
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy and design of amino acid-based radical enzymes using unnatural amino acids
Feiyan
Yuan
a,
Binbin
Su
a,
Yang
Yu
 *a and
Jiangyun
Wang
*b
*a and
Jiangyun
Wang
*b
aInstitute of Biochemical Engineering, Key Laboratory of Medical Molecule Science and Pharmaceutical Engineering, Ministry of Industry and Information Technology, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 102488, China. E-mail: yang_yu@outlook.com
bLaboratory of RNA Biology, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China. E-mail: jwang@ibp.ac.cn
First published on 18th May 2023
Abstract
Radical enzymes harness the power of reactive radical species by placing them in a protein scaffold, and they are capable of catalysing many important reactions. New native radical enzymes, especially those with amino acid-based radicals, in the category of non-heme iron enzymes (including ribonucleotide reductases), heme enzymes, copper enzymes, and FAD-radical enzymes have been discovered and characterized. We discussed recent research efforts to discover new native amino acid-based radical enzymes, and to study the roles of radicals in processes such as enzyme catalysis and electron transfer. Furthermore, design of radical enzymes in a small and simple scaffold not only allows us to study the radical in a well-controlled system and test our understanding of the native enzymes, but also allows us to create powerful enzymes. In the study and design of amino acid-based radical enzymes, the use of unnatural amino acids allows precise control of pKa values and reduction potentials of the residue, as well as probing the location of the radical through spectroscopic methods, making it a powerful research tool. Our understanding of amino acid-based radical enzymes will allow us to tailor them to create powerful catalysts and better therapeutics.
Introduction
Free radicals are chemical species with an unpaired electron. Their high activity makes the radical species either good catalysts or detrimental contaminants in reactions.1 Nature harnesses active radical species by placing them in protein scaffolds, to afford efficient radical enzymes. Radical enzymes can be seen in the process of photosynthesis, O2 reduction, natural product biosynthesis and other important reactions in the cell. The radicals in radical enzymes can reside on the protein scaffold, in which amino acid residues form radicals during catalysis, or on organic/metal cofactors associated with the enzyme.Here we discussed recent research efforts to discover new native tyrosine or tryptophan-based radical enzymes, study the roles of radicals in processes such as enzyme catalysis and electron transfer, and design and engineering of new radical enzymes, with emphasis on using unnatural amino acids (UAAs) as mechanistic study probes and design tools.
There are many other reviews from major players in the field, either providing an account of certain amino acid-based radical enzymes, such as ribonucleotide reductase (RNR),2–6 heme copper oxidase,7,8 non-heme iron oxidases,9,10 and glycyl radical enzymes (GREs),11 or certain aspects of amino acid-based radical enzymes.12–14 Furthermore, as we are focusing on radicals residing on amino acid sidechains, radicals appearing on organic cofactors (e.g., radical S-adenosyl-L-methionine and flavin), metal ions or complexes (e.g., porphyrin radicals in heme enzymes and cobalamin) are beyond the scope of this review. Readers interested in these topics can refer to related reviews.15–21
Radical enzymes in catalysis
The redox and spectroscopic properties of amino acids
Although metal ions or metallo-cofactors in proteins are usually responsible for redox reactions, protein scaffolds are not redox-inert. Sidechains of Tyr, Trp, Cys, and Met are vulnerable to oxidation. Additionally, glycine and other amino acids can be oxidized to generate radical species. The protein-based radicals can be a measure to contain and control oxidative damage cause by reactive oxygen species (ROS), where radicals formed on backbone or sidechains of amino acid residues can propagate on peptide chain and reduced by antioxidants such as GSH, Cys and ascorbic acid.22Among all the 20 natural amino acids, Tyr forms relatively stable radical in the protein environment and thus are well characterized. Tyr has a pKa value of about 10 in the free form ref. 23 and the value is 11.3 in a protein environment.13,24 As a result, the thermodynamics of single electron oxidation of tyrosine highly favours formation of a neutral radical, from tyrosinate anion, with a reduction potential of 0.71 V, while formation of cationic radical has a theoretical reduction potential of 1.4 V.23 The reduction potential of Tyr is dependent on its protonation state, which is influenced by its environment (buffer pH, hydrogen bonding). Due to the transient nature of the oxidized species, only the oxidative potential was observed on cyclic voltammogram, and this is termed peak potential. The peak potential may differ from the formal reduction potential, which is obtained under fully reversible conditions. Tommos and coworkers placed Tyr into the model protein α3, and obtained the formal reduction potential (980 mV vs. NHE at pH 7.0) of Tyr in a protein environment through square-wave voltammetry (Table 1).24
| Amino acid | Free amino acid | In model protein (α3) | ||
|---|---|---|---|---|
| pKa | E p (Y˙/Y−, mV vs. NHE) | pKa |
E°′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (mV) (pH) b (mV) (pH) |
|
| a Peak potential measured by differential pulse voltammetry or cyclic voltammetry, typically at pH 13. b Formal reduction potential measured by square-wave voltammetry. | ||||
| Tyr | 9.8 | 642 | 11.3 | 1065 (5.53 ± 0.05) |
| Trp | 16.7 | — | 16.7 | 1095 ± 4 (7.0) |
| 3,4-Dihydroxyphenylalanine 1 | 9.7 | 570 | — | — |
| 3-Aminotyrosine 2 | ∼10 | 640 | — | — |
| 3-Nitrotyrosine 3 | 7.2 | 1020 | — | — |
| 3-Fluorotyrosine 4 | 8.4 | 705 | — | — |
| 3,5-Difluorotyrosine 5 | 7.2 | 755 | 8.0 | 1040 ± 3 (5.49 ± 0.03) |
| 2,3-Difluorotyrosine 6 | 7.7 | 810 | 8.6 | 1136 ± 2 (5.57 ± 0.09) |
| 2,3,5-Trifluorotyrosine 7 | 6.4 | 853 | 7.2 | 1104 ± 2 (5.54 ± 0.05) |
| 2,3,6-Trifluorotyrosine 8 | 6.9 | 911 | 7.9 | 1200 ± 3 (5.54 ± 0.05) |
| 3-Chlorotyrosine 10 | 8.1 | 734 | — | — |
| 3,5-Dichlorotyrosine 12 | 6.3 | 808 | — | — |
| 3-Methoxytyrosine 11 | 9.9 | 480 | — | — |
A critical feature for radical enzymes is their distinct electron paramagnetic resonance (EPR) spectra from the unpaired electron. A typical tyrosyl radical gives exquisite hyperfine splitting within the g ≈ 2 signal. While continuous wave (CW) X-band EPR readily detects tyrosyl radical, combination of multi-frequency (X, Q, D, and W-band) CW-EPR and CW/pulse electron–nuclear double resonance (ENDOR) allows more accurate characterization of the radical species.6 The g tensors are further resolved with high frequency CW-EPR methods, while ENDOR spectroscopy reveals the distribution of the electron density on the phenol ring.32,33 Due to the high expression level of RNR and readily formed and relatively stable radical, the tyrosyl radical of RNR inside E. coli cells can be directly detected by EPR, making it possible to perform in situ study under physiological conditions.34
In addition to direct characterization of the Tyr residue, it can be mutated to other aromatic amino acid residues, that is, Trp and Phe. The advent of genetic code expansion also allows incorporation of UAAs into a given protein. More than 200 UAAs have been incorporated into protein using this method.36,37 Among them, there are several structurally similar to Tyr (Fig. 1). These Tyr analogues, with varying pKa values and reduction potentials (Table 1), serve as a toolbox to study or design amino acid-based radical enzymes. Additionally, halogenated UAAs alter EPR spectrum of radical, making them a good spectroscopic probe (Table 2).
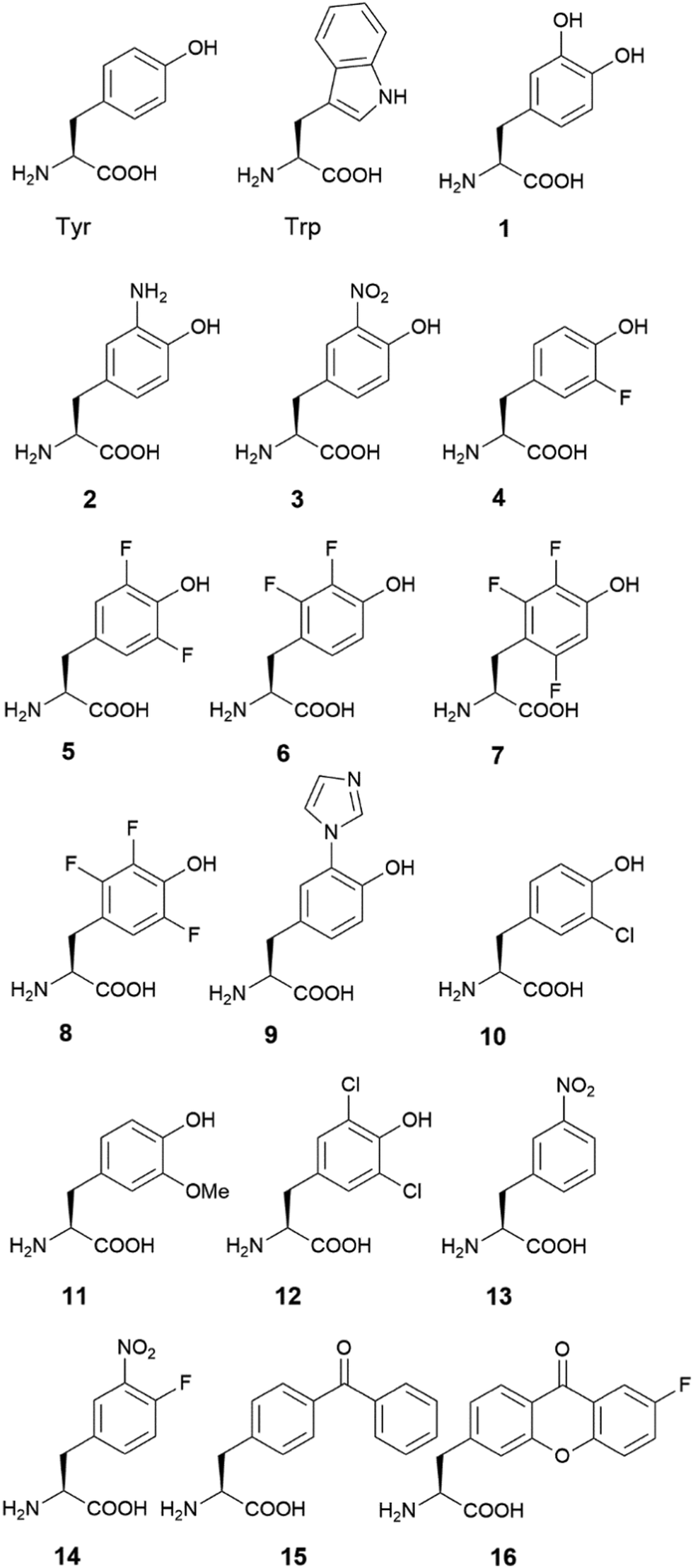 | ||
| Fig. 1 Structures of Tyr, Trp and Tyr analogues. | ||
| Amino acid at the 122nd position of RNRb | g values | Nucleus | A (MHz) | ||||
|---|---|---|---|---|---|---|---|
| g x | g y | g z | A x | A y | A z | ||
| a Please refer to ref. 30 and 35 for other parameters and for parameters of other UAAs. b From ref. 35. c From ref. 30. Only major species shown. | |||||||
| Tyr | 2.00912 | 2.00454 | 2.00219 | 1Hβ1 | +59 | +52 | +55 |
| 1Hβ2 | +2.1 | −5.0 | −4.0 | ||||
| 2-H | +5 | +7.6 | +2.1 | ||||
| 3-H | −26.7 | −8.4 | −19.6 | ||||
| 5-H | −26.7 | −8.4 | −19.6 | ||||
| 6-H | 5 | 7.6 | 2.1 | ||||
| (3,5) F2Y | 2.00828 | 2.00500 | 2.00196 | 1Hβ1 | +56 | +50 | +52 |
| 1Hβ2 | −0.5 | −0.5 | +3 | ||||
| 2-H | +5.3 | +5.8 | +8.5 | ||||
| 3-F | −24 | −10 | +157 | ||||
| 5-F | −24 | −10 | +157 | ||||
| 6-H | +5.3 | +6.3 | +3 | ||||
| Amino acid at the 33rd position of CuBMbc | g x | g y | g z | Nucleus | A x /MHz | A y /MHz | A z /MHz |
|---|---|---|---|---|---|---|---|
| Tyr | 2.0091 | 2.0044 | 2.0021 | 3-H† | 26 | 11 | 19 |
| 5-H† | 26 | 11 | 19 | ||||
| 1Hβ1 | 51 | 60 | 59 | ||||
| 1Hβ2 | 24 | 8 | 16 | ||||
| 3,5-F2Tyr | 2.0128 | 2.0073 | 2.0036 | 3-F | 44 | 27 | 161 |
| 5-F | 35 | 17 | 144 | ||||
| 1Hβ1 | 63 | 52 | 56 | ||||
| 1Hβ2 | 37 | 20 | 15 | ||||
Native enzymes
An ever-increasing number of native enzymes are being discovered with amino acid radicals involved in their reaction. Based on the nature of the radical species and the associated cofactors, they can be grouped into non-heme iron radical enzymes (including Type I RNR), heme-radical enzymes, copper-radical enzymes, FAD-radical enzymes, and GREs. There are also heme-copper oxidases (HCOs) and photosystem II, which cannot be grouped in either category but are important radical enzymes. Native enzymes with tyrosyl radicals and tryptophanyl radicals will be discussed in the following section.Among different types of RNRs, the Class Ia RNRs are the most studied and serve as a model system for amino acid-based radical enzymes. A class Ia RNR consists of two subunits, α and β, and contains a di-iron center for radical initiation and a thiyl radical at the active site. The di-iron center at the β subunit reacts with O2 and oxidizes a nearby Tyr residue (Tyr122, numbering from E. coli RNR Ia, the same numbering was applied to Tyr356, Cys439, Tyr730, and Tyr731 in RNR) to generate a radical. As mentioned above, the single electron oxidation of tyrosine is usually accompanied by proton transfer in a thermodynamically favourable process.23 The radical on Tyr122 is transferred over 35 Å to Cys439 in a rare and classical long-range PCET process.
The radical in subunit α of RNR readily forms after purification, the majority of which is on Tyr122. It can be further enriched by addition of ferrous iron, which, combined with its stability (stable at room temperature), make RNR an ideal system to study protein-based radicals.42 Based on structural analysis, a series of Tyr residues and other amino acid residues (Trp48 and Tyr356 on the β subunit, Tyr731 and Tyr730 on the α subunit) between Tyr122 from the β subunit and Cys439 from the α subunit, some of which on the interface between α and β subunits, are believed to participate in the relay of radicals through a PCET process (Fig. 2A and B).43,44
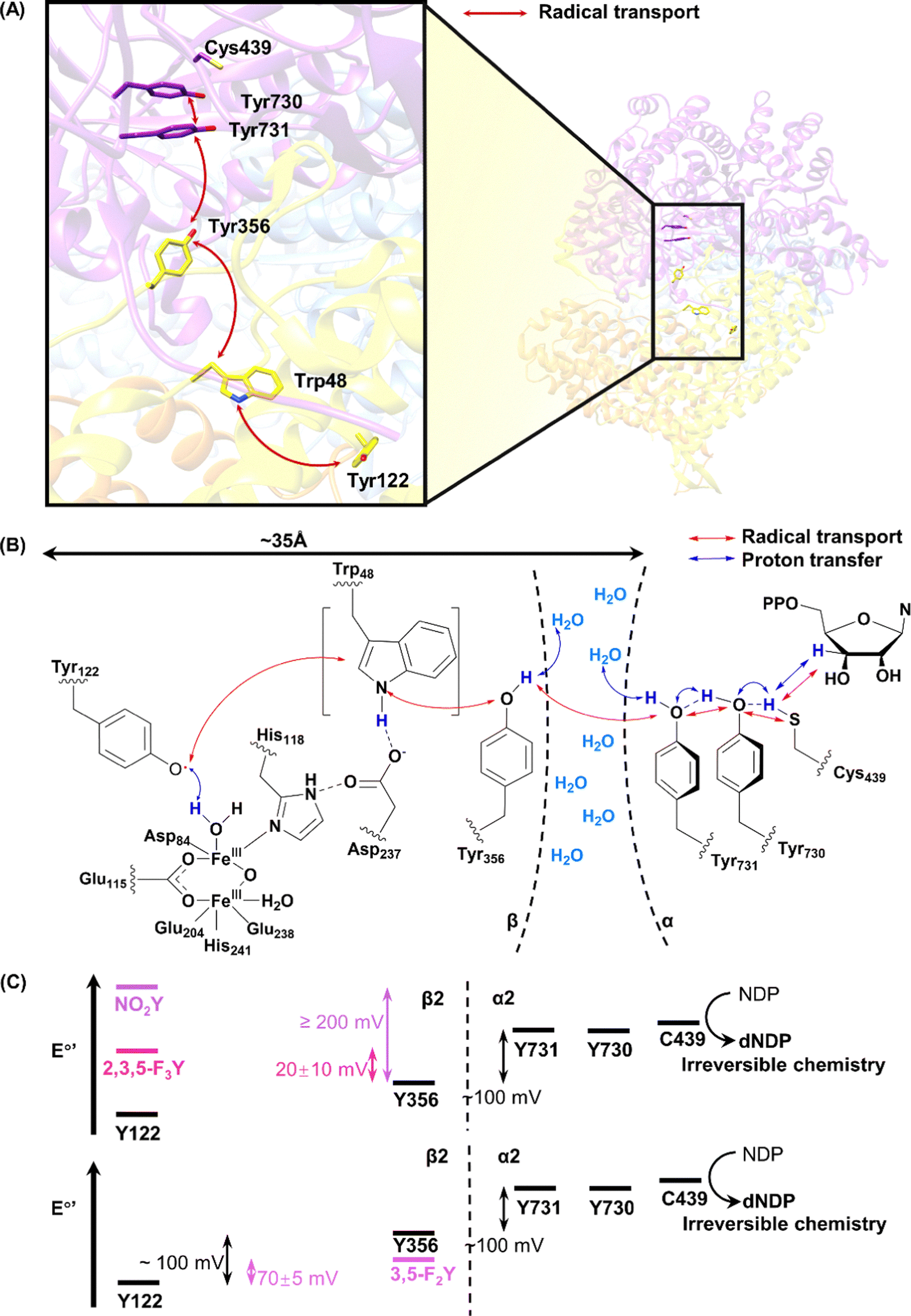 | ||
| Fig. 2 The radical transport process in RNR. Pathway residues identified in radical transport of type Ia RNR in the atomic structure (PDB ID 6W4X) (A) and the scheme of radical transport (B). (C) Energy landscape of radical transport in RNR. Figure (C) produced from ref. 45 with permission from the American Chemical Society. | ||
Long range radical transfer is a fascinating phenomenon in RNR. The radical transfer pathway and its energy landscape are deciphered through incorporation of UAAs in RNR. Stubbe, Nocera and coworkers started using native chemical ligation, and then switched to the more popular genetic codon expansion method to study the tyrosyl radical with UAAs.46,47 A series of tyrosine analogues, including 3-hydroxyltyrosine (DOPA, 1), 3-aminotyrosine (NH2-Tyr, 2), 3-nitrotyrosine (NO2-Tyr, 3), and fluorinated tyrosines (FnY, 4–8), differ from the natural tyrosine by a few atoms, yet have different pKa values and reduction potentials (Fig. 1 and Table 1).27,48–55 Replacing the native Tyr residues in RNR with these Tyr analogues causes minimal structure perturbation, and allows systematic variation of certain properties of the enzyme.
The altered reduction potentials of Tyr analogues, after being incorporated into RNR, lead to the altered distribution of pathway radicals. Due to the less positive potentials of DOPA and NH2-Tyr, radicals are trapped on the Tyr analogues during the PCET process. Stubbe, Nocera, and coworkers systematically probed possible residues on the radical transfer pathway using Tyr analogues, and identified Tyr356 (from subunit β), Tyr730, and Tyr731(from subunit α) in the process.48,50,51,53
In addition to the position of the pathway radicals, the distribution of radicals on the pathway can be deconvoluted using UAAs. Through incorporation of 3-nitrotyrosine, Stubbe and coworkers proved that the radical observed on EPR spectrum is a mixture of radicals on different residues, with the majority (85–90%) residing on Tyr356.56 A previous ENDOR study on RNR revealed that the electron density on the phenol ring of the tyrosyl radical mainly resides on the 3- and 5-C. Replacing the 3- and 5-H with a halogen, such as fluorine and chlorine, leads to altered EPR spectra of tyrosyl radicals. Stubbe and coworkers showed that the fluorinated tyrosyl radicals produced different splitting patterns, making it possible to distinguish FnY and Tyr radicals (Table 2).27 An efficient system with a multi-purpose aminoacyl-tRNA synthetase for fluorinated tyrosines (FnY, 4–8) incorporation was later developed, enabling deconvolution of EPR spectrum and determination of the radical distribution on different positions.47 At equilibrium, the free energy difference can be calculated from the relative abundance of Tyr and FnY radicals, which can be calculated based on the EPR spectrum. Sequential incorporation of FnY on the pathway residues led to the mapping of the reduction potentials of the residues on the radical transfer pathway (Fig. 2C).45 The result showed that the PCET process between residues on the same subunit is thermodynamics favourable, with less positive reduction potentials from upstream to downstream of the pathway. However, the process is thermodynamically uphill at the α/β subunit interface, where the reduction potentials of the downstream residues, Tyr730 and Tyr731 on the α subunit are more positive than that of the upstream Tyr356 on the β subunit. The inverted reduction potentials lead to the hypothesis that the enzymatic activity is controlled at the interface. The elucidation of pathway residues and energy landscape using UAAs in RNR not only provides us with a deeper understanding of the enzyme, but it also creates a toolkit for other radical enzymes and sets an example for study (Fig. 2).
The crystal structures of the α and β subunits of RNR were first reported in the 1990s.43,44 The structures served as an important reference for identifying the pathway residues. However, the α2β2 holoenzyme complex quickly disassembles after the PCET process, making the molecular details of the cross-subunit PCET unclear. Until recently, the holoenzyme structure, solved by cryo-electron microscopy (cryo-EM), was reported by Drennan, Stubbe, and coworkers.57 The holoenzyme structure indicated that the PCET distance from Tyr122 to Cys439 is 32.4 Å, close to previous estimations. In the structure, the elusive C terminal part of the β subunit, due to its flexibility outside the complex, was first revealed and found in the active site cavity of the α subunit. Tyr356 on this part of the protein is close to Tyr730 on the α subunit, again confirming previous calculations. The structure revealed the intricate mechanism of the subunit interaction and how long-range PCET works.
With biochemical and structural analysis mapping out the PCET pathway, much attention is on how radical is transported over the α/β subunit interface. Nocera, Stubbe and coworkers labelled a Re complex in close proximity to Tyr356 on the β subunit.58 The Re complex works as a photo-trigger, which upon excitation, oxidized Tyr356 and initiates the radical transport process.59 As the electron goes to the Re complex in the “photoRNR”, the proton transfer process during Tyr356 radical generation can be studied separately. Transient absorption and emission spectroscopy on the PhotoRNR and the Glu52Gln mutant showed that the glutamate is a crucial step for dissipating the proton to the outside water, and the proton transfer is crucial for the PCET process.60
In addition to the well-studied class Ia RNR, Hogbom and coworkers reported a new class of metal-free RNR.61 An operon similar to the ones containing a class I RNR was discovered through genome analysis of Mesoplasma florum and other human pathogens. The protein encoding the β subunit of RNR, MfR2, shows high sequence similarity to that of a class Ib RNR. However, the putative RNR lacks three out of six metal coordinating residues and presents an active site without any metal ions in the crystal structure of the active MfR2. More surprisingly, a DOPA residue appears at the position of Tyr122 and forms a radical based on the EPR spectrum. The new class of RNR, termed class Ie RNR, showcases the ability of microorganisms to adapt to a metal-deprived environment. At the same time, it raises questions about the mechanism of radical generation, as well as the energetics of radical transfer, as DOPA, with a less positive reduction potential relative to Tyr, can hardly be reduced by tyrosine residues downstream of the PCET pathway.61
Mononuclear iron-α-ketoglutarate (α-KG) enzymes form a major class of non-heme iron enzymes.62 Fe(II)/α-KG enzymes couple the oxidative decarboxylation of α-KG to the oxidation of a substrate, through a putative Fe(IV)-oxo species. Fe(II)/α-KG enzymes can activate the inert C–H bonds on the substrate to perform hydroxylation, halogenation, ring formation, desaturation and other types of reactions.9 Que, Hausinger and coworkers discovered that the reaction of a non-heme iron enzyme, TfdA, in the absence of its substrate, leads to the formation of a high-spin Fe(III) species. Further tandem mass spectrometry study revealed that the Trp112, a residue close to the iron center, was hydroxylated. One of the proposed self-hydroxylation mechanisms involves the formation of a Trp radical.63 Hausinger and coworkers later worked on TauD, a model non-heme iron enzyme, and identified a transient yellow species with an absorption maximum at 408 nm, which was characteristic of a tyrosyl radical.64 Further EPR experiments confirmed that the transient species contains a radical with g ≈ 2, which is likely a tyrosyl radical. Kinetics experiments and isotopic labelling showed that the radical species was not related to the taurine oxidation reaction, but led to self-hydroxylation to catechol. The occurrence of uncoupled self-hydroxylation and the formation Tyr and Trp radicals during the reaction were proposed to be a mechanism to prevent irreversible damage of the enzyme.64
In addition to being a side reaction intermediate, Tyr in non-heme iron enzymes can directly participate in the reactions. Carbapenem synthase, CarC is a bifunctional enzyme that catalyses the epimerization of C5 and desaturation of the C2–C3 bond in the biosynthesis of carbapenem antibiotics.65 The stereo-inversion of C5 is redox neutral, which is not common for non-heme iron enzymes. Based on density functional theory calculations, Maya et al. proposed that the epimerization proceeds with an initial hydrogen atom abstraction by the high-valent iron-oxo species, followed by hydrogen rebound, possibly from another reductant.66 Borowski and coworkers further identified the additional reductant as the Tyr67 by a computational study.67 Townsend and coworkers developed a high-throughput screening method for β-lactam antibiotics based on the detection of the accumulation of cell wall component N-acetyl pentapeptide as a result of carbapenem's inhibition of cell wall biosynthesis.68 Such a system allowed the same group to perform a systematic mutational study of CarC.69 They performed saturation mutagenesis on six active site residues and four additional in the second sphere, and they identified the mutability of the residues for either epimerization and desaturation reactions, or the desaturation reaction alone. Based on the different mutation tolerance profiles, they proposed that the epimerization and desaturation reactions occur in a stepwise fashion rather than consecutively. Tyr67 was confirmed to be a key residue for the enzyme. Bollinger, Krebs, Boal, Chang and coworkers approached the reaction mechanism problem through the combination of structural and spectroscopic methods. They determined the crystal structure of CarC in complex with Fe(II), α-KG, and 3S,5S-carbapenam, the initial substrate of CarC (Fig. 3).70 The tertiary complex showed the relative orientation of the substrate to the enzyme and revealed that Tyr165, a residue missing in other crystal structures, was positioned at the opposite side of the substrate relative to the Fe(II), and the tentative ferryl intermediate, thus being the dedicated hydrogen atom donor. They first observed a species with 410 nm absorption, characteristic of tyrosyl radicals, reaching its maximum intensity at 3 s on a stopped flow apparatus. The EPR spectrum of reaction intermediates showed a g ≈ 2 radical signal and a g = 6.95 Fe(III) signal. The radical signal was broader than the typical tyrosyl radical signal at 10 K, due to spin coupling to Fe(III). Combining the evidence from the stopped flow spectroscopy and Mössbauer spectroscopy, the authors proposed a mechanism for CarC, in which the C5 hydrogen is abstracted by a ferryl species, and Tyr165 on the opposite side of the substrate donated a hydrogen atom to complete the stereo-inversion of C5 (Fig. 3).70
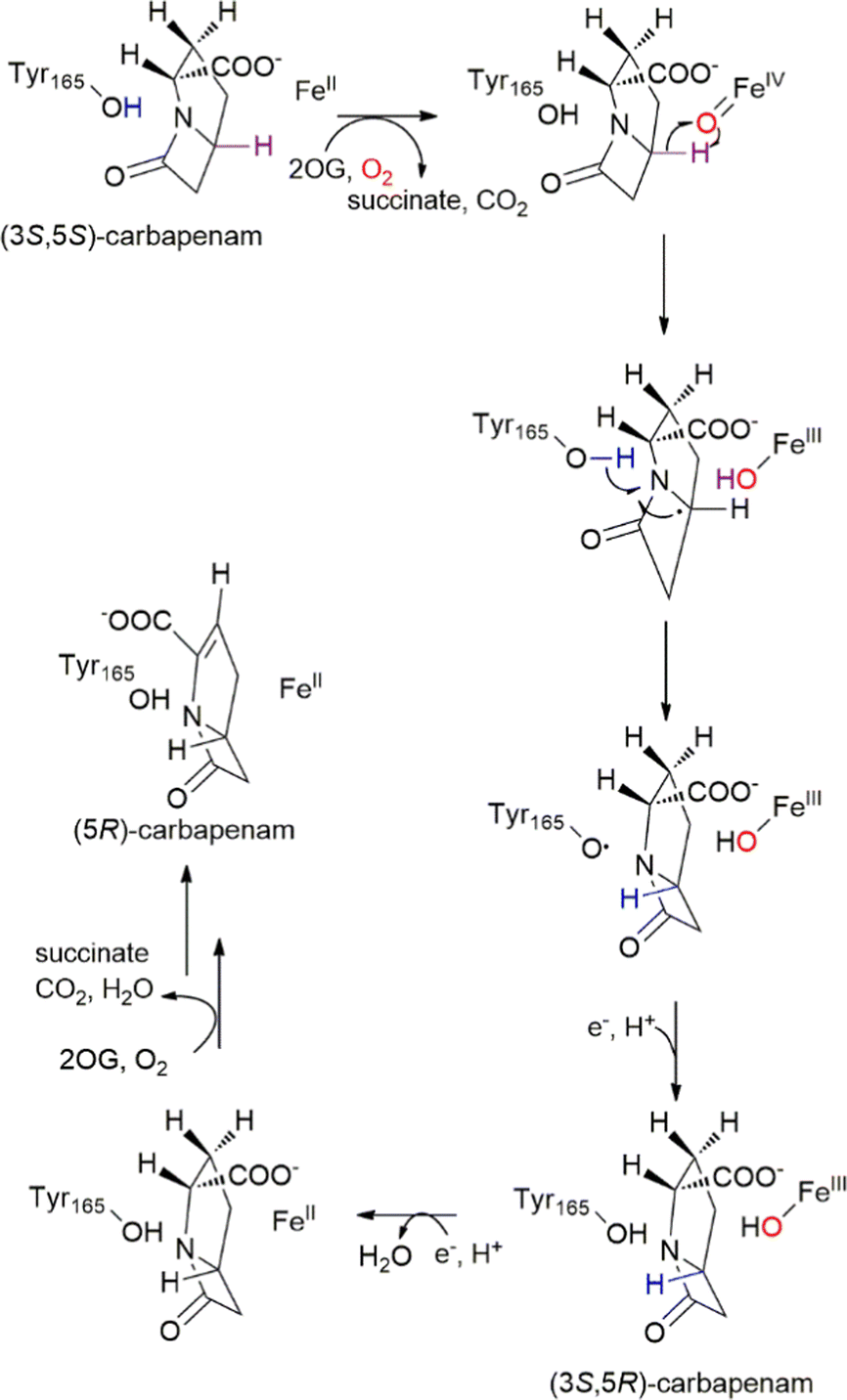 | ||
| Fig. 3 Mechanism of the CarC mediated, stoichiometric stereoinversion of (3S,5S)-carbapenam to (3S,5R)-carbapenam, involving abstraction of H˙ from C5 by a ferryl complex and H˙ donation by Tyr165.70 Adapted from ref. 70 with permission from AAAS. | ||
A non-heme iron enzyme, fumitremorgin oxidase (FtmOx1) was identified and characterized as an endoperoxide forming enzyme.71 Endoperoxide is critical for the bioactivity of many natural products, including the anti-malaria drug artemisinin. A tyrosyl radical was first identified in FtmOx1 with a lifetime of ∼3 s based on the 410 nm absorption and a g ≈ 2 radical EPR signal.72,73 Based on the crystal structure whose electron density in the active site was mistakenly assigned to the substrate, and a mutational study, the source of the radical was initially assigned to Tyr224. A mechanism that involves tyrosyl radical formation and H abstraction from C21 was also proposed.
Bollinger and coworkers examined the product of FtmOx1 and found a deprenylation product due to oxygen rebound.74 The appearance of the side product contradicts the proposed cyclooxygenase (COX)-like mechanism, in which the ferryl species abstracts H from the tyrosine residue instead of the substrate. They also examined the crystal structure data and did not find enough evidence for the existence of the substrate, fumitremorgin B, which led to a question about the true configuration of the active site relative to the substrate. Their investigation of the structure revealed that in addition to Tyr224, three additional tyrosine residues could participate in the H atom transfer reaction. Mutating these four residues individually to Phe revealed that only the Y68F mutant lost its activity, suggesting that Tyr68 was the hydrogen donor. Based on Tyr68 being the H donor, a computational study by Bollinger and coworkers with a docked substrate supports a CarC-like mechanism, in which ferryl species abstract hydrogen from C21 of the substrate directly, and Tyr68 subsequently transfers a hydrogen to the substrate radical. The mechanism and position of the tyrosyl radical was further confirmed by a structural and computational study by Zhou, Wang, Cen, and coworkers.75 The crystal structure of the ternary complex of FtmOx1, α-KG, and the substrate, fumitremorgin B, revealed that Tyr68 is 4 Å away from C26 of the substrate, while both Tyr68 and Tyr224 are over 10 Å from the Fe, which makes direct hydrogen transfer from Tyr to Fe(IV)-oxo unlikely. Recently, Bollinger and coworkers replaced Tyr68 and Tyr224 with fluorinated tyrosine analogues using the genetic codon expansion method. The altered radical EPR spectra of the Tyr68 mutants confirmed that Tyr68 is the hydrogen donor to C26 of the substrate radical.76
Liu and coworkers further argued that the COX-like mechanism is more feasible.77 FtmOx1 is a tri-functional enzymes with endoperoxidation, alcohol dehydrogenation and dealkylation activities, which complicates mechanism investigation. By designing and synthesis of a substrate analogue, 13-oxo-fumitremorgin, and use it for crystallization and reaction, they can decouple the activities of FtmOx1 and showed structures and reactions that support rotation of Y224 during the reaction, favouring the original COX-like mechanism.
Ferritin is a ubiquitous, iron-storage protein. The bacterial and archaeal ferritins, and the eukaryote H-type ferritin contain a di-iron catalytic site with ferroxidase activity.78 The reaction converts the ferrous ion, the common oxidation state in a cell, to the oxidized ferric form, which further undergoes biomineralization to the storage form. Early studies of the human H chain ferritin using EPR spectroscopy and site-directed mutagenesis suggested that a tyrosine residue close to the di-iron center forms a radical.79 Mutation of the corresponding Tyr to Phe in ferritin would stop the radical formation, but does not eliminate the ferroxidase activity, leading to the hypothesis that the radical formation serves as a side reaction to detoxify superoxide. A series of recent studies in archaeal and bacterial ferritin revealed the direct participation of tyrosine in the reaction. Hagen and coworkers studied the kinetics of the ferroxidase reaction in human heavy chain ferritin (HuHF) and an archaeal ferritin from Pyrococcus furiosus (PfFtn). A blue reaction intermediate that exhibited a 625 nm absorption was captured and identified as a peroxodiferric species. An additional 410 nm feature, characteristic of tyrosyl radical, was identified in the stopped flow spectra of the PfFtn reaction. A mutational study indicated that Tyr24 near the active site is crucial for the activity, serving as a “capacitor”.80 Le Brun and coworkers studied bacterial ferritin from E. coli (EcBFR). Mutations of three aromatic residues 4.0–9.7 Å from the di-iron center, Tyr25, Tyr58, and Trp133, to Phe slowed the ferrous ion oxidation. However, only Tyr25 is associated with a radical intermediate observed on the EPR spectra.81 The same group also identified a similar tyrosyl radical in a cyanobacterial ferritin and located the radical to Tyr40 using site-directed mutagenesis. They also found that the reaction led to a mixed-valent Fe2+/Fe3+ center, instead of the di-ferric center in other forms of ferritins, showcasing the diversity of the reaction of di-iron enzymes.82
One such example is the prostaglandin H synthase, also known as COX. The enzyme catalyses the conversion of arachidonic acid to prostaglandin G2 in the cyclooxygenase reaction, then to prostaglandin H in a peroxidase reaction.84 In the initial reaction, the ferryl species generated after Fe(II) reacts with O2 oxidizes Tyr358 (numbering in COX1), yielding Fe(IV)-oxo and a tyrosyl radical. The tyrosyl radical further abstracts the hydrogen from the substrate, arachidonic acid for the cyclooxygenation reaction.85
Cytochrome c peroxidase (CcP) catalyses the reduction of H2O2 using electrons from Cyt c. The reaction of the heme enzyme with H2O2 goes through a “shunt” pathway relative to the O2 activation process, in which the ferric heme reacts with H2O2 to form the ferryl species or compound I.83 While the ferryl species in cytochrome P450 or HCO are in the form of Fe(IV)-oxo porphyrin radical or Fe(IV)-oxo tyrosyl radical, the ferryl species in CcP is in the form of Fe(IV)-oxo Trp radical, where the radical resides on Trp191 (yeast numbering). The Trp191 radical itself has been extensively characterized before.86,87 Due to the stability of the protein and its radical intermediate, CcP is a good platform to compare the radical biology between Tyr and Trp residues (vide infra).
Tyr and Trp have similar reduction potentials to their respective cation radicals, with modelled E°′(TyrO˙+/Tyr) of 1510 mV and E°′(Trp˙+/Trp) at 1293 mV in a model protein system.31 Trp191Tyr mutation on CcP, without much structural perturbation, yields an inactive enzyme with a stable tyrosyl radical.88 Crane and coworkers systematically investigated the mutant by using unnatural Tyr analogues and constructing H bonds to the Tyr191 residue.89 They uncovered that the reduction potential of the Tyr or Tyr analogues is directly related the reactivity of CcP. Mutant 2,3,5-trifluorotyrosine has a higher activity than the Trp191Tyr mutant, while the mutant with 2,3-difluorotyrosine has a lower activity. Introducing a His or Glu at the Leu232 position will add a hydrogen bond to Tyr191, according to the crystal structure, CW-EPR, ENDOR and electron spin echo envelope modulation (ESEEM) spectroscopy. The mutations effectively add a conjugated base nearby and changes the protic environment of Tyr191. The electron transfer rates of the mutants are increase 30-fold compared to that of the Trp191Tyr mutant. Further modelling suggested that adding a hydrogen bond to Tyr191 in the Leu232Glu/Trp191Tyr mutant increases the reduction potential as much as 200 mV, leading to the dramatic activity enhancement.89 Such a strong correlation of the hydrogen bonding environment and activity shed light on the Tyr radical tuning in the photosystem II, in which YZ is known to have a strong hydrogen bonding partner.90
Other than these two enzymes, Roth and coworkers discovered a new heme enzyme, RαO, a fatty acid α-(di)oxygenase, uses tyrosyl radical to abstract hydrogen atom to initiate the reaction.91,92 Kinetic isotope effect of the reaction indicates that the homolysis of the substrate C–H bond occurred by nuclear tunnelling.
Researchers have observed a set of intermediates from transient spectroscopy after the treating the Fe(III) enzymes with H2O2, presumably through the peroxo shunt pathway. One of the intermediates, PM, is similar to the ferryl species. The origin of the radical signal from the PM intermediate is under debate.95 Gerfen and coworkers captured the PM intermediate by treating CcO with H2O2 and freeze quenching the reaction mixture.96,97 Through a combination of multi-wavelength EPR (X- and D-band) and quantum mechanical calculations, they deconvoluted the signal to a wide signal (46 Gauss, or 128.9 MHz) and a narrow signal (12 Gauss, or 33.6 MHz). The narrow (12 Gauss) radical species was likely from the Tyr244 in the active site with a post-translational modification, while the wide one with more hyperfine patterns was postulated to be from a Tyr residue involving in the radical transfer to Tyr244. Solomon, Gennis and coworkers characterized the PM intermediate from the E. coli ubiquinone oxidase through MCD spectroscopy. Simulation showed the existence of a coupled three-spin system, indicating the coexistence of Fe(IV)-oxo, Cu(II), and tyrosyl radical and they are electronically coupled.98
The Cys–Tyr cofactor, universal in GO and related glyoxal oxidase, are important for the function of the enzyme, but its synthesis process was elusive previously. Liu and coworkers established a methodology for cofactor biogenesis study using in UAA replacement and crystal reaction monitoring.102 Cysteine dioxygenase is a mononuclear non-heme iron protein with a Tyr–Cys crosslink crucial for its enzymatic activity. but with no sign of forming a radical during the reaction. Liu and coworkers replaced the C-3 and C-5 hydrogens with halogens, including Cl and F, and found the Cys–Tyr crosslink forms, resulting in the loss of one halogen atom. Further mass spectrometry and structural characterization point to a rare oxidative dehalogenation process, mediated by the mononuclear iron. The biosynthesis of the cofactor was captured by an in-crystal reaction.103 The same team moved to GO, which contains the same Cys–Tyr cofactor participating in the reaction as a radical.104 Replacing the tyrosine residue with 3,5-dihalogenated tyrosine also leads to the formation of the cross-link, with the loss of one halogen atom. EPR and crystal structure analysis also suggested the formation of Cys–Tyr cofactor involves C–Cl/F bond cleavage, possibly mediated by the mononuclear copper in GO.104 The Cys–Tyr cross-link also appears in other enzymes, such as glyoxal oxidase, sulphite reductase and nitrite reductase.
The Cys–Tyr crosslink is believed to fine-tune the properties of Tyr residue for optimized reaction. Such role in catalysis was probed by Wang and coworkers, by installing an UAA, methylthiotyrosine, in myoglobin for better hydroxylamine reduction activity.105
Another pair of conserved tyrosine residues exists in the symmetrical branched photosynthetic reaction center, which are believed to modulate the initial charge separation in the photo-induced electron transfer (PET). Although they do not form radical during the electron transfer process, thus strictly not within the realm of radical enzymes, their function of charge separation is important and relevant to many tyrosyl radicals discussed in the review. Boxer and coworkers first realized genetic codon expansion in a model organism to produce photosynthetic reaction center, Rhodobacter sphaeroides.108 Later they replaced one of the active-site tyrosine residues, termed M210, with a set of tyrosine analogues. As shown in other enzymes, the replacement did not cause major structural perturbations, based on the structure data. They used ultrafast transient absorption spectroscopy, accompanied by Stark spectroscopy, redox titrations, and other measurements, to reveal the kinetics of the charge separation of the wild-type enzyme and the enzymes with tyrosine analogues. The free energy change of charge separation intermediates indicates the role of M210 in tuning primary electron transfer in the photosynthetic reaction center.109
DNA photolyase is a Flavin adenine dinucleotide (FAD)-containing enzyme responsible for the repair of pyrimidine dimers using a reduced FADH− cofactor. Three conserved tryptophan residues (Trp306, Trp359, and Trp382 in E. coli photolyase numbering) participate in the electron transfer process, and the Trp382 is believed to be the ultimate electron donor. The distance from Trp382 to the FAD cofactor is 14 Å, consistent with another long-range electron transfer.
The kinetics of DNA photolyase activation has been extensively studied using transient spectroscopy and other methods.110,111 The enzyme turns over at a time scale of 1 μs, and the cofactor activation is at a similar time scale. The FADH˙ cofactor will abstract an electron from a nearby Trp, yielding a tryptophan cation radical (TrpH˙+), which has a characteristic absorption centred at 540 nm. The electrons from the external reductant transfer through several Trp residues, which form similar cation radicals and relay electron to the FAD cofactor. The electron transfer process occurs in 30 ps, and the final deprotonation of Trp382 takes 300 ns. The role of the three Trp residues in the electron transfer was further probed by direct mutational studies. Mutating these residues to Phe confirmed their involvement in the electron transfer, and the sequence of electron transfer is determined to be FAD → Trp382 → Trp359 → Trp306.111
An obstacle remains to prevent further elucidation of the electron transfer kinetics between the three Trp residues: although the Trp cation radical has a pronounced 540 nm absorption, the transfer of electrons between two Trp residues will generate little spectral change. Although chemically equivalent, the three Trp residues have different orientations relative to the FAD cofactor in the protein environment. Brettel and coworkers developed polarized transient absorption spectroscopy to distinguish between the three Trp residues in the electron transfer pathway.110 They used a polarized laser to excite a sub-population of DNA photolyases, which had FADH˙ transition dipole moments parallel to the laser-flash polarization direction. In analogy to fluorescence spectroscopy, the anisotropy of the absorption can be calculated based on absorption parallel or perpendicular to the excitation light. The contribution to the anisotropy from different Trp residues (Trp359 and Trp306) and the FAD cofactor can be deconvoluted to provide the kinetics of Trp radical generation. By combining the mutation to Phe, the full electron transfer kinetics were determined.111
A blue light sensor using FAD (BLUF) protein is a class of FAD-containing proteins used to sense light and trigger structural rearrangement for downstream biological effects. First discovered in 2002, the BLUF domain can be potentially used for optogenetics.18 Among the flavin-containing photoreceptors, including the photolyase/cryptochromes, the light-oxygen-voltage (LOV) domain proteins, and BLUF proteins, BLUF proteins are the only family of photoreceptors known to show photo-induced PCET.18 Upon blue light excitation, electron transfer through a PCET (light adapted state) or sequential proton transfer after electron transfer (dark-adapted state) leads to the generation of a FAD/Tyr neutral radical pair, which leads to the structural rearrangement of effector domains. The radical formation can be studied by transient spectroscopy methods.18 Mino and coworkers carried out EPR and ENDOR of the radical intermediate, which had a g value of 2.0045 with a separation of 85 Gauss or 238.5 MHz.112 The so-called Pake's pattern of magnetic dipole–dipole interactions was indicative of a neutral radical pair. ENDOR study determined that the distance between the radicals is 6.9 Å, closer that that observed in the crystal structure in the dark state.112 Meech, Tonge and coworkers have replaced the tyrosine in the active site of AppA (Tyr21) and PixD (Tyr8) with fluorinated Tyr analogues with altered pKa values and reduction potentials, thus changing the energy landscape of the PET process. They showed that the two BLUF domains react differently to the UAA replacement: in some AppA mutants, the radical intermediate is absent, while in PixD the activity is largely affected by the Tyr replacement, indicating its essential role.113,114
Designer enzymes
Studies of the native radical enzymes often face challenges of complicated sample preparation processes, interference from other cofactors and unstable intermediates. Structure–function relationship can be established from examination of knowledge accumulated from native enzyme studies, further facilitate engineering of radical enzymes for synthetic applications.21,115,116 General computational methods such as molecular dynamics, quantum mechanics–molecular mechanics simulation, and more specialized tool to calculate radical stabilization energies also greatly facilitates the engineering process.117–119 Integration of these methods leads to a workflow for radical enzyme identification and validation proposed by Jäger and Croft.116De novo design120–122 or redesign123,124 of radical enzymes can create radical-containing models with a simple scaffold and/or mimic the structural features and function of the native enzymes, making it an complementary approach to study the native radical enzymes.
Replacing the 32nd residue in the middle of the protein with Trp or Tyr does not perturb the structure of the protein,24 and allows the study the reduction of these residues in a protein environment at different pH.126 Tommos and coworkers further replaced the Tyr32 with a set of Tyr analogues, and the reduction potential of these Tyr analogues were measured by DPV in the protein environment (Table 2).55 The data was used to calculate the reduction potentials of pathway Tyr residues in RNR.45 Tommos, Hammarström and coworkers further labelled the protein with a Ru complex as a photo-oxidant. Study of the radical formation under different pH values indicates additional proton acceptor, possibly water molecules in the interior of the protein or a Glu residue in H-bonding distance.127
Attaching a Ru or Re-based photosensitizer to a protein to realize PET is a common practice.58,134 Realization of charge separation and substrate reduction, as the photosystem II does, poses challenge to our understanding of radical enzymes and photosystem II. Wang and coworkers created several artificial PET systems in photon-sensitive proteins, such as green fluorescent protein (GFP) and flavin-containing proteins. A Cu(II) ion is introduced into fluorescence protein with the help of two metal-chelating amino acids, (S)-2-amino-3-[4-hydroxy-3-(1H-pyrazol-1-yl)phenyl]propanoic acid (pyTyr) and 2-amino-3-(8-hydroxyquinolin-5-yl)propanoic acid (HqAla). The GFP mutants showed fluorescence quenching upon Cu(II) binding, and Cu(I) is generated upon photo-irradiation, suggesting PET in GFP mutants. The distance between the chromophore of the fluorescent protein and the Cu(II) changes as the UAA was placed at the different positions on the protein, and the electron transfer rate, in the 109 s−1 range, can be modulated by the distance change, in accordance to the Marcus equation.
The reduction potential of Cu(II), at 168 mV, hinders the electron transfer. Two UAAs, 3-nitrophenylalanine (NO2Phe, 13) and 4-fluoro-3-nitrophenylalanine (FNO2Phe, 14), with peak potentials of −310 and −470 mV, respectively, extend the reduction potential range of amino acids. These amino acids were incorporated into different positions of GFP, and showed an electron transfer rate of (9.09 ± 0.45) × 1010 s−1. The rate is faster than the ET rate between P700* and A0 in photosystem I, suggesting that by careful design and optimization, efficient electron transfer could be realized in a designer protein.135
A long-lived excited state is needed for efficient charge transfer and energy conversion. Wang and coworkers introduced benzophenone-alanine (BPA, 15) into the chromophore of a fluorescent protein (superfolder yellow fluorescent protein, sfYFP). The resulting protein displays colour change upon irradiation by a 405 nm laser, suggestion that a photochemical reaction happens, thus named as photosensitizer protein (PSP). Additional mutations (Tyr203Asp, His148Glu) were added on the original PSP protein to give PSP2. PSP2 is able to generate an organic radical with a reduction potential lower than −1.1 V in presence of sacrificial reductant, indicating efficient charge transfer. Covalently attaching a Ni-terpyridine complex to PSP enables photo-catalysed CO2 reduction to CO with 2.6% quantum yield, and a TON of 120.136
Wang, Xia, Liu and coworkers placed a Tyr residue close to the chromophore, and showed by transient absorption spectroscopy that the Tyr residue transferred electron to the excited chromophore with benzophenone in 1 ps, a rate comparable to the electron transfer rate from YZ to the OEC in photosystem II.
Constructing a protein-based photon energy harvesting mechanism allows further engineering to realize genetically encoded chemical energy storage. Wang and coworkers replaced the Ni complex in the original PSP2 was replaced by a protein fragment with Fe–S clusters, to create a miniature photocatalytic CO2-reducing enzyme (mPCE). The Fe–S clusters accept electron from the benzophenone-containing chromophore and catalyse the reduction of CO2 to formate with a quantum efficiency of 1.43%, which can further be converted to more complex carbon-containing molecules or provide electron for reduction reactions.137
Recently, a xanthone-containing UAA (16) was synthesized and incorporated into protein. The designer protein allows C–H bond activation of amino acid sidechain and formation of crosslink. The photo-triggered process was captured by X-ray free-electron lasers via time-resolved serial femtosecond crystallography, with a bi-radical intermediate state proposed in the reaction.138
Discussion
Function of amino acid-based radical enzymes
Amino acid-based radicals are widely distributed in enzymes, and their roles in catalysis are diverse. Davies and coworkers argued that protein radicals formed under oxidant attack propagate along peptide chain and get repaired by cellular antioxidants, which can be a general defense mechanism against ROS.22 In many cases, Tyr and Trp, which cycle between the radical and the reduced form, scavenge highly reactive species and protect the enzyme from oxidative damage, such as in the case of non-heme iron enzymes, TfdA and TauD. Due to the transient nature of amino acid-based radicals, this kind of protection could be largely undetected and more common in oxygen activating enzymes. Tyr and Trp have reduction potentials at ∼1 V, at a higher range of biologically accessible potentials.23 Gray and Wrinkler surveyed the structures and sequences of different classes of enzymes, and found over three Try and Trp tend to appear on the same strand in oxidoreductases, possibly dissipating the holes to the periphery of the enzymes and protecting the enzyme active sites.139 Beratan and coworkers further mapped the route in cytochrome P450, cytochrome c peroxidase, and benzylsuccinate synthase, the latter being a glycyl radical enzyme.140With the biologically accessible reduction potentials, Tyr and Trp residues can work in tandem to form a wire for electron or hole transfer. Such cofactor-free long-range transfer is illustrated in RNR and in artificial systems such as metal cofactor labelled cytochrome c or azurin, where the transfer could occur over a range of several nm and even across protein–protein interfaces. Nature uses metal cofactors, such as heme for electron transfer in general. Cytochromes or multi-heme proteins could be wired by protein assembly, capable of transferring electrons over a micrometer scale.141 DNA is also known to conduct charge transfer over a long range.142 Compared with the metallo-cofactors, amino acid residues are more economic and versatile, and the reduction potentials of amino acid residues are on the higher end of the full spectrum. A gating mechanism could be implemented at the protein interface, posing tight control of the enzyme activity. However, it is worth exploring whether the electron/charge transfer can occur over longer ranges between amino acid residues, just like that mediated by hemes or nucleic acids.
The amino acid-based radicals sometimes do not directly interact with the substrate of the enzyme, but rather serve as a “capacitor”.80 In the case of RNR, the radical is first generated on Tyr122 and transfers to Cys439. The segregation of radical generation and catalysis protects the vulnerable Cys from oxidative damage. In the case of CcO, Tyr244 provides the fourth electron for the concerted four-electron reduction of O2, along with metal cofactors such as heme and copper. Amino acid-based radicals also appear in other multi-electron reactions, such as in photosystems and ferritin.
Radical enzymes can readily interact with light or magnetism, making it possible to use light to drive the reaction, or control the reaction with light or magnetism. Light-driven radical generation can be found in the photosystem or some artificial systems, such as photo-RNR. Amino acid-based radicals can work in concert with flavin radicals, forming a di-radical pair for light or magnetism sensing. The mechanism can be applied to design photo or magnetic switch for enzyme reactions or signalling cascades.
Study of radical enzymes using UAAs
In order to probe the radical species, which are usually transient and unstable in the reaction, sensitive detection methods and precise computational methods are needed. There are well established experimental methods, such as ultraviolet-visible (UV-vis) spectroscopy, stopped flow, freeze quenching, continuous-wave EPR and more advanced EPR methods, and X-ray crystallography. Furthermore, the use of some emerging techniques provides additional insight in the identification and mechanistic study of radical enzymes. For example, mining from genome data leads to the discovery of a new sub-class of RNR,61 and the sequence similarity network analysis allows fast identification of new glycyl radical enzymes.143 Cryo-EM provides atomic structure details of the organization of large protein complexes, such as the holo enzyme of RNR.57 Development of transient absorption spectroscopy allows the study of the kinetics in the ps time scale, from IR to visible region, even with capabilities to perform two-dimensional data collection.111,144In our opinion, application of UAAs in the mechanistic study and design of radical enzymes deserves special attention. Schultz and others developed and optimized the genetic codon expansion method to incorporate UAAs in a site-specific fashion.145,146 Once a specific amino acyl-tRNA synthetase and tRNA orthogonal pair for a specific UAA is developed and the UAA is available, any lab with basic biochemistry knowledge could use put the UAA in any protein of interest.36
Over 200 UAAs with different structures and functional groups can be introduced into protein through genetic codon expansion, after two decades of research.37 Bio-orthogonal groups, photo-reactive or photo-caging groups, post-translational modification groups, metal coordination groups and spectroscopic probes can be designed in UAAs, for protein labelling, protein–protein interaction study, signal transduction study, and enzyme engineering.36,147 Several aforementioned abilities of UAAs are of particular interest for radical enzyme study, including being spectroscopic probes, mimicking post-translational modifications, and tuning redox properties of the amino acids.37,148,149 At the same time, UAAs can be structurally similar to the native amino acids, minimally perturbing the structure of the target protein.
The unpaired electron on the radical produces a signal on the EPR spectrum, making EPR an important tool for radical enzyme study. The tyrosyl radical is influenced by nuclei from protons on the beta and ring carbon atoms, creating a distinct splitting pattern. Although hyperfine splitting can distinguish tyrosyl radicals from other organic radicals, it is difficult to determine which tyrosine residue from the protein is responsible for radical generation.150 The hydrogen atoms on the phenyl ring of Tyr can be readily replaced by other atoms, such as F or Cl, to change the hyperfine splitting pattern on the radical. Stubbe, Nocera and coworkers applied the fluorinated Tyr analogues in RNR, to illustrate their role as EPR probes.27,56 By replacing a specific Tyr with the Tyr analogues, they can identify the position of the radical based on change of radical EPR signal.56 The EPR probes were further expanded to tyrosine analogues with chlorine or deuterium substitutions.30 The set of UAAs was used in other systems, such as in FtmOx1, BLUF protein (AppA and PixD), and a Mb-based oxidase model to identify the position of the radical as well, indicating that it could be a general method.30,76,113,114,131 A set of Tyr analogues, especially the fluorinated tyrosines, are useful for the identification of the position of the radical in an enzyme.148,151 Due to the high specificity and sensitivity of the labelling by UAAs, they can be used in more complex systems, such as study the action of radical enzymes in vivo.
Among all 20 natural amino acids, only radicals based on Tyr/Trp/Cys/Gly are trapped and characterized. In addition to modulate the local protein environment and hydrogen bonding network, nature has evolved various post-translational modifications to directly fine-tune the properties of radical-generating residues for better catalytic performance. Liu and coworkers replaced the Tyr in GO with F2Tyr to probe the formation of the cross-linked cofactor.104 We have developed imiTyr and methylthiotyrosine to mimic the naturally occurring modified Tyr residues in CcO130 and GO.105 A DOPA residue is formed by post-translational modification at the active site for radical initiation on a recently discovered new subclass of RNR.61 The exact residue can be installed using the genetic codon expansion method, to study the mechanism of radical formation and reaction in this new RNR.
The pKa and reduction potential of Tyr residues in a radical enzyme are often fine-tuned through post-translational modification and/or by the local protein environment. The thermodynamics of the electron and/or proton transfer process are key to the enzyme activity. A series of tyrosine analogues, with substitutions on the phenyl ring, have a range of pKa values and reduction potentials. They can be applied to alter the enzyme activity or probe the electron/proton transfer process. Fluorinated tyrosines have both altered reduction potentials, altered pKa values, and a different radical EPR signal. This allows researchers to introduce perturbations to the PCET process and determine the ratio of radicals trapped on a particular residue at the same time.148,151 Stubbe, Nocera, and coworkers used the fluorinated tyrosines to map the “thermodynamic landscape” for the PCET process in RNR.45
These works have demonstrated the utility of UAAs for radical enzyme study, but most UAAs used are tyrosine analogues. There is a great demand to develop UAAs for the study of amino acid-based radicals centred on other amino acid residues than Tyr. UAAs with other functionalities, such as photo-caging, could be used to decipher the mechanisms of these enzymes.152
Conclusions
Nature uses protein scaffolds to contain and protect radicals, as well as fine-tune the position and electronic structure of the cofactors for some of the most fascinating reactions, from providing energy to drive the whole bio-system, to making essential biomolecules. After 50 years of research on radical enzymes, we are still discovering new radical enzymes and generating new questions. We are still expecting method development to help solve current problems, and explore the diverse family of radical enzymes. At the same time, with current knowledge and tools, we should be able to tailor radical enzymes for human needs, such as driving chemical synthesis and harvesting energy from sunlight.Author contributions
J. W. and Y. Y. conceived the project and drafted the outline, F. Y. and Y. Y. drafted the manuscript, B. S. revised the manuscript. All authors have read and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Key Research and Development Program of China under awards 2018YFA0903300, and the National Science Foundation of China under award 21878020, 21890743, 21837005.Notes and references
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- M. Kolberg, K. R. Strand, P. Graff and K. Kristoffer Andersson, Biochim. Biophys. Acta, 2004, 1699, 1–34 CrossRef CAS PubMed.
- P. Nordlund and P. Reichard, Annu. Rev. Biochem., 2006, 75, 681–706 CrossRef CAS PubMed.
- J. A. Cotruvo and J. Stubbe, Annu. Rev. Biochem., 2011, 80, 733–767 CrossRef CAS PubMed.
- B. L. Greene, G. Kang, C. Cui, M. Bennati, D. G. Nocera, C. L. Drennan and J. Stubbe, Annu. Rev. Biochem., 2020, 89, 45–75 CrossRef CAS PubMed.
- J. Stubbe and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 13463–13472 CrossRef CAS PubMed.
- S. Ferguson-Miller and G. T. Babcock, Chem. Rev., 1996, 96, 2889–2908 CrossRef CAS PubMed.
- P. Brzezinski and R. B. Gennis, J. Bioenerg. Biomembr., 2008, 40, 521–531 CrossRef CAS PubMed.
- S. Martinez and R. P. Hausinger, J. Biol. Chem., 2015, 290, 20702–20711 CrossRef CAS PubMed.
- E. I. Solomon, D. E. DeWeese and J. T. Babicz, Jr., Biochemistry, 2021, 60, 3497–3506 CrossRef CAS PubMed.
- L. R. F. Backman, M. A. Funk, C. D. Dawson and C. L. Drennan, Crit. Rev. Biochem. Mol. Biol., 2017, 52, 674–695 CrossRef CAS PubMed.
- S. Y. Reece and D. G. Nocera, Annu. Rev. Biochem., 2009, 78, 673–699 CrossRef CAS PubMed.
- C. Tommos, Annu. Rev. Biophys., 2022, 51, 453–471 CrossRef PubMed.
- K. J. Koebke, T. B. J. Pinter, W. C. Pitts and V. L. Pecoraro, Chem. Rev., 2022, 122, 12046–12109 CrossRef CAS PubMed.
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC.
- E. N. G. Marsh and G. D. R. Meléndez, Biochim. Biophys. Acta, 2012, 1824, 1154–1164 CrossRef CAS PubMed.
- J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Chem. Rev., 2014, 114, 4229–4317 CrossRef CAS PubMed.
- S. Y. Park and J. R. H. Tame, Biophys. Rev., 2017, 9, 169–176 CrossRef CAS PubMed.
- T. H. Yosca, A. P. Ledray, J. Ngo and M. T. Green, J. Biol. Inorg. Chem., 2017, 22, 209–220 CrossRef CAS PubMed.
- W. E. Broderick and J. B. Broderick, J. Biol. Inorg. Chem., 2019, 24, 769–776 CrossRef CAS PubMed.
- Y. Nicolet, Nat. Catal., 2020, 3, 337–350 CrossRef CAS.
- C. L. Hawkins and M. J. Davies, Biochim. Biophys. Acta, Bioenerg., 2001, 1504, 196–219 CrossRef CAS PubMed.
- J. J. Warren, J. R. Winkler and H. B. Gray, FEBS Lett., 2012, 586, 596–602 CrossRef CAS PubMed.
- M. C. Martínez-Rivera, B. W. Berry, K. G. Valentine, K. Westerlund, S. Hay and C. Tommos, J. Am. Chem. Soc., 2011, 133, 17786–17795 CrossRef PubMed.
- M. Balón, M. C. Carmona, M. A. Muñoz and J. Hidalgo, Tetrahedron, 1989, 45, 7501–7504 CrossRef.
- C. Tommos, J. J. Skalicky, D. L. Pilloud, A. J. Wand and P. L. Dutton, Biochemistry, 1999, 38, 9495–9507 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, S. Y. Reece, D. G. Nocera and J. Stubbe, J. Am. Chem. Soc., 2006, 128, 1569–1579 CrossRef CAS PubMed.
- K. R. Ravichandran, L. Liang, J. Stubbe and C. Tommos, Biochemistry, 2013, 52, 8907–8915 CrossRef CAS PubMed.
- E. C. Minnihan, D. G. Nocera and J. Stubbe, Acc. Chem. Res., 2013, 46, 2524–2535 CrossRef CAS PubMed.
- Y. Yu, X. Lv, J. Li, Q. Zhou, C. Cui, P. Hosseinzadeh, A. Mukherjee, M. J. Nilges, J. Wang and Y. Lu, J. Am. Chem. Soc., 2015, 137, 4594–4597 CrossRef CAS PubMed.
- S. D. Glover, R. Tyburski, L. Liang, C. Tommos and L. Hammarström, J. Am. Chem. Soc., 2018, 140, 185–192 CrossRef CAS PubMed.
- G. J. Gerfen, B. F. Bellew, S. Un, J. M. Bollinger, J. Stubbe, R. G. Griffin and D. J. Singel, J. Am. Chem. Soc., 1993, 115, 6420–6421 CrossRef CAS.
- M. Bennati, C. T. Farrar, J. A. Bryant, S. J. Inati, V. Weis, G. J. Gerfen, P. Riggs-Gelasco, J. Stubbe and R. G. Griffin, J. Magn. Reson., 1999, 138, 232–243 CrossRef CAS PubMed.
- S. L. Meichsner, Y. Kutin and M. Kasanmascheff, Angew. Chem., Int. Ed., 2021, 60, 19155–19161 CrossRef CAS PubMed.
- P. H. Oyala, K. R. Ravichandran, M. A. Funk, P. A. Stucky, T. A. Stich, C. L. Drennan, R. D. Britt and J. Stubbe, J. Am. Chem. Soc., 2016, 138, 7951–7964 CrossRef CAS PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- Y. Yu, C. Hu, L. Xia and J. Wang, ACS Catal., 2018, 8, 1851–1863 CrossRef CAS.
- W. Jiang, D. Yun, L. Saleh, E. W. Barr, G. Xing, L. M. Hoffart, M.-A. Maslak, C. Krebs and J. M. Bollinger Jr, Science, 2007, 316, 1188–1191 CrossRef CAS PubMed.
- A. K. Boal, J. A. Cotruvo, J. Stubbe and A. C. Rosenzweig, Biochemistry, 2012, 51, 3861–3871 CrossRef CAS PubMed.
- K. Grāve, J. J. Griese, G. Berggren, M. D. Bennett and M. Högbom, J. Biol. Inorg. Chem., 2020, 25, 571–582 CrossRef PubMed.
- J. A. Cotruvo, Jr., T. A. Stich, R. D. Britt and J. Stubbe, J. Am. Chem. Soc., 2013, 135, 4027–4039 CrossRef PubMed.
- J. M. Bollinger, D. E. Edmondson, B. H. Huynh, J. Filley, J. R. Norton and J. Stubbe, Science, 1991, 253, 292–298 CrossRef CAS PubMed.
- P. Nordlund, B.-M. Sjöberg and H. Eklund, Nature, 1990, 345, 593–598 CrossRef CAS PubMed.
- U. Uhlin and H. Eklund, Nature, 1994, 370, 533–539 CrossRef CAS PubMed.
- K. R. Ravichandran, A. T. Taguchi, Y. Wei, C. Tommos, D. G. Nocera and J. Stubbe, J. Am. Chem. Soc., 2016, 138, 13706–13716 CrossRef CAS PubMed.
- C. S. C. S. Yee, M. R. M. R. Seyedsayamdost, M. C. Y. M. C. Y. Chang, D. G. D. G. Nocera and J. Stubbe, Biochemistry, 2003, 42, 14541–14552 CrossRef CAS PubMed.
- E. C. Minnihan, D. D. Young, P. G. Schultz and J. Stubbe, J. Am. Chem. Soc., 2011, 133, 15942–15945 CrossRef CAS PubMed.
- M. R. Seyedsayamdost and J. Stubbe, J. Am. Chem. Soc., 2006, 128, 2522–2523 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, C. S. Yee, S. Y. Reece, D. G. Nocera and J. Stubbe, J. Am. Chem. Soc., 2006, 128, 1562–1568 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, C. T. Y. Chan, V. Mugnaini, J. Stubbe and M. Bennati, J. Am. Chem. Soc., 2007, 129, 15748–15749 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, J. Xie, C. T. Y. Chan, P. G. Schultz and J. Stubbe, J. Am. Chem. Soc., 2007, 129, 15060–15071 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, C. S. Yee and J. Stubbe, Nat. Protoc., 2007, 2, 1225–1235 CrossRef CAS PubMed.
- K. Yokoyama, U. Uhlin and J. Stubbe, J. Am. Chem. Soc., 2010, 132, 15368–15379 CrossRef CAS PubMed.
- E. C. Minnihan, N. Ando, E. J. Brignole, L. Olshansky, J. Chittuluru, F. J. Asturias, C. L. Drennan, D. G. Nocera and J. Stubbe, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3835–3840 CrossRef CAS PubMed.
- K. R. Ravichandran, A. B. Zong, A. T. Taguchi, D. G. Nocera, J. Stubbe and C. Tommos, J. Am. Chem. Soc., 2017, 139, 2994–3004 CrossRef CAS PubMed.
- K. Yokoyama, A. A. Smith, B. Corzilius, R. G. Griffin and J. Stubbe, J. Am. Chem. Soc., 2011, 133, 18420–18432 CrossRef CAS PubMed.
- G. Kang, A. T. Taguchi, J. Stubbe and C. L. Drennan, Science, 2020, 368, 6794 Search PubMed.
- S. Y. Reece, M. R. Seyedsayamdost, J. Stubbe and D. G. Nocera, J. Am. Chem. Soc., 2007, 129, 13828–13830 CrossRef CAS PubMed.
- A. Pizano Arturo, A. Lutterman Daniel, G. Holder Patrick, S. Teets Thomas, J. Stubbe and G. Nocera Daniel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 39–43 CrossRef CAS PubMed.
- C. Cui, B. L. Greene, G. Kang, C. L. Drennan, J. Stubbe and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 176–183 CrossRef CAS PubMed.
- V. Srinivas, H. Lebrette, D. Lundin, Y. Kutin, M. Sahlin, M. Lerche, J. Eirich, R. M. M. Branca, N. Cox, B.-M. Sjoberg and M. Hogbom, Nature, 2018, 563, 416–420 CrossRef CAS PubMed.
- M. S. Islam, T. M. Leissing, R. Chowdhury, R. J. Hopkinson and C. J. Schofield, Annu. Rev. Biochem., 2018, 87, 585–620 CrossRef CAS PubMed.
- A. Liu, R. Y. N. Ho, L. Que, M. J. Ryle, B. S. Phinney and R. P. Hausinger, J. Am. Chem. Soc., 2001, 123, 5126–5127 CrossRef CAS PubMed.
- M. J. Ryle, A. Liu, R. B. Muthukumaran, R. Y. N. Ho, K. D. Koehntop, J. McCracken, L. Que and R. P. Hausinger, Biochemistry, 2003, 42, 1854–1862 CrossRef CAS PubMed.
- M. C. Sleeman and C. J. Schofield, J. Biol. Chem., 2004, 279, 6730–6736 CrossRef CAS PubMed.
- M. Topf, G. M. Sandala, D. M. Smith, C. J. Schofield, C. J. Easton and L. Radom, J. Am. Chem. Soc., 2004, 126, 9932–9933 CrossRef CAS PubMed.
- T. Borowski, E. Broclawik, C. J. Schofield and P. E. M. Siegbahn, J. Comput. Chem., 2006, 27, 740–748 CrossRef CAS PubMed.
- R. M. Phelan, B. J. DiPardo and C. A. Townsend, ACS Chem. Biol., 2012, 7, 835–840 CrossRef CAS PubMed.
- R. M. Phelan and C. A. Townsend, J. Am. Chem. Soc., 2013, 135, 7496–7502 CrossRef CAS PubMed.
- W.-C. Chang, Y. Guo, C. Wang, S. E. Butch, A. C. Rosenzweig, A. K. Boal, C. Krebs and J. M. Bollinger, Science, 2014, 343, 1140–1144 CrossRef CAS PubMed.
- N. Steffan, A. Grundmann, S. Afiyatullov, H. Ruan and S.-M. Li, Org. Biomol. Chem., 2009, 7, 4082–4087 RSC.
- W. Yan, H. Song, F. Song, Y. Guo, C.-H. Wu, A. Sae Her, Y. Pu, S. Wang, N. Naowarojna, A. Weitz, M. P. Hendrich, C. E. Costello, L. Zhang, P. Liu and Y. Jessie Zhang, Nature, 2015, 527, 539–543 CrossRef CAS PubMed.
- W. Yan, H. Song, F. Song, Y. Guo, C.-H. Wu, A. S. Her, Y. Pu, S. Wang, N. Naowarojna, A. Weitz, M. P. Hendrich, C. E. Costello, L. Zhang, P. Liu and Y. J. Zhang, Nature, 2021, 593, 612 CrossRef CAS PubMed.
- N. P. Dunham, J. M. Del Río Pantoja, B. Zhang, L. J. Rajakovich, B. D. Allen, C. Krebs, A. K. Boal and J. M. Bollinger, J. Am. Chem. Soc., 2019, 141, 9964–9979 CrossRef CAS PubMed.
- L. Wu, Z. Wang, Y. Cen, B. Wang and J. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202112063 CAS.
- C.-Y. Lin, A. L. Muñoz Hernández, T. N. Laremore, A. Silakov, C. Krebs, A. K. Boal and J. M. Bollinger, Jr., ACS Catal., 2022, 12, 6968–6979 CrossRef CAS.
- G. Zhu, W. Yan, X. Wang, R. Cheng, N. Naowarojna, K. Wang, J. Wang, H. Song, Y. Wang, H. Liu, X. Xia, C. E. Costello, X. Liu, L. Zhang and P. Liu, JACS Au, 2022, 2, 1686–1698 CrossRef CAS PubMed.
- G. Jutz, P. van Rijn, B. Santos Miranda and A. Böker, Chem. Rev., 2015, 115, 1653–1701 CrossRef CAS PubMed.
- Y. Chen-Barrett, P. M. Harrison, A. Treffry, M. A. Quail, P. Arosio, P. Santambrogio and N. D. Chasteen, Biochemistry, 1995, 34, 7847–7853 CrossRef CAS PubMed.
- K. H. Ebrahimi, P.-L. Hagedoorn and W. R. Hagen, ChemBioChem, 2013, 14, 1123–1133 CrossRef CAS PubMed.
- J. M. Bradley, D. A. Svistunenko, T. L. Lawson, A. M. Hemmings, G. R. Moore and N. E. Le Brun, Angew. Chem., Int. Ed., 2015, 54, 14763–14767 CrossRef CAS PubMed.
- J. M. Bradley, D. A. Svistunenko, J. Pullin, N. Hill, R. K. Stuart, B. Palenik, M. T. Wilson, A. M. Hemmings, G. R. Moore and N. E. Le Brun, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2058–2067 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- W. A. van der Donk, A.-L. Tsai and R. J. Kulmacz, Biochemistry, 2002, 41, 15451–15458 CrossRef CAS PubMed.
- A.-L. Tsai and R. J. Kulmacz, Arch. Biochem. Biophys., 2010, 493, 103–124 CrossRef CAS PubMed.
- S. Mohanram, D. B. Goodin, S. Michael and B. M. Hoffman, Science, 1989, 245, 738–740 CrossRef PubMed.
- J. E. Huyett, P. E. Doan, R. Gurbiel, A. L. P. Houseman, M. Sivaraja, D. B. Goodin and B. M. Hoffman, J. Am. Chem. Soc., 1995, 117, 9033–9041 CrossRef CAS.
- T. M. Payne, E. F. Yee, B. Dzikovski and B. R. Crane, Biochemistry, 2016, 55, 4807–4822 CrossRef CAS PubMed.
- E. F. Yee, B. Dzikovski and B. R. Crane, J. Am. Chem. Soc., 2019, 141, 17571–17587 CrossRef CAS PubMed.
- C. Tommos, X.-S. Tang, K. Warncke, C. W. Hoganson, S. Styring, J. McCracken, B. A. Diner and G. T. Babcock, J. Am. Chem. Soc., 1995, 117, 10325–10335 CrossRef CAS.
- A. Gupta, A. Mukherjee, K. Matsui and J. P. Roth, J. Am. Chem. Soc., 2008, 130, 11274–11275 CrossRef CAS PubMed.
- A. Mukherjee, A. M. Angeles-Boza, G. S. Huff and J. P. Roth, J. Am. Chem. Soc., 2011, 133, 227–238 CrossRef CAS PubMed.
- P. Brzezinski and R. B. Gennis, J. Bioenerg. Biomembr., 2008, 40, 521–531 CrossRef CAS PubMed.
- D. A. Proshlyakov, D. A. Proshlyakov, M. A. Pressler, C. DeMaso, J. F. Leykam, D. L. DeWitt and G. T. Babcock, Science, 2000, 290, 1588–1591 CrossRef CAS PubMed.
- S. E. J. S. E. J. Rigby, S. Jünemann, P. R. P. R. Rich and P. Heathcote, Biochemistry, 2000, 39, 5921–5928 CrossRef CAS PubMed.
- M. A. Yu, T. Egawa, K. Shinzawa-Itoh, S. Yoshikawa, S.-R. Yeh, D. L. Rousseau and G. J. Gerfen, Biochim. Biophys. Acta, 2011, 1807, 1295–1304 CrossRef CAS PubMed.
- M. A. Yu, T. Egawa, K. Shinzawa-Itoh, S. Yoshikawa, V. Guallar, S.-R. Yeh, D. L. Rousseau and G. J. Gerfen, J. Am. Chem. Soc., 2012, 134, 4753–4761 CrossRef CAS PubMed.
- A. Jose, A. W. Schaefer, A. C. Roveda, W. J. Transue, S. K. Choi, Z. Ding, R. B. Gennis and E. I. Solomon, Science, 2021, 373, 1225–1229 CrossRef CAS PubMed.
- M. S. Rogers and D. M. Dooley, Curr. Opin. Chem. Biol., 2003, 7, 189–196 CrossRef CAS PubMed.
- J. W. Whittaker, Arch. Biochem. Biophys., 2005, 433, 227–239 CrossRef CAS PubMed.
- Y.-K. Lee, M. M. Whittaker and J. W. Whittaker, Biochemistry, 2008, 47, 6637–6649 CrossRef CAS PubMed.
- Y. Wang and A. Liu, Chem. Soc. Rev., 2020, 49, 4906–4925 RSC.
- J. Li, W. P. Griffith, I. Davis, I. Shin, J. Wang, F. Li, Y. Wang, D. J. Wherritt and A. Liu, Nat. Chem. Biol., 2018, 14, 853–860 CrossRef CAS PubMed.
- J. Li, I. Davis, W. P. Griffith and A. Liu, J. Am. Chem. Soc., 2020, 142, 18753–18757 CrossRef CAS PubMed.
- Q. Zhou, M. Hu, W. Zhang, L. Jiang, S. Perrett, J. Zhou and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 1203–1207 CrossRef CAS PubMed.
- K. V. Lakshmi, C. S. Coates, S. Smith and R. Chatterjee, in The Biophysics of Photosynthesis, ed. J. Golbeck and A. van der Est, Springer New York, New York, NY, 2014, pp. 299–320 Search PubMed.
- F. Rappaport, A. Boussac, D. A. Force, J. Peloquin, M. Brynda, M. Sugiura, S. Un, R. D. Britt and B. A. Diner, J. Am. Chem. Soc., 2009, 131, 4425–4433 CrossRef CAS PubMed.
- J. B. Weaver and S. G. Boxer, ACS Synth. Biol., 2018, 7, 1618–1628 CrossRef CAS PubMed.
- J. B. Weaver, C.-Y. Lin, K. M. Faries, I. I. Mathews, S. Russi, D. Holten, C. Kirmaier and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2116439118 CrossRef CAS PubMed.
- M. Byrdin, A. P. M. Eker, M. H. Vos and K. Brettel, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8676–8681 CrossRef CAS PubMed.
- A. Lukacs, A. P. M. Eker, M. Byrdin, K. Brettel and M. H. Vos, J. Am. Chem. Soc., 2008, 130, 14394–14395 CrossRef CAS PubMed.
- H. Nagai, Y. Fukushima, K. Okajima, M. Ikeuchi and H. Mino, Biochemistry, 2008, 47, 12574–12582 CrossRef CAS PubMed.
- A. A. Gil, A. Haigney, S. P. Laptenok, R. Brust, A. Lukacs, J. N. Iuliano, J. Jeng, E. H. Melief, R.-K. Zhao, E. Yoon, I. P. Clark, M. Towrie, G. M. Greetham, A. Ng, J. J. Truglio, J. B. French, S. R. Meech and P. J. Tonge, J. Am. Chem. Soc., 2016, 138, 926–935 CrossRef CAS PubMed.
- A. A. Gil, S. P. Laptenok, J. N. Iuliano, A. Lukacs, A. Verma, C. R. Hall, G. E. Yoon, R. Brust, G. M. Greetham, M. Towrie, J. B. French, S. R. Meech and P. J. Tonge, J. Am. Chem. Soc., 2017, 139, 14638–14648 CrossRef CAS PubMed.
- C. M. Jäger and A. K. Croft, Chem. Bio Eng. Rev., 2018, 5, 143–162 Search PubMed.
- C. M. Jäger and A. K. Croft, Biochemistry, 2023, 62, 241–252 CrossRef PubMed.
- W. Zhu, Y. Liu and R. Zhang, Theor. Chem. Acc., 2013, 132, 1385 Search PubMed.
- C. J. Suess, F. L. Martins, A. K. Croft and C. M. Jäger, J. Chem. Inf. Model., 2019, 59, 5111–5125 CrossRef CAS PubMed.
- M. J. Toda, A. P. Ghosh, S. Parmar and P. M. Kozlowski, in Methods Enzymol., ed. E. N. G. Marsh, Academic Press, 2022, 669, pp. 119–150 Search PubMed.
- C. W. Hoganson and C. Tommos, Biochim. Biophys. Acta, 2004, 1655, 116–122 CrossRef CAS PubMed.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495–3578 CrossRef CAS PubMed.
- M. J. Chalkley, S. I. Mann and W. F. DeGrado, Nat. Rev. Chem., 2022, 6, 31–50 CrossRef CAS PubMed.
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855–862 CrossRef CAS PubMed.
- F. Yuan, Y. Yu and C. Li, Synth. Biol. J., 2020, 1, 685–696 Search PubMed.
- K. Westerlund, B. W. Berry, H. K. Privett and C. Tommos, Biochim. Biophys. Acta, Bioenerg., 2005, 1707, 103–116 CrossRef CAS PubMed.
- W. Berry Bruce, C. Martínez-Rivera Melissa and C. Tommos, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9739–9743 CrossRef CAS PubMed.
- A. Nilsen-Moe, C. R. Reinhardt, S. D. Glover, L. Liang, S. Hammes-Schiffer, L. Hammarström and C. Tommos, J. Am. Chem. Soc., 2020, 142, 11550–11559 CrossRef CAS PubMed.
- K. D. Miner, A. Mukherjee, Y.-G. Gao, E. L. Null, I. D. Petrik, X. Zhao, N. Yeung, H. Robinson and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 5589–5592 CrossRef CAS PubMed.
- A. P. Ledray, S. Dwaraknath, K. Chakarawet, M. R. Sponholtz, C. Merchen, C. Van Stappen, G. Rao, R. D. Britt and Y. Lu, Biochemistry, 2022, 62, 388–395 CrossRef PubMed.
- X. Liu, Y. Yu, C. Hu, W. Zhang, Y. Lu and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 4312–4316 CrossRef CAS PubMed.
- Y. Yu, Q. Zhou, L. Wang, X. Liu, W. Zhang, M. Hu, J. Dong, J. Li, X. Lv, H. Ouyang, H. Li, F. Gao, W. Gong, Y. Lu and J. Wang, Chem. Sci., 2015, 6, 3881–3885 RSC.
- Y. Yu, C. Cui, X. Liu, I. D. Petrik, J. Wang and Y. Lu, J. Am. Chem. Soc., 2015, 137, 11570–11573 CrossRef CAS PubMed.
- D. Zheng, Y. Zhang, X. Liu and J. Wang, Photosynth. Res., 2019, 143, 221–231 CrossRef PubMed.
- H. B. Gray and J. R. Winkler, Q. Rev. Biophys., 2003, 36, 341–372 CrossRef CAS PubMed.
- X. Lv, Y. Yu, M. Zhou, C. Hu, F. Gao, J. Li, X. Liu, K. Deng, P. Zheng, W. Gong, A. Xia and J. Wang, J. Am. Chem. Soc., 2015, 137, 7270–7273 CrossRef CAS PubMed.
- X. Liu, F. Kang, C. Hu, L. Wang, Z. Xu, D. Zheng, W. Gong, Y. Lu, Y. Ma and J. Wang, Nat. Chem., 2018, 10, 1201–1206 CrossRef CAS PubMed.
- F. Kang, L. Yu, Y. Xia, M. Yu, L. Xia, Y. Wang, L. Yang, T. Wang, W. Gong, C. Tian, X. Liu and J. Wang, ACS Catal., 2021, 11, 5628–5635 CrossRef CAS.
- X. Liu, P. Liu, H. Li, Z. Xu, L. Jia, Y. Xia, M. Yu, W. Tang, X. Zhu, C. Chen, Y. Zhang, E. Nango, R. Tanaka, F. Luo, K. Kato, Y. Nakajima, S. Kishi, H. Yu, N. Matsubara, S. Owada, K. Tono, S. Iwata, L.-J. Yu, J.-R. Shen and J. Wang, Nat. Chem., 2022, 14, 1054–1060 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2015, 2015, 201512704 Search PubMed.
- D. Teo Ruijie, R. Wang, R. Smithwick Elizabeth, A. Migliore, J. Therien Michael and N. Beratan David, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15811–15816 CrossRef CAS PubMed.
- X. Jiang, J. H. van Wonderen, J. N. Butt, M. J. Edwards, T. A. Clarke and J. Blumberger, J. Phys. Chem. Lett., 2020, 11, 9421–9425 CrossRef CAS PubMed.
- A. R. Arnold, M. A. Grodick and J. K. Barton, Cell Chem. Biol., 2016, 23, 183–197 CrossRef CAS PubMed.
- B. J. Levin, Y. Y. Huang, S. C. Peck, Y. Wei, A. Martínez-del Campo, J. A. Marks, E. A. Franzosa, C. Huttenhower and E. P. Balskus, Science, 2017, 355, eaai8386 CrossRef PubMed.
- C. Cristina, A. Gerald, V. M. Frank and C. Majed, Science, 2013, 339, 1586–1589 CrossRef PubMed.
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS PubMed.
- T. S. Young, I. Ahmad, J. A. Yin and P. G. Schultz, J. Mol. Biol., 2010, 395, 361–374 CrossRef CAS PubMed.
- A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Chem. Sci., 2015, 6, 50–69 RSC.
- C. Hu, Y. Yu and J. Wang, Chem. Commun., 2017, 53, 4173–4186 RSC.
- Y. Yu, X. Liu and J. Wang, Acc. Chem. Res., 2019, 52, 557–565 CrossRef CAS PubMed.
- D. A. Svistunenko and C. E. Cooper, Biophys. J., 2004, 87, 582–595 CrossRef CAS PubMed.
- C. Hu, S. I. Chan, E. B. Sawyer, Y. Yu and J. Wang, Chem. Soc. Rev., 2014, 43, 6498–6510 RSC.
- A. Deiters, D. Groff, Y. Ryu, J. Xie and P. G. Schultz, Angew. Chem., Int. Ed., 2006, 45, 2728–2731 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |