
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRiPP enzyme heterocomplex structure-guided discovery of a bacterial borosin α-N-methylated peptide natural product†
K. K.
Crone‡
 a,
T.
Jomori‡§
b,
F. S.
Miller
a,
J. A.
Gralnick
bc,
M. H.
Elias
ab and
M. F.
Freeman
*ab
a,
T.
Jomori‡§
b,
F. S.
Miller
a,
J. A.
Gralnick
bc,
M. H.
Elias
ab and
M. F.
Freeman
*ab
aDepartment of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota – Twin Cities, St. Paul 55108, USA. E-mail: mffreema@umn.edu
bThe BioTechnology Institute, University of Minnesota – Twin Cities, St. Paul 55108, USA
cDepartment of Plant and Microbial Biology, University of Minnesota – Twin Cities, St. Paul 55108, USA
First published on 21st August 2023
Abstract
Amide peptide backbone methylation is a characteristic post-translational modification found in a family of ribosomally synthesized and post-translationally modified peptide natural products (RiPPs) called borosins. Previously, we bioinformatically identified >1500 putative borosin pathways in bacteria; however, none of the pathways were associated with a known secondary metabolite. Through in-depth characterization of a borosin pathway in Shewanella oneidensis MR-1, we have now identified a bacterially derived borosin natural product named Shewanellamide A. Borosin identification was facilitated by the creation and analysis of a series of precursor variants and crystallographic interrogation of variant precursor and methyltransferase complexes. Along with assaying two proteases from S. oneidensis, probable boundaries for proteolytic maturation of the metabolite were projected and confirmed via comparison of S. oneidensis knockout and overexpression strains. All in all, the S. oneidensis natural product was found to be a 16-mer linear peptide featuring two backbone methylations, establishing Shewanellamide A as one of the few borosin metabolites yet identified, and the first from bacteria.
Introduction
Borosins are a recently described family of ribosomally synthesized and post-translationally modified peptide (RiPP) natural products signified by α-N-methylations incorporated into the amide backbone.1 α-N-Methylations are pharmacologically relevant modifications shown to enhance the pharmaceutical properties of peptides through mechanisms such as increased membrane permeability, oral bioavailability, and resistance to proteolytic degradation.2 While borosin pathways were initially described in fungi,3–5 we recently discovered and characterized the first borosin RiPP system in bacteria.6 Subsequently, we and others reported the identification of >1500 putative borosin pathways encoded in a variety of bacteria and select archaea.7,8Borosin pathways are categorized based on the overall protein architecture(s) of encoded α-N-methyltransferases and their cognate natural product coding precursors. Type I–III borosins, predominantly found in fungi, harbour unusually long RiPP precursors that encode iterative autocatalytic α-N-methyltransferases fused to a C-terminal natural product core peptide sequence.5 Type IV ‘split’ borosin pathways, thus far found only in bacteria and a few archaea, encode the simplest precursor–methyltransferase architectures and follow canonical RiPP biosynthetic logic.7 In RiPP biosynthesis, the natural product backbone is directly encoded in the core peptide of a precursor protein that also often encodes an N-terminal leader peptide. After translation, the leader peptide is recognized by one or more tailoring enzymes, which then bind to the core peptide and install post-translational modifications (PTMs). For nearly all borosin pathways studied to date, α-N-methylations are iteratively installed in an N- to C-terminally directed fashion on the core peptide, which are presumed to then be proteolytically cleaved to release the final natural products (Fig. 1(a)). In addition to type IV borosin pathways, a diverse array of bacterial α-N-methyltransferases and precursors are found with different architectures.7,8
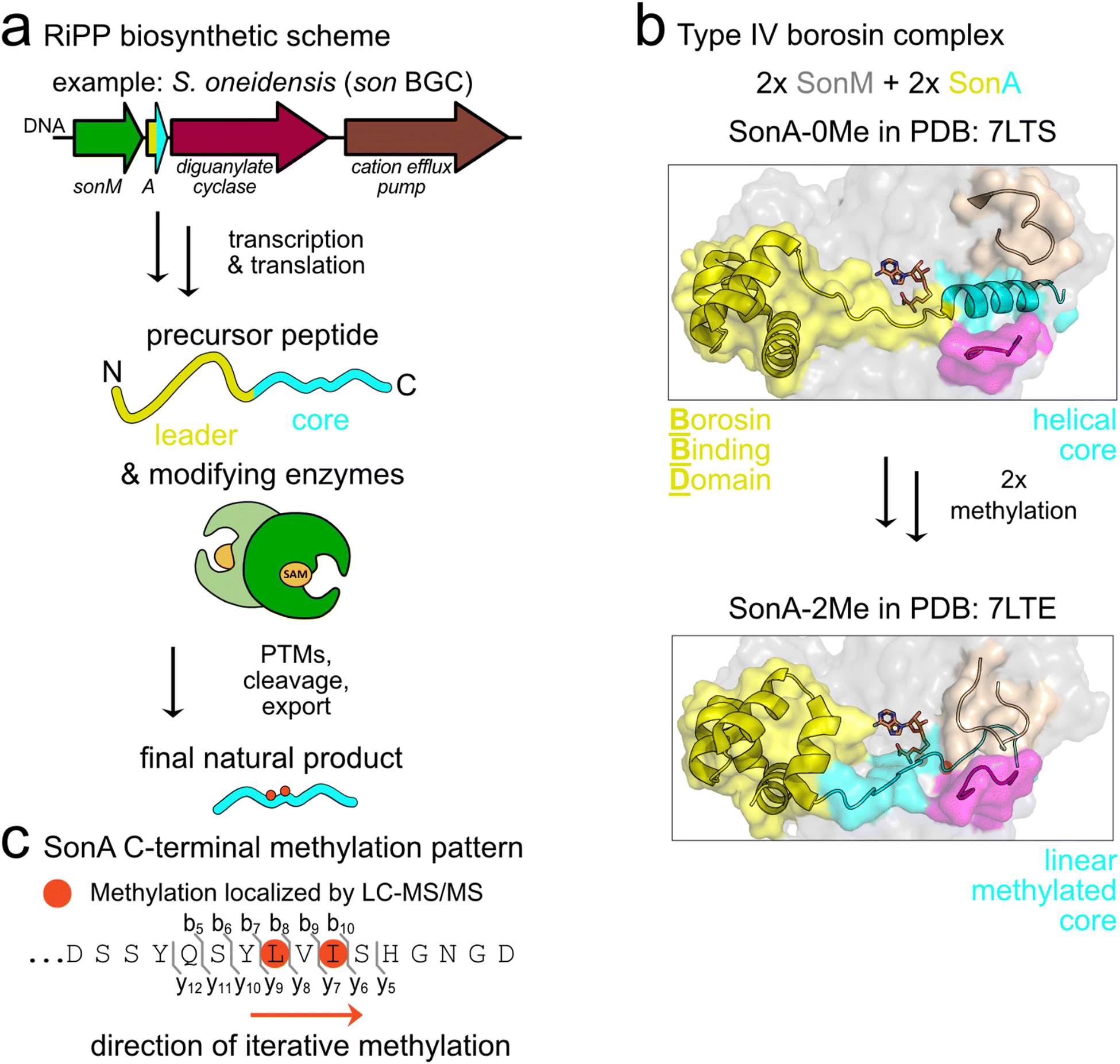 | ||
| Fig. 1 Type IV borosin biosynthesis in S. oneidensis. (a) Schematic illustrating canonical RiPP maturation of the type IV borosin precursor peptide, SonA, by the α-N-methyltransferase enzyme, SonM. (b) Co-crystal structures of R67A-SonM-SonA-0Me-SAH (PDB: 7LTS) and SonM-SonA-2Me-SAH (PDB: 7LTE) highlighting the conformational rearrangements of the core peptide (cyan), metamorphic helix 5 of the borosin binding domain (yellow), and enzyme active site clamps (magenta and beige) that are implicated in substrate positioning for catalysis. α-N-Methylated (Me) residues are circled (orange). (c) The amino acid sequence of the SonA C-terminus with iteratively installed α-N-methylated residues, SonA-L63 and SonA-I65, highlighted with orange circles. | ||
The most thoroughly characterized bacterial borosin pathway is the type IV system from Shewanella oneidensis MR-1.6 We have previously reported multiple crystal structures along with kinetic analyses detailing the interactions between the α-N-methyltransferase (SonM) and precursor peptide (SonA). The SonA leader peptide encodes an N-terminal five-helical bundle, termed the borosin binding domain (BBD), which accounts for most of the binding affinity between SonA and SonM. In the majority of solved crystal structures, we observe the fully resolved BBD (SonA 1–53) and loosely defined linker and core peptide regions (SonA 54–67) with two α-N-methylations (Me) installed on L63 and I65. A co-crystal structure of an inactive SonM variant and SonA shows considerable secondary structural changes in unmethylated SonA as compared to the bismethylated species. Here, the unmethylated SonA C-terminus is bound in the active site of SonM as an α-helix, with a seemingly compensatory unwinding of helix 5 in the BBD of SonA. We hypothesize these conformational changes may guide the two iteratively installed methylations (Fig. 1(b) and (c)).
While fungal borosin pathways have been linked to the production of the omphalotins, gymnopeptides, lentinulins, and dendrothelins,3,5,9 none of the prokaryotic borosin biosynthetic gene clusters (BGC) have associated final natural products. The S. oneidensis son BGC is composed of four genes encoding the borosin α-N-methyltransferase SonM (SO1478; UNIPROTKB: Q8EGW3), precursor peptide SonA (SO1479; UNIPROTKB: Q8EGW2), a GGDEF domain-containing protein (SO1480; UNIPROTKB: Q8EGW1), and a putative cation efflux pump (SO1481; UNIPROTKB: Q8EGW0). Notably, there are no other predicted modifying enzymes or proteases found in the gene cluster to aid in identifying the final metabolite. The colocalization of these four genes is well-conserved throughout the Shewanella genus, however their native role in the organism remains elusive.6
Here we report the identification of the first bacterial borosin metabolite and link it to the type IV split borosin pathway in S. oneidensis. The bismethylated 16 amino acid fragment, DSSYQSYLMeVIMeSHGNGD (the subscript ‘Me’ refers to α-N-methylation), was identified in wildtype S. oneidensis cultures as well as through overexpression in the native host. Identification of the low-abundance natural product was facilitated through interrogation of a series of SonA truncation variants and a small set of methyltransferase-precursor co-crystal structures to better define the sequence boundaries of the core peptide. Additional experiments with candidate proteases encoded in S. oneidensis gave further credence to the hypothesis that the helical non-methylated core peptide observed in the R67A variant SonM–SonA crystal structure designates the boundaries of the mature natural product. Trace detection of the borosin metabolite in S. oneidensis led to larger-scale production of the natural product in Escherichia coli that allowed for structural elucidation via mass spectrometry, NMR, and Marfey's analysis.
Results and discussion
SonA metamorphic helix 5 and N-terminal linker region variants do not alter SonM specificity
To gain insight towards the boundaries of the putative core peptide, we first probed the sequence requirements of the SonA precursor for the methyltransferase SonM, namely the 5th “metamorphic” helix (SonA M47-L53) of the BBD as well as the linker region (SonA 54-62). From the previously reported co-crystal structures SonM—SonA-2Me—SAH (PDB code: 7LTE) and SonM-R67A—SonA-0Me—SAH (PDB code: 7LTS), a large secondary structural change is observed in the C-terminal region of SonA (Fig. 1).6 Concomitantly, residues M47-L53 in the BBD are unwound in the SonM-R67A—SonA-0Me—SAH structure and were proposed to help position the C-terminus of SonA, pre-methylation, in the active site.We constructed a series of SonA variants that include deletions or duplications in the targeted BBD region SonA 47–53 and linker SonA 54–62 (Fig. 2). SonM relative rates of each SonA variant were compared to the rate of wildtype (wt) using our previously optimized coupled-enzyme kinetics assay.6 In parallel, methylation patterns were analysed via LC–MS/MS to observe the relative abundance of methylated species and any shifts in the location of the backbone methylations due to the mutations. The variants assessed included a variety of deletions or duplications of the metamorphic helix. SonM activity was resilient towards this set of six variants with minimal effects on reaction rate. Complete deletion of the metamorphic helix (SonA_M47-L53del) showed the largest decrease at ∼50% rate of wt (Fig. 2(a)). Moreover, >90% of bismethylated species with the native N-methylation pattern on L63 and I65 was observed after SonM overnight reactions for all six variants (Fig. 2(a) and Fig. S1, ESI†). These results indicate the BBD metamorphic helix is not required for SonM activity nor critical for determining the sites of methylation.
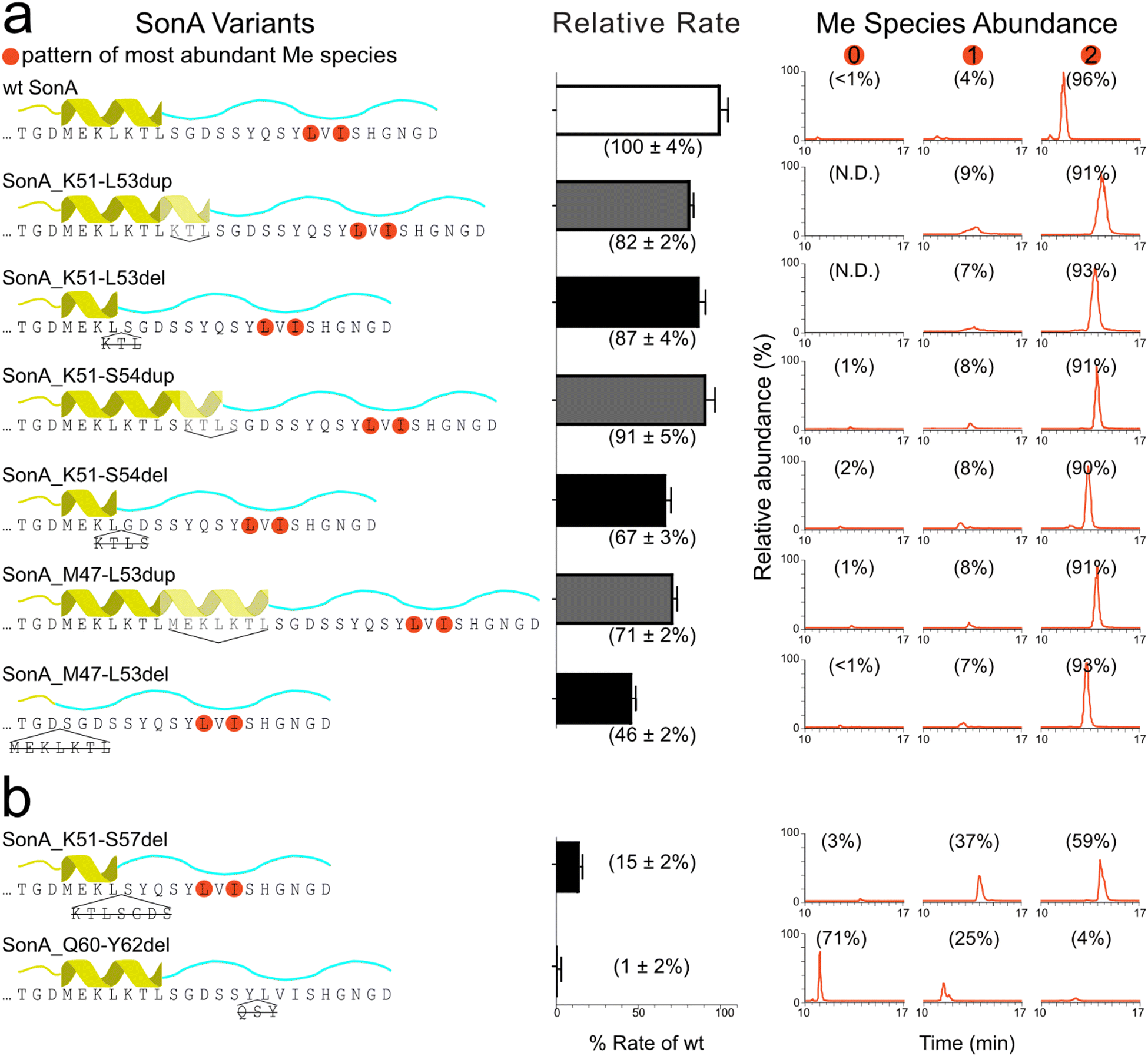 | ||
| Fig. 2 Relative rates and methylation patterns for SonA variants. The left column contains the amino acid sequence of each SonA variant with a cartoon representation of the region duplicated or deleted. Methylated residues are circled in orange for the most abundant methylated species observed via LC–MS/MS. The middle column shows the relative rate of each SonA variant normalized to the rate of wt SonA and wt SonM. The mean and standard deviations from three replicates is displayed in parentheses. For each variant, the right column contains the MS1 extracted ion chromatogram with the relative abundance of each methylated species (0, 1, or 2) written as a percentage above the peak. (a) SonA variants with duplications or deletions made within the metamorphic helix. (b) SonA variants with deletions in different regions of the linker. | ||
With this information, we decided to probe two variants with deletions closer to the site of methylation. The first variant extended the deletion of SonA_K51-S54del to also delete SonA residues 55–57 (sequence GDS) in the SonA_K51-S57del variant. SonA_K51-S57del saw a more drastic decrease in rate at 15% compared to wt and produced only ∼60% bismethylated species after overnight incubation with SonM (Fig. 2(b)). While most of the fragmentation data supports the native pattern for the bismethylated species, some lower abundance off-target methylation was also observed (Fig. S1, ESI†). Secondly, SonA_Q60-Y62del was created to remove three residues N-terminally adjacent to the first site of methylation in the core peptide. The rate of reaction of wt SonM with SonA_Q60-Y62del was undetectable, within error, via our coupled-enzyme assay. Overnight incubation with SonM revealed predominantly unmethylated species (71%), along with multiple mono-methylated species exhibiting the largest fraction methylated on I65, followed by V64, and trace amounts on L63 (Fig. S1, SI). The large decrease in rate observed with the ‘GDS’ and ‘QSY’ sequence deletions and shift in methylation patterns are notable. Due to the sensitivity of SonM to these sequence regions, we hypothesized that the borosin core peptide boundary would be likely found between G55 and Y62. More specifically, the ‘QSY’ sequence appeared to be critical for core recognition and methylation pattern fidelity. We reasoned that since the Q60-Y62 sequence is either an important SonM recognition sequence or part of the core peptide, further structural investigation of the Q60-Y62del variant was warranted.
Crystallographic insights into core peptide mutational effects
The current set of available co-crystal structures of SonM and SonA do not show evidence of specific side chain interactions near the site of methylation that could be influencing the regiospecificity of methylation. To gain understanding of the role of Q60-Y62 on SonM activity and fidelity, we chose to co-crystallize wt SonM with SonA_Q60-Y62del and S-adenosylmethionine (SAM) or S-adenosylhomocysteine (SAH). The structure of the SonM—SonA_Q60-Y62del variant bound to SAH (PDB code: 8T1S) was determined to 2.00 Å. In this structure, as in the previously resolved wt SonM–SonA structure,6 SonM and SonA form dimers of heterodimers (Fig. S2, ESI†). Overall, the structure is very similar to the wt structure bound to SAH (PDB code: 7LTE, r.m.s.d. 0.21 Å over 626 atoms). In both heterodimers, the linker region of SonA is disordered and could not be completely modelled. Yet, the electron density clearly shows the core peptide is bound and ordered in one of the heterodimers (SonM chain A; it is disordered and not fully modelled in SonM chain C). For the first time, we observe mono-methylated SonA in the active site on I65 (Fig. 3(a)). However, the core peptide adopts a near identical conformation to that of SonA-2Me in the wt structure (Fig. 3(b)). Unique to these most recently solved structures, we see electron density for a metal bound at the dimer interface of both complexes (Fig. S2, ESI†). We have modelled in zinc as the bound metal based on ICP-OES data (Fig. S3, ESI†). Future work will delve deeper into this phenomenon and the possible effects of this bound metal.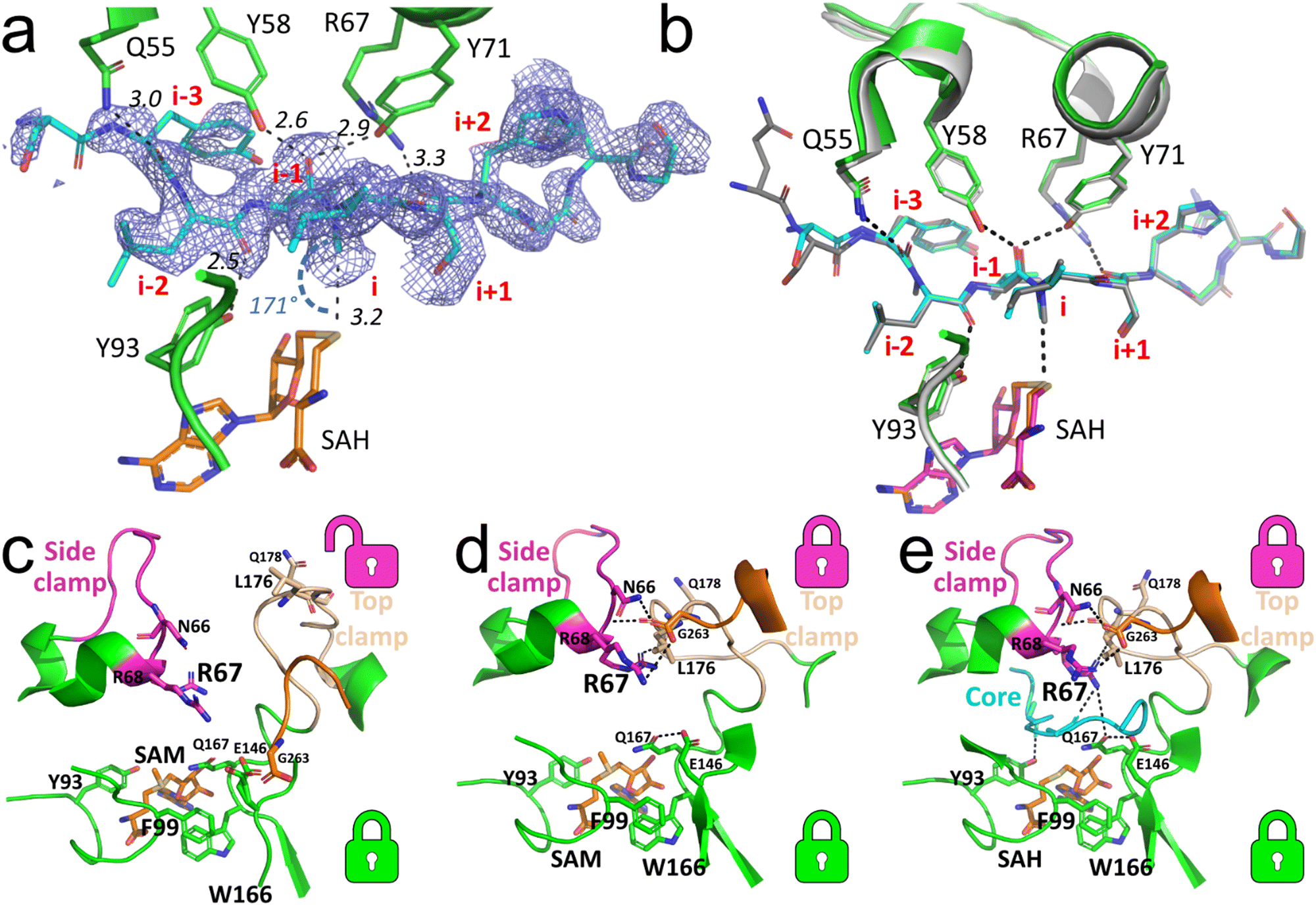 | ||
| Fig. 3 Core peptide and loop positioning for the SonM—SonA_Q60-Y62del complexes. (a) Simulated annealing Fourier difference electron density map (Fobs − Fcalc; blue mesh; contoured at 2σ) from the SonM—SonA_Q60-Y62del—SAH complex (PDB code: 8T1S) is shown for the SonA core peptide (SonA; chain B; occupancy set to 0.9). While the observation of the density reveals the presence of an α-N-methylation at the residue SonA-I65 (i), no methylation is observed at the residue SonA-L63 (i-2). Here, the position “i” relates to the core peptide SonA residue that is in line with the cofactor donor group; negative “i” values are for residues towards the SonA N-terminus, positive “i” values are for residues towards the SonA C-terminus. (b) Superposition of the structure of SonM—SonA_Q60-Y62del—SAH with the wt structure of SonM—SonA-2Me—SAH (PDB code: 7LTE). For the wt structure, SonM, SonA and the bound SAH are shown as light gray, dark gray, and pink cartoons, respectively. For the Q60-Y62del structure, SonM, SonA, and the bound SAH are shown as green, cyan, and orange cartoons, respectively. (c) The open conformation in the SonM—SonA_Q60-Y62del—SAM complex (PDB code: 8T1T) with SAM (orange sticks) bound in the active site. The π-stacking of SonM-F99 and SonM-W166 forms the bottom lock, while the top lock between the side and top clamp is open. (d) The other heterodimer of the SAM-bound complex structure showcases closed top and bottom locks. The hydrogen-bonding network involving SonM-R67, SonM-N66, and SonM-R68 (with SonM C-termini (G263) from the other molecule) closes the top lock in the active site. (e) The SonM—SonA_Q60-Y62del—SAH complex with SAH (orange sticks) reveals both the top and bottom locks closed, using a similar network of interactions seen in (d) but with the core peptide (cyan cartoon) present. Key hydrogen bonds and distances are depicted as black dashed lines. SonM C-termini (G263) from the other monomer is shown as orange sticks in all three panels. Top (magenta and beige) and bottom (green) locks are illustrated with corresponding color-coded icons next to each motif. | ||
The structure of the SonM—SonA_Q60-Y62del-SAM complex (PDB code: 8T1T) was determined at 1.55 Å resolution. Contrary to previously solved structures, the core peptide of SonA is entirely unresolved in the electron density maps and could not be modelled. Yet, several other features differ from previously reported SonM–SonA complexes. Interestingly, from the SonM—SonA_Q60-Y62del-SAM, we can observe SAM bound in both active sites, and the previously defined bottom lock, π-stacking interaction between SonM-F99 and SonM-W166, closed (Fig. 3(c)). However, the top and side clamps of each heterodimer active site are in a different conformation relative to each other, closed and open. This results in a closed top lock and open top lock for each heterodimer, respectively (Fig. 3(c) and (d)). The heterodimer with the open top lock displays the active site clamps in a similar position to the SonM—R67A—SonA-0Me—SAH structure (Fig. 3(e)), providing evidence that wt SonM can adopt a similar conformation to the inactive variant SonM_R67A. Furthermore, this indicates that each monomer can act independently, as our data has alluded to in the past.6
While the core peptide is not observed in the SonM—SonA_Q60-Y62del-SAM structure, the bounds of where we see electron density at SonA-G55 would indicate a high degree of disorder from SonA-D56 to the C-terminus. Of note, RiPP proteases often have specificity for glycine in the P1 position.10,11 In addition, the unmethylated C-terminus of SonA in the SonM-R67A—SonA-0Me—SAH structure is an α-helix that begins after residue SonA-G55. These observations are consistent with our hypothesis that the core peptide boundary exists in this region of SonA. Thus, we were intrigued with the possibility that the helical SonA C-terminal sequence DSSYQSYLVISHGNGD found in SonM-R67A—SonA-0Me—SAH marks the boundaries of the final natural product.
In vitro functional assessment of two proteases
In an attempt to further define the putative core peptide region, we mined the S. oneidensis genome for candidate proteases that could process SonA. Like with many RiPPs, there are no encoded proteases in the son BGC. Our two main criteria for selecting protease candidates were (1) homology to known RiPP-active proteases, and (2) matching protease expression profiles to those known for the son BGC. Genes sonM and sonA are expressed under aerobic conditions. In 2005, Gralnick et al. reported that sonA in S. oneidensis MR-1 is one of the most negatively regulated genes by the Arc system, an oxygen-sensing global regulator.12 In S. oneidensis, ArcA is responsible for regulating the switch from aerobic to anaerobic metabolism; therefore, negative regulation by ArcA results in loss of expression during an aerobic to anaerobic growth transition.Based on these criteria, we selected two putative S. oneidensis proteases negatively regulated by ArcA13 that also bore homology to RiPP maturating proteases. The first candidate was the putative S8 family serine protease LanP homolog SO0867 (UNIPROTKB: Q8EIH2). After recombinantly expressing and purifying SO0867 in E. coli, exposure of SO0867 to SonA-2Me resulted in non-specific degradation of SonA-2Me as assessed by LC–MS/MS (data not shown) and was therefore excluded from further consideration.
The second protease candidate, SO1059 (UNIPROTKB: Q8EHZ5), is predicted to be an M1 family metalloprotease. This putative periplasmic alanyl aminopeptidase has homology to the recently identified bi-functional Zn-dependent peptidase LabP (UNIPROTKB: V9PX19; 32.3% identity) that is active on class III lanthipeptides.14 LabP is a cytosolic enzyme that acts as both an endo- and exo-protease on the RiPP precursor LabA. As SO1059 is predicted to be localized to the periplasm and SonA is found in the cytosol, an intermediary proteolytic step on SonA would likely be necessary if SO1059 was involved in maturation of the son BGC natural product. Nonetheless, gene so1059 was also found to be down-regulated by ArcA,13 and therefore was selected for heterologous expression in E. coli.
Gene so1059 was expressed encoding a C-terminal His6-tag and an N-terminal periplasmic signal sequence (DsbAss from E. coli).15 After obtaining soluble protein, the protease was first assayed against full-length SonA-2Me, but did not display endo-proteolytic activity. Next, we tested SO1059 for more general proteolytic activity with alanine-p-nitro-aniline as previously described,14 and the enzyme was highly active on the small model substrate (Fig. S4, ESI†). We then assessed SO1059 activity with SonA-2Me core fragments, since LabP also acts as an exo-protease on RiPP precursors. SonA-2Me was first trypsinized to yield the SonA fragment 52-TLSGDSSYQSYLMeVIMeSHGNGD-71. Upon incubation with SO1059, the highest abundance fragment observed for this sequence was 56-DSSYQSYLMeVIMeSHGNGD-71 as measured by LC-MS/MS, suggesting SO1059 has some preferential exo-proteolytic activity on SonA precursor fragments (Fig. S5, ESI†). While certain RiPP systems require multiple proteolytic maturation steps during their biosynthesis,16,17 it is currently unclear whether this holds true for the son BGC or whether SO1059 is involved in the maturation of the encoded borosin metabolite.
Metabolomic analysis identifies the son natural product
With the putative SonA core region further defined through biochemical and structural interrogation, we set out to identify the son BGC natural product in the native host. Given that no reports exist detailing α-N-methylated peptides from S. oneidensis, a well-studied model organism, we anticipated low titres from the son BGC. Therefore, we created a plasmid-based sonM/sonA homologous overexpression strain as well as an in-frame precursor knockout strain (ΔsonA) to aid in the identification of the putative S. oneidensis borosin natural product. All three S. oneidensis strains (wt, ΔsonA, and sonM/sonA overexpression) were cultured in LB or Shewanella minimal medium (SBM)18 at 30 °C for 12 hours under aerobic conditions. RT-PCR results revealed that both wt and arabinose-induced sonM/sonA overexpression strains transcribed sonM and sonA under these culturing conditions. As expected, the ΔsonA negative control strain did not reveal detectable transcription of sonA (Fig. S6, ESI†).To identify the natural product produced by the son BGC, metabolomic analysis was conducted via LC–MS/MS of extracts from the three strains described above (wt, ΔsonA, and the sonM/sonA overexpression strain). The strains were cultured in SBM for 24 hours at 30 °C. Culture supernatants were directly desalted through a C18 Sep-Pak cartridge, while cell-pellets were extracted with methanol prior to C18 Sep-Pak cartridge desalting. LC–MS/MS analysis of the various extracts revealed the bismethylated peptide fragment DSSYQSYLMeVIMeSHGNGD in the cell pellet extracts from wt and sonM/sonA overexpression strains (Fig. 4 and Fig. S7, ESI†). Extracted ion chromatogram peak integration for the modified SonA core peptide in the overexpression strain displayed a twelve-fold increase in metabolite production compared to wt. SonA peptide fragments were not observed in ΔsonA supernatant or pellet fractions, nor were they found in the supernatant fractions for the wt and overexpression strains. We also created a ΔsonM strain as an additional control and did not find SonA peptide fragments in any fractions (Fig. 4(a)). In order to determine whether any other metabolites related to the son BGC were being produced, we used Global Natural Product Social Molecular Networking (GNPS) molecular networking19 to tease out any additional sequence variants or unexpected post-translational modifications on the SonA core peptide. No additional masses were detected in LC–MS/MS data from the strains grown in SBM for 24 hours using GNPS.
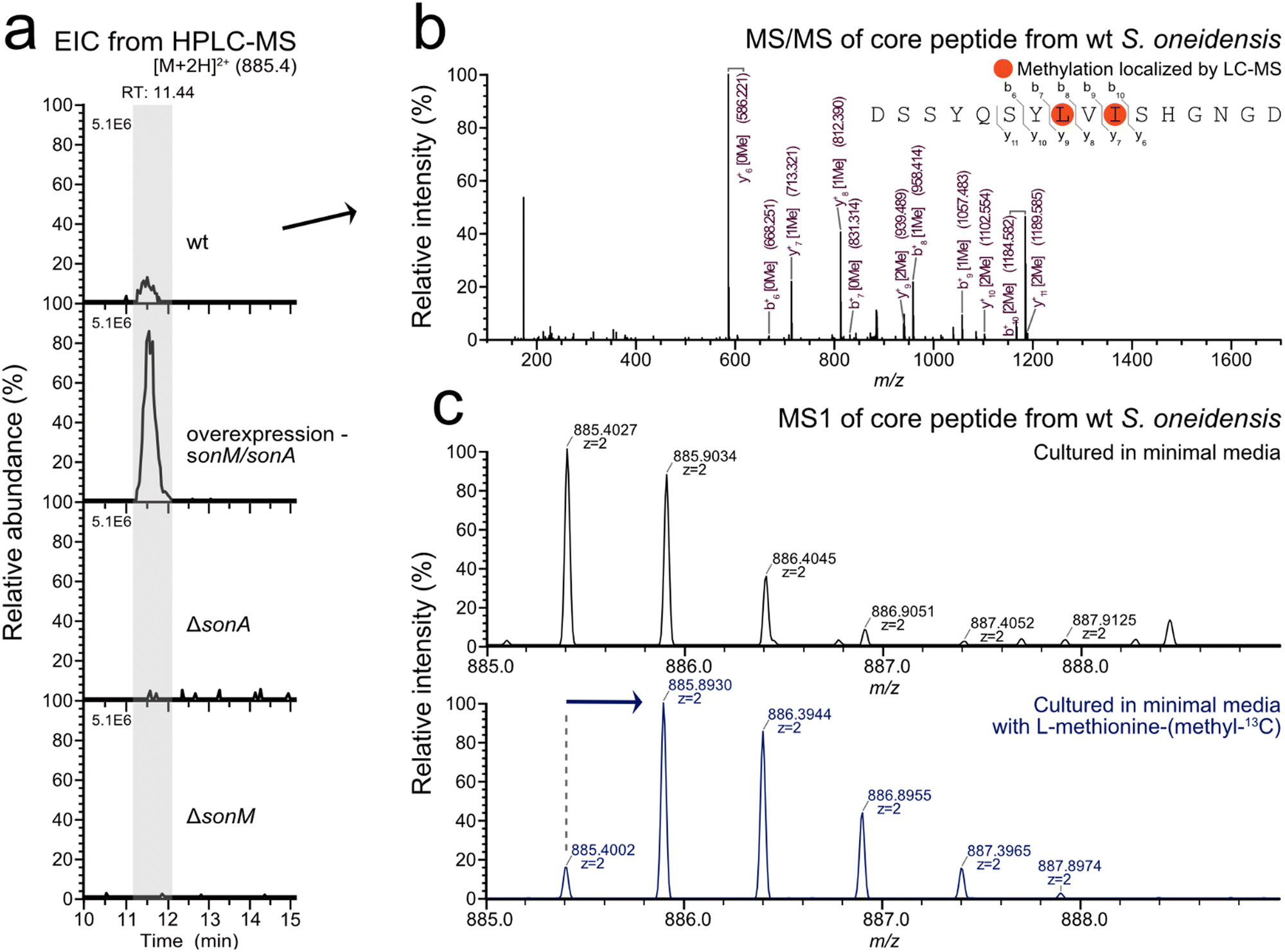 | ||
| Fig. 4 Identification of the SonA core peptide by LC–MS/MS. (a) MS1 extracted ion chromatogram (EIC) for the mass of the SonA core peptide from wt, sonM/sonA overexpression, and knockout strains of S. oneidensis. Strains were cultured for 24 hours and metabolites were extracted with methanol and desalted prior to analysis by LC–MS/MS. (b) MS2 spectra of the core peptide identified from the wt organism. The amino acid sequence is written above the spectra with localized methylations circled orange. Fragments identified in the spectra are labelled above (b-ions) and below (y-ions) the sequence and divided by gray lines. The masses for the respective ion fragments in the spectra are labelled in burgundy in parentheses. Fragments with mass shifts corresponding to methylated residues have “Me” denoted in brackets. (c) The top panel shows the MS1 isotopic distribution of the identified SonA core peptide from wt S. oneidensis cultured for 24 hours in minimal media. The bottom panel shows the shift in the MS1 isotopic distribution of the SonA core peptide when the organism is cultured for 24 hours in minimal media labelled with L-methionine-(methyl-13C). | ||
As a final verification of our LC–MS/MS data, we repeated aerobic cultures of wt S. oneidensis and our overexpression strain grown in SBM supplemented with L-methionine-(methyl-13C). As expected, a concomitant shift in the isotopic distribution pattern for the mass corresponding to the peptide DSSYQSYLMeVIMeSHGNGD was observed, indicating 13C-labeled α-N-methyl incorporation into the natural product (Fig. 4(c)). Based on LC–MS peak integrations, methyl-13C incorporation was approximated using the NRC isotope enrichment calculator.20,21 The bis-N-methylated natural product showed 85% methyl-13C incorporation for one position in wt S. oneidensis under these conditions. In sum, our data strongly suggest that DSSYQSYLMeVIMeSHGNGD is the borosin natural product produced by the son BGC in S. oneidensis.
Structure elucidation of Shewanellamide A, a bacterial borosin natural product
Despite scale-up and use of the sonM/sonA overexpression strain, we were unable to purify sufficient quantities of DSSYQSYLMeVIMeSHGNGD from S. oneidensis for structural analysis. Therefore, to verify the structure, we prepared 1 (DSSYQSYLMeVIMeSHGNGD) from heterologously co-expressed sonM and sonA in E. coli. In this co-expression construct, a Factor Xa cleavage site was inserted between encoded SonA residues 55 and 56 for ease of purification (SonA-Xa). After heterologous co-expression of SonA-Xa with SonM, SonA-Xa was affinity-purified and digested with Factor Xa protease followed by HPLC purification. The retention time of 1 was identical to the metabolite (m/z = 885.403) detected in extracts from wt S. oneidensis by LC–MS/MS. In addition, co-injection of S. oneidensis wt extract with 1 also eluted as a single peak (Fig. S8, ESI†). Thus, we conclude that the natural product of the son BGC in S. oneidensis MR-1 is 1 (DSSYQSYLMeVIMeSHGNGD), and we name this metabolite Shewanellamide A.A total of 0.9 mg of 1 was purified, and NMR experiments were carried out in H2O/D2O = 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix solvent. 1H, 13C, HSQC, HMBC, COSY, 2D-TOCSY, ROESY NMR spectra were collected concomitant with MS/MS spectra (Fig. S9, ESI†). Carbon signals were assigned from HSQC and HMBC correlations. The 1H, HSQC, 2D TOCSY and COSY correlations allowed us to assign each of 16 amino acid residues as indicated in Fig. 5. The interconnections between the amino acids were determined by HMBC and ROESY correlations (Fig. 5) and were consistent with the sequence DSSYQSYLMeVIMeSHGNGD inferred by LC–MS/MS data from the extracts of S. oneidensis (wt and the sonM/sonA overexpression strain). HMBC correlations from N-methyl protons δH 2.91 to the carbonyl C-1 (δC 175.7) of Y7 suggested that the α-nitrogen of L8 is methylated. Moreover, L8 α-N-methylation was supported by ROESY cross-peaks of the methyl group (δH 2.91) with H-5,9 (δH 7.12) of Y7 and H-2 (δH 4.48) of V9. Another α-N-methyl group (δH 3.17) observed HMBC correlations to carbonyl carbon C-1 (δC 177.7) of V9 and ROESY cross-peaks to H-3 (δH 2.04) of I10 and CH3-4 (δH 0.86). This suggested that α-N-methylation at I10 is also present. The absolute configuration of all amino acid units in 1 were determined to be in the L-configuration by Marfey's analysis (Fig. S10, ESI†). From the NMR and MS/MS spectra, we determined the planar structure of 1 as in Fig. 5; i.e. DSSYQSYLMeVIMeSHGNGD. Initial bioactivity for 1 was assayed against bacteria (E. coli TOP10, S. oneidensis MR-1, Pseudomonas putida KT2440, Bacillus subtilis WB800N) and yeast (Saccharomyces cerevisiae YPH499, Pichia pastoris X-33), however no antibiotic activity was observed (Fig. S11, ESI†). Further ongoing work will be necessary to determine the biological role(s) of Shewanellamide A.
:1 mix solvent. 1H, 13C, HSQC, HMBC, COSY, 2D-TOCSY, ROESY NMR spectra were collected concomitant with MS/MS spectra (Fig. S9, ESI†). Carbon signals were assigned from HSQC and HMBC correlations. The 1H, HSQC, 2D TOCSY and COSY correlations allowed us to assign each of 16 amino acid residues as indicated in Fig. 5. The interconnections between the amino acids were determined by HMBC and ROESY correlations (Fig. 5) and were consistent with the sequence DSSYQSYLMeVIMeSHGNGD inferred by LC–MS/MS data from the extracts of S. oneidensis (wt and the sonM/sonA overexpression strain). HMBC correlations from N-methyl protons δH 2.91 to the carbonyl C-1 (δC 175.7) of Y7 suggested that the α-nitrogen of L8 is methylated. Moreover, L8 α-N-methylation was supported by ROESY cross-peaks of the methyl group (δH 2.91) with H-5,9 (δH 7.12) of Y7 and H-2 (δH 4.48) of V9. Another α-N-methyl group (δH 3.17) observed HMBC correlations to carbonyl carbon C-1 (δC 177.7) of V9 and ROESY cross-peaks to H-3 (δH 2.04) of I10 and CH3-4 (δH 0.86). This suggested that α-N-methylation at I10 is also present. The absolute configuration of all amino acid units in 1 were determined to be in the L-configuration by Marfey's analysis (Fig. S10, ESI†). From the NMR and MS/MS spectra, we determined the planar structure of 1 as in Fig. 5; i.e. DSSYQSYLMeVIMeSHGNGD. Initial bioactivity for 1 was assayed against bacteria (E. coli TOP10, S. oneidensis MR-1, Pseudomonas putida KT2440, Bacillus subtilis WB800N) and yeast (Saccharomyces cerevisiae YPH499, Pichia pastoris X-33), however no antibiotic activity was observed (Fig. S11, ESI†). Further ongoing work will be necessary to determine the biological role(s) of Shewanellamide A.
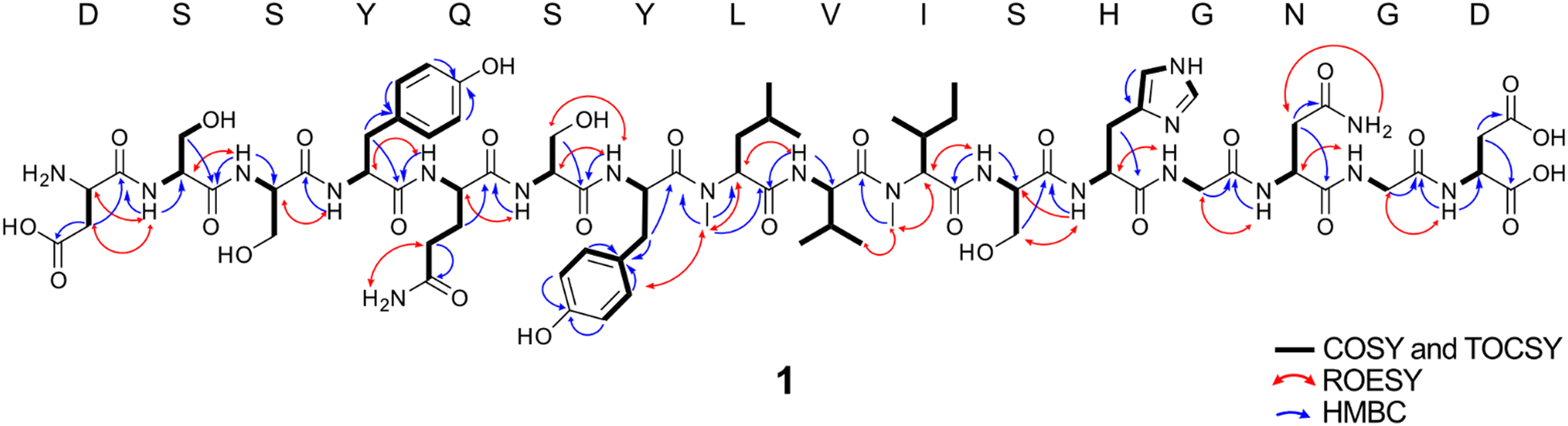 | ||
| Fig. 5 Structure of Shewanellamide A (1) as determined by NMR. Through bond connectivity of individual amino acids determined by COSY and TOCSY are shown as bolded black lines. Multiple bond correlations determined by HMBC are blue arrows. Through space proton correlations determined by ROESY are red arrows. | ||
Conclusions
Here we have presented the discovery of a linear split borosin natural product from the metal respiring bacterium S. oneidensis MR-1. Identification of Shewanellamide A (1) was facilitated through the creation and analysis of a suite of precursor variants and heterocomplex crystal structures probing the boundaries between the SonA leader and core peptides. Modifications that tailor the peptide backbone, including amide backbone N-methylation, alter the structure and properties of peptides. N-to-C cyclized α-N-methylated peptides, like the blockbuster drug cyclosporin, are known for their ability to passively diffuse biological membranes and to favour the formation of type VI β-turns.22 Linear α-N-methylated peptides, on the other hand, often harbour distinct characteristics compared to their cyclized counterparts. For example, linear α-N-methylated peptides are known to form β-hairpins in solution23 or to exhibit β-strand character in the trans configuration when displaying alternating methylated residues.24 As a result, linear α-N-methylated peptides have been designed to effectively cap β-sheet protofilaments as potential treatments in β-amyloid associated diseases.25–27 As such, borosin BGCs hold great promise in natural product discovery as well as providing mutable platforms to engineer custom α-N-methylated peptides.Experimental
General procedures
All constructs for the heterologous expression of SonM (Uniprot Q8EGW3) and SonA (Uniprot Q8EGW2) proteins in E. coli were made using the genes previously cloned out of S. oneidensis MR-1. Constructs of individual his6-sonA and his6-sonM as well as the co-expression vector containing sonM-his6-sonA were made as described previously and used as templates in creating the variants.6 All primer sequences, plasmid names, and gene sequences used in this study can be found in Table S1 (ESI†).sonA variant expression constructs
Metamorphic helix and linker region variants, including the Factor Xa cleavage site variant of SonA, were constructed in the sonM and sonA co-expression and sonA individual expression backgrounds using site-directed mutagenesis. Primers prTJ1054-prTJ1055 and prKC1035-prKC1048 were used in appropriate pairs to amplify the entire plasmid under standard Q5 (NEB) reaction conditions: initial denaturation 30s at 98°C; denature 98°C 10s, anneal (temperature varied) for 20s, extension 72°C 3min for 30 cycles; final extension 72°C 2min. The PCR reactions were digested with DpnI (NEB) overnight followed by PCR clean-up using a kit (Thermo Scientific). Purified DNA was treated with T4 polynucleotide kinase (Thermo Scientific), cleaned-up again using the same kit, and ligated with T4 ligase (NEB) according to manufacturer's instructions. Ligated plasmids were transformed into E. coli TOP10 cells for propagation and submitted for sequencing (ACGT).
Protease expression constructs
For the SO1059 and SO0867 candidate proteases, the corresponding genes were cloned from isolated S. oneidensis MR-1 gDNA.6 Standard Q5 polymerase reaction conditions were followed. The so1059 insert was prepared with a two-stage PCR to add both an N-terminal E. coli periplasmic signal sequence and a C-terminal his-tag, first using primer pairs prKKC1000 and prKKC1002, followed by using a PCR clean-up kit and a successive PCR with primer pairs prKKC1004 and prKKC1001. The pET28b backbone was amplified with prKKC1005 and prKKC1003, DpnI digested, and purified. For so0867, the insert was amplified from gDNA using the primer pair prTJ1042 and prTJ1043. The pET28b backbone was amplified with prTJ1044-prTJ1045, DpnI digested, and purified. The respective backbone and insert were assembled with HiFi DNA Assembly Master Mix (NEB), transformed into TOP10 cells, and sequence verified.Protein over-production and purification
Either E. coli LOBSTR BL21(DE3) or BL21(DE3) cells were transformed with the pET28b expression plasmids and cultured overnight with 50 μg mL−1 kanamycin at 37 °C. A 10 mL overnight culture was added to 1 L Terrific Broth with 50 μg mL−1 kanamycin in 2.5 L baffled flasks (Thomson Scientific) with foam stoppers. The 1 L culture was grown to an optical density at 600 nm (OD600) of approximately 2.0. At this time, the cultures were cold-shocked on ice for 30 min followed by induction with 200 μM IPTG (final concentration). After induction, cultures were incubated at 16 °C for 24 h shaking at 240 rpm. Cells were harvested by centrifugation at 5000 × g for 30 min at 4 °C. Cell pellets were resuspended in ice-cold lysis buffer (50 mM HEPES pH 8, 300 mM NaCl, 10% (v/v) glycerol) with 20 mM imidazole, lysed using 1 mg mL−1 (final concentration) lysozyme for 30 min at 4 °C, and sonicated (QSonica). The resultant lysate was clarified by centrifugation at 15000 × g for 30 min at 4 °C. The supernatant was incubated with Ni-NTA resin (Gold Biotechnology) on a rotator at 4 °C for 1 h. For the SonA variants and protease candidates, resin was washed with lysis buffer and then eluted with lysis buffer containing 250 mM imidazole. For the SonM_his6-SonA_Xa variant, resin was washed with ten column volumes of lysis buffer with 8 M urea (to dissociate SonM from SonA) and protein was eluted with lysis buffer containing 250 mM imidazole. Protein-containing fractions were collected and concentrated using Amicon Ultrafilters (3 kDa MWCO) and dialyzed at 4 °C to remove imidazole. For the SonM—SonAQ60-Y62del complex, size exclusion chromatography was performed using an AKTA FPLC on a HiLoad® 16/600 Superdex column (Cytiva) with a flow rate of 1 mL min−1. Only fractions with the highest purity of the target proteins were pooled and concentrated for crystallization.
Relative rate determination of SonA variants
The relative rate of each SonA variant was assayed with wt SonM using a published coupled-enzyme assay as previously optimized by our lab.6,28 Briefly, the reactions were performed in volumes of 100 μL in a 96-well flat-bottomed plate (Sarstedt). Absorbance at 340 nm was recorded using SpectraMax iD5 (Molecular Devices, Inc.) plate reader. All components of the coupled-enzyme assay with the addition of 80 μM wt his6-SonA (or variant) and 1000 μM SAM were allowed to equilibrate for ∼10 min. The reaction was started by the addition of 5 μM his6-SonM. Reads were taken every 30–40 s. The slope over the linear range was measured as the rate of the reaction and used to compare between wt and variants. Measured rates were normalized to the rate of wt SonA at 100% in GraphPad Prism 8.Preparation of SonA variants for analysis of methylation patterns by LC–MS/MS
Heterologously expressed and purified protein was prepared for mass spectrometric analysis using an in-gel digest method. Briefly, the band corresponding to his6-SonA (or variant) was cut out of an SDS-PAGE gel, cut into ∼2 mm × 2 mm pieces, and placed in 1.5 mL low protein binding tubes (Thermo Scientific). Gel pieces were then washed with a 1:1 ratio of 100 mM ammonium bicarbonate (ABC):acetonitrile (ACN) to remove the dye. Once clear, they were dehydrated in 100% ACN until semi-opaque (∼30 s) and the ACN was discarded. Gel pieces were rehydrated in digest buffer (50 mM ABC and 1:50 AspN protease or Trypsin protease (Promega)), placed on ice for 15 min, and then transferred to a 37 °C incubator overnight. Digested peptides were extracted from the gel pieces with 60 μL of 50% ACN and 0.3% formic acid (FA) and incubated at room temperature (RT) for 15 min. The supernatant was recovered in a fresh low protein binding tube. This extraction was repeated with 60 μL of 80% ACN and 0.3% FA, followed by 95% ACN and 0.1% FA. The pooled peptide extractions were frozen at −80 °C for 30 min to inactivate the protease. After freezing, the extracted peptides were dried using a SpeedVac (Eppendorf), reconstituted in 0.1% FA, and purified/desalted using C18 ZipTips according to the manufacturer's instructions. Purified and desalted peptides were again dried by SpeedVac, reconstituted in 15–30 μL of 20% ACN, 0.1% FA, and transferred to an autosampler vial (Agilent) for MS analysis.
LC–MS/MS of SonA variants
LC–MS/MS measurements of digested peptides were obtained on a Thermo Scientific Fusion mass spectrometer with a Dionex Ultimate 3000 UHPLC system with a nLC column (200 mm × 75 μm) packed with Vydac 5 μm particles of 300 Å pore size (Hichrom Limited). The LC method used the following solvents: 0.1% FA in water (solvent A) and 0.1% FA in acetonitrile (solvent B). After a 4.5 min equilibration of 20% solvent B at a flow rate of 1 μL min−1, the flow rate was dropped to 0.3 μL min−1 over 0.5 min. The sample was then injected and the gradient run was as follows: solvent B at 20–85% for 32 min, 85% for 2 min. Mass spectra were acquired in positive-ion mode. Full MS was done at a resolution of 60000 [automatic gain control (AGC) target, 4 × 105; maximum ion trap (IT), 50 ms; range, 150–1800 m/z], and data-dependent and targeted MS/MS were both performed at a resolution of 15000 [AGC target, 5 × 105; maximum IT, 500 ms; isolation window, 2.2] using higher-energy collisional dissociation (HCD) fragmentation. HCD collision energy was 18% with steps of ±3% during LC–MS/MS measurements. Data were processed and analysed using Thermo Fisher Xcalibur 3.0.63 and MaxQuant v1.6.10.
Crystallizing the SonM—SonAQ60-Y62del complex
The purified protein complex was dialyzed into 10 mM HEPES pH 8 and concentrated to 20 mg mL−1. Trays were set up based on previously established conditions for the wt SonM—SonA complex. After a modified screen, conditions producing diffraction quality crystals were 240 mM sodium malonate pH 5–5.5, 8–12% PEG 3350 at a 1:1 or 2:1 ratio of protein to mother liquor. For crystals co-crystallized with SAM or SAH, either 1 mM SAM or 0.5 mM SAH (final concentration) was added to the protein solution prior to setting up the trays. Diffraction quality crystals were formed within 5 days at 292 K. For co-crystallization with SAH, seed trays were set up using a 1:1 ratio of mother liquor (240 mM sodium malonate pH 5, 12% PEG) to protein solution. A seed solution was made at a 1:20 dilution and crystals sheared by pipetting up and down ∼10×. A seeding tool (Hampton Research) was used to transfer seeds to the drop. Crystals were cryoprotected by transferring to a drop consisting of 240 mM sodium malonate at the pH of mother liquor, 17–20% PEG 3350, and 20% glycerol, and then immediately flash-frozen in liquid nitrogen.
Data collection, structure resolution, and refinement
X-ray diffraction datasets were collected at 100 K using synchrotron radiation on the 23-IDB beamline at the Advanced Photon Source (APS, Argonne, Illinois, USA). The X-ray diffraction data were integrated and scaled using the XDS package version Mar 15, 2019.29 Molecular replacement was performed using the wt SonM—SonA-2Me—SAH structure as a model (PDB: 7LTE) and the MOLREP software.30 Model construction was done using Coot 0.8.9.247.31 Cycles of refinement were performed using REFMAC 5.8.023848.32 Simulated annealing omit Fourier difference map was calculated using Phenix.33 Ramachandra statistics were calculated using Sfcheck.34 Clashscore (number of serious steric overlaps per 1000 atoms) were determined using MolProbity.35 Final refinement statistics are shown in Table S2 (ESI†).Assays with SO1059
After purification, SO1059 was assessed for activity with the small peptidase substrate L-alanine 4-nitroanilide hydrochloride (Sigma-Aldrich). Assays were performed in a Spectramax iD5 plate reader monitoring absorbance at 405 nm. Reaction conditions were 50 mM HEPES pH 8, 1 mM L-alanine 4-nitroanilide hydrochloride and 1 μM SO1059 (excluded for the negative control). In vitro assays with SonA-2Me were performed in 50 mM HEPES buffer pH 8 at RT for 40 h with 50 μM SonA-2Me and 5 μM of SO1059, however no cleavage was observed by MS. For trypsinized SonA-2Me, SonA-2Me was incubated overnight with trypsin in 50 mM HEPES buffer pH 8, centrifuged through a 10 kDa Nanosep centrifugal filter (Pall) to remove trypsin, and then 10-fold less SO1059 (or equal volume of buffer for the negative control) was added and incubated for ∼50 h. The reactions were quenched with 6 M urea, vacuum-concentrated, and then C18 zip-tipped as previously described for analysis by LC–MS/MS.Bacterial strain construction
Shewanella strains were constructed as previously described.36The plasmid pSMV336 was used to generate clean in-frame deletions in S. oneidensis MR-1. Regions of approximately 1 kb up- and downstream of the gene to be deleted were cloned into the pSMV3 backbone. The ligated plasmids were transformed into electrocompetent UQ950 cells, grown overnight at 37 °C on LB agar with 50 μg mL−1 kanamycin, and screened by colony PCR for successful ligations.
The pBBAD18K37 plasmid was used in the construction of the sonM/sonA overexpression strain. Amplified inserts were assembled into the prepared backbone by Gibson assembly, transformed into electrocompetent TOP10 cells, plated onto LB agar containing 50 μg mL−1 kanamycin, and incubated at 37 °C until colonies formed.
Sequence-verified pSMV3 or pBBAD18K plasmids for the knockout and overexpression strains, respectively, were transformed into electrocompetent WM3064 E. coli cells for subsequent conjugation into S. oneidensis MR-1. Transformed WM3064 cells were grown on LB agar with 50 μg mL−1 kanamycin and 3 μM diaminopimelic acid (DAP) overnight at 37 °C until colonies formed. Separately, S. oneidensis MR-1 was streaked from a frozen glycerol stock onto LB agar and grown at 30 °C until colonies formed. A fresh colony of the plasmid-harbouring WM3064 cell was then patched onto a fresh colony of S. oneidensis MR-1 on LB agar supplemented with 3 μM DAP and grown overnight at 30 °C until a lawn formed. A sterile pipette tip was swiped across the lawn and streaked onto an LB agar plate containing 50 μg mL−1 kanamycin. The plate was incubated at 30 °C. The sonM/sonA overexpression strain was complete after conjugation. For the pSMV3-conjugated ΔsonM and ΔsonA knockout strains, sucrose selection was used to identify clones with a double-crossover event. Several merodiploid clones (either as fresh colonies or glycerol stocks) were streaked onto LB agar plates (with 15% sucrose and no salt) and allowed to grow at 30 °C until colonies formed (up to two days). Individual colonies were screened by colony PCR to verify the correct genomic location of the deletion and the PCR products were subsequently sequence-verified.
Shewanella growth conditions
For metabolomics experiments, bacterial strains of S. oneidensis MR-1 (wt, ΔsonA, ΔsonM, or sonM/sonA overexpression strains) were cultured on LB agar plates (with 50 μg mL−1 kanamycin only for the sonM/sonA overexpression strain) overnight at 30 °C. A single colony was picked and inoculated into 10 mL of LB media. Overnight cultures were centrifuged at 4000 × g for 5 min and the cell pellets were washed with 10 mL of SBM,18 containing 20 mM lactate, three times to remove residual LB media. The resuspended cells in SBM were inoculated into 40 mL of SBM, containing 20 mM lactate, in 50 mL falcon tubes (growth start OD600 = 0.05). For the sonM/sonA overexpression strain, sonM and sonA on pBBAD were induced by addition of 0.1 mg mL−1 of L-arabinose (final concentration) in SBM. The bacteria were cultured at 30 °C for 24 h with shaking at 220 rpm.RT-PCR of sonA and sonM
To confirm whether sonM and sonA were transcribed in S. oneidensis MR-1 under aerobic conditions, cells were cultured in 200 mL of either LB or SBM for 18 h at 30 °C and 220 rpm. RNA was isolated from four strains of S. oneidensis MR-1 (wt, ΔsonA, ΔsonM, and the sonM/sonA overexpression strain), using the RNeasy Mini Kit (Qiagen). Briefly, a 2 mL aliquot from each culture was taken and the cells were harvested by centrifugation. The cell pellet was resuspended in TE buffer containing 15 mg mL−1 of lysozyme at a concentration of ∼1.25 × 109 cells per mL. Resuspended cells were incubated at RT for 15 min with vortexing every 2 min. After lysis, 200 μL of cell lysate was taken and used in the remaining steps following the manufacturer's protocol. Eluted RNA was further treated with a DNA digest, followed by an additional purification step with the RNeasy Mini Kit. cDNA was reverse transcribed from the RNA sample with random hexamer primers and SuperScript® IV Reverse Transcriptase (Thermo Fisher) following the manufacturer's protocol. Transcription of sonM and sonA in each strain was verified by PCR amplification of generated cDNA using Q5 DNA polymerase and primers (sonA_fwd/sonA_rev, and sonM_fwd/sonM_rev).Metabolite purification
Cells of S. oneidensis MR-1 from 40 mL cultures were harvested by centrifugation at 4000 × g for 10 min at 4 °C. Cells were resuspended in 10 mL of 50% MeOH and were extracted with sonication followed by addition of 90 mL of ddH2O. The cell extracts were applied onto a Sep-Pak cartridge equilibrated with 5% methanol aq., washed with 5 mL of 20% methanol twice, and metabolites were eluted with 3 mL of 80% methanol twice (supernatants of cultures were also desalted using C18 Sep-Pak). The eluates were evaporated in vacuo using an evaporator, and then reconstituted in 100 μL of 5% methanol aq. and transferred to 1.5 mL glass vials for LC–MS/MS analysis.Metabolomic analysis using LC–MS/MS
Data were obtained on a Thermo Scientific Fusion mass spectrometer as described above using the same solvents with a modified LC gradient. After 4.5 min equilibration of 20% solvent B at a flow rate of 1 μL min−1, the flow rate was dropped to 0.3 μL min−1 over 0.5 min. The sample was then injected, and the gradient run was as follows: solvent B at 20–40% for 15 min, 40–90% for 1 min, 90% 4 min. All other parameters were the same. HCD collision energies from 18% with steps of ±4% were used during LC–MS/MS measurements. Data processing and analysis was performed using the same software packages as noted above. The GNPS METABOLOMICS-SNETS-V219 was run under default conditions for the “small data pre-set” with a tolerance of 0.2 Da. All peptide masses searched for and identified in the wt data are shown in Table S3 (ESI†).Preparation and purification of bis-N-methylated core peptide standard (1)
After metal-affinity purification, his6-SonA_Xa was concentrated to approximately 8 mg mL−1 and dialyzed into cleavage buffer (20 mM tris–HCl pH 8.0, 100 mM NaCl, 2 mM CaCl2) to remove imidazole. Samples were divided into 250 μL aliquots, flash-frozen in liquid nitrogen, and stored at −80 °C until needed. For the cleavage reaction, 2.5 μL of 1 mg mL−1 Factor Xa Protease (NEB) was added to a 250 μL aliquot. After incubation for 4 h at RT, an additional 2.5 μL of Factor Xa protease was added to the aliquot and incubated for 12 h at RT. Before purification with HPLC, digested peptides were desalted using Sep-Pak(R) Plus C18 Environmental Cartridges (Waters). The digested proteins were loaded onto Sep-Pak equilibrated with 5% methanol aq., washed with 5 mL of 20% methanol twice, and digested peptides were eluted with 3 mL of 60% methanol twice. Desalted peptides were evaporated in vacuo, and then reconstituted in 15–30 μL of 5% methanol aq. and transferred to glass vials for HPLC purification. The desalted peptide fraction was further purified by ODS HPLC on a Kinetex(R) C18 LC column (150 × 4.6 mm, 100 Å), eluted isocratically with 35% MeOH aq. containing 0.1% formic acid, to afford bismethylated core peptide.NMR general experimental procedures
0.9 mg of 1 was dissolved in solvent (H2O:D2O = 9:1). All 1H, COSY, TOCSY and ROESY NMR spectra were recorded using a Bruker 600 MHz spectrometer, equipped with a 5 mm cryoprobe, at 25 °C. HSQC and HMBC NMR spectra were recorded using a Bruker 900 MHz spectrometer, equipped with a 5 mm cryoprobe, at 25 °C.
Marfey's analysis
0.2 mg of 1 was placed in a 1 mL reaction vial, where 200 μL of 6 N HCl was added. The compound was hydrolysed for 24 h at 100 °C and then dried under vacuum. The hydrolysed compound was dissolved in 50 μL of water and 100 μL of 1% (w/v) FDAA and 20 μL of 1 M sodium bicarbonate. The reaction was heated at 40 °C for 1 h with intermittent mixing. After removal from heat, the reaction was quenched with 20 μL of 2 N HCl, and the hydrolysate was compared to derivatized standards prepared in the identical manner by LC–MS. 1 μL of a 1:10000 diluted sample was injected. The same solvents were used as in previous LC–MS methods. After equilibration and sample injection, a gradient of 10–55% solvent B over 20 min was used with a flow rate of 0.3 μL min−1.
Peptide activity testing
The minimum inhibitory concentrations (MICs) against yeast Saccharomyces cerevisiae YPH499, Pichia pastoris X-33, bacteria E. coli TOP10, Pseudomonas putida KT2440, Shewanella oneidensis MR-1, and Bacillus subtilis WB800N were determined using optical density at 600 nm (OD600). Compound 1 was dissolved in H2O, and the activity was evaluated by preparing a 2-fold dilution series from 50 μM (final concentration). Overnight cultures of yeast and bacteria were used to inoculate 100 μL cultures (OD600 = 0.05) of YPD and LB media, respectively, in a 96-well plate. The serially diluted compound was added to the 96-well plate. The cultures were incubated standing at 30 °C for 12 h, where the OD600 was monitored to determine the MIC values. The reproducibility of data was confirmed by three independent experiments.Data availability
Protein crystallographic data collected for this study has been deposited in the PDB under codes: 8T1T [https://doi.org/10.2210/pdb8T1T/pdb] (SonM—SonA_Q60-Y62del—SAM) and 8T1S [https://doi.org/10.2210/pdb8t1s/pdb] (SonM—SonA_Q60-Y62del—SAH). The mass spectrometry, NMR and plate reader raw data files have been uploaded to the Data Repository for the University of Minnesota [https://doi.org/10.13020/egtn-6b03]. Additional data relevant to this study can be found in the ESI.† Any further information can be requested from the corresponding author.Author contributions
K. K. C, T. J., M. H. E., J. A. G. and M. F. F. designed experiments. K. K. C., T. J., and F. S. M. performed experiments. M. H. E. solved crystal structures, K.K. C., T. J., M. H. E. and M. F. F. wrote the manuscript. All authors edited the manuscript. M. F. F. supervised the work.Conflicts of interest
M. F. F. is an inventor on patent WO2017EP58327, US20190112583A1, WO2021168399, and US-20230090771-A1. All other authors declare no competing interests.Acknowledgements
We thank S. Shaw for technical assistance and advice concerning crystallography, and the Advanced Photon Source (APS, Argonne, Illinois) and staff for time on the beamline. We are grateful to the Research Analytical Laboratory at the University of Minnesota for running the ICP-OES experiments on our samples. This work was funded by the National Institutes of Health (R35 GM133475 to M. F. F.), the University of Minnesota BioTechnology Institute (M. H. E., J. A. G., and M. F. F.), the University of Minnesota Graduate School (K. K. C.), and the Daiichi Sankyo Foundation of Life Science (T. J.). The table of contents figure was created with graphics from BioRender.com.References
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- J. Chatterjee, C. Gilon, A. Hoffman and H. Kessler, Acc. Chem. Res., 2008, 41, 1331–1342 CrossRef CAS PubMed.
- N. S. van der Velden, N. Kälin, M. J. Helf, J. Piel, M. F. Freeman and M. Künzler, Nat. Chem. Biol., 2017, 13, 833–835 CrossRef CAS PubMed.
- S. Ramm, B. Krawczyk, A. Mühlenweg, A. Poch, E. Mösker and R. D. Süssmuth, Angew. Chem., Int. Ed., 2017, 56, 9994–9997 CrossRef CAS PubMed.
- M. R. Quijano, C. Zach, F. S. Miller, A. R. Lee, A. S. Imani, M. Künzler and M. F. Freeman, J. Am. Chem. Soc., 2019, 141, 9637–9644 CrossRef CAS PubMed.
- F. S. Miller, K. K. Crone, M. R. Jensen, S. Shaw, W. R. Harcombe, M. H. Elias and M. F. Freeman, Nat. Commun., 2021, 12, 5355 CrossRef CAS PubMed.
- A. S. Imani, A. R. Lee, N. Vishwanathan, F. De Waal and M. F. Freeman, ACS Chem. Biol., 2022, 17, 908–917 CrossRef CAS PubMed.
- H. Cho, H. Lee, K. Hong, H. Chung, I. Song, J.-S. Lee and S. Kim, Biochemistry, 2022, 61, 183–194 CrossRef CAS PubMed.
- E. Matabaro, H. Kaspar, P. Dahlin, D. L. V. Bader, C. E. Murar, F. Staubli, C. M. Field, J. W. Bode and M. Künzler, Sci. Rep., 2021, 11, 3541 CrossRef CAS PubMed.
- Y. Kotake, S. Ishii, T. Yano, Y. Katsuoka and H. Hayashi, Biochemistry, 2008, 47, 2531–2538 CrossRef CAS PubMed.
- M. J. Helf, M. F. Freeman and J. Piel, J. Ind. Microbiol. Biotechnol., 2019, 46, 551–563 CrossRef CAS PubMed.
- J. A. Gralnick, C. T. Brown and D. K. Newman, Mol. Microbiol., 2005, 56, 1347–1357 CrossRef CAS PubMed.
- H. Gao, X. Wang, Z. K. Yang, T. Palzkill and J. Zhou, BMC Genomics, 2008, 9, 42 CrossRef PubMed.
- S. Chen, B. Xu, E. Chen, J. Wang, J. Lu, S. Donadio, H. Ge and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2533–2538 CrossRef CAS PubMed.
- S. Han, S. Machhi, M. Berge, G. Xi, T. Linke and R. Schoner, AMB Express, 2017, 7, 93 CrossRef PubMed.
- T. Caetano, J. M. Krawczyk, E. Mösker, R. D. Süssmuth and S. Mendo, Chem. Biol., 2011, 18, 90–100 CrossRef CAS PubMed.
- J. Wang, L. Zhang, K. Teng, S. Sun, Z. Sun and J. Zhong, Appl. Environ. Microbiol., 2014, 80, 2633–2643 CrossRef PubMed.
- H. H. Hau, A. Gilbert, D. Coursolle and J. A. Gralnick, Appl. Environ. Microbiol., 2008, 74, 6880–6886 CrossRef CAS PubMed.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W.-T. Liu, M. Crüsemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderón, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C.-C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C.-C. Liaw, Y.-L. Yang, H.-U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. Boya P, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O’Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodríguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P.-M. Allard, P. Phapale, L.-F. Nothias, T. Alexandrov, M. Litaudon, J.-L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D.-T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Müller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. Ø. Palsson, K. Pogliano, R. G. Linington, M. Gutiérrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed.
- V. Mallia, S. Uhlig, C. Rafuse, J. Meija and C. O. Miles, Mar. Drugs, 2019, 17, 643 CrossRef CAS PubMed.
- E. M. Mudge, J. Meija, S. Uhlig, A. Robertson, P. McCarron and C. O. Miles, Toxicon, 2022, 211, 11–20 CrossRef CAS PubMed.
- J. G. Beck, J. Chatterjee, B. Laufer, M. U. Kiran, A. O. Frank, S. Neubauer, O. Ovadia, S. Greenberg, C. Gilon, A. Hoffman and H. Kessler, J. Am. Chem. Soc., 2012, 134, 12125–12133 CrossRef CAS PubMed.
- D. Ghosh, P. Lahiri, H. Verma, S. Mukherjee and J. Chatterjee, Chem. Sci., 2016, 7, 5212–5218 RSC.
- S. Zhang, S. Prabpai, P. Kongsaeree and P. I. Arvidsson, Chem. Commun., 2006, 497–499 RSC.
- P. Soto, M. A. Griffin and J. E. Shea, Biophys. J., 2007, 93, 3015 CrossRef CAS PubMed.
- D. J. Gordon, R. Tappe and S. C. Meredith, J. Pept. Res., 2002, 60, 37–55 CrossRef CAS PubMed.
- B. Schwarze, A. Korn, C. Höfling, U. Zeitschel, M. Krueger, S. Roßner and D. Huster, Sci. Rep., 2021, 11, 23767 CrossRef CAS PubMed.
- S. Duchin, Z. Vershinin, D. Levy and A. Aharoni, Epigenet. Chromatin, 2015, 8, 56 CrossRef PubMed.
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- A. Vagin and A. Teplyakov, J. Appl. Crystallogr., 1997, 30, 1022–1025 CrossRef CAS.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- D. Liebschner, P. V. Afonine, M. L. Baker, G. Bunkoczi, V. B. Chen, T. I. Croll, B. Hintze, L. W. Hung, S. Jain, A. J. McCoy, N. W. Moriarty, R. D. Oeffner, B. K. Poon, M. G. Prisant, R. J. Read, J. S. Richardson, D. C. Richardson, M. D. Sammito, O. V. Sobolev, D. H. Stockwell, T. C. Terwilliger, A. G. Urzhumtsev, L. L. Videau, C. J. Williams and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2019, 75, 861–877 CrossRef CAS PubMed.
- A. A. Vaguine, J. Richelle and S. J. Wodak, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 191–205 CrossRef CAS PubMed.
- C. J. Williams, J. J. Headd, N. W. Moriarty, M. G. Prisant, L. L. Videau, L. N. Deis, V. Verma, D. A. Keedy, B. J. Hintze, V. B. Chen, S. Jain, S. M. Lewis, W. B. Arendall, J. Snoeyink, P. D. Adams, S. C. Lovell, J. S. Richardson and D. C. Richardson, Protein Sci., 2018, 27, 293–315 CrossRef CAS PubMed.
- C. W. Saltikov and D. K. Newman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10983–10988 CrossRef CAS PubMed.
- R. Sukchawalit, P. Vattanaviboon, R. Sallabhan and S. Mongkolsuk, FEMS Microbiol. Lett., 1999, 181, 217–223 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00093a |
| ‡ These authors contributed equally to this work. |
| § Current address: Department of Chemistry, Biology and Marine Science, University of the Ryukyus, Okinawa 903-0213, Japan. |
| This journal is © The Royal Society of Chemistry 2023 |