis. However, in order to create NRP-like compounds from ribosomally translated precursors, RiPP biosynthetic enzymes need to be combined from different pathways.213–215 The following sections describe the synthetic biology considerations and methods available to enable this ultimate goal of combinatorial natural product assembly for the generation of NRP mimics by RiPP biosynthetic machineries. This section thereby provides some guidelines of how several different novel NRP analogues can be biosynthesised with the biosynthetic tools described in the previous sections, some early successful examples of NRP mimicry by RiPP tools, and the challenges that lie ahead.3.1.1.1 Hybrid leader design for RiPP combinatorial biosynthesis. As PTM enzymes recognise specific structural elements or amino acid sequences in their cognate leaders, combinatorial biosynthesis of unrelated pathways requires the design of hybrid or chimeric leaders.216 Protein–protein interactions involved in PTMs are commonly mediated through a so-called RiPP recognition element (RRE) comprising a secondary structural motif of N-terminal helices and a three-stranded β-sheet.216,217 Canonically, a 5–6 amino acid stretch of the precursor leader binds in a cleft between the third helix and third β-strand of the RRE to make a four-stranded, antiparallel β-sheet. RREs can be embedded as a subdomain of the maturase enzymes or act as discrete proteins to facilitate interactions between the maturase and precursor leader.
Over the past few years, much knowledge has become available on the minimal requirements of leader recognition for several RiPP classes.104,218–221 In a key example of precursor engineering, utilising knowledge of such minimal recognition motifs, Burkhart and co-workers combined the required leader elements from two different RiPP enzyme classes to rationally design a hybrid substrate.222 By using E. coli as the heterologous expression host, they were able to introduce a thiazoline, a lanthionine, and a D-amino acid into a single core peptide. Similarly, two thiazolines and sactionines (sulphur-to-α-carbon thioether bridges) were introduced into the same core peptide using a redesigned hybrid leader, showing the versatility of this approach (Fig. 10A).
 | ||
| Fig. 10 Leader design considerations. (A) The combination of leader recognition sequences in one chimeric leader allows for modification of enzymes from different RiPP classes on one core peptide. (B) Alternatively, a sortase (SrtA) approach can be used to exchange leaders required for different modification enzymes by adding a small sequence in the C-terminal part of the leader (dark blue) for SrtA recognition. (C) Leader sequences from gene clusters that naturally display a wide variety of PTMs on the core peptide do not require chimeric leader design. (D) Besides these approaches, the leader can also be provided in trans (as a separate part not connected to the core peptide) or in cis (the modification enzyme is altered to contain the attached leader peptide). (E) Further important design considerations are biosynthetic timing and promiscuity, as the active site of the modification enzyme might only tolerate certain pre-installed core peptide PTMs. | ||
Designer leader peptides have also been created for the in vitro flexizyme-enabled biosynthesis of macrocyclic thiopeptides. Flexizyme technology223 allows the reprogramming of codons to accept unnatural amino acids in vitro using aptamers to charge tRNAs. In this study, flexizyme-incorporated unnatural Se-phenylselenocysteine residues were chemically converted to dehydroalanine as an alternative to lanthionine dehydratases.224 The dehydroalanines act as substrates for macrocyclisation by the pyridine synthase TclM from thiocillin biosynthesis.225 Thiazoles were incorporated by the cumulative action of the cyclodehydratase LynD from aesturamide biosynthesis and the leader-independent azoline-oxidase TbtE from thiomuracin biosynthesis.224 In this hybrid chemical and biocatalysis approach, the leader recognition sequences from the unrelated TclM and LynD RiPP enzymes were combined in a chimeric leader to facilitate the generation of thiocillin-type thiopeptide scaffolds.
As an alternative approach to hybrid leader design, Franz and Koehnke exchanged full-length leader peptides for a shared core by a sortase A-mediated transpeptidation (Fig. 10B).226 As a proof of principle for the method, the in vitro maturation of the core with enzymes from unrelated pathways by both the cyclodehydratase LynD and the ω-ester-bond installing heterocyclase MdnC from microviridin J biosynthesis was realised.226 This approach could potentially be useful for combinatorial biosynthesis using RiPP enzymes that have strict leader peptide requirements or are not compatible with hybrid leader design, as is the case for the fused borosins, where the backbone N-methylating enzyme OphMA comprises the enzyme and the substrate as parts of a single polypeptide chain (see Section 2.7 N-Methylamides).194 Notably, all of the aforementioned chimeric and hybrid leader design and leader swapping examples used natural core sequences as substrates for the PTM enzymes to date.
Non-native core peptides have also been tested as substrates for various RiPP maturases to elucidate the biosynthetic potential of RiPP maturases.194,227,228 Often these studies include peptide libraries based on the natural core peptide yielding peptides still resembling native core sequences (with multiple variations).42,229,230 For example, testing the rSAM peptide epimerase OspD in a library approach revealed that OspD is an extraordinarily promiscuous maturase that does not require specific amino acid moieties or positions, and the epimerisation pattern is dictated by the peptide sequence.42 However, until recently, few attempts have been made to use RiPP enzymes to introduce modifications into designer peptides that are completely unrelated to native core sequences. Evidence for the combinatorial use of different RiPP enzymes on such non-native substrates is even more sparse. Indeed, the design of a hybrid leader is not required in some cases, as the RiPP modification enzymes required already stem from pathways with PTM enzymes that act on the same precursor leader type and have promiscuity for diverse core sequences (Fig. 10C).44,46,231 The application of such a pathway for NRP-mimicking is exemplified by landornamide. The wild-type precursor protein OspA is natively modified by three enzymes: peptide epimerase OspD, lanthionine synthetase OspM, and peptide arginase OspR to introduce D-amino acids, lanthionine bridges, and ornithines, respectively.107 Modifications by OspR and OspD were combined on several NRP-mimicking substrates fused to the native OspA precursor leader, successfully yielding a brevicidine-mimetic with both ornithines and a D-amino acid.108
3.1.1.2 Engineered enzymes providing leader peptides. Despite the importance of the leader peptide for activity of many RiPP enzymes, the leader is dispensable in some cases. Leader-independent activity with direct recognition of the core peptide for modification, has also been shown.232–235 Furthermore, successful examples of the leader being provided in an alternative fashion also exist, such as in trans as a separate protein or as a fusion protein to the PTM enzyme (Fig. 10D). However, leader-in trans and leader-independent activity, when possible, tends to be less efficient toward the native substrates and has mostly been demonstrated toward natural core substrate sequences in vitro, where protein concentrations are artificially high compared to natural production by the host.
For example, the LynD heterocyclase converts multiple Cys residues to thiazolines within a peptide substrate with broad promiscuity to the Cys-flanking residues, making it an interesting enzyme to install thiazolines within non-native substrates. These heterocycles are a recurring motif in bioactive secondary metabolites (see Section 2.4 Heterocyclisations), including some important NRPs, such as bleomycin and bacitracin. Thiazoline-installing enzymes such as LynD are potentially interesting to mimic these NRPs. LynD can be activated by providing the leader peptide in trans or by an engineered LynD enzyme with a leader fusion, thereby eliminating the requirement for designing chimeric leader peptides.236 As an added benefit, free-peptide substrates do not require proteolytic cleavage of the leader and may facilitate combinatorial biosynthesis. Similarly engineered leader-fusions were created for the LctM lantibiotic synthetase from lacticin 481 antibiotic biosynthetic pathway234 and the graspetide ATP-grasp ligases.233,237
3.1.1.3 Leader-independent PTM enzyme activity. In contrast to the leader-dependent enzymes mentioned above, many RiPP tailoring enzymes do not require a leader peptide at all, although they most often do recognise a certain motif or a previously installed modification in the core peptide. For example, the oxidation of thiazolines and oxazolines to thiazoles and oxazoles, respectively, can be performed by the azoline-oxidase TbtE from thiomuracin biosynthesis in a leader-independent manner.238 The generation of these heterocycles can thus be achieved by coupling the cognate leader peptide of heterocyclase LynD to the NRP-mimicking core peptide, which might be matured sequentially by LynD and TbtE to install thiazoles and oxazoles without the need of leader engineering.224 Leader-independent in vitro activity has also been observed for a variety of other RiPP enzymes. This includes common NRP modifications such as D-amino acids, side-chain crosslinks, methylations, and lipidations.45,48,239–242 Although in vitro leader-independent activity eliminates the need of designing a hybrid leader, in vivo modification might require such, where low-expressed peptide cores may be rapidly degraded or below the relevant concentration for efficient protein–protein recognition needed for modification. Indeed, certain RiPP modifications are naturally leader independent, requiring the proteolytic removal of the leader to install their modifications. This is the case for N-terminal peptide modifications that utilise a free N-terminal amine as substrate, such as methylation by the CypM methyltransferase in cypemycin biosynthesis240 and for the attachment of the dimethylguanidino fatty acid in microvionin biosynthesis.104,105 As another example, prenyltransferases from cyanobactin RiPPs act on the released cyclic peptide following leader removal and macrocyclisation of the core peptide. The cyclases as well as the prenyltransferases from various cyanobactin pathways and the CypM and MonM methyltransferases show high promiscuity toward alternative core sequences and are amenable to peptide engineering.82,243 Besides, two recently published studies have shown that RiPP enzymes can be redirected to proteins and install heterocyclisation and β-amino acid formation in protein backbones.244,245
Furthermore, for some of these tailoring maturases, the enzyme might require the instalment of a previous PTM in order to introduce the next one, where the leader might be necessary for the initial modification (Fig. 10E). For example, in duramycin biosynthesis, a lysoalanine-crosslink is formed by DurN242 that distantly resembles the crosslink found in the nonribosomal antibiotic bacitracin. DurN does not require a leader peptide for its activity, but uses a substrate-assisted mechanism that involves a hydroxylated Asp residue that is previously installed by a different duramycin biosynthetic enzyme.242 Similarly, for the incorporation of D-amino acids by the hydrogenase LtnJ, an α,β-dehydrated Ser or Thr residue is required that can be installed by the leader-dependent NisBC machinery.246 In landornamide biosynthesis, the activity of the arginase OspR, the rSAM epimerase OspD, and the lanthionine synthetase OspM is coordinated to yield a product containing two ornithines, two D-amino acids, and two lanthionine rings. Investigations on the progression of biosynthetic events of these enzymes on the native core peptide indicated that OspD and OspM are co-dependent: neither enzyme catalysed complete conversion of the core peptide when the activity of the other enzyme was absent.107 In addition, instead of a leader peptide, modification enzymes might require a certain “recognition” motif in the core peptide or structural feature such as a macrocycle to install its modification.166,239
Two of the most promiscuous RiPP enzymes described to date are macrocyclases: for example, the macrocyclases from cyanobactins, such as PatG and PagG,82,228,249,250 and the macrocyclases from some lanthipeptides pathways, such as ProcM and SyncM.251,252 The ProcM lanthionine synthetase naturally accepts 29 different substrate precursor proteins encoded in the host genome to generate a mixture of RiPP products collectively known as prochlorosins (or cyanotins as the collective name for products from similar pathways).251 Although lanthionine bridges are not present in any NRP described to date, lanthionine ring structures might provide similar proteolytic stability to those observed for lactones, lactams and other common NRP macrocycles.201 Head-to-tail cyclisation, such as the reaction catalysed by cyanobactin macrocyclases, is a more commonly observed modification in NRPs (see Section 2.2 Macrocycles) and is present in potent antibiotics such as gramicidin S and tyrocidines.228 The ProcM lanthionine synthetases and cyanobactin macrocyclases both have an exceptionally high tolerance for variable core sequences that have been applied to generate libraries of non-native products, including macrocycles with D-amino acids and other nonproteinogenic amino acids.229,250 Furthermore, core substrate requirements are minimal for these cyclising enzymes: lanthionine generation by ProcM requires only a Cys and a Ser/Thr to form the lanthionine bridge in the core peptide, and in cyanobactin biosynthesis, a conserved C-terminal recognition element flanking the core peptide, which is excised upon N-to-C cyclisation, and an azoline (heterocycle) or Pro at the N-terminus of the released core are all that is required.82,227 Similarly, macrocyclisation by the RiPP enzymes involved in lactazole biosynthesis, as well as macrolactamisation in lasso peptides, were shown to be flexible enzymes for heavily mutated native substrates. However, compared to ProcM and PagG, these enzymes call for more specific core motifs and thus, might be less amenable for NRP-mimicry.218,253,254 Nonetheless, since macrocycles are a commonly observed modification in NRPs, the broad substrate specificity of these enzymes show promise to generate NRP-like products.
Further interesting promiscuous enzymes for NRP mimicry are for example D-amino acid-installing enzymes, such as LtnJ and OspD. LtnJ-type hydrogenases from lanthipeptide-type pathways convert dehydrated Ser or Thr residues to D-Ala or D-ethylglycine, respectively. The rSAM peptide epimerase OspD from the proteusin landornamide pathway has been shown to install D-amino acids in highly diverse unnatural core sequences at many different proteinogenic residues and in different patterns (see Section 2.1 D-Amino acids).42 Core-swap experiments using chimeric precursors as substrates for various OspD-like proteusin epimerases suggested that core elements rather than epimerase identity largely direct the regiospecificity of modifications to control the pattern of installed D-amino acids.43 However, the core peptide elements that direct the site(s) of epimerisation are not yet defined and are therefore difficult to predict at this time.42
Thus, an important challenge is how to guide the PTM enzymes towards modifying the desired amino acid, as the substitution of certain amino acids can alter the pattern and the extent of modification.45,255–257 For example, the NRPS product cyclosporine A has been closely mimicked by replacing the fused borosin OphMA core peptide with a cyclosporine A-resembling one. Subsequent backbone N-methylation by OphMA and macrocyclisation by OphP yielded products with methylation patterns that did not fully correspond to the pattern of the cyclosporine A natural product.194 Although the NRP has structurally been closely mimicked, in this case, the position of the methyl groups of cyclosporine A is very likely to be important for bioactivity. Nonetheless, this study shows the possibility of using OphMA for introducing N-methyl groups in non-native substrates. In addition, the cyanobactin prenyltransferases have been shown to act on a variety of linear and macrocyclic peptides, together with a high regio- and stereospecificity. The broad substrate selectivity includes hydroxyl and phenolic forward and reverse O-prenylation, as well as C- and N-termini, Arg and Trp prenylation, thereby highlighting this broad RiPP toolset for peptide alkylation.111,113–116,258–260 Interestingly, the prenyltransferase PagF was also shown to prenylate a lanthipeptide, highlighting its broad substrate specificity and potential use for the generation of NRP lipopeptide mimics or to boost the bioactivity or pharmacokinetics of peptides.110,117,261
Besides natural promiscuity of many RiPP enzymes, certain limitations of their substrate selectivity can be circumvented by the search for novel analogues and by engineering the enzyme itself.82 For example, although the cyanobacterial prenyltransferases can function on a diverse range of acceptor substrates, they show strict specificity for the prenyl donor. However, this prenyl donor specificity was altered by changing a single amino acid in the isoprene-binding pocket of the PagF enzyme, highlighting the ability to further engineer RiPP enzymes by site-directed mutagenesis to increase donor promiscuity.117 Similarly, adapted substrate specificity for the NisB dehydratase was obtained.262
Altogether, it is possible to engineer RiPP combinatorial pathways that act on NRP-mimicking core substrates. This however requires the careful consideration and design of hybrid leaders, engineered RiPP enzymes, biosynthetic timing, and installation of other substrate requirements. Another unique challenge herein is to maintain efficient conversion of the non-native core by several RiPP enzymes to obtain sufficient production of the multiple-modified product. Also, specific selection methods must be put into place to select products of interest from the heterologous product mixtures that are generated. When applied and designed successfully, RiPP combinatorial pathways can generate libraries of novel NRP-like products. The production, screening and selection methods of these libraries will be addressed in the following section.
3.2 Generation and screening of combinatorial RiPP libraries for the production of bioactive NRP-mimics
The broad variety of peptides that can be synthesised by combinatorial biosynthesis call for elaborate screening and production methods to accelerate the pipeline towards bioactive NRP-mimicking peptides. Although the current body of literature regarding the generation of NRP-like compounds specifically via RiPP biosynthetic enzymes is sparse, in this section we will summarise different methods for generating and screening peptide libraries modified by RiPP enzymes. The production methods for these combinatorial biosynthetic pathways each possess various advantages and drawbacks, which will be addressed to highlight the considerations involved in going towards the generation of bioactive NRP-mimicking RiPPs.An important factor for the successful engineering of NRP mimics is the production method. Although heterologous expression and native expression of a variety of RiPP enzymes in vivo is well-established,263–267 the in vitro generation of NRP-mimicking substance libraries by combinatorial biosynthesis offers several advantages over in vivo systems. First, as the core peptide is often not completely modified in vivo, combinatorial biosynthesis with an NRP-mimicking substrate will generate a heterologous mix with the desired peptide produced only at low amounts. In vitro systems are advantageous as they allow more control over reaction conditions. For example, monitoring reaction times, optimising substrate-to-enzyme ratios, and exerting temporal control (e.g., the stepwise addition of enzymes) can reduce the amount of non-modified or partially modified peptide and mitigate the product mixtures caused by competition of multiple enzymes for the same functional group.238 Second, a cell-free production framework can avoid issues encountered in in vivo heterologous expression systems, such as product toxicity, poor gene expression, pathway enzyme inactivity, or other host incompatibilities. Even more, noncanonical amino acids present in many NRPs—including D-amino acids, β-amino acids and N-methylated amino acids (see Chapter 2)—have been introduced in vitro using methods that do not rely on PTMs but rather on incorporation of the noncanonical amino acid by engineered ribosomes that charge tRNAs with noncanonical amino acid monomers.268,269 One could imagine using these technologies to generate modified precursor proteins that are further combined with RiPP modifying enzymes in vitro to generate hybrid modified peptides. One advantage of in vivo production approaches is that any biosynthetic enzyme, including difficult-to-purify enzymes, can be utilised to install PTMs. Moreover, the most noteworthy advantage of using native or heterologous hosts is that it allows for large-scale production of RiPP analogues and the possibility for direct bioactivity screening as compared to in vitro methods.
Flexizyme-assisted biosynthesis is another in vitro approach that has been applied to study a variety of RiPP enzymes.101,224,235,253,274 Flexizymes (flexible tRNA acylation ribozymes) are aptamers developed to condense a wide array of amino acid esters with tRNAs of choice, thereby eliminating the requirement of structural similarity to the canonically loaded amino acid as is needed for amino acyl-tRNA synthetase-based loading. Flexizymes thus facilitate reprogramming of the genetic code to express peptides containing various nonproteinogenic amino acids.223 This in vitro production platform has been exploited for the incorporation of multiple, noncanonical amino acids within a thiopeptide scaffold, which would be highly challenging using solely RiPP combinatorial biosynthesis.253 In addition, chemical handles, such as the noncanonical amino acid Se-phenylselenocysteine (SecPh) can be incorporated, which allow for the introduction of dehydroalanines by oxidative elimination with H2O2, thereby circumventing the use of a lanthipeptide dehydratase and consequently reducing the number of RiPP enzymes in a combinatorial pathway to allow for a less heterologous mix of products.224 Though this approach can therefore in theory generate a plenitude of NRP-mimics, and is not interfered by proteolytic stability issues (as one might observe with in vivo systems and cell-free expression systems), it requires the successful purification of the RiPP enzymes of interest and generates low production titres relative to other technologies. Low production titres can hereby pose a significant challenge, as it complicates the characterisation of the heterologous product mixture and requires sufficient knowledge on the promiscuity and efficiency of the RiPP enzymes used to be able to shift the reactions to a more homologous product.
Combinatorial pathways from in vitro libraries have also been integrated with high-throughput screening, such as by mRNA display.275–277 mRNA display is a peptide display technology in which peptides are associated to their own mRNA during in vitro ribosomal translation via a puromycin linker. By panning this mRNA-peptide fusion library after reverse transcription of the mRNA tag against the target of interest, the peptide of interest can be isolated by several means of selection (Fig. 11A), after which sequence information of the peptide is obtained by sequencing of the mRNA tag.278 As this technique yields a linear peptide, modifications can be introduced either during in vitro ribosomal translation via the previously mentioned FIT system (flexible in vitro translation facilitated by flexizymes) or via PTMs. For the latter, mRNA display has shown to be compatible with the lactazole and pantocin RiPP machineries.276,277 The mRNA display method was hereby used as a biochemical tool to study substrate recognition by the RiPP enzyme PaaA, an enzyme that catalyses the double dehydration/decarboxylation of two Glu residues in its peptide substrate PaaP to form a fused-bicyclic core. The modification of Glu allowed for the selection of modified from unmodified substrate analogues by a degradation step with the Glu-specific GluC protease and subsequent enrichment by a streptavidin pulldown assay. A broader applicability for the technique with other RiPP enzymes looks promising. Recent advances in the combination of incorporating multiple noncanonical amino acids through the FIT-system with mRNA display (referred to as the RaPID system for ![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif)
![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) ndom nonstandard
ndom nonstandard ![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) eptides
eptides ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) ntegrated
ntegrated ![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif) iscovery) theoretically already allow for mimicking of natural products, though screening for bioactivity is still a barrier to be overcome.276,277,279
iscovery) theoretically already allow for mimicking of natural products, though screening for bioactivity is still a barrier to be overcome.276,277,279
 | ||
| Fig. 11 Selected in vitro vs. in vivo RiPP library screening methods. (A) mRNA display method for studying the modification of a pantocin RiPP library by PaaA. The pantocin RiPP library is generated by N-terminal biotinylation (in green) and the addition of a C-terminal puromycin linker (in red) that is attached to the mRNA of the peptide. Glu residues are indicated in orange and are modified by PaaA. Mutations highlighted in dark blue do not result in modification of the Glu residues. Subsequent GluC treatment selects modified from unmodified peptides. Next, modified peptides can be pulled down by a streptavidin assay, after which the mRNA tag allows for sequencing of the modified peptides. (B) Schematic depiction of the in vivo split-intein system for the detection of RiPP peptides binding to a single target protein. The system consists of a split σ factor that is attached to a split intein from Nostoc punctiforme PCC 73102 (Npu) and the target protein of interest (from N- to C-terminus) that is complemented by the RiPP peptide attached to the other half of the split intein and σ factor. Upon interaction between the target protein and the RiPP peptide, the split intein splices together the σ factor that leads to downstream transcription of a fluorescent reporter. | ||
Together, in vitro synthesis approaches are highly compatible for screening a variety of RiPP peptides bearing several noncanonical amino acids, given that RiPP enzyme(s) can be rendered active in these cell-free systems and an appropriate selection method is in place to exclude unmodified variants from the RiPP library. Interesting tailoring reactions catalysed by oxygen-sensitive rSAM enzymes might herein not yet be accessible for cell-free platforms or will require special reaction conditions in vitro in order to pave the way for combinatorial biosynthesis.280 Next to making use of the RiPP biosynthetic enzymes at hand, the in vitro synthesis methods described can be combined with synthetic chemistry through hybrid chemical reactions to further tailor the final NRP-emulating product.
Besides antimicrobial discovery screening assays—phage, yeast, and bacterial display techniques also have been adapted to allow for screening of large RiPP libraries—mainly using lanthipeptide biosynthetic enzymes (LctM, HalM, ProcM, NisBC).283–285 These screening methods were largely successful due to the high conversion rates of alternative peptide sequences to cyclic structures and the likely lack of activity for unmodified linear lanthipeptides. Aside from cell surface display techniques of modified peptides, successful intracellular lanthipeptide libraries have been generated as well. Such an intracellular lanthipeptide library was generated using the previously described promiscuous ProcM enzyme in E. coli, thereby identifying a bicyclic lanthipeptide that was able to successfully disrupt the protein–protein interaction crucial for HIV budding from infected cells. This lanthipeptide, displaying novel biological activity, was selected by coupling the lanthipeptide library to a bacterial reverse two-hybrid system, where inhibition of the viral budding particle protein–protein interaction caused reporter genes for cell survival to be expressed.286 Recently another in vivo peptide selection strategy was developed to identify RiPPs that bind to a single target of interest, in this case the SARS-CoV-2 spike receptor binding domain. In this study, a precursor peptide library was fused to half of a split intein and split σ factor, with the target of interest fused to the other half of the split intein and σ factor. Upon binding of the modified precursor peptide to the target of interest, the fusion of the split inteins releases the σ factor that turns on a promoter for a selectable marker (Fig. 11B).287 In contrast to most other reporter RiPP peptide libraries that are based on lanthipeptide biosynthesis, the cyclic peptide library herein is generated through rSAM mediated biosynthesis from the freyrasin family,288 in which a thioether macrocycle is formed between a Cys and Asp or Glu. This highlights the possibility of using rSAM enzymes for the generation of in vivo RiPP libraries, which can—as described in the previous sections—mimic many different NRP features.
In conclusion, several library generation and screening techniques have been developed for different classes of RiPPs, for peptides generated by both in vivo and in vitro techniques. Recent advances in cell-free protein synthesis and flexible in vitro translation (FIT)-systems have significantly expanded the construction of vast peptide libraries, as they allow for the incorporation of several noncanonical amino acids and, depending on the desirable bioactivity of the final peptide product, coupling to high-throughput assays for screening of new drug leads. Although most of these techniques have been developed using a single RiPP class, i.e., lanthipeptides, they are in theory amenable to combinatorial biosynthesis, thereby taken into consideration that the tags for mRNA, phage and yeast display that are often necessary for selection are compatible with the promiscuity of the enzyme and leader peptide design. New interesting lead-peptides can subsequently be synthesised in vivo by combinatorial biosynthesis.
4 Conclusion
In this review, we highlighted the PTMs catalysed by RiPP biosynthetic enzymes that mimic structural features of NRPs—a class of natural products known for their diverse structures and potent bioactivities, which have been exploited for human use as drugs. Citing a few successful examples, we further discuss the potential of using diverse sets of RiPP enzymes in a combinatorial manner to engineer NRP-like peptides. While significant advances have been made in re-engineering NRPS modules to produce novel NRP analogues (and chemical synthesis and chemoenzymatic methods), the ribosomal approach to peptide engineering offers several advantages, such as the facile generation of large peptide libraries and the coupling of peptide generation with cell-based screening. Aided by a significant increase of biochemical RiPP biosynthetic enzyme understanding, the biotechnological tools are in place to achieve the goal of generating novel peptide natural products at will, although many challenges lie ahead.As reviewed here, mimicking NRPs by RiPP biosynthesis enzymes requires a sufficient promiscuity of RiPP enzymes to install modifications in the NRP-mimicking core template, correct biosynthetic timing, optional leader design, choice of in vitro or in vivo expression and coupling synthesis to efficient screening methods. As the biochemical mechanisms and astounding biosynthetic plasticity of several RiPP pathways are being deciphered, the opportunities and limitations for the creation of rationally engineered products start to be discerned.260,289 Though vast libraries of NRP-mimicking peptides can be generated, the promiscuity of RiPP enzymes involved remains a bottleneck and is not limitless. RiPP enzymes from many different classes are known to disfavour certain amino acids in the core peptide such as negatively charged Asp and Glu residues in lactazole and PagG macrocycle biosynthesis, or do not accept nonproteinogenic amino acids, such as D-amino acids for OphMA.82,253 Moreover, despite their promiscuity, RiPP enzymes might require certain motifs, preformed secondary structures or scaffolds in their core peptide that are indispensable or highly contribute to efficient modification, thereby limiting the NRP-mimicking variants that could be synthesised in practice.166,253,290–292 Recently, a number of studies have indeed indicated at least partial recognition of the RiPP enzyme resides on the core peptide. For example, highly specific substrate recognition of the side chain-to-side chain macrolactam- and macrolactone-catalysing graspetide macrocyclase PsnB for its core substrate was shown.292 Another example is the leader-independent installation of α-keto-β-amino acids in protein backbones; the rSAM splicease PlpXY recognises an 11-residue splice tag from the natural core peptide in diverse proteins.245 Mechanistic studies on ProcM also suggest that its extreme substrate tolerance is a result of the substrate sequence, rather than the enzyme determining the outcome of the cyclisation process.251,256,257 Indeed, some enzymatic limitations can be overcome by engineering of individual RiPP enzymes or by genome mining efforts in search for more promiscuous variants. Another possible bottleneck for NRP-mimicking is that in novel combinatorial biosynthesis, a certain desired order or combination of modifications might still not be possible even after optimisation of the novel biosynthetic pathway, as in certain RiPP pathways RiPP enzymes are proposed to communicate with each other via the substrate or require a certain cooperativity, and interactions between installed PTMs influence the assembly process considerably.242,293,294
This brings us to another pending question concerning the degree of flexibility of NRP scaffolds themselves to exert their biological action. When using combinatorial biosynthesis, the chance is high that the NRP “scaffold” will need to be adapted in order to ensure modification by two different enzymes. Even though the aim is not to mirror the NRP but to structurally mimic it, will structural mimicking of the NRP be sufficient to create novel drug leads? Structure-activity relationship (SAR) studies have, for example, shown that replacing the thiazolidine ring in the novel anti-MRSA peptide lugdunin with several similar structures diminished the activity of the peptide, while the activity was retained for its enantiomer.295 Further important questions to create NRP analogues are whether all the biosynthetic tools are at hand to mimic the NRP pharmacophore, in addition to whether these RiPP enzymes are compatible with other biosynthetic enzymes (when necessary) and the availability of appropriate screening methods. Moreover, are NRP analogues possibly more readily attainable through chemo-enzymatic methods? The choice of the NRP to mimic and knowledge from SAR studies on essential motifs for bioactivities will therefore have to be carefully considered for the successful rational design of these new-to-nature peptides. Notably, the RaPID system that integrates genetic code reprogramming by flexizymes with mRNA display technology is advancing rapidly. The specific introduction of several noncanonical amino acids and ring structures with or without the addition of RiPP enzymes that structurally mimic NRPs, is already possible.279 If sufficient amounts of peptides and a suitable method to screen for bioactive peptides are available, this in vitro chemo-enzymatic technology is another viable strategy for the generation of novel NRP-emulating drug leads.
Despite these challenges, some newly discovered RiPP enzymes are highly interesting to create NRP mimics, as their modifications are challenging to install by synthetic approaches, or their modifications are essential for the bioactivity of certain NRPs. Examples are the hybrid PKS-RiPPs lipolanthine and goadvionin enzymes to create lipopeptides (essential for many NRP lipopeptide antibiotics), glycosyltransferases to create glycosylated peptides that are abundant in NRPS products together with the bicyclic structures in cittilins resembling glycopeptide antibiotics vancomycin and teicoplanin, as well as backbone N-methylation to improve pharmacological properties of the peptide.103,105,150,296–298 However, the biotechnological applicability of many of these enzymes for NRP-mimicking mostly remains unexplored to date, as the enzymes have not yet been characterised in detail.
All in all, the generation of novel NRP-mimics by RiPP enzymes is still in early development, although a few proofs-of-principle for the partial structural mimicking of brevicidine, daptomycin, gramicidin S, and cyclosporine A have already been demonstrated.108,194,201 Coupling these early NRP engineering efforts to combinatorial biosynthesis and library screening will potentially yield many interesting novel lead drug scaffolds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. was supported by a Feodor-Lynen fellowship of the Alexander-von-Humboldt foundation and A. L. V.'s research is supported by a Novartis FreeNovation Grant (FN20-0000000194). In addition, this work was partly funded by a grant to F. R. and O. P. K. by the Dutch Research Council (NWO), grant no. ALWOP.415.References
- C. T. Walsh, Science, 2004, 303, 1805–1810 CrossRef CAS PubMed.
- J. Grünewald and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2006, 70, 121 CrossRef.
- C. T. Walsh, H. Chen, T. A. Keating, B. K. Hubbard, H. C. Losey, L. Luo, C. G. Marshall, D. A. Miller and H. M. Patel, Curr. Opin. Chem. Biol., 2001, 5, 525–534 CrossRef CAS.
- M. Inoue, N. Shinohara, S. Tanabe, T. Takahashi, K. Okura, H. Itoh, Y. Mizoguchi, M. Iida, N. Lee and S. Matsuoka, Nat. Chem, 2010, 2, 280–285 CrossRef CAS.
- L. C. Wieland Brown, M. G. Acker, J. Clardy, C. T. Walsh and M. A. Fischbach, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2549–2553 CrossRef CAS PubMed.
- W. L. Kelly, L. Pan and C. Li, J. Am. Chem. Soc., 2009, 131, 4327–4334 CrossRef CAS PubMed.
- R. P. Morris, J. A. Leeds, H. U. Naegeli, L. Oberer, K. Memmert, E. Weber, M. J. LaMarche, C. N. Parker, N. Burrer, S. Esterow, A. E. Hein, E. K. Schmitt and P. Krastel, J. Am. Chem. Soc., 2009, 131, 5946–5955 CrossRef CAS.
- M. F. Freeman, C. Gurgui, M. J. Helf, B. I. Morinaka, A. R. Uria, N. J. Oldham, H.-G. Sahl, S. Matsunaga and J. Piel, Science, 2012, 338, 387–390 CrossRef CAS.
- C. M. Ireland, A. R. Durso, R. A. Newman and M. P. Hacker, J. Org. Chem., 1982, 47, 1807–1811 CrossRef CAS.
- E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315–7320 CrossRef CAS.
- S. Ramm, B. Krawczyk, A. Mühlenweg, A. Poch, E. Mösker and R. D. Süssmuth, Angew. Chem., Int. Ed., 2017, 56, 9994–9997 CrossRef CAS.
- S. C. Kessler, X. Zhang, M. C. McDonald, C. L. M. Gilchrist, Z. Lin, A. Rightmyer, P. S. Solomon, B. G. Turgeon and Y.-H. Chooi, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 24243–24250 CrossRef CAS PubMed.
- W. Gevers, H. Kleinkauf and F. Lipmann, Proc. Natl. Acad. Sci. U. S. A., 1969, 63, 1335–1342 CrossRef CAS PubMed.
- T. Montavon and S. Bruner, Compr. Nat. Prod. II Chem. Biol., 2010, 5, 619–655 Search PubMed.
- B. K. Scholz-Schroeder, J. D. Soule and D. C. Gross, Mol. Plant. Microbe Interact., 2003, 16, 271–280 CrossRef CAS PubMed.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef.
- R. E. Wirawan, K. M. Swanson, T. Kleffmann, R. W. Jack and J. R. Tagg, Microbiology, 2007, 153, 1619–1630 CrossRef CAS.
- H. Mathur, D. Field, M. Upton and P. D. Cotter, Front. Microbiol., 2021, 12, 695081 CrossRef PubMed.
- D. Sardar, Z. Lin and E. W. Schmidt, Chem. Biol., 2015, 22, 907–916 CrossRef CAS PubMed.
- H. L. Condurso and S. D. Bruner, Nat. Prod. Rep., 2012, 29, 1099–1110 RSC.
- C. T. Walsh, Nat. Prod. Rep., 2016, 33, 127–135 RSC.
- M. McErlean, J. Overbay and S. Van Lanen, J. Ind. Microbiol. Biotechnol., 2019, 46, 493–513 CrossRef CAS PubMed.
- M. Dell, K. L. Dunbar and C. Hertweck, Nat. Prod. Rep., 2022, 39, 453–459 RSC.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2012, 30, 108–160 RSC.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- S. L. Clugston, S. A. Sieber, M. A. Marahiel and C. T. Walsh, Biochemistry, 2003, 42, 12095–12104 CrossRef CAS PubMed.
- C. Rausch, I. Hoof, T. Weber, W. Wohlleben and D. H. Huson, BMC Evol. Biol., 2007, 7, 78 CrossRef PubMed.
- M. J. Wheadon and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2026017118 CrossRef CAS PubMed.
- K. D. Patel, F. B. d’Andrea, N. M. Gaudelli, A. R. Buller, C. A. Townsend and A. M. Gulick, Nat. Commun., 2019, 10, 3868 CrossRef.
- T. A. Ronnebaum and A. L. Lamb, Curr. Opin. Struct. Biol., 2018, 53, 1–11 CrossRef CAS PubMed.
- K. Hoffmann, E. Schneider-Scherzer, H. Kleinkauf and R. Zocher, J. Biol. Chem., 1994, 269, 12710–12714 CrossRef CAS PubMed.
- G. Shi, N. Shi, Y. Li, W. Chen, J. Deng, C. Liu, J. Zhu, H. Wang and Y. Shen, ACS Chem. Biol., 2016, 11, 876–881 CrossRef CAS PubMed.
- P. C. Montecucchi, R. de Castiglione, S. Piani, L. Gozzini and V. Erspamer, Int. J. Pept. Protein Res, 1981, 17, 275–283 CrossRef CAS PubMed.
- M. Kuwada, T. Teramoto, K. Y. Kumagaye, K. Nakajima, T. Watanabe, T. Kawai, Y. Kawakami, T. Niidome, K. Sawada and Y. Nishizawa, Mol. Pharmacol., 1994, 46, 587–593 CAS.
- O. Buczek, D. Yoshikami, M. Watkins, G. Bulaj, E. C. Jimenez and B. M. Olivera, FEBS J., 2005, 272, 4178–4188 CrossRef CAS PubMed.
- O. Buczek, D. Yoshikami, G. Bulaj, E. C. Jimenez and B. M. Olivera, J. Biol. Chem., 2005, 280, 4247–4253 CrossRef CAS PubMed.
- C. Ollivaux, D. Soyez and J.-Y. Toullec, J. Pept. Sci., 2014, 20, 595–612 CrossRef CAS PubMed.
- P. D. Cotter, P. M. O’Connor, L. A. Draper, E. M. Lawton, L. H. Deegan, C. Hill and R. P. Ross, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18584–18589 CrossRef CAS PubMed.
- C. T. Lohans, J. L. Li and J. C. Vederas, J. Am. Chem. Soc., 2014, 136, 13150–13153 CrossRef CAS PubMed.
- M. Xu, F. Zhang, Z. Cheng, G. Bashiri, J. Wang, J. Hong, Y. Wang, L. Xu, X. Chen, S.-X. Huang, S. Lin, Z. Deng and M. Tao, Angew. Chem., Int. Ed., 2020, 59, 18029–18035 CrossRef CAS.
- B. I. Morinaka, A. L. Vagstad, M. J. Helf, M. Gugger, C. Kegler, M. F. Freeman, H. B. Bode and J. Piel, Angew. Chem., Int. Ed., 2014, 53, 8503–8507 CrossRef CAS.
- M. Korneli, S. W. Fuchs, K. Felder, C. Ernst, L. V. Zinsli and J. Piel, ACS Synth. Biol., 2021, 10, 236–242 CrossRef CAS PubMed.
- B. I. Morinaka, M. Verest, M. F. Freeman, M. Gugger and J. Piel, Angew. Chem., Int. Ed., 2017, 56, 762–766 CrossRef CAS PubMed.
- A. Bhushan, P. J. Egli, E. E. Peters, M. F. Freeman and J. Piel, Nat. Chem., 2019, 11, 931–939 CrossRef CAS PubMed.
- A. Parent, A. Benjdia, A. Guillot, X. Kubiak, C. Balty, B. Lefranc, J. Leprince and O. Berteau, J. Am. Chem. Soc., 2018, 140, 2469–2477 CrossRef CAS PubMed.
- M. F. Freeman, M. J. Helf, A. Bhushan, B. I. Morinaka and J. Piel, Nat. Chem., 2017, 9, 387–395 CrossRef CAS PubMed.
- A. Benjdia, A. Guillot, P. Ruffié, J. Leprince and O. Berteau, Nat. Chem., 2017, 9, 698–707 CrossRef CAS PubMed.
- A. L. Vagstad, T. Kuranaga, S. Püntener, V. R. Pattabiraman, J. W. Bode and J. Piel, Angew. Chem., Int. Ed., 2019, 58, 2246–2250 CrossRef CAS PubMed.
- A. Sikandar, L. Franz, S. Adam, J. Santos-Aberturas, L. Horbal, A. Luzhetskyy, A. W. Truman, O. V. Kalinina and J. Koehnke, Nat. Chem. Biol., 2020, 16, 1013–1018 CrossRef CAS PubMed.
- Z. Feng, Y. Ogasawara and T. Dairi, Chem. Sci., 2021, 12, 2567–2574 RSC.
- Z. Shang, J. M. Winter, C. A. Kauffman, I. Yang and W. Fenical, ACS Chem. Biol., 2019, 14, 415–425 CrossRef CAS PubMed.
- W. Xiao, Y. Satoh, Y. Ogasawara and T. Dairi, ChemBioChem, 2022, 23, e202100705 CAS.
- M. R. Naylor, A. T. Bockus, M.-J. Blanco and R. S. Lokey, Curr. Opin. Chem. Biol., 2017, 38, 141–147 CrossRef CAS PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- F. P. Tally and M. F. DeBruin, J. Antimicrob. Chemother., 2000, 46, 523–526 CrossRef CAS PubMed.
- M. Heidary, A. D. Khosravi, S. Khoshnood, M. J. Nasiri, S. Soleimani and M. Goudarzi, J. Antimicrob. Chemother., 2017, 73, 1–11 CrossRef PubMed.
- G. F. Gause and M. G. Brazhnikova, Nature, 1944, 154, 703 CrossRef.
- H. D. Mootz and M. A. Marahiel, J. Bacteriol., 1997, 179, 6843–6850 CrossRef CAS PubMed.
- M. Dreyfuss, E. Härri, H. Hofmann, H. Kobel, W. Pache and H. Tscherter, Eur. J. Appl. Microbiol. Biotechnol., 1976, 3, 125–133 CrossRef CAS.
- N. Fusetani, S. Matsunaga, H. Matsumoto and Y. Takebayashi, J. Am. Chem. Soc., 1990, 112, 7053–7054 CrossRef CAS.
- M. C. Wilson, T. Mori, C. Rückert, A. R. Uria, M. J. Helf, K. Takada, C. Gernert, U. A. E. Steffens, N. Heycke, S. Schmitt, C. Rinke, E. J. N. Helfrich, A. O. Brachmann, C. Gurgui, T. Wakimoto, M. Kracht, M. Crüsemann, U. Hentschel, I. Abe, S. Matsunaga, J. Kalinowski, H. Takeyama and J. Piel, Nature, 2014, 506, 58–62 CrossRef CAS PubMed.
- C. Keller-Juslén, M. Kuhn, H. R. Loosli, T. J. Petcher, H. P. Weber and A. von Wartburg, Tetrahedron Lett., 1976, 17, 4147–4150 CrossRef.
- M. Deleu, H. Razafindralambo, Y. Popineau, P. Jacques, P. Thonart and M. Paquot, Colloids Surf., A, 1999, 152, 3–10 CrossRef CAS.
- N. Vanittanakom, W. Loeffler, U. Koch and G. Jung, J. Antibiot., 1986, 39, 888–901 CrossRef CAS PubMed.
- A. Kakinuma, H. Sugino, M. Isono, G. Tamura and K. Arima, Agric. Biol. Chem., 1969, 33, 973–976 CrossRef CAS.
- T. Golakoti, W. Y. Yoshida, S. Chaganty and R. E. Moore, J. Nat. Prod., 2001, 64, 54–59 CrossRef CAS PubMed.
- J. E. Becker, R. E. Moore and B. S. Moore, Gene, 2004, 325, 35–42 CrossRef CAS PubMed.
- D. Thibaut, D. Bisch, N. Ratet, L. Maton, M. Couder, L. Debussche and F. Blanche, J. Bacteriol., 1997, 179, 697–704 CrossRef CAS PubMed.
- N. Pulsawat, S. Kitani and T. Nihira, Gene, 2007, 393, 31–42 CrossRef CAS PubMed.
- T. Schwecke, J. F. Aparicio, I. Molnár, A. König, L. E. Khaw, S. F. Haydock, M. Oliynyk, P. Caffrey, J. Cortés and J. B. Lester, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7839 CrossRef CAS PubMed.
- M. A. Gregory, S. Gaisser, R. E. Lill, H. Hong, R. M. Sheridan, B. Wilkinson, H. Petkovic, A. J. Weston, I. Carletti, H.-L. Lee, J. Staunton and P. F. Leadlay, Angew. Chem., Int. Ed., 2004, 43, 2551–2553 CrossRef CAS PubMed.
- M. J. Bertin, A. F. Roduit, J. Sun, G. E. Alves, C. W. Via, M. A. Gonzalez, P. V. Zimba and P. D. R. Moeller, Mar. Drugs, 2017, 15, 206 CrossRef PubMed.
- Y. Kawai, R. Kemperman, J. Kok and T. Saito, Curr. Protein Pept. Sci., 2004, 5, 393–398 CrossRef CAS PubMed.
- R. Kemperman, M. Jonker, A. Nauta, O. P. Kuipers and J. Kok, Appl. Environ. Microbiol., 2003, 69, 5839–5848 CrossRef CAS PubMed.
- C. Gabrielsen, D. A. Brede, I. F. Nes and D. B. Diep, Appl. Environ. Microbiol., 2014, 80, 6854–6862 CrossRef PubMed.
- R. H. Perez, T. Zendo and K. Sonomoto, Front. Microbiol., 2018, 9, 2085 CrossRef PubMed.
- O. Saether, D. J. Craik, I. D. Campbell, K. Sletten, J. Juul and D. G. Norman, Biochemistry, 1995, 34, 4147–4158 CrossRef CAS PubMed.
- S. Gunasekera, N. L. Daly, M. A. Anderson and D. J. Craik, IUBMB Life, 2006, 58, 515–524 CrossRef CAS PubMed.
- K. Amagai, H. Ikeda, J. Hashimoto, I. Kozone, M. Izumikawa, F. Kudo, T. Eguchi, T. Nakamura, H. Osada, S. Takahashi and K. Shin-ya, Sci. Rep., 2017, 7, 3382 CrossRef PubMed.
- K. Sivonen, N. Leikoski, D. P. Fewer and J. Jokela, Appl. Microbiol. Biotechnol., 2010, 86, 1213–1225 CrossRef CAS PubMed.
- J. Koehnke, A. Bent, W. E. Houssen, D. Zollman, F. Morawitz, S. Shirran, J. Vendome, A. F. Nneoyiegbe, L. Trembleau, C. H. Botting, M. C. M. Smith, M. Jaspars and J. H. Naismith, Nat. Struct. Mol. Biol., 2012, 19, 767–772 CrossRef CAS PubMed.
- S. Sarkar, W. Gu and E. W. Schmidt, ACS Catal., 2020, 10, 7146–7153 CrossRef CAS PubMed.
- E. Matabaro, H. Kaspar, P. Dahlin, D. L. V. Bader, C. E. Murar, F. Staubli, C. M. Field, J. W. Bode and M. Künzler, Sci. Rep., 2021, 11, 3541 CrossRef CAS PubMed.
- H. Luo, S.-Y. Hong, R. M. Sgambelluri, E. Angelos, X. Li and J. D. Walton, Chem. Biol., 2014, 21, 1610–1617 CrossRef CAS PubMed.
- G. Yim, M. N. Thaker, K. Koteva and G. Wright, J. Antibiot., 2014, 67, 31–41 CrossRef CAS PubMed.
- A. Greule, T. Izoré, D. Iftime, J. Tailhades, M. Schoppet, Y. Zhao, M. Peschke, I. Ahmed, A. Kulik, M. Adamek, R. J. A. Goode, R. B. Schittenhelm, J. A. Kaczmarski, C. J. Jackson, N. Ziemert, E. H. Krenske, J. J. De Voss, E. Stegmann and M. J. Cryle, Nat. Commun., 2019, 10, 2613 CrossRef PubMed.
- L. M. Repka, J. R. Chekan, S. K. Nair and W. A. van der Donk, Chem. Rev., 2017, 117, 5457–5520 CrossRef CAS PubMed.
- L. Flühe and M. A. Marahiel, Curr. Opin. Chem. Biol., 2013, 17, 605–612 CrossRef PubMed.
- G. A. Hudson, B. J. Burkhart, A. J. DiCaprio, C. J. Schwalen, B. Kille, T. V. Pogorelov and D. A. Mitchell, J. Am. Chem. Soc., 2019, 141, 8228–8238 CrossRef CAS PubMed.
- D. J. Craik, N. L. Daly, T. Bond and C. Waine, J. Mol. Biol., 1999, 294, 1327–1336 CrossRef CAS PubMed.
- S. M. Rowe and D. R. Spring, Chem. Soc. Rev., 2021, 50, 4245–4258 RSC.
- A. Caruso, R. J. Martinie, L. B. Bushin and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2019, 141, 16610–16614 CrossRef CAS PubMed.
- K. A. Clark, L. B. Bushin and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2019, 141, 10610–10615 CrossRef CAS PubMed.
- T. Q. N. Nguyen, Y. W. Tooh, R. Sugiyama, T. P. D. Nguyen, M. Purushothaman, L. C. Leow, K. Hanif, R. H. S. Yong, I. Agatha, F. R. Winnerdy, M. Gugger, A. T. Phan and B. I. Morinaka, Nat. Chem., 2020, 12, 1042–1053 CrossRef CAS PubMed.
- J. A. Hutchinson, S. Burholt and I. W. Hamley, J. Pept. Sci., 2017, 23, 82–94 CrossRef CAS PubMed.
- N. Roongsawang, K. Washio and M. Morikawa, Int. J. Mol. Sci., 2010, 12, 141–172 CrossRef PubMed.
- A. M. Gulick, ACS Chem. Biol., 2009, 4, 811–827 CrossRef CAS PubMed.
- M. A. T. Blaskovich, K. A. Hansford, M. S. Butler, Z. Jia, A. E. Mark and M. A. Cooper, ACS Infect. Dis., 2018, 4, 715–735 CrossRef CAS PubMed.
- C. J. Balibar, A. R. Howard-Jones and C. T. Walsh, Nat. Chem. Biol., 2007, 3, 584–592 CrossRef CAS PubMed.
- N. Ziemert, K. Ishida, A. Liaimer, C. Hertweck and E. Dittmann, Angew. Chem., Int. Ed., 2008, 47, 7756–7759 CrossRef CAS PubMed.
- T. Ozaki, K. Yamashita, Y. Goto, M. Shimomura, S. Hayashi, S. Asamizu, Y. Sugai, H. Ikeda, H. Suga and H. Onaka, Nat. Commun., 2017, 8, 14207 CrossRef CAS PubMed.
- C. Zong, W. L. Cheung-Lee, H. E. Elashal, M. Raj and A. J. Link, Chem. Commun., 2018, 54, 1339–1342 RSC.
- V. Wiebach, A. Mainz, M.-A. J. Siegert, N. A. Jungmann, G. Lesquame, S. Tirat, A. Dreux-Zigha, J. Aszodi, D. Le Beller and R. D. Süssmuth, Nat. Chem. Biol., 2018, 14, 652–654 CrossRef CAS PubMed.
- V. Wiebach, A. Mainz, R. Schnegotzki, M.-A. J. Siegert, M. Hügelland, N. Pliszka and R. D. Süssmuth, Angew. Chem., Int. Ed., 2020, 59, 16777–16785 CrossRef CAS PubMed.
- R. Kozakai, T. Ono, S. Hoshino, H. Takahashi, Y. Katsuyama, Y. Sugai, T. Ozaki, K. Teramoto, K. Teramoto, K. Tanaka, I. Abe, S. Asamizu and H. Onaka, Nat. Chem., 2020, 12, 869–877 CrossRef CAS PubMed.
- F. Hubrich, N. M. Bösch, C. Chepkirui, B. I. Morinaka, M. Rust, M. Gugger, S. L. Robinson, A. L. Vagstad and J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2113120119 CrossRef CAS PubMed.
- N. M. Bösch, M. Borsa, U. Greczmiel, B. I. Morinaka, M. Gugger, A. Oxenius, A. L. Vagstad and J. Piel, Angew. Chem., Int. Ed., 2020, 59, 11763–11768 CrossRef PubMed.
- S. Mordhorst, B. I. Morinaka, A. L. Vagstad and J. Piel, Angew. Chem., Int. Ed., 2020, 59, 21442–21447 CrossRef CAS PubMed.
- Y. Zheng, Y. Cong, E. W. Schmidt and S. K. Nair, Acc. Chem. Res., 2022, 55, 1313–1323 CrossRef CAS PubMed.
- Y. Zhang, K. Hamada, D. T. Nguyen, S. Inoue, M. Satake, S. Kobayashi, C. Okada, K. Ogata, M. Okada, T. Sengoku, Y. Goto and H. Suga, Nat. Catal., 2022, 5, 682–693 CrossRef CAS.
- C.-S. Phan, K. Matsuda, N. Balloo, K. Fujita, T. Wakimoto and T. Okino, J. Am. Chem. Soc., 2021, 143, 10083–10087 CrossRef CAS PubMed.
- D. Sardar, Y. Hao, Z. Lin, M. Morita, S. K. Nair and E. W. Schmidt, J. Am. Chem. Soc., 2017, 139, 2884–2887 CrossRef CAS PubMed.
- A. Mattila, R.-M. Andsten, M. Jumppanen, M. Assante, J. Jokela, M. Wahlsten, K. M. Mikula, C. Sigindere, D. H. Kwak, M. Gugger, H. Koskela, K. Sivonen, X. Liu, J. Yli-Kauhaluoma, H. Iwaï and D. P. Fewer, ACS Chem. Biol., 2019, 14, 2683–2690 CrossRef CAS PubMed.
- Y. Hao, E. Pierce, D. Roe, M. Morita, J. A. McIntosh, V. Agarwal, T. E. Cheatham, E. W. Schmidt and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14037–14042 CrossRef CAS PubMed.
- A. Parajuli, D. H. Kwak, L. Dalponte, N. Leikoski, T. Galica, U. Umeobika, L. Trembleau, A. Bent, K. Sivonen, M. Wahlsten, H. Wang, E. Rizzi, G. De Bellis, J. Naismith, M. Jaspars, X. Liu, W. Houssen and D. P. Fewer, Angew. Chem., Int. Ed., 2016, 55, 3596–3599 CrossRef CAS PubMed.
- L. Dalponte, A. Parajuli, E. Younger, A. Mattila, J. Jokela, M. Wahlsten, N. Leikoski, K. Sivonen, S. A. Jarmusch, W. E. Houssen and D. P. Fewer, Biochemistry, 2018, 57, 6860–6867 CrossRef CAS PubMed.
- P. Estrada, M. Morita, Y. Hao, E. W. Schmidt and S. K. Nair, J. Am. Chem. Soc., 2018, 140, 8124–8127 CrossRef CAS PubMed.
- M. Okada, I. Sato, S. J. Cho, H. Iwata, T. Nishio, D. Dubnau and Y. Sakagami, Nat. Chem. Biol., 2005, 1, 23–24 CrossRef CAS PubMed.
- R. Sinha Roy, A. M. Gehring, J. C. Milne, P. J. Belshaw and C. T. Walsh, Nat. Prod. Rep., 1999, 16, 249–263 RSC.
- D. L. Boger and H. Cai, Angew. Chem., Int. Ed., 1999, 38, 448–476 CrossRef CAS PubMed.
- T. A. Keating, C. G. Marshall and C. T. Walsh, Biochemistry, 2000, 39, 15513–15521 CrossRef CAS PubMed.
- M. D. McMahon, J. S. Rush and M. G. Thomas, J. Bacteriol., 2012, 194, 2809–2818 CrossRef CAS PubMed.
- B. Kunze, R. Jansen, L. Pridzun, E. Jurkiewicz, G. Hunsmann, G. Höfle and H. Reichenbach, J. Antibiot., 1993, 46, 1752–1755 CrossRef CAS PubMed.
- K. J. Weissman and R. Müller, Bioorg. Med. Chem., 2009, 17, 2121–2136 CrossRef CAS PubMed.
- D. A. Miller, L. Luo, N. Hillson, T. A. Keating and C. T. Walsh, Chem. Biol., 2002, 9, 333–344 CrossRef CAS PubMed.
- L. E. N. Quadri, T. A. Keating, H. M. Patel and C. T. Walsh, Biochemistry, 1999, 38, 14941–14954 CrossRef CAS PubMed.
- H. M. Patel and C. T. Walsh, Biochemistry, 2001, 40, 9023–9031 CrossRef CAS PubMed.
- D. Konz, A. Klens, K. Schörgendorfer and M. A. Marahiel, Chem. Biol., 1997, 4, 927–937 CrossRef CAS PubMed.
- N. Gaitatzis, A. Hans, R. Müller and S. Beyer, J. Biochem., 2001, 129, 119–124 CrossRef CAS PubMed.
- L. Du, C. Sánchez, M. Chen, D. J. Edwards and B. Shen, Chem. Biol., 2000, 7, 623–642 CrossRef CAS PubMed.
- Y.-M. Li, J. C. Milne, L. L. Madison, R. Kolter and C. T. Walsh, Science, 1996, 274, 1188 CrossRef CAS PubMed.
- J. K. Lewis, A. S. Jochimsen, S. J. Lefave, A. P. Young, W. M. Kincannon, A. G. Roberts, M. T. Kieber-Emmons and V. Bandarian, Biochemistry, 2021, 60, 3347–3361 CrossRef CAS PubMed.
- S. V. Moradi, W. M. Hussein, P. Varamini, P. Simerska and I. Toth, Chem. Sci., 2016, 7, 2492–2500 RSC.
- P. Varamini, F. M. Mansfeld, J. T. Blanchfield, B. D. Wyse, M. T. Smith and I. Toth, J. Med. Chem., 2012, 55, 5859–5867 CrossRef CAS PubMed.
- A. R. Costa, M. E. Rodrigues, M. Henriques, R. Oliveira and J. Azeredo, Crit. Rev. Biotechnol., 2014, 34, 281–299 CrossRef CAS PubMed.
- E. Uchida, K. Morimoto, N. Kawasaki, Y. Ahmed, A. Said and T. Hayakawa, Free Radical Res., 1997, 27, 311–323 CrossRef CAS PubMed.
- R. Polt, M. Dhanasekaran and C. M. Keyari, Med. Res. Rev., 2005, 25, 557–585 CrossRef CAS PubMed.
- H. H. Ho, M. T. Gilbert, D. R. Nussenzveig and M. C. Gershengorn, Biochemistry, 1999, 38, 1866–1872 CrossRef CAS PubMed.
- G. E. Norris and M. L. Patchett, Curr. Opin. Struct. Biol., 2016, 40, 112–119 CrossRef CAS PubMed.
- S. I. Elshahawi, K. A. Shaaban, M. K. Kharel and J. S. Thorson, Chem. Soc. Rev., 2015, 44, 7591–7697 RSC.
- C. M. Harris, H. Kopecka and T. M. Harris, J. Am. Chem. Soc., 1983, 105, 6915–6922 CrossRef CAS.
- J. C. J. Barna, D. H. Williams, D. J. M. Stone, T. W. C. Leung and D. M. Doddrell, J. Am. Chem. Soc., 1984, 106, 4895–4902 CrossRef CAS.
- M. P. Singh, P. J. Petersen, W. J. Weiss, J. E. Janso, S. W. Luckman, E. B. Lenoy, P. A. Bradford, R. T. Testa and M. Greenstein, Antimicrob. Agents Chemother., 2003, 47, 62–69 CrossRef CAS PubMed.
- X. Fu, C. Albermann, J. Jiang, J. Liao, C. Zhang and J. S. Thorson, Nat. Biotechnol., 2003, 21, 1467–1469 CrossRef CAS PubMed.
- X. Fu, C. Albermann, C. Zhang and J. S. Thorson, Org. Lett., 2005, 7, 1513–1515 CrossRef CAS PubMed.
- P. Peltier-Pain, K. Marchillo, M. Zhou, D. R. Andes and J. S. Thorson, Org. Lett., 2012, 14, 5086–5089 CrossRef CAS PubMed.
- A. Kaunietis, A. Buivydas, D. J. Čitavičius and O. P. Kuipers, Nat. Commun., 2019, 10, 1115 CrossRef PubMed.
- H. Ren, S. Biswas, S. Ho, W. A. van der Donk and H. Zhao, ACS Chem. Biol., 2018, 13, 2966–2972 CrossRef CAS PubMed.
- M. Iorio, O. Sasso, S. I. Maffioli, R. Bertorelli, P. Monciardini, M. Sosio, F. Bonezzi, M. Summa, C. Brunati, R. Bordoni, G. Corti, G. Tarozzo, D. Piomelli, A. Reggiani and S. Donadio, ACS Chem. Biol., 2014, 9, 398–404 CrossRef CAS PubMed.
- F. J. Ortiz-López, D. Carretero-Molina, M. Sánchez-Hidalgo, J. Martín, I. González, F. Román-Hurtado, M. de la Cruz, S. García-Fernández, F. Reyes, J. P. Deisinger, A. Müller, T. Schneider and O. Genilloud, Angew. Chem., Int. Ed., 2020, 59, 12654–12658 CrossRef PubMed.
- T. Zyubko, M. Serebryakova, J. Andreeva, M. Metelev, G. Lippens, S. Dubiley and K. Severinov, Chem. Sci., 2019, 10, 9699–9707 RSC.
- Y. Amano, H. Tsubouchi, H. Shinohara, M. Ogawa and Y. Matsubayashi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18333 CrossRef CAS PubMed.
- H. Wang, T. J. Oman, R. Zhang, C. V. Garcia De Gonzalo, Q. Zhang and W. A. van der Donk, J. Am. Chem. Soc., 2014, 136, 84–87 CrossRef CAS PubMed.
- Y. Ding, Y. Yu, H. Pan, H. Guo, Y. Li and W. Liu, Mol. Biosyst., 2010, 6, 1180–1185 RSC.
- F. Kudo, A. Miyanaga and T. Eguchi, Nat. Prod. Rep., 2014, 31, 1056–1073 RSC.
- H. Aoki, H.-I. Sakai, M. Kohsaka, T. Konomi, J. Hosoda, Y. Kubochi, E. Iguchi and H. Imanaka, J. Antibiot., 1976, 29, 492–500 CrossRef CAS PubMed.
- D. W. Woolley, R. B. Pringle and A. C. Braun, J. Biol. Chem., 1952, 197, 409–417 CrossRef CAS PubMed.
- K. Tahlan and S. E. Jensen, J. Antibiot., 2013, 66, 401–410 CrossRef CAS PubMed.
- U. Galm, E. Wendt-Pienkowski, L. Wang, S.-X. Huang, C. Unsin, M. Tao, J. M. Coughlin and B. Shen, J. Nat. Prod., 2011, 74, 526–536 CrossRef CAS PubMed.
- S. Meyer, A. Mainz, J.-C. Kehr, R. D. Süssmuth and E. Dittmann, ChemBioChem, 2017, 18, 2376–2379 CrossRef CAS PubMed.
- S. Rachid, D. Krug, K. J. Weissman and R. Müller, J. Biol. Chem., 2007, 282, 21810–21817 CrossRef CAS PubMed.
- J. Cieślak, A. Miyanaga, R. Takaku, M. Takaishi, K. Amagai, F. Kudo and T. Eguchi, Proteins Struct. Funct. Bioinf., 2017, 85, 1238–1247 CrossRef PubMed.
- N. A. Magarvey, P. D. Fortin, P. M. Thomas, N. L. Kelleher and C. T. Walsh, ACS Chem. Biol., 2008, 3, 542–554 CrossRef CAS PubMed.
- S. G. Van Lanen, S. Lin, P. C. Dorrestein, N. L. Kelleher and B. Shen, J. Biol. Chem., 2006, 281, 29633–29640 CrossRef CAS PubMed.
- F. Kudo, A. Miyanaga and T. Eguchi, J. Ind. Microbiol. Biotechnol., 2019, 46, 515–536 CrossRef CAS PubMed.
- B. I. Morinaka, E. Lakis, M. Verest, M. J. Helf, T. Scalvenzi, A. L. Vagstad, J. Sims, S. Sunagawa, M. Gugger and J. Piel, Science, 2018, 359, 779–782 CrossRef CAS PubMed.
- J. J. Barkei, B. M. Kevany, E. A. Felnagle and M. G. Thomas, ChemBioChem, 2009, 10, 366–376 CrossRef CAS PubMed.
- M. G. Thomas, Y. A. Chan and S. G. Ozanick, Antimicrob. Agents Chemother., 2003, 47, 2823–2830 CrossRef CAS PubMed.
- Z. D. Aron, P. C. Dorrestein, J. R. Blackhall, N. L. Kelleher and C. T. Walsh, J. Am. Chem. Soc., 2005, 127, 14986–14987 CrossRef CAS PubMed.
- D. Tillett, E. Dittmann, M. Erhard, H. von Döhren, T. Börner and B. A. Neilan, Chem. Biol., 2000, 7, 753–764 CrossRef CAS PubMed.
- E. H. Duitman, L. W. Hamoen, M. Rembold, V. Gerard, H. Seitz, W. Saenger, F. Bernhard, R. Reinhardt, M. Schmidt, C. Ullrich, T. Stein, F. Leenders and J. Vater, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13294–13299 CrossRef CAS PubMed.
- T. A. Scott, M. Verest, J. Farnung, C. C. Forneris, S. L. Robinson, X. Ji, F. Hubrich, C. Chepkirui, D. U. Richter, S. Huber, P. Rust, A. B. Streiff, Q. Zhang, J. W. Bode and J. Piel, Chem, 2022, 8, 2659–2677 CAS.
- M. Issac, M. Aknin, A. Gauvin-Bialecki, N. De Voogd, A. Ledoux, M. Frederich, Y. Kashman and S. Carmeli, J. Nat. Prod., 2017, 80, 1110–1116 CrossRef CAS PubMed.
- N. Fusetani, T. Sugawara, S. Matsunaga and H. Hirota, J. Am. Chem. Soc., 1991, 113, 7811–7812 CrossRef CAS.
- A. Plaza, K. Viehrig, R. Garcia and R. Müller, Org. Lett., 2013, 15, 5882–5885 CrossRef CAS PubMed.
- M. Kimura, T. Wakimoto, Y. Egami, K. C. Tan, Y. Ise and I. Abe, J. Nat. Prod., 2012, 75, 290–294 CrossRef CAS PubMed.
- J. Z. Acedo, I. R. Bothwell, L. An, A. Trouth, C. Frazier and W. A. van der Donk, J. Am. Chem. Soc., 2019, 141, 16790–16801 CrossRef CAS PubMed.
- A. Ökesli, L. E. Cooper, E. J. Fogle and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 13753–13760 CrossRef PubMed.
- X.-D. Kong, J. Moriya, V. Carle, F. Pojer, L. A. Abriata, K. Deyle and C. Heinis, Nat. Biomed. Eng., 2020, 4, 560–571 CrossRef CAS PubMed.
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS PubMed.
- K. Komatsu, H. Shigemori and J. Kobayashi, J. Org. Chem., 2001, 66, 6189–6192 CrossRef CAS PubMed.
- A. Haese, M. Schubert, M. Herrmann and R. Zocher, Mol. Microbiol., 1993, 7, 905–914 CrossRef CAS PubMed.
- S. D. Jolad, J. J. Hoffman, S. J. Torrance, R. M. Wiedhopf, J. R. Cole, S. K. Arora, R. B. Bates, R. L. Gargiulo and G. R. Kriek, J. Am. Chem. Soc., 1977, 99, 8040–8044 CrossRef CAS PubMed.
- T. Sasaki, M. Takagi, T. Yaguchi, S. Miyadoh, T. Okada and M. Koyama, J. Antibiot., 1992, 45, 692–697 CrossRef CAS PubMed.
- L. Li, L. W. MacIntyre, T. Ali, R. Russo, B. Koirala, Y. Hernandez and S. F. Brady, J. Nat. Prod., 2021, 84, 1056–1066 CrossRef CAS PubMed.
- J. E. Coleman, E. Dilip de Silva, F. Kong, R. J. Andersen and T. M. Allen, Tetrahedron, 1995, 51, 10653–10662 CrossRef CAS.
- G. R. Pettit, Y. Kamano, C. L. Herald, A. A. Tuinman, F. E. Boettner, H. Kizu, J. M. Schmidt, L. Baczynskyj, K. B. Tomer and R. J. Bontems, J. Am. Chem. Soc., 1987, 109, 6883–6885 CrossRef CAS.
- D. A. Miller, C. T. Walsh and L. Luo, J. Am. Chem. Soc., 2001, 123, 8434–8435 CrossRef CAS PubMed.
- M. Welker and H. Von Döhren, FEMS Microbiol. Rev., 2006, 30, 530–563 CrossRef CAS PubMed.
- A. Lawen and R. Zocher, J. Biol. Chem., 1990, 265, 11355–11360 CrossRef CAS PubMed.
- T. Velkov and A. Lawen, J. Biol. Chem., 2003, 278, 1137–1148 CrossRef CAS PubMed.
- A. Billich and R. Zocher, Biochemistry, 1987, 26, 8417–8423 CrossRef CAS.
- W. Weckwerth, K. Miyamoto, K. Iinuma, M. Krause, M. Glinski, T. Storm, G. Bonse, H. Kleinkauf and R. Zocher, J. Biol. Chem., 2000, 275, 17909–17915 CrossRef CAS PubMed.
- N. S. van der Velden, N. Kälin, M. J. Helf, J. Piel, M. F. Freeman and M. Künzler, Nat. Chem. Biol., 2017, 13, 833–835 CrossRef CAS PubMed.
- H. Song, N. S. van der Velden, S. L. Shiran, P. Bleiziffer, C. Zach, R. Sieber, A. S. Imani, F. Krausbeck, M. Aebi, M. F. Freeman, S. Riniker, M. Künzler and J. H. Naismith, Sci. Adv., 2018, 4, eaat2720 CrossRef CAS PubMed.
- M. R. Quijano, C. Zach, F. S. Miller, A. R. Lee, A. S. Imani, M. Künzler and M. F. Freeman, J. Am. Chem. Soc., 2019, 141, 9637–9644 CrossRef CAS PubMed.
- F. S. Miller, K. K. Crone, M. R. Jensen, S. Shaw, W. R. Harcombe, M. H. Elias and M. F. Freeman, Nat. Commun., 2021, 12, 5355 CrossRef CAS PubMed.
- L. Paoli, H.-J. Ruscheweyh, C. C. Forneris, F. Hubrich, S. Kautsar, A. Bhushan, A. Lotti, Q. Clayssen, G. Salazar, A. Milanese, C. I. Carlström, C. Papadopoulou, D. Gehrig, M. Karasikov, H. Mustafa, M. Larralde, L. M. Carroll, P. Sánchez, A. A. Zayed, D. R. Cronin, S. G. Acinas, P. Bork, C. Bowler, T. O. Delmont, J. M. Gasol, A. D. Gossert, A. Kahles, M. B. Sullivan, P. Wincker, G. Zeller, S. L. Robinson, J. Piel and S. Sunagawa, Nature, 2022, 607, 111–118 CrossRef CAS PubMed.
- S. Caboche, V. Leclère, M. Pupin, G. Kucherov and P. Jacques, J. Bacteriol., 2010, 192, 5143–5150 CrossRef CAS PubMed.
- O. Lazos, M. Tosin, A. L. Slusarczyk, S. Boakes, J. Cortés, P. J. Sidebottom and P. F. Leadlay, Chem. Biol., 2010, 17, 160–173 CrossRef CAS PubMed.
- X. Zhao, Z. Li and O. P. Kuipers, Cell Chem. Biol., 2020, 27, 1262–1271.e4 CrossRef CAS PubMed.
- K. A. Clark, B. C. Covington and M. R. Seyedsayamdost, Nat. Chem, 2022 DOI:10.1038/s41557-022-01063-3.
- M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317–347 RSC.
- R. Iacovelli, R. A. L. Bovenberg and A. J. M. Driessen, J. Ind. Microbiol. Biotechnol., 2021, 48, kuab045 CrossRef CAS PubMed.
- K. A. Bozhüyük, J. Micklefield and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 88–96 CrossRef PubMed.
- C. Beck, J. F. G. Garzón and T. Weber, Biotechnol. Bioprocess Eng., 2020, 25, 886–894 CrossRef CAS.
- A. Camus, G. Truong, P. R. E. Mittl, G. Markert and D. Hilvert, J. Am. Chem. Soc., 2022, 144, 17567–17575 CrossRef CAS PubMed.
- D. L. Niquille, D. A. Hansen, T. Mori, D. Fercher, H. Kries and D. Hilvert, Nat. Chem., 2018, 10, 282–287 CrossRef CAS PubMed.
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C.-P. Niesert and H. B. Bode, Nat. Chem., 2018, 10, 275–281 CrossRef PubMed.
- K. A. J. Bozhüyük, A. Linck, A. Tietze, J. Kranz, F. Wesche, S. Nowak, F. Fleischhacker, Y.-N. Shi, P. Grün and H. B. Bode, Nat. Chem., 2019, 11, 653–661 CrossRef PubMed.
- K. A. J. Bozhüyük, J. Watzel, N. Abbood and H. B. Bode, Angew. Chem., Int. Ed., 2021, 60, 17531–17538 CrossRef PubMed.
- H.-M. Huang, P. Stephan and H. Kries, Cell Chem. Biol., 2021, 28, 221–227.e7 CrossRef CAS PubMed.
- J. A. McIntosh, M. S. Donia and E. W. Schmidt, Nat. Prod. Rep., 2009, 26, 537–559 RSC.
- F. Ruijne and O. P. Kuipers, Biochem. Soc. Trans., 2021, 49, 203–215 CrossRef CAS PubMed.
- C. Wu and W. A. van der Donk, Curr. Opin. Biotechnol., 2021, 69, 221–231 CrossRef CAS PubMed.
- B. J. Burkhart, G. A. Hudson, K. L. Dunbar and D. A. Mitchell, Nat. Chem. Biol., 2015, 11, 564–570 CrossRef CAS PubMed.
- A. M. Kloosterman, K. E. Shelton, G. P. van Wezel, M. H. Medema and D. A. Mitchell, mSystems, 2020, 5, e00267–20 CrossRef CAS PubMed.
- J. R. Chekan, C. Ongpipattanakul and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 24049–24055 CrossRef CAS PubMed.
- T. J. Oman and W. A. Van Der Donk, Nat. Chem. Biol., 2010, 6, 9–18 CrossRef CAS PubMed.
- I. R. Rahman, J. Z. Acedo, X. R. Liu, L. Zhu, J. Arrington, M. L. Gross and W. A. van der Donk, ACS Chem. Biol., 2020, 15, 1473–1486 CrossRef CAS PubMed.
- S. W. Fuchs, G. Lackner, B. I. Morinaka, Y. Morishita, T. Asai, S. Riniker and J. Piel, Angew. Chem., Int. Ed., 2016, 55, 12330–12333 CrossRef CAS PubMed.
- B. J. Burkhart, N. Kakkar, G. A. Hudson, W. A. van der Donk and D. A. Mitchell, ACS Cent. Sci., 2017, 3, 629–638 CrossRef CAS PubMed.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
- S. R. Fleming, T. E. Bartges, A. A. Vinogradov, C. L. Kirkpatrick, Y. Goto, H. Suga, L. M. Hicks and A. A. Bowers, J. Am. Chem. Soc., 2019, 141, 758–762 CrossRef CAS PubMed.
- W. J. Wever, J. W. Bogart, J. A. Baccile, A. N. Chan, F. C. Schroeder and A. A. Bowers, J. Am. Chem. Soc., 2015, 137, 3494–3497 CrossRef CAS PubMed.
- L. Franz and J. Koehnke, Chem. Commun., 2021, 57, 6372–6375 RSC.
- J. Lee, J. McIntosh, B. J. Hathaway and E. W. Schmidt, J. Am. Chem. Soc., 2009, 131, 2122–2124 CrossRef CAS PubMed.
- J. A. McIntosh, C. R. Robertson, V. Agarwal, S. K. Nair, G. W. Bulaj and E. W. Schmidt, J. Am. Chem. Soc., 2010, 132, 15499–15501 CrossRef CAS PubMed.
- D. E. Ruffner, E. W. Schmidt and J. R. Heemstra, ACS Synth. Biol., 2015, 4, 482–492 CrossRef CAS PubMed.
- S. Schmitt, M. Montalbán-López, D. Peterhoff, J. Deng, R. Wagner, M. Held, O. P. Kuipers and S. Panke, Nat. Chem. Biol., 2019, 15, 437–443 CrossRef CAS PubMed.
- H. Saad, S. Aziz, M. Gehringer, M. Kramer, J. Straetener, A. Berscheid, H. Brötz-Oesterhelt and H. Gross, Angew. Chem., Int. Ed., 2021, 60, 16472–16479 CrossRef CAS PubMed.
- R. Khusainov and O. P. Kuipers, ChemBioChem, 2012, 13, 2433–2438 CrossRef CAS PubMed.
- E. Reyna-González, B. Schmid, D. Petras, R. D. Süssmuth and E. Dittmann, Angew. Chem., Int. Ed., 2016, 55, 9398–9401 CrossRef PubMed.
- T. J. Oman, P. J. Knerr, N. A. Bindman, J. E. Velásquez and W. A. van der Donk, J. Am. Chem. Soc., 2012, 134, 6952–6955 CrossRef CAS PubMed.
- Y. Goto, Y. Ito, Y. Kato, S. Tsunoda and H. Suga, Chem. Biol., 2014, 21, 766–774 CrossRef CAS PubMed.
- J. Koehnke, G. Mann, A. F. Bent, H. Ludewig, S. Shirran, C. Botting, T. Lebl, W. E. Houssen, M. Jaspars and J. H. Naismith, Nat. Chem. Biol., 2015, 11, 558–563 CrossRef CAS PubMed.
- K. P. Patel, L. M. Silsby, G. Li and S. D. Bruner, J. Org. Chem., 2021, 86, 11212–11219 CrossRef CAS PubMed.
- Z. Zhang, G. A. Hudson, N. Mahanta, J. I. Tietz, W. A. van der Donk and D. A. Mitchell, J. Am. Chem. Soc., 2016, 138, 15511–15514 CrossRef CAS PubMed.
- J. A. McIntosh, M. S. Donia, S. K. Nair and E. W. Schmidt, J. Am. Chem. Soc., 2011, 133, 13698–13705 CrossRef CAS PubMed.
- Q. Zhang and W. A. van der Donk, FEBS Lett., 2012, 586, 3391–3397 CrossRef CAS PubMed.
- X. Yang and W. A. van der Donk, J. Am. Chem. Soc., 2015, 137, 12426–12429 CrossRef CAS PubMed.
- L. An, D. P. Cogan, C. D. Navo, G. Jiménez-Osés, S. K. Nair and W. A. van der Donk, Nat. Chem. Biol., 2018, 14, 928–933 CrossRef CAS PubMed.
- F. Wang, W. Wei, J. Zhao, T. Mo, X. Wang, X. Huang, S. Ma, S. Wang, Z. Deng, W. Ding, Y. Liang and Q. Zhang, CCS Chem., 2020, 3, 1049–1057 CrossRef.
- J. A. Walker, N. Hamlish, A. Tytla, D. D. Brauer, M. B. Francis and A. Schepartz, ACS Cent. Sci., 2022, 8, 473–482 CrossRef CAS PubMed.
- E. Lakis, S. Magyari and J. Piel, Angew. Chem., Int. Ed., 2022, 61, e202202695 CrossRef CAS PubMed.
- A. J. van Heel, D. Mu, M. Montalbán-López, D. Hendriks and O. P. Kuipers, ACS Synth. Biol., 2013, 2, 397–404 CrossRef CAS PubMed.
- A. Parent, A. Guillot, A. Benjdia, G. Chartier, J. Leprince and O. Berteau, J. Am. Chem. Soc., 2016, 138, 15515–15518 CrossRef CAS PubMed.
- N. Mahanta, Z. Zhang, G. A. Hudson, W. A. van der Donk and D. A. Mitchell, J. Am. Chem. Soc., 2017, 139, 4310–4313 CrossRef CAS PubMed.
- E. Oueis, M. Jaspars, N. J. Westwood and J. H. Naismith, Angew. Chem., Int. Ed., 2016, 55, 5842–5845 CrossRef CAS PubMed.
- E. Oueis, B. Nardone, M. Jaspars, N. J. Westwood and J. H. Naismith, ChemistryOpen, 2017, 6, 11–14 CrossRef CAS PubMed.
- T. Le and W. A. van der Donk, Trends Chem., 2021, 3, 266–278 CrossRef CAS.
- P. Arias-Orozco, M. Inklaar, J. Lanooij, R. Cebrián and O. P. Kuipers, ACS Synth. Biol., 2021, 10, 2579–2591 CrossRef CAS PubMed.
- A. A. Vinogradov, M. Shimomura, Y. Goto, T. Ozaki, S. Asamizu, Y. Sugai, H. Suga and H. Onaka, Nat. Commun., 2020, 11, 2272 CrossRef CAS PubMed.
- Y. Si, A. M. Kretsch, L. M. Daigh, M. J. Burk and D. A. Mitchell, J. Am. Chem. Soc., 2021, 143, 5917–5927 CrossRef CAS PubMed.
- H. Song, J. Fahrig-Kamarauskaitè, E. Matabaro, H. Kaspar, S. L. Shirran, C. Zach, A. Pace, B.-A. Stefanov, J. H. Naismith and M. Künzler, ACS Chem. Biol., 2020, 15, 1901–1912 CrossRef CAS PubMed.
- J. D. Hegemann, S. C. Bobeica, M. C. Walker, I. R. Bothwell and W. A. van der Donk, ACS Synth. Biol., 2019, 8, 1204–1214 CrossRef CAS PubMed.
- T. Le, K. Jeanne Dit Fouque, M. Santos-Fernandez, C. D. Navo, G. Jiménez-Osés, R. Sarksian, F. A. Fernandez-Lima and W. A. van der Donk, J. Am. Chem. Soc., 2021, 143, 18733–18743 CrossRef CAS PubMed.
- M. Morita, Y. Hao, J. K. Jokela, D. Sardar, Z. Lin, K. Sivonen, S. K. Nair and E. W. Schmidt, J. Am. Chem. Soc., 2018, 140, 6044–6048 CrossRef CAS PubMed.
- M. Purushothaman, S. Sarkar, M. Morita, M. Gugger, E. W. Schmidt and B. I. Morinaka, Angew. Chem., Int. Ed., 2021, 60, 8460–8465 CrossRef CAS PubMed.
- W. Gu, S.-H. Dong, S. Sarkar, S. K. Nair and E. W. Schmidt, in Methods in Enzymology, ed. B. S. Moore, Academic Press, 2018, vol. 604, pp. 113–163 Search PubMed.
- N. Mupparapu, Y.-H. C. Lin, T. H. Kim and S. I. Elshahawi, Chem. – Eur. J., 2021, 27, 4176–4182 CrossRef CAS PubMed.
- X. Zhao, R. Cebrián, Y. Fu, R. Rink, T. Bosma, G. N. Moll and O. P. Kuipers, ACS Synth. Biol., 2020, 9, 1468–1478 CrossRef CAS PubMed.
- W. Tang and W. A. van der Donk, Biochemistry, 2012, 51, 4271–4279 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, S. Zhu, D. Klug and M. A. Marahiel, Pept. Sci., 2013, 100, 527–542 CrossRef CAS PubMed.
- J. D. Hegemann and W. A. van der Donk, J. Am. Chem. Soc., 2018, 140, 5743–5754 CrossRef CAS PubMed.
- T. E. Smith, C. D. Pond, E. Pierce, Z. P. Harmer, J. Kwan, M. M. Zachariah, M. K. Harper, T. P. Wyche, T. K. Matainaho, T. S. Bugni, L. R. Barrows, C. M. Ireland and E. W. Schmidt, Nat. Chem. Biol., 2018, 14, 179–185 CrossRef CAS PubMed.
- Y. Zhang, M. Chen, S. D. Bruner and Y. Ding, Front. Microbiol., 2018, 9, 1801 CrossRef PubMed.
- R. Maini, S. Umemoto and H. Suga, Curr. Opin. Chem. Biol., 2016, 34, 44–52 CrossRef CAS PubMed.
- Y. Goto and H. Suga, Curr. Opin. Chem. Biol., 2018, 46, 82–90 CrossRef CAS PubMed.
- A. D. Silverman, A. S. Karim and M. C. Jewett, Nat. Rev. Genet., 2020, 21, 151–170 CrossRef CAS PubMed.
- Z. Cui, W. A. Johnston and K. Alexandrov, Front. Bioeng. Biotechnol., 2020, 8, 1031 CrossRef PubMed.
- R. Liu, Y. Zhang, G. Zhai, S. Fu, Y. Xia, B. Hu, X. Cai, Y. Zhang, Y. Li, Z. Deng and T. Liu, Adv. Sci., 2020, 7, 2001616 CrossRef CAS PubMed.
- Y. Si, A. M. Kretsch, L. M. Daigh, M. J. Burk and D. A. Mitchell, J. Am. Chem. Soc., 2021, 143, 5917–5927 CrossRef CAS PubMed.
- Y. Goto and H. Suga, ChemBioChem, 2020, 21, 84–87 CrossRef CAS PubMed.
- Y. Huang, M. M. Wiedmann and H. Suga, Chem. Rev., 2019, 119, 10360–10391 CrossRef CAS PubMed.
- S. R. Fleming, P. M. Himes, S. V. Ghodge, Y. Goto, H. Suga and A. A. Bowers, J. Am. Chem. Soc., 2020, 142, 5024–5028 CrossRef CAS PubMed.
- A. A. Vinogradov, E. Nagai, J. S. Chang, K. Narumi, H. Onaka, Y. Goto and H. Suga, J. Am. Chem. Soc., 2020, 142, 20329–20334 CrossRef CAS PubMed.
- M. S. Newton, Y. Cabezas-Perusse, C. L. Tong and B. Seelig, ACS Synth. Biol., 2020, 9, 181–190 CrossRef CAS PubMed.
- Y. Goto and H. Suga, Acc. Chem. Res., 2021, 54, 3604–3617 CrossRef CAS PubMed.
- M. E. Boyer, J. A. Stapleton, J. M. Kuchenreuther, C. Wang and J. R. Swartz, Biotechnol. Bioeng., 2008, 99, 59–67 CrossRef CAS PubMed.
- S. J. Pan and A. J. Link, J. Am. Chem. Soc., 2011, 133, 5016–5023 CrossRef CAS PubMed.
- T. Si, Q. Tian, Y. Min, L. Zhang, J. V. Sweedler, W. A. van der Donk and H. Zhao, J. Am. Chem. Soc., 2018, 140, 11884–11888 CrossRef CAS PubMed.
- T. Bosma, A. Kuipers, E. Bulten, L. de Vries, R. Rink and G. N. Moll, Appl. Environ. Microbiol., 2011, 77, 6794–6801 CrossRef CAS PubMed.
- J. H. Urban, M. A. Moosmeier, T. Aumüller, M. Thein, T. Bosma, R. Rink, K. Groth, M. Zulley, K. Siegers, K. Tissot, G. N. Moll and J. Prassler, Nat. Commun., 2017, 8, 1500 CrossRef PubMed.
- K. J. Hetrick, M. C. Walker and W. A. van der Donk, ACS Cent. Sci., 2018, 4, 458–467 CrossRef CAS PubMed.
- X. Yang, K. R. Lennard, C. He, M. C. Walker, A. T. Ball, C. Doigneaux, A. Tavassoli and W. A. van der Donk, Nat. Chem. Biol., 2018, 14, 375–380 CrossRef CAS PubMed.
- A. M. King, D. A. Anderson, E. Glassey, T. H. Segall-Shapiro, Z. Zhang, D. L. Niquille, A. C. Embree, K. Pratt, T. L. Williams, D. B. Gordon and C. A. Voigt, Nat. Commun., 2021, 12, 6343 CrossRef CAS PubMed.
- T. W. Precord, N. Mahanta and D. A. Mitchell, ACS Chem. Biol., 2019, 14, 1981–1989 CrossRef CAS PubMed.
- W. Gu and E. W. Schmidt, Acc. Chem. Res., 2017, 50, 2569–2576 CrossRef CAS PubMed.
- S. C. Bobeica, L. Zhu, J. Z. Acedo, W. Tang and W. A. van der Donk, Chem. Sci., 2020, 11, 12854–12870 RSC.
- S. C. do Amaral, P. R. Monteiro, J. da, S. P. Neto, G. M. Serra, E. C. Gonçalves, L. P. Xavier and A. V. Santos, Mar. Drugs, 2021, 19, 17 CrossRef CAS PubMed.
- I. Song, Y. Kim, J. Yu, S. Y. Go, H. G. Lee, W. J. Song and S. Kim, Nat. Chem. Biol., 2021, 17, 1123–1131 CrossRef CAS PubMed.
- J. R. Chekan, C. Ongpipattanakul and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 24049–24055 CrossRef CAS PubMed.
- A. A. Vinogradov, M. Shimomura, N. Kano, Y. Goto, H. Onaka and H. Suga, J. Am. Chem. Soc., 2020, 142, 13886–13897 CrossRef CAS PubMed.
- N. A. Schilling, A. Berscheid, J. Schumacher, J. S. Saur, M. C. Konnerth, S. N. Wirtz, J. M. Beltrán-Beleña, A. Zipperer, B. Krismer, A. Peschel, H. Kalbacher, H. Brötz-Oesterhelt, C. Steinem and S. Grond, Angew. Chem., Int. Ed., 2019, 58, 9234–9238 CrossRef CAS PubMed.
- J. J. Hug, J. Dastbaz, S. Adam, O. Revermann, J. Koehnke, D. Krug and R. Müller, ACS Chem. Biol., 2020, 15, 2221–2231 CrossRef CAS PubMed.
- H. Song, A. J. Burton, S. L. Shirran, J. Fahrig-Kamarauskaitė, H. Kaspar, T. W. Muir, M. Künzler and J. H. Naismith, Angew. Chem., Int. Ed., 2021, 60, 14319–14323 CrossRef CAS PubMed.
- D. Fujinami, C. V. Garcia de Gonzalo, S. Biswas, Y. Hao, H. Wang, N. Garg, T. Lukk, S. K. Nair and W. A. van der Donk, Cell Chem. Biol., 2021, 28, 1740–1749.e6 CrossRef CAS PubMed.
Footnotes |
| † Present address: Pharmaceutical Institute, Department of Pharmaceutical Biology, University of Tübingen, Auf der Morgenstelle 8, 72076 Tübingen, Germany. |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |