
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of multidentate tyrosyl-DNA phosphodiesterase 1 (TDP1) inhibitors that simultaneously access the DNA, protein and catalytic-binding sites by oxime diversification†
Xue Zhi
Zhao
 *a,
Wenjie
Wang
b,
George T.
Lountos
c,
Evgeny
Kiselev
b,
Joseph E.
Tropea
d,
Danielle
Needle
d,
Yves
Pommier
b and
Terrence R.
Burke
Jr.
a
*a,
Wenjie
Wang
b,
George T.
Lountos
c,
Evgeny
Kiselev
b,
Joseph E.
Tropea
d,
Danielle
Needle
d,
Yves
Pommier
b and
Terrence R.
Burke
Jr.
a
aChemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, MD, USA. E-mail: xuezhi.zhao@nih.gov
bDevelopmental Therapeutics Branch & Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
cBasic Science Program, Frederick National Laboratory for Cancer Research, Frederick, MD, USA
dCenter for Structural Biology, Center for Cancer Research, National Cancer Institute, Frederick, MD, USA
First published on 27th March 2023
Abstract
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is a member of the phospholipase D family that can downregulate the anticancer effects of the type I topoisomerase (TOP1) inhibitors by hydrolyzing the 3′-phosphodiester bond between DNA and the TOP1 residue Y723 in the critical stalled intermediate that is the foundation of TOP1 inhibitor mechanism of action. Thus, TDP1 antagonists are attractive as potential enhancers of TOP1 inhibitors. However, the open and extended nature of the TOP1–DNA substrate-binding region has made the development of TDP1 inhibitors extremely challenging. In this study, starting from our recently identified small molecule microarray (SMM)-derived TDP1-inhibitory imidazopyridine motif, we employed a click-based oxime protocol to extend the parent platform into the DNA and TOP1 peptide substrate-binding channels. We applied one-pot Groebke–Blackburn–Bienayme multicomponent reactions (GBBRs) to prepare the needed aminooxy-containing substrates. By reacting these precursors with approximately 250 aldehydes in microtiter format, we screened a library of nearly 500 oximes for their TDP1 inhibitory potencies using an in vitro florescence-based catalytic assay. Select hits were structurally explored as their triazole- and ether-based isosteres. We obtained crystal structures of two of the resulting inhibitors bound to the TDP1 catalytic domain. The structures reveal that the inhibitors form hydrogen bonds with the catalytic His-Lys-Asn triads (“HKN” motifs: H263, K265, N283 and H493, K495, N516), while simultaneously extending into both the substrate DNA and TOP1 peptide-binding grooves. This work provides a structural model for developing multivalent TDP1 inhibitors capable of binding in a tridentate fashion with a central component situated within the catalytic pocket and extensions that project into both the DNA and TOP1 peptide substrate-binding regions.
Introduction
Topoisomerases (TOPs) correct DNA topological stress by creating DNA–protein crosslinks (DPCs) during the cleavage of one DNA strand (TOP1 and TOP3) or both strands (TOP2). TOP1 is essential for genomic stability by resolving DNA supercoiling or knotting that could otherwise result in alternate DNA structures.1 TOP1 achieves its effects through nucleophilic attack of its Y723 hydroxyl onto a DNA 3′-phosphodiester bond, resulting in the formation of a covalent TOP1–DNA bond with concomitant strand breakage. This generates topoisomerase cleavage complexes (TOPCCs), which are quite transient and rapidly relegated with release of the TOP1 following decatenation of the DNA. In quiescent cells the TOP1CCs are not inherently harmful, however, in highly dividing cells they can be converted into cytotoxic irreversible TOP1CCs and ultimately lead to DNA damaging double-strand breaks.2 TOP1 inhibitors, such as the alkaloid camptothecin (CPT), bind at the TOP1–DNA interface, stall relegation of TOPCCs and induce cell death in mitotic cells.3,4 The water-soluble camptothecin derivatives topotecan and irinotecan are potent TOP1 inhibitors used for the treatment of ovarian and lung cancers and colorectal cancers, respectively.5 However, dose-limiting myelosuppression in combination with additional side effects can reduce the clinical effectiveness of TOP1 inhibitors.6 Cytotoxicity of TOP1 inhibitors can also be reduced by cancer cell resistance mediated by DNA repair pathways.7The human tyrosyl-DNA phosphodiesterase 1 (TDP1) is a DNA repair enzyme that promiscuously processes 3′-DNA end blocking lesions using a wide range of synthetic DNA adducts as substrates.8 The enzyme is found at low levels in most tissues, but its expression is elevated in nearly all tumors.9 TDP1 can rescue the genome from damage arising from persistent TOP1CCs by hydrolysing the Y723-DNA phosphodiester linkage and liberating DNA with a free 3′-phosphate group. Down-regulating TDP1 can enhance the toxicity of phosphodiester-linked DNA adducts, such as TOP1CCs (Fig. 1(A)).10–13 TDP1 antagonists may be viewed as TOP1 inhibitor chemosensitizers with a potential to lessen dosage-related toxicity and side effects. Accordingly, TDP1 is recognized as a target for new anticancer therapeutics that could be used in combination with TOP1 inhibitors.14–17 Significant effort devoted to developing TDP1 inhibitors has resulted in the discovery of a spectrum of structurally diverse inhibitors (recent examples18–21). However, the understanding of the molecular protein–ligand interactions of most of these inhibitors with TDP1 is unclear and clinical agents have yet to result.12,22–24 This raises concerns related to potentially promiscuous mechanisms of action that could impact the rational design of new analogs.25,26
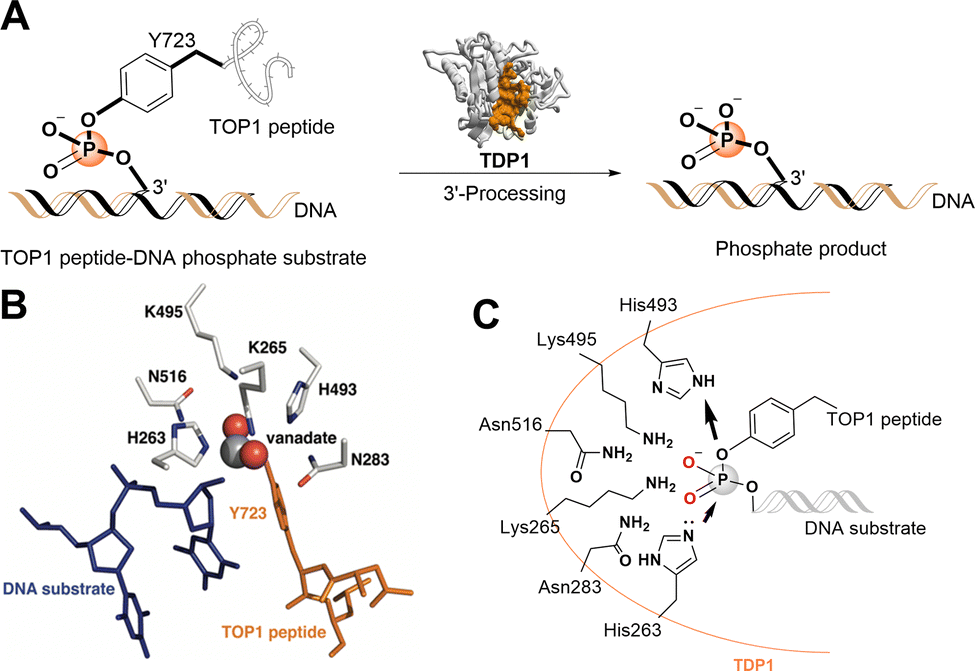 | ||
| Fig. 1 TDP1 3′-processing reaction and substrates. (A) TDP1 hydrolyses the phosphotyrosyl ester bond between the Y723 residue of the TOP1-derived peptide and the 3′-end of the DNA substrate to generate a 3′-processed phosphate containing product. (B) Structure of a TDP1-vanadate-TOP1 peptide complex. TDP1 (carbon atoms are shown in gray, nitrogen atoms are shown in blue, and oxygen atoms are shown in red) is complexed with a single DNA strand (blue sticks) and TOP1-derived peptide (orange sticks) with the phosphate mimetic vanadate highlighted in a sphere representation (PDB code: 1NOP). (C) Hydrolytic mechanism of the TOP1CC phosphodiester hydrolysis by TDP1 HKN motifs. | ||
The catalytic mechanisms of TDP1 have been studied using vanadate and tungstate as phosphate mimetics. These can replicate aspects of phosphorylase and phosphatase catalytic transition states in crystal structures and thereby clarify mechanisms of these enzymes.27–32 Preliminary insights into how catalytic residues engage the DNA 3′-phosphonamide linkage have been provided by X-ray crystal structures of TDP1 without33 and with vanadate or tungstate transition state mimetics bound within the catalytic site and with single-strand DNA and TOP1-derived substrates (Fig. 1(B))34–36 as well as crystal structures of TDP1 in complex with double-stranded DNA.37 As a member of the phospholipase D superfamily of enzymes, the TDP1 catalytic pocket contains paired histidine and lysine residues within two conserved histidine-lysine-asparagine catalytic motifs [HxKx(n)N, with x being any amino acid].38,39 These residues are arranged to hydrolyse the TOP1CC phosphodiester linkage in a two-step process (Fig. 1(C)).40 In the first step, a H263 imidazole nitrogen executes a nucleophilic attack on the 3′-phosphotyrosyl linkage. This releases the tyrosyl residue and associated protein while forming a covalent 3′-phosphonamide linkage with the DNA. The second signature histidine (H493) then donates a proton to the leaving nucleophilic phenoxy anion of the liberated tyrosyl residue.41 This converts H493 into a basic nucleophile that activates a water molecule to attack the DNA 3′-phosphonamide linkage to H263, thereby releasing the DNA from TDP1. The K265 and N283 residues of the first HKN motif form hydrogen bonds with the 3′-phosphoryl group, while the K495 and N516 residues of the second HKN motif stabilize the substrate and function as a relay to protonate/deprotonate the H493 residue.
Progress in the development of TDP1 inhibitors could be facilitated by maximizing interactions of ligands with specific features of the protein catalytic apparatus. TDP1's topographical features are organized to accommodate substrate and execute phosphodiester hydrolysis. Protein–DNA adducts, such as TOP1CCs, present three distinct components: the protein adducted to DNA, the DNA polynucleotide chain and the 3′-phosphodiester crosslink between the two. Accordingly, interaction of the trimeric TOP1CC with TDP1 occurs within three functional regions. The tyrosyl-3′-phosphodeoxyribose ester binds in a well-formed pocket containing the catalytic pocket, while a narrow (approximately 8 Å wide) positively charged channel extends from one side of the catalytic pocket to accommodate the negatively charged DNA polynucleotide chain. A wider, more neutral concave region extends in the opposite direct from the catalytic pocket to hold and align the TOP1-derived peptide moiety. The width of this region expands from 8 Å near the active site to as wide as 20 Å.40
The design of TDP1-binding small-molecule ligands could benefit by taking advantage of the tripartite nature of the interaction of TDP1 with TOP1CC substrates, exploiting structural components capable of accessing the catalytic pocket as well as the DNA- and peptide-binding channels. The TDP1-binding affinities of the resulting multivalent ligands could potentially be significantly enhanced relative to those of monovalent ligands due to thermodynamic considerations.42,43 In spite of the fact that increases in molecular size and complexity required to realize these multidentate interactions may place such compounds outside parameters defined by the “Lipinski rule of 5”,44 this does not necessarily limit their suitability as drug candidates.45
Although the catalytic pocket is organized to recognize phosphate esters, phosphates suffer from several disadvantages that render them unattractive for ligand design. For this reason, bioisosteres are frequently used to replicate biological interactions of phosphates.46 Recently, we employed an X-ray crystallographic screen of TDP-binding small molecule fragments, which lead to the discovery of phthalic acids and quinolone-based motifs that engage the catalytic core of TDP1 in a fashion similar to the previously described vanadate phosphate mimetics.47 We then employed Alexa Fluor 647 (AF647)-tagged TDP1(148-608) as a fluorescent probe and evaluated its ability to bind to a small-molecule microarray (SMM) against 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 drug-like small molecules.48 In this way we identified the imidazopyrazine nucleus as a TDP1-binding motif. Further structural variation of the core heterocycle skeleton using one-pot Groebke-Blackburn-Bienayme multicomponent reactions (GBBRs) identified 2-phenylimidazo[1,2-a]pyridines (1 and 2) having higher inhibitory potencies (Fig. 2(A)). The crystal structure of 3 bound to TDP1 (PDB code: 6W7K) showed that it replicates important interactions with the catalytic phosphate-binding pocket as previously shown by our structurally more simple phthalic acid-containing inhibitors, such as 4-aminophthalic acid.47 However, while the phthalic acid moiety anchors the SMM-derived inhibitor within the phosphate-binding pocket, the additional 2-phenylimidazo[1,2-a]pyridine nucleus is situated above the phthalic acid phosphate mimic, with the pyridine ring directed toward the DNA substrate-binding region and the 2-phenyl group directed toward the more open TOP-derived peptide-binding channel (Fig. 2(B)).
000 drug-like small molecules.48 In this way we identified the imidazopyrazine nucleus as a TDP1-binding motif. Further structural variation of the core heterocycle skeleton using one-pot Groebke-Blackburn-Bienayme multicomponent reactions (GBBRs) identified 2-phenylimidazo[1,2-a]pyridines (1 and 2) having higher inhibitory potencies (Fig. 2(A)). The crystal structure of 3 bound to TDP1 (PDB code: 6W7K) showed that it replicates important interactions with the catalytic phosphate-binding pocket as previously shown by our structurally more simple phthalic acid-containing inhibitors, such as 4-aminophthalic acid.47 However, while the phthalic acid moiety anchors the SMM-derived inhibitor within the phosphate-binding pocket, the additional 2-phenylimidazo[1,2-a]pyridine nucleus is situated above the phthalic acid phosphate mimic, with the pyridine ring directed toward the DNA substrate-binding region and the 2-phenyl group directed toward the more open TOP-derived peptide-binding channel (Fig. 2(B)).
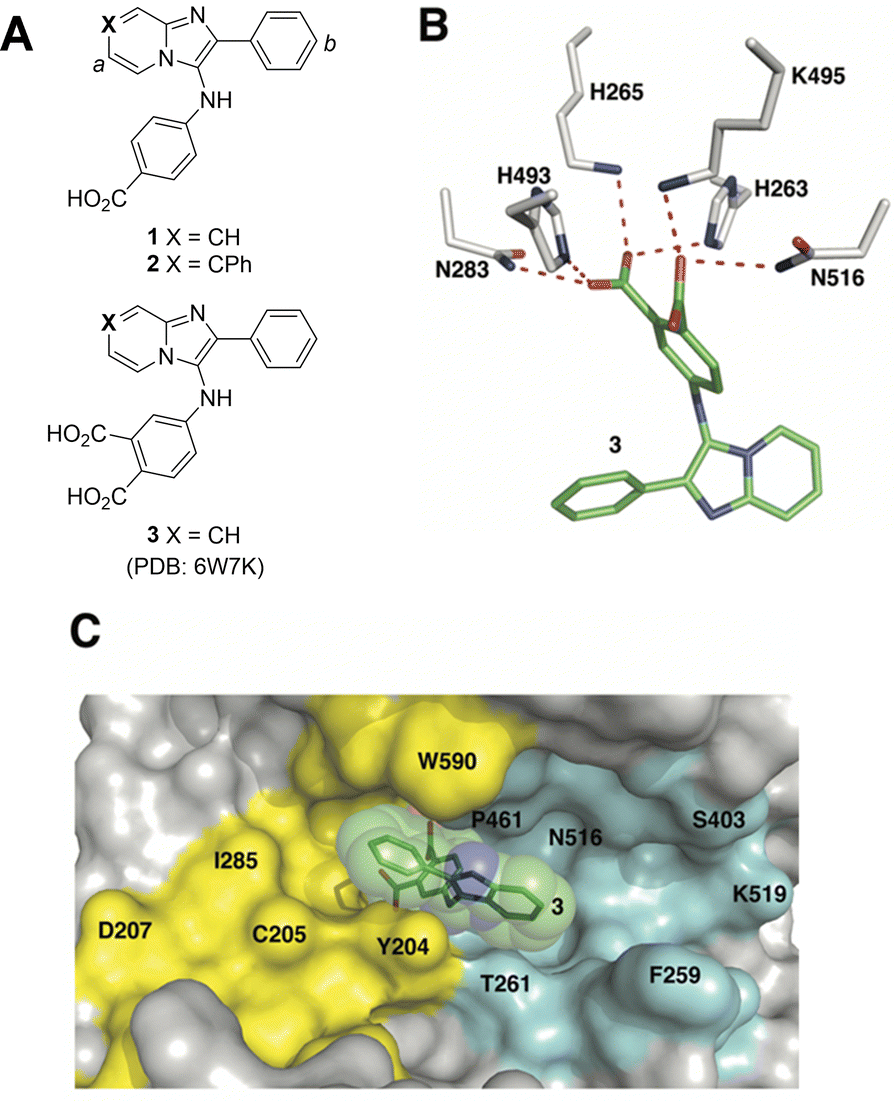 | ||
| Fig. 2 Structures of TDP1 inhibitors and their binding modes. (A) Structures of the lead TDP1 inhibitors 1–3. (B) Structure of TDP1 (HKN motif residues are shown as sticks with carbon atoms in gray) complexed to small molecule inhibitor 3 (carbon atoms in green) with hydrogen bonding interactions shown as red dotted lines (PDB code: 6W7K). (C) Small molecule TDP1 inhibitor 3 (carbon atoms are shown in green with transparent green sphere) in the binding regions of the DNA substrate (cyan surface) and a TOP1-derived peptide (in yellow surface) (PDB code: 1NOP). | ||
The 2-phenylimidazo[1,2-a]pyridine nucleus represents a platform for construction of trivalent ligands that could potentially simultaneously access the catalytic phosphate-binding pocket and the neighbouring DNA and peptide-binding channels (Fig. 2(C)). This is significant, since multivalency affords a well-known means of achieving significant enhancement in binding affinity.42,43 The objective of our current study was to install functionality into the parent platform that would extend into the substrate-binding channels. We were particularly interested in utilizing approaches that would be amenable to parallel structural diversification in microtiter format. “Click chemistry” was coined by Sharpless more than 20 years ago to describe a genre of high-yield reactions capable of facilely ligating building blocks using simple reactions, benign solvents and readily available starting materials to produce modular products that require simple isolation techniques.49,50 Oxime ligation is a form of click chemistry in which aminooxy-containing molecules react with aldehyde or ketone-containing molecules to join two reactants together through oxime bonds. The oxime bonds are typically biocompatible and stable at neutral pH.51–54 We have found that oxime diversification strategies are powerful approaches to rapidly generate libraries of tethered fragments.55–58 In contrast to imines, which can undergo rapid hydrolysis in aqueous solutions,59 oxime products are chemically stable and can be stored for extended periods without degradation.52,54,55 In our current paper, we report application of an oxime-based diversification strategy to prepare a microtiter library of more than 500 analogs having functionality appended onto the 2-phenyl and 7-phenyl rings of the parent imidazo[1,2-a]pyridine nucleus (Ra and Rb groups in general structure 4, Fig. 3(A)). We directly screened the reaction products at two concentrations in in vitro gel-based TDP1 inhibition assays. Secondary assays having more data points were run for promising analogs. The added new functionality introduces projections into the DNA and peptide substrate-binding channels.
 | ||
| Fig. 3 Schematic overview of oxime-diversification workflow. (A) Three steps of diversification are indicated in blue: (1) diversification of aminooxy-labelled compounds (5 and 6) by parallel conversion to oximes (5-Y and 6-Y) by reacting with aldehydes (Y-CHO) in a 96-well plate format; (2) screening of the oxime library (5-Y and 6-Y) using a in vitro gel-based TDP1 fluorescence assay to identify lead oximes (5-Y′ and 6-Y′); (3) conversion of lead oximes to triazoles (7-Y′) and ether-based isosteres (8-Y′). (B) Inhibitory screen of oximes at a single concentration of 100 μM in an in vitro gel-based TDP1 fluorescence assay. A total of 19 oximes in the 5-Y series library and 47 oximes in the 6-Y series library showed greater than 90% inhibition (in orange). (C) Structures of hit aldehyde precursors from primary screen. | ||
This work resulted in the identification of oximes that exhibit submicromolar TDP1 inhibitory potencies. X-ray crystal structures of lead compounds bound to TDP1 show that these small molecules can form hydrogen bonds with the catalytic HKN motifs while simultaneously extending into both the DNA substrate- and TOP1 peptide-binding grooves. This validates our original design hypothesis and provides the first trivalent platforms that can access the catalytic site and DNA- and TOP1 peptide substrate-binding regions. The multivalent nature of these interactions provides a model for developing more efficient TDP1-binding ligands.
Results and discussion
Overview of the oxime-diversification strategy
We employed a click-based oxime diversification similar to what we have previously successfully applied to optimize ligands directed at a variety of biological targets (Fig. 3).55–58 Keys to this approach are that reactions can be run in DMSO with a slight (5-fold) molar excess of acetic acid and that products are formed in high yield of sufficient purity for direct biological evaluation without purification. This required the availability of the aminooxy-labelled imidazopyridines 5 and 6, which we prepared using one-pot GBBR synthetic protocols.60–63 Synthetic details are presented in the ESI† (Schemes S1 and S2). We anticipated that positioning of aminooxy handles on 5 and 6 would allow the product oxime fragments to project into the substrate-binding channels. The aminophthalic acid moiety appended to the 3-amino substituent on the imidazo[1,2-a]pyridine nucleus should nestle securely within the central phosphate-binding catalytic pocket (Fig. 2(C)).Oxime ligation of the aminooxy substituents was accomplished using the following strategy: (1) The aminooxy-labelled precursors (5 and 6, 30 mM in DMSO) were reacted with a library of aldehydes A1-T12 (Y-CHO, 30 mM in DMSO; approximately 250 aldehydes as shown in Table S1, ESI†) in the presence of acetic acid (150 mM) to yield a final library of oximes (5-Y and 6-Y, 10 mM in DMSO), which were directly evaluated against TDP1 without purification (Fig. 3(A)). (2) All library members were diluted by adding TDP1 reaction buffer (50 mM tris–HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg mL−1 bovine serum albumin and 0.01% Tween 20) to yield 100 μM solutions and then screened in an in vitro gel-based fluorescence assay to measure inhibitory activity against TDP1 (1 nM DNA Cy5N14Y, 40 pM TDP1).47,48 Compounds that showed promising inhibition (>90% inhibition at 100 μM) were subjected to re-evaluation at three different concentrations (1 μM, 10 μM, 100 μM) (Fig. 3(B), Tables S2, S3 and Fig. S1, S2, ESI†). A total of 19 oximes in the 5-Y series library and 47 oximes in the 6-Y series library showed good (>90%) inhibition. Notably, the oxime products in both 5- and 6-series resulting from reactions with the same aldehydes having 2-phenylpyridine moieties (D1, H1 and T12) showed reproducibly good TDP1 inhibition (>90%) in repeat tests (Fig. 3(C)). Oximes 6-B7 and 6-E6 also showed good inhibition (>90%). We conducted a secondary screen of the lead oximes at three different drug concentrations (1 μM, 10 μM, and 100 μM) in gel-based TDP1 inhibition assays (Fig. S2, ESI†). Oximes from aldehydes B7, D1, E6, M10, and P3 showed TDP1 inhibition between 10 μM and 100 μM in this secondary screen (Fig. S2, ESI†). (3) A sub-group of oximes (5-Y′ and 6-Y′) was then subjected to HPLC purification and structure confirmation by NMR and mass spectrometry. For a select set of promising compounds as based on the structures of aldehyde hits (Fig. 3(C)), we replaced the oxime groups with isosteric triazole or ether moieties (7-Y′ and 8-Y′, respectively, Fig. 3(A)).
Biological evaluation of oxime leads
Based on the results of the preliminary screens, we prepared the most promising oximes on a larger scale, and the products were subjected to HPLC purification (5-D1, 5-P3, 6-D1, 6-E6, 6-B7, 6-P3, 6-M10; Table 1 and Fig. S3, ESI†). During the HPLC purification, we identified two products having different retention times but with identical molecular weights as shown by liquid chromatography–mass spectrometry (LC–MS). The major and minor products were consistently present in an approximate 95:5 ratio based on absorbance at 254 nm. We assigned the major product as the (E)-oxime isomer and the minor product as the (Z)-oxime isomer due to anticipated thermodynamic stabilities of the oxime double bond (Table 1). We evaluated the inhibitory potencies of both isomers using TDP1 fluorescence inhibition assays (Table 1).64–66 Compounds 5-D1, 6-D1, 6-E6, and 6-B7 showed single-digital micromolar TDP1 inhibitory potencies. For the 5-series oximes, the major (E)-5-D1 isomer (IC50 = 17.4 ± 3.2 μM) showed greater potency than the minor (Z)-5-D1 isomer (IC50 = 50.3 ± 10.7 μM). However, this represents an approximate 2-fold loss in potency relative to that of the parent compound 1 (IC50 = 7.87 ± 2.24 μM). A minor structural change to yield 5-P3 (IC50 > 100 μM) led to a significant decrease in TDP1 inhibitory potency. Among the 6-series oximes, the (E)-isomers of 6-D1 and 6-E1 showed approximately 10-fold higher inhibitory potencies than the corresponding (Z)-isomers. In the 6-series oximes 6-B7, 6-P3 and 6-M10, the (Z)-isomers showed slightly better inhibitory potencies than their corresponding (E)-isomers. Several oximes, including (E)-6-D1, (Z)-6-D1, (E)-6-E6 and (Z)-6-B7, showed single-digital inhibitory potencies, with (E)-6-E6 exhibiting an IC50 value of 3.1 ± 0.5 μM. The lead oxime (E)-6-D1 displayed a potency (IC50 = 0.38 ± 0.06 μM) that was 10-fold greater than that of the parent compound 2 (IC50 = 2.98 ± 0.24 μM). Based on these results, we selected (E)-6-D1 for isostere replacement.
|
|
||||
|---|---|---|---|---|
| Compound | X | Structure (–CH2ON![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHY′) CHY′) |
Isomer | TDP1 IC50b (μM) |
| a TDP1 inhibition of unpurified reaction mixtures (see Tables S2 and S3, ESI) and HPLC-purified oxime isomers. b The half maximal inhibitory concentration (IC50) values were evaluated with an in vitro gel-based TDP1 fluorescence assay. | ||||
| 5-D1 | CH |
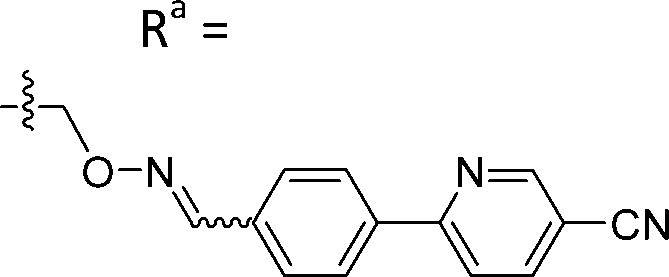
|
Mixturea | 94% |
| E | 17.4 ± 3.2 | |||
| Z | 50.3 ± 10.7 | |||
| 5-P3 | CH |
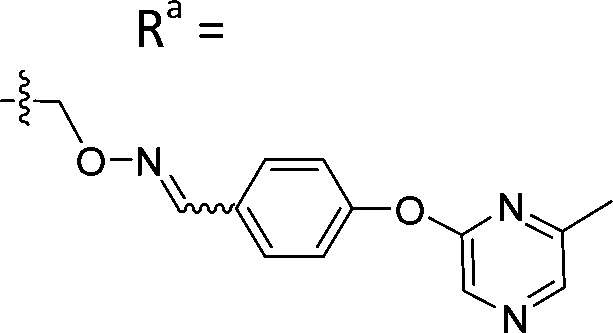
|
Mixturea | 111% |
| E | >100 | |||
| Z | >100 | |||
| 6-D1 | CPh |
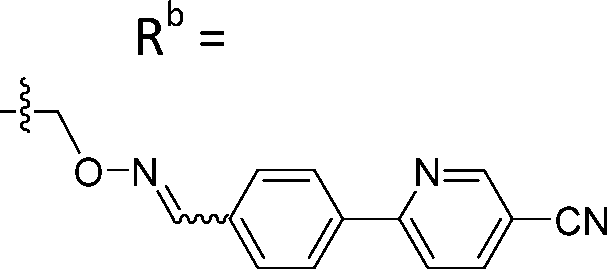
|
Mixturea | 99% |
| E | 0.38 ± 0.06 | |||
| Z | 5.65 ± 3.29 | |||
| 6-E6 | CPh |
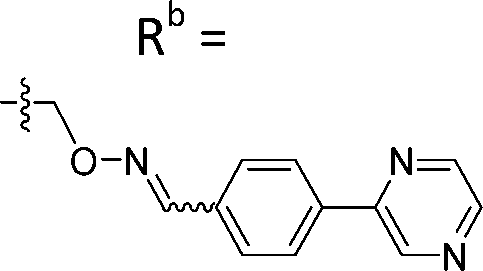
|
Mixturea | 102% |
| E | 3.1 ± 0.5 | |||
| Z | 65.3 ± 17.8 | |||
| 6-B7 | CPh |
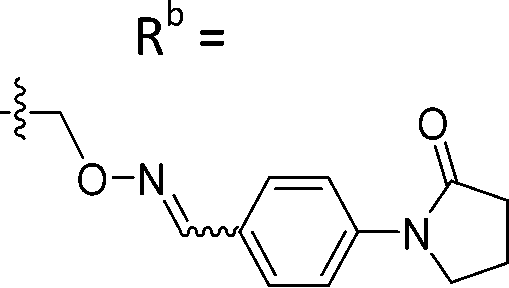
|
Mixturea | 91% |
| E | 10.2 ± 0.33 | |||
| Z | 7.67 ± 2.3 | |||
| 6-P3 | CPh |
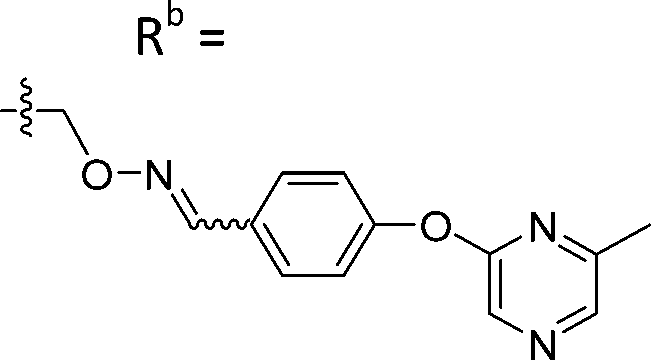
|
Mixturea | 111% |
| E | 76.3 ± 41.9 | |||
| Z | 54.2 ± 18.7 | |||
| 6-M10 | CPh |
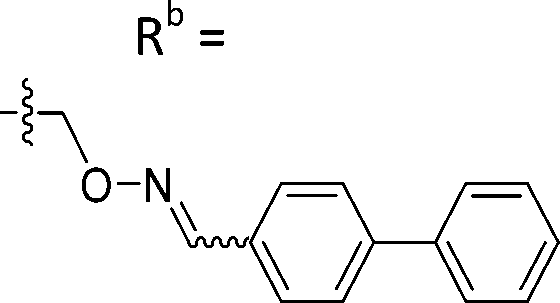
|
Mixturea | 36% |
| E | 67.7 ± 17.6 | |||
| Z | 63.1 ± 27.5 | |||
| 6-X | CPh |
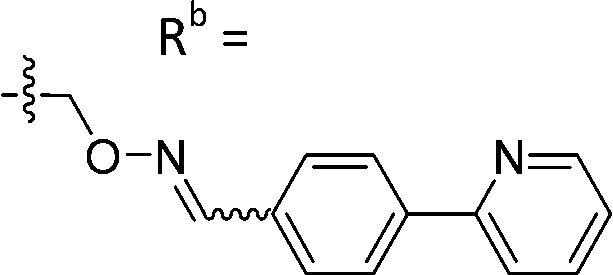
|
Mixturea of E/Z = 6/1 | 73 ± 11 |
| 1 | CH | Ra = Rb = H | — | 8.72 ± 1.81 |
| 2 | CPh | Ra = Rb = H | — | 2.98 ± 0.24 |
Isosteric replacement of the linker functionality
Having identified preferred aryl fragments (Ra and Rb, Table 1), we replaced oxime-based linker segments with triazole and ether functionalities (Table 2). Heterocycles can serve as useful isosteres and triazoles have particular utility, because of their chemical stability and the fact that they can be readily formed by Husigen copper catalysed alkyne-azide [3+2] cycloaddition (CuAAC) reactions.49,67,68 To replace the (E)-oxime linkage in lead (E)-6-D1, we prepared a series of triazole-linked analogs 7a–e using CuAAC reactions (Scheme S3, ESI†).69 We also prepared the ether-linked analogs 8a, b and the phthalic acid-containing imidazopyridines 9a–c with and without the ether-linked biaryl ring (synthetic details in Schemes S3–S5, ESI† and Table 2).|
|
||||
|---|---|---|---|---|
| Compound | X | Y | Structure | TDP1 IC50a (μM) |
| a Half maximal inhibitory concentration (IC50) values were evaluated with an in vitro gel-based TDP1 fluorescence assay. | ||||
| 7a | CH | H |
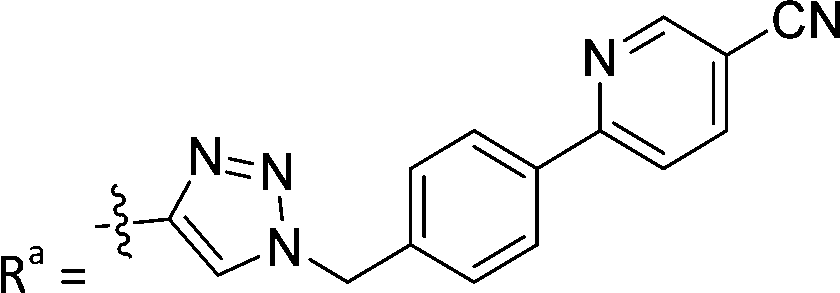
|
66.4 ± 3.7 |
| 7b | CH | H |
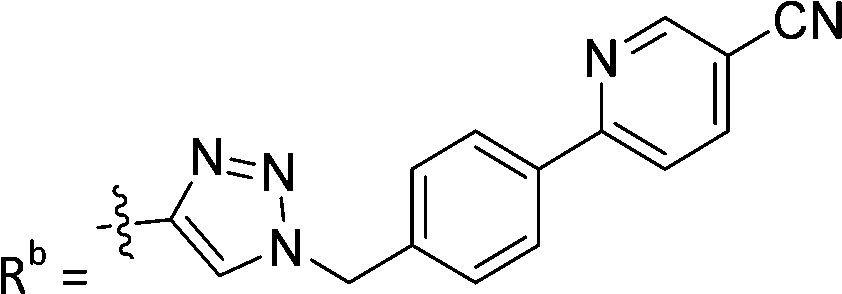
|
21.6 ± 5.0 |
| 7c | CH | H |
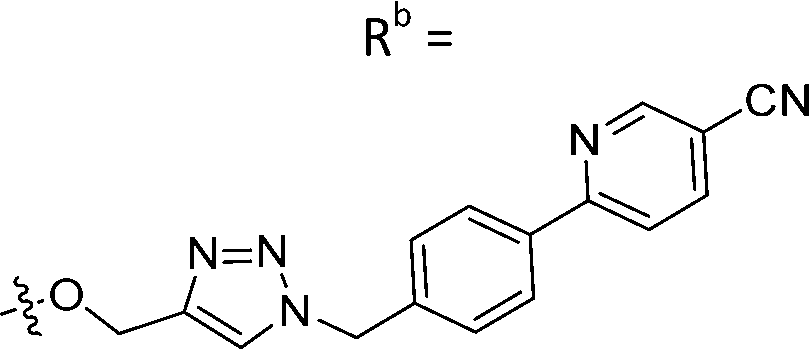
|
60 ± 17 |
| 7d | CPh | H |
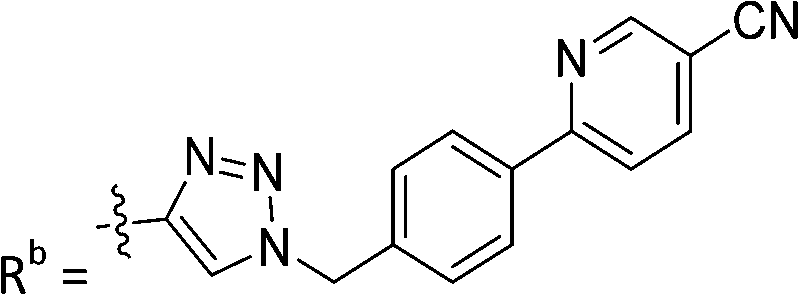
|
3.1 ± 0.2 |
| 7e | CPh | H |
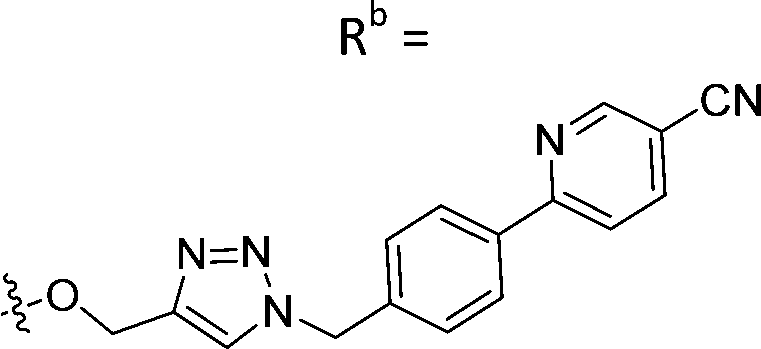
|
23.5 ± 3.2 |
| 8a | CH | H |
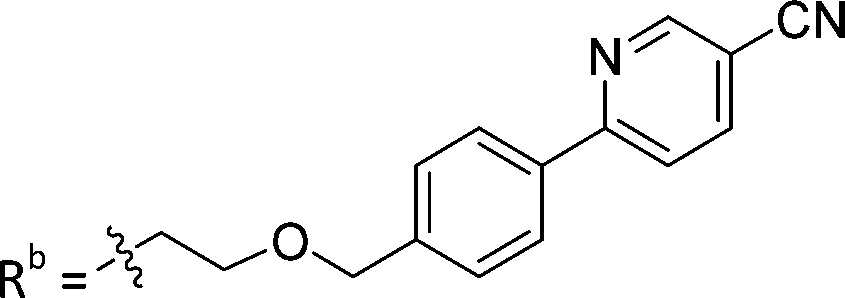
|
25.1 ± 1.25 |
| 8b | CPh | H |
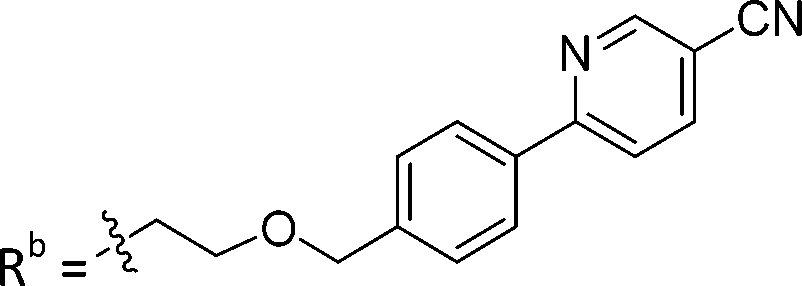
|
2.75 ± 0.25 |
| 9a | CPh | CO2H | Rb =H | >100 |
| 9b | CPh | CO2H |
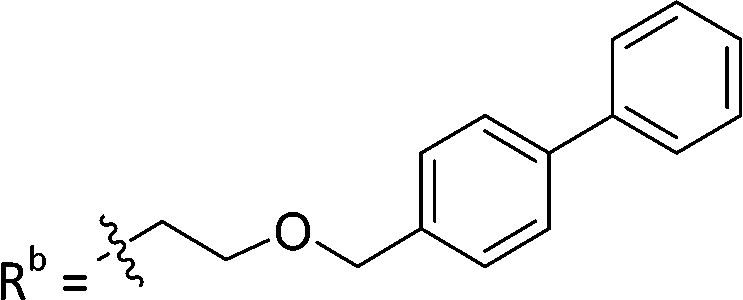
|
11.3 ± 1.93 |
| 9c | CPh | CO2H |
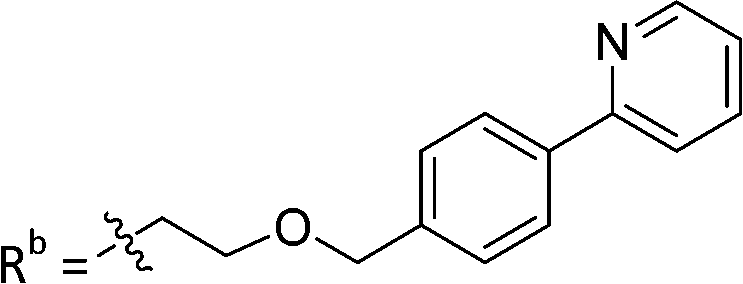
|
19.8 ± 1.35 |
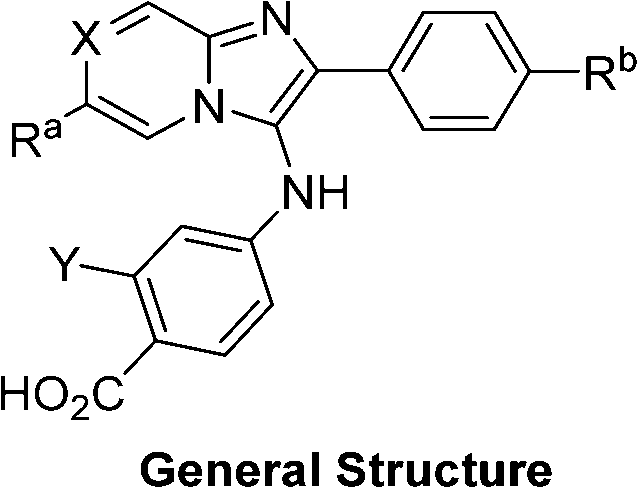
While keeping the phenylpyridine skeleton from the oxime lead (E)-6-D1, we prepared a series of analogs with triazole (7a–e) and ether (8a, b) linkers (Table 2, Schemes S3, S4 and Fig. S4, ESI†). Among compounds with triazole linkers on the “left side” (Ra), 7a showed poor TDP1 inhibition (IC50 = 66.4 ± 3.7 μM). Placement of the triazole on the “right side” (Rb) gave 7b (IC50 = 21.6 ± 5.0 μM), indicating that the compound is 3-fold more potent than 7a. Increasing the linker length in 7b reduced inhibitory potency (7c, IC50 = 60 ± 17 μM) compared to that of 7b, while inclusion of a 4-phenyl group further increased the inhibitory potency (7d, IC50 = 3.3 ± 0.2 μM) compared to that of 7b. Additional extension of the linker length in 7d by one methylene unit resulted in an 8-fold decrease in potency (7e, IC50 = 23.5 ± 3.2 μM). Replacement of the triazole linker in 7b with an ether linker of similar extension length (8a, IC50 = 25.1 ± 1.25 μM) had little effect on inhibitory potency (7b, IC50 = 21.6 ± 5.0 μM). Replacement of the triazole linker in 7d with an ether linker of similar extension length (8b, IC50 = 2.75 ± 0.25 μM) also had little effect on inhibitory potency (7d, IC50 = 3.3 ± 0.2 μM). Overall, we found that compounds with analogous linker lengths exhibited comparable inhibitory potencies, whereas increasing the linker extension length tended to reduce inhibitory potency. Meanwhile, the inclusion of a phenyl group at the Ra position enhanced inhibitory potency. Lead compounds, including oxime (E)-6-D1, triazole 7d and ether 8b, were selected to evaluate their TDP1 selectivity over TDP2 using in vitro gel-based assays (Table S5 and Fig. S5, ESI†). Oxime (E)-6-D1 shows 74-fold greater inhibitory potency against TDP1 than against TDP2. Ether 8b is also 36-fold more potent against TDP1 than against TDP2. Potencies of triazole 7d are similar, however an 8-fold enhancement was observed against TDP1 relative to TDP2. In the HCT116 human colon cancer cell line, oxime (E)-6-D1, triazole 7d and ether 8b showed cytotoxicity CC50 values of 4.8 μM, 136 μM, and 4.3 μM, respectively (Fig. S6, ESI†). The CC50 values are higher than the TDP1 in vitro IC50 values. At concentrations below their CC50 values, compounds (E)-6-D1, 7d and 8b were shown to act synergistically with the TOP1 inhibitor CPT using HCT116 cells (Fig. S6 and S7, ESI†). These data are consistent with the compounds selectively targeting TDP1.
X-ray crystallography
As reported above, through an oxime diversification strategy, we arrived at molecules with TDP1 inhibitory potencies in the low micromolar to nanomolar range (6-D1, IC50: 0.38 μM; 7d, IC50: 3.3 μM; 8b, IC50: 2.75 μM). Obtaining crystal structures of small molecule ligands bound to TDP1 has been proven to be extremely challenging. We have previously observed that the crystallization of complexes consisting of ligands bound within the TDP1 catalytic pocket is favored by an aryl 3,4-dicarboxyl substituent pattern.47,48 Therefore, to enhance crystallization, we added a second carboxyl group to the phenyl-imidazo[1,2-a]pyridine platform to yield the parent phthalic acid-containing compound 9a. The corresponding ether linker-modified analogs 9b and 9c were also prepared (Table 2 and Scheme S5, ESI†). We found that while analogs 9b and 9c exhibited micromolar TDP1 inhibitory potencies (IC50 = 11.3 ± 1.93 μM and 19.8 ± 1.35 μM, respectively) the parent compound 9a lacking the linked biaryl fragment was significantly less potent (IC50 > 100 μM, Table 2).We obtained high-resolution crystal structures of the TDP1 catalytic domain in complex with compounds 9a and 9c at 1.65 Å and 1.81 Å resolution, respectively (Fig. 4 and Table S4, ESI†). As observed in our previous crystal structures of TDP1 bound to imidazopyridine-based inhibitors containing dicarboxylate functionalities,47,48 we found that compound 9a binds to the active site pocket with the carboxylate groups engaged in direct hydrogen bonds with the catalytic residues H263, K265, N283, H493, K495, and N516 (Fig. 4(A)). An additional hydrogen bond interaction is observed with the side chain oxygen atom of S399. A bound water molecule, Wa1019, mediates a water-bridged hydrogen bond between the oxygen atom of one carboxylate group on compound 9a and the amine nitrogen of the K265 side chain. The phenylimidazopyridine platform of 9a is positioned such that the phenyl rings at the 3- and 7-positions protrude into the peptide- and DNA-binding pockets, respectively. Y204 interacts with the inhibitor via a hydrogen bond between the side-chain hydroxyl oxygen atom and the amine nitrogen linker of compound 9a, and the side chain is positioned such that it also forms an edge-to-face (“T-shape”)70–72 π–π interaction (4.3 Å) with the phenyl ring at the 3-position of the imidazopyridine, which is positioned within the peptide-binding pocket of TDP1. Typically, the preferred π–π interaction between Trp and the phenyl ring of Phe occurs through the T-shape, with the phenyl ring being edge-to-face with the indole ring.73,74 An additional T-shape π–π interaction (4.8 Å) is observed between the side chain phenyl ring of W590 and the 3-position phenyl ring of the imidazopyridine. The phenyl ring at the 7-position of the imidazopyridine of compound 9c extends into the DNA-binding pocket of TDP1 and interacts with the side chain phenyl ring of F259 via a parallel-displaced π–π interaction (5.3 Å).75 Serendipitously, a MOPS buffer molecule from the crystallization solution is also bound in the active site and is nestled between the imidazopyridine moiety and residues P461 and W590.
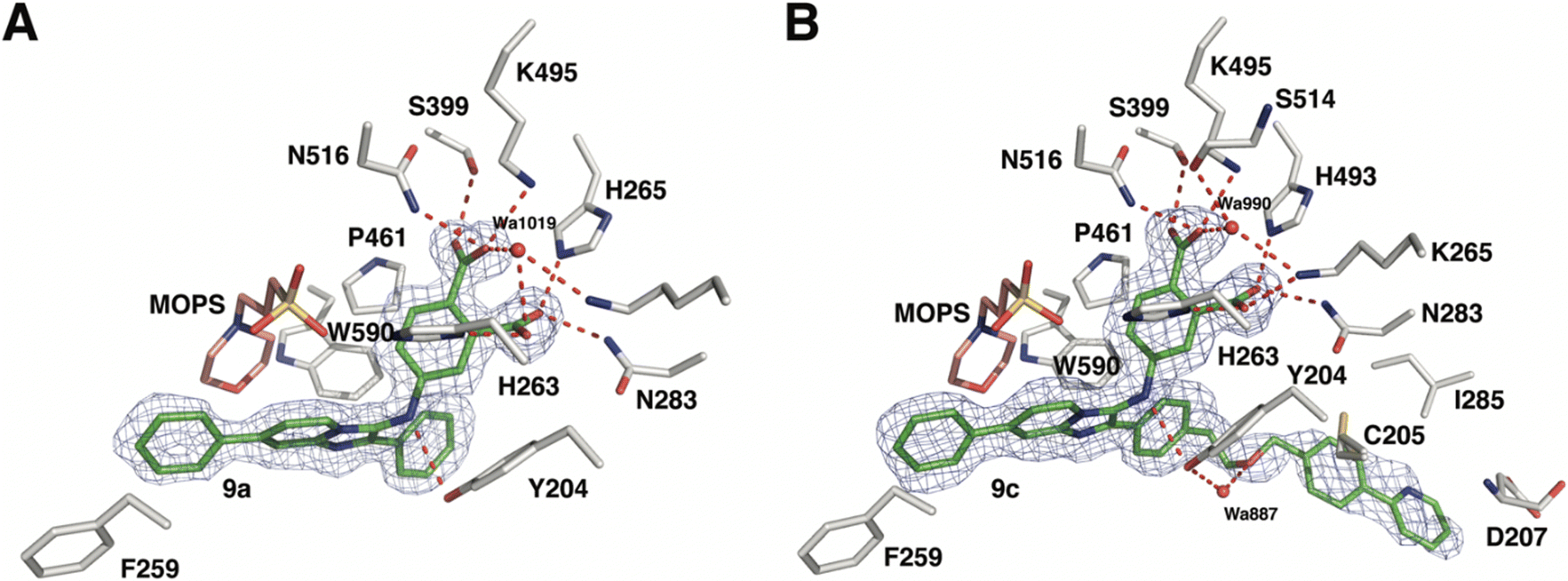 | ||
| Fig. 4 Crystal structures of TDP1 bound to compounds 9a and 9c. (A) The active site of TDP1 (gray sticks) from the crystal structure of TDP1 bound to compound 9a (PDB code: 8CVQ). The fit of compound 9a (green sticks) to the final 2Fo − Fc electron density map (blue mesh, 1.65 Å resolution, contoured at 1.0 rmsd) is shown. (B) The active site of TDP1 (gray sticks) from the crystal structure of TDP1 bound to compound 9c (PDB code: 8CW2). The fit of compound 9c (green sticks) to the final 2Fo − Fc electron density map (blue mesh, 1.81 Å resolution, contoured at 0.7 rmsd) is shown. | ||
Clear electron density was observed in the active site of chain A in the crystal structure of TDP1 soaked with compound 9c that allowed the placement of the entire molecule of 9c. However, only partial electron density for compound 9c was observed in chain B. Chain A-bound 9c interacts in the same manner as compound 9a with the catalytic residues. In addition, Wa990 mediates water-bridged hydrogen bonds between the oxygen of one of the carboxylates of compound 9c with the backbone carbonyl oxygen atom of S399 and the amide nitrogen of the K265 side chain. The π–π interactions between residues Y204, P461 and W590 and the imidazopyridine are also maintained and the side chain of Y204 forms a hydrogen bond to the ether oxygen of 9c. The appended ether-linked phenylpyridine moiety extends into the peptide-binding pocket and interacts with the residues lining this pocket, primarily via hydrophobic interactions with the side chains of the nonpolar residues C205 and I285 and the aliphatic portion of the D207 side chain. The side chain thiol of C205 also interacts with the phenyl of the phenylpyridine via a thiol–π interaction (3.2 Å).76 The backbone amide of D207 forms an amide–π interaction77 with the pyridine of the phenylpyridine. The carboxylic acid side chain of D207 is also within close distance (3.3 Å) to the 5-carbon of the terminal pyridine, which provides a potential hydrogen bond, possibly between the nitrile in (E)-6-D1 and D207.
Structural basis of inhibition
To gain insights into the structural basis by which compounds 9a and 9c exert their inhibitory effect, the coordinates for each complex were superimposed onto the coordinates of TDP1 bound to a TOP1-derived peptide, vanadate, and a DNA substrate; this structure mimics the transition state in the first step of the TDP1 catalytic reaction (Fig. 5). The active site structures of TDP1 bound to 9a and 9c are highly similar. For simplicity, only the residues from the TDP1–9c complex are shown superimposed onto the 1NOP coordinates (Fig. 5). The core phenyl-dicarboxylate and phenyl-imidazopyridine scaffold of compounds 9a and 9c are essentially in the same position in the overlay. Rotamer shifts are observed for the catalytic residues H263, K265, and K495 when compound 9c is bound, which results in proper positioning for hydrogen bonding interactions to the dicarboxylate head group. The 7-phenyl ring of the imidazopyridine platform extends into the DNA binding pocket and as a result there is a rotamer shift of residues F259 in order to accommodate the biding of the phenyl ring. In the 1NOP coordinates, F259 is involved in a base stacking interaction with the guanine 804. The appended phenyl ring at the 3-position induces rotamer shifts of the side chains of the neighboring residues Y204 and W590 to optimize their side chain positions for proper hydrogen bonding and π–π interactions with 9c. Extension of the ether-linked phenylpyridine moiety into the peptide-binding pocket allows for several stabilizing hydrophobic interactions.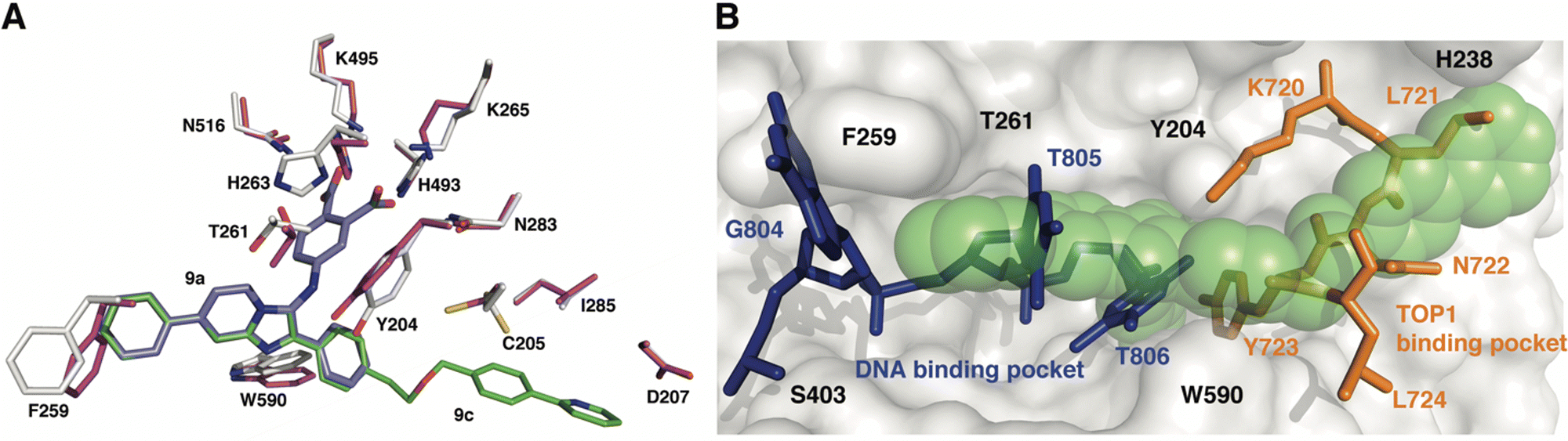 | ||
| Fig. 5 Structure comparison of TDP1–9c complex with the TDP1–vanadate complex. (A) Overlay of the structure of the TDP1–9c complex (PDB code: 8CW2), carbon atoms in gray, compound 9c (carbon atoms in green) onto the coordinates of TDP1 (carbon atoms in magenta) bound to a TOP1-derived peptide and DNA (PDB code: 1NOP). The position of compound 9a from the TDP1-9a complex (PDB code: 8CVQ) is shown in cyan sticks. (B) Surface representation of the active site of TDP1 bound to compound 9c (represented in green spheres). The position of the DNA substrate (blue sticks) and the TOP1-derived peptide (orange sticks) is shown based on superimposition onto the 1NOP coordinates. | ||
We previously demonstrated that the phenyl-dicarboxylate headgroup of the bound compound 9c (green spheres) mimics the position of the phosphoryltyrosine substrate (Fig. 5(B)). The imidazopyridine and phenyl ring at the 7-position occupy the positions of thymidine bases 805 and 806, thus highlighting the importance of this moiety in blocking the binding of the DNA substrate in the active site. The 3-phenyl ring and the ether-linked phenylpyridine moiety, which extend into the peptide-binding pocket, overlap with residues Y721, N722, Y723, and L724 residues of the TOP1-derived peptide. This highlights the structural importance of this moiety in blocking the binding of the peptide residues. The X-ray structure of the TDP1–9c complex provides structural evidence that compound 9c serves as a tridentate inhibitor of TDP1.
Conclusions
Starting with small molecule microarray-originated imidazopyridine-based TDP1 inhibitors, we utilized a “click”-based oxime diversification protocol to screen a library of aldehyde fragments. This resulted in an initial set of structures that extend the parent imidazopyridine platform to the DNA substrate- and TOP1 peptide-binding channels. The lead oxime (E)-6-D1 shows submicromolar TDP1 inhibition. For a subset of analogs, we replaced oxime linkages with triazole and ether isosteres. Lead compounds, including oxime (E)-6-D1, triazole 7d and ether 8b inhibit TDP1 with selectivity over TDP2 in gel-based in vitro assays and they synergize with the TOP1 inhibitor CPT in assays using HCT116 cells. X-ray crystal structures of TDP1 bound to the lead compounds 9a and 9c reveal that these small molecules can form hydrogen bonds with the catalytic HKN motifs, while simultaneously extending into both the DNA substrate- and TOP1 peptide-binding grooves. Our findings identify new ways in which small molecules can engage the catalytic pocket, while simultaneously interacting with the DNA substrate- and TOP1 peptide-binding channels. The trivalent nature of these interactions may provide a basis for developing more efficient multivalent TDP1 inhibitors, which could serve as a new genre of anticancer chemotherapeutics.Author contributions
XZ and TB conceived the study. XZ designed and synthesized the compounds. WW and EK performed the biological studies. GL performed the X-ray crystallography work. DN performed protein expression work. JT performed protein purification. XZ, WW, GL, EK, YP and TB interpreted the data. XZ, GL, and TB took the lead in writing the manuscript. All authors have provided critical feedback and approved the final manuscript.Conflicts of interest
Aspects of the work presented are contained within one or more patent applications. The authors declare that there is no conflict of interest.Acknowledgements
This work was supported in part by Staff Scientist/Staff Clinician Research Award (SSSC-RA) and the NIH Intramural Program, Center for Cancer Research, National Cancer Institute, National Institutes of Health (Z01-BC 006150 and Z01-BC 006198) and the Frederick National Laboratory for Cancer Research, National Institutes of Health under contract 75N91019D00024 (this contract number represents work performed within the scope of the severable FFRDC Bridge contract). We thank James A. Kelley and Christopher C. Lai (Chemical Biology Laboratory, NCI, NIH) for HRMS data collection. We thank the Biophysics Resource in the Center for Structural Biology, NCI at Frederick for use of the LC/ESMS instrument. X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory. SER-CAT is supported by its member institutions and equipment grants (S10_RR25528, S10_RR028976 and S10_OD027000) from the National Institutes of Health. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. W-31-109-Eng-38. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products or organizations imply endorsement by the US Government.Notes and references
- Y. Pommier, Y. Sun, S.-Y. N. Huang and J. L. Nitiss, Nat. Rev. Mol. Cell Biol., 2016, 17, 703–721 CrossRef CAS PubMed.
- F. Goldwasser, I. Bae, M. Valenti, K. Torres and Y. Pommier, Cancer Res., 1995, 55, 2116–2121 CAS.
- Y. Pommier, Nat. Rev. Cancer, 2006, 6, 789–802 CrossRef CAS PubMed.
- S. M. Cuya, M.-A. Bjornsti and R. C. A. M. van Waardenburg, Cancer Chemother. Pharmacol., 2017, 80, 1–14 CrossRef CAS PubMed.
- Y. Pommier and M. Cushman, Mol. Cancer Ther., 2009, 8, 1008–1014 CrossRef CAS PubMed.
- A. Thomas and Y. Pommier, Clin. Cancer Res., 2019, 25, 6581 CrossRef CAS PubMed.
- M. Alagoz, D. C. Gilbert, S. El-Khamisy and A. J. Chalmers, Curr. Med. Chem., 2012, 19, 3874–3885 CrossRef CAS PubMed.
- J. Heo, J. Li, M. Summerlin, A. Hays, S. Katyal, P. J. McKinnon, K. C. Nitiss, J. L. Nitiss and L. A. Hanakahi, DNA Repair, 2015, 30, 28–37 CrossRef CAS PubMed.
- H. K. Fam, M. K. Chowdhury, C. Walton, K. Choi, C. F. Boerkoel and G. Hendson, J. Mol. Histol., 2013, 44, 481–494 CrossRef CAS PubMed.
- S.-W. Yang, A. B. Burgin, B. N. Huizenga, C. A. Robertson, K. C. Yao and H. A. Nash, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11534–11539 CrossRef CAS PubMed.
- J. Murai, S.-y N. Huang, B. B. Das, T. S. Dexheimer, S. Takeda and Y. Pommier, J. Biol. Chem., 2012, 287, 12848–12857 CrossRef CAS PubMed.
- A. S. Kawale and L. F. Povirk, Nucleic Acids Res., 2018, 46, 520–537 CrossRef CAS PubMed.
- A. Zakharenko, N. Dyrkheeva and O. Lavrik, Med. Res. Rev., 2019, 39, 1427–1441 CrossRef CAS PubMed.
- T. S. Dexheimer, S.-y N. Huang, S. Antony, C. Marchand and Y. Pommier, Adv. Anticancer Agents Med. Chem., 2013, 2, 444–471 CAS.
- S. S. Laev, N. F. Salakhutdinov and O. I. Lavrik, Bioorg. Med. Chem., 2016, 24, 5017–5027 CrossRef CAS PubMed.
- E. J. Brettrager and R. C. A. M. van Waardenburg, Cancer Drug Resist., 2019, 2, 1153–1163 Search PubMed.
- E. Leung, J. Patel, J. A. Hollywood, A. Zafar, P. Tomek, D. Barker, L. I. Pilkington, M. van Rensburg, R. J. Langley, N. A. Helsby, C. J. Squire, B. C. Baguley, W. A. Denny, J. Reynisson and I. K. H. Leung, Oncol. Ther., 2021, 9, 541–556 CrossRef PubMed.
- Y. Zhang, Y. Li, C. Sun, X. Chen, L. Han, T. Wang, J. Liu, X. Chen and D. Zhao, Cancers, 2021, 13, 4002 CrossRef CAS PubMed.
- D.-X. Hu, W.-L. Tang, Y. Zhang, H. Yang, W. Wang, K. Agama, Y. Pommier and L.-K. An, J. Med. Chem., 2021, 64, 7617–7629 CrossRef CAS PubMed.
- O. V. Salomatina, I. I. Popadyuk, A. L. Zakharenko, O. D. Zakharova, A. A. Chepanova, N. S. Dyrkheeva, N. I. Komarova, J. Reynisson, R. O. Anarbaev, N. F. Salakhutdinov, O. I. Lavrik and K. P. Volcho, Steroids, 2021, 165, 108771 CrossRef CAS PubMed.
- N. S. Dyrkheeva, A. S. Filimonov, O. A. Luzina, A. L. Zakharenko, E. S. Ilina, A. A. Malakhova, S. P. Medvedev, J. Reynisson, K. P. Volcho, S. M. Zakian, N. F. Salakhutdinov and O. I. Lavrik, Biomolecules, 2021, 11, 973 CrossRef CAS PubMed.
- J. B. Baell, L. Ferrins, H. Falk and G. Nikolakopoulos, Aust. J. Chem., 2013, 66, 1483–1494 CrossRef CAS.
- D. A. Erlanson, J. Med. Chem., 2015, 58, 2088–2090 CrossRef CAS PubMed.
- J. B. Baell and J. W. M. Nissink, ACS Chem. Biol., 2018, 13, 36–44 CrossRef CAS PubMed.
- S. L. McGovern, E. Caselli, N. Grigorieff and B. K. Shoichet, J. Med. Chem., 2002, 45, 1712–1722 CrossRef CAS PubMed.
- J. L. Dahlin, J. W. M. Nissink, J. M. Strasser, S. Francis, L. Higgins, H. Zhou, Z. G. Zhang and M. A. Walters, J. Med. Chem., 2015, 58, 2091–2113 CrossRef CAS PubMed.
- R. N. Lindquist, J. L. Lynn and G. E. Lienhard, J. Am. Chem. Soc., 1973, 95, 8762–8768 CrossRef CAS PubMed.
- V. Lopez, T. Stevens and R. N. Lindquist, Arch. Biochem. Biophys., 1976, 175, 31–38 CrossRef CAS PubMed.
- W. Plass, Angew. Chem., Int. Ed., 1999, 38, 909–912 CrossRef CAS PubMed.
- D. R. Davies and W. G. J. Hol, FEBS Lett., 2004, 577, 315–321 CrossRef CAS PubMed.
- D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev., 2004, 104, 849–902 CrossRef CAS PubMed.
- A. Peck, F. Sunden, L. D. Andrews, V. S. Pande and D. Herschlag, J. Mol. Biol., 2016, 428, 2758–2768 CrossRef CAS PubMed.
- D. R. Davies, H. Interthal, J. J. Champoux and W. G. J. Hol, Structure, 2002, 10, 237–248 CrossRef CAS PubMed.
- D. R. Davies, H. Interthal, J. J. Champoux and W. G. J. Hol, J. Mol. Biol., 2002, 324, 917–932 CrossRef CAS PubMed.
- D. R. Davies, H. Interthal, J. J. Champoux and W. G. J. Hol, Chem. Biol., 2003, 10, 139–147 CrossRef CAS PubMed.
- D. R. Davies, H. Interthal, J. J. Champoux and W. G. J. Hol, J. Med. Chem., 2004, 47, 829–837 CrossRef CAS PubMed.
- F. J. Flett, E. Ruksenaite, L. A. Armstrong, S. Bharati, R. Carloni, E. R. Morris, C. L. Mackay, H. Interthal and J. M. Richardson, Nat. Commun., 2018, 9, 1–13 CrossRef CAS PubMed.
- H. Interthal, J. J. Pouliot and J. J. Champoux, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12009–12014 CrossRef CAS PubMed.
- N. Dyrkheeva, R. Anarbaev, N. Lebedeva, M. Kuprushkin, A. Kuznetsova, N. Kuznetsov, N. Rechkunova and O. Lavrik, Front. Cell. Develop. Biol., 2020, 8, 1427 Search PubMed.
- E. Q. Comeaux and R. C. A. M. van Waardenburg, Drug Metab. Rev., 2014, 46, 494–507 CrossRef CAS PubMed.
- A. C. Raymond, M. C. Rideout, B. Staker, K. Hjerrild and A. B. Burgin, J. Mol. Biol., 2004, 338, 895–906 CrossRef CAS PubMed.
- V. M. Krishnamurthy, L. A. Estrofi and G. M. Whitesides, in Fragment-based Approaches in Drug Discovery, ed. W. J. A. D. A. Erlanson, Wiley-VCH GmbH & Co., Weinheim, DE, 2006, pp. 11–53 Search PubMed.
- C. Chittasupho, Therap. Deliv., 2012, 3, 1171–1187 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- I. V. Hartung, B. R. Huck and A. Crespo, Nat. Rev. Chem., 2023, 7, 3–4 CrossRef.
- T. S. Elliott, A. Slowey, Y. Ye and S. J. Conway, MedChemComm, 2012, 3, 735–751 RSC.
- G. T. Lountos, X. Z. Zhao, T. R. Burke, E. Kiselev, Y. Pommier, J. E. Tropea, D. Needle and D. S. Waugh, Nucleic Acids Res., 2019, 47, 10134–10150 CrossRef CAS PubMed.
- X. Z. Zhao, E. Kiselev, G. T. Lountos, W. Wang, J. E. Tropea, D. Needle, T. A. Hilimire, J. S. Schneekloth, D. S. Waugh, Y. Pommier and T. R. Burke, Chem. Sci., 2021, 12, 3876–3884 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. King and A. Wagner, Bioconjugate Chem., 2014, 25, 825–839 CrossRef CAS PubMed.
- A. Bruylants and E. Feytmants-de Medicis, Carbon–Nitrogen Double Bonds, 1970, 465–504, DOI:10.1002/9780470771204.ch10.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem. – Eur. J., 2014, 20, 34–41 CrossRef CAS PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- M. Bahta, F. Liu, S.-E. Kim, A. G. Stephen, R. J. Fisher and T. R. Burke, Jr., Nat. Protoc., 2012, 7, 686–702 CrossRef CAS PubMed.
- X. Z. Zhao, D. Hymel and T. R. Burke, Jr., Bioorg. Med. Chem. Lett., 2016, 26, 5009–5012 CrossRef CAS PubMed.
- X. Z. Zhao, D. Hymel and T. R. Burke, Jr., Bioorg. Med. Chem., 2017, 25, 5041–5049 CrossRef CAS PubMed.
- X. Z. Zhao, T. R. Burke, Jr. and F. Liu, Molecules, 2020, 25, 2807 CrossRef CAS PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- H. Bienaymé and K. Bouzid, Angew. Chem., Int. Ed., 1998, 37, 2234–2237 CrossRef.
- C. Blackburn, B. Guan, P. Fleming, K. Shiosaki and S. Tsai, Tetrahedron Lett., 1998, 39, 3635–3638 CrossRef CAS.
- K. Groebke, L. Weber and F. Mehlin, Synlett, 1998, 661–663 CrossRef CAS.
- S. Shaaban and B. F. Abdel-Wahab, Mol. Diversity, 2016, 20, 233–254 CrossRef CAS PubMed.
- C. G. McCarty, Carbon–Nitrogen Double Bonds, 1970, pp. 363–464 DOI:10.1002/9780470771204.ch9.
- H. Sharghi and M. H. Sarvari, Synlett, 2001, 0099–0101 CrossRef CAS.
- J. Dhuguru, E. Zviagin and R. Skouta, Pharmaceuticals, 2022, 15, 66 CrossRef CAS PubMed.
- N. A. Meanwell, in Tactics in Contemporary Drug Design, ed. N. A. Meanwell, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 283–381 DOI:10.1007/7355_2013_29.
- M. S. Malik, S. A. Ahmed, I. I. Althagafi, M. A. Ansari and A. Kamal, RSC Med. Chem., 2020, 11, 327–348 RSC.
- R. P. Wurz, K. Dellamaggiore, H. Dou, N. Javier, M.-C. Lo, J. D. McCarter, D. Mohl, C. Sastri, J. R. Lipford and V. J. Cee, J. Med. Chem., 2018, 61, 453–461 CrossRef CAS PubMed.
- G. B. McGaughey, M. Gagné and A. K. Rappé, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS PubMed.
- S. E. Wheeler and K. N. Houk, Mol. Phys., 2009, 107, 749–760 CrossRef CAS PubMed.
- T. Chen, M. Li and J. Liu, Cryst. Growth Des., 2018, 18, 2765–2783 CrossRef CAS.
- U. Samanta, D. Pal and P. Chakrabarti, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, D55, 1421–1427 CrossRef CAS PubMed.
- A. Sengupta, R. Mahalakshmi, N. Shamala and P. Balaram, J. Pept. Res., 2005, 65, 113–129 CrossRef CAS PubMed.
- M. Brylinski, Chem. Biol. Drug Des., 2018, 91, 380–390 CrossRef CAS PubMed.
- C. R. Forbes, S. K. Sinha, H. K. Ganguly, S. Bai, G. P. A. Yap, S. Patel and N. J. Zondlo, J. Am. Chem. Soc., 2017, 139, 1842–1855 CrossRef CAS PubMed.
- M. W. Krone, C. R. Travis, G. Y. Lee, H. J. Eckvahl, K. N. Houk and M. L. Waters, J. Am. Chem. Soc., 2020, 142, 17048–17056 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00230b |
| This journal is © The Royal Society of Chemistry 2023 |